
Src-Family Protein Kinase Inhibitors Suppress MYB Activity in a p300-Dependent Manner

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Library Screening
2.3. Expression Vectors
2.4. Electrophoretic Mobility Shift Assays (EMSA)
2.5. Transfections
2.6. Flow Cytometry
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
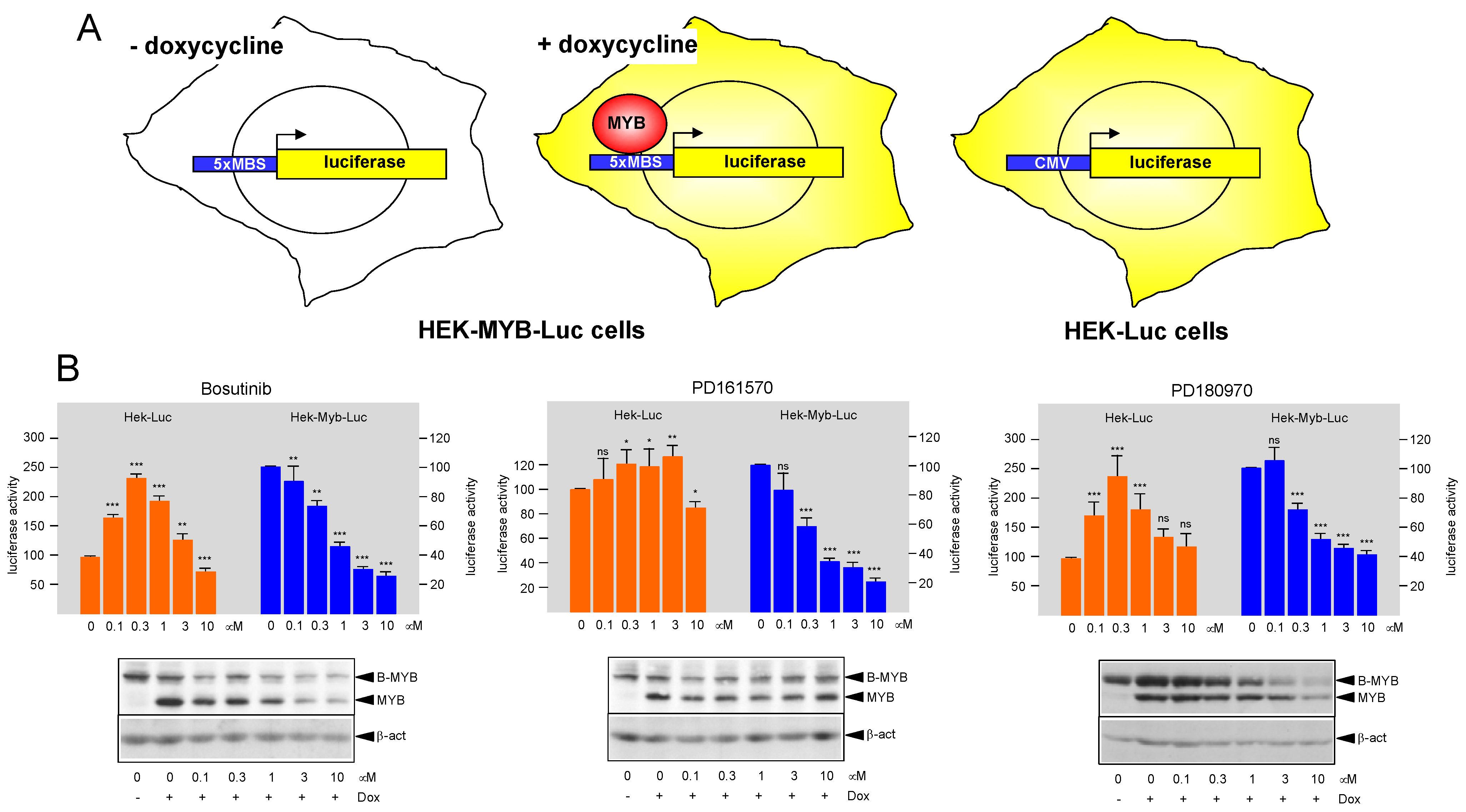
3.1. Identification of Protein Kinase Inhibitors Bosutinib, PD161570, and PD180970 as MYB-Inhibitory Agents
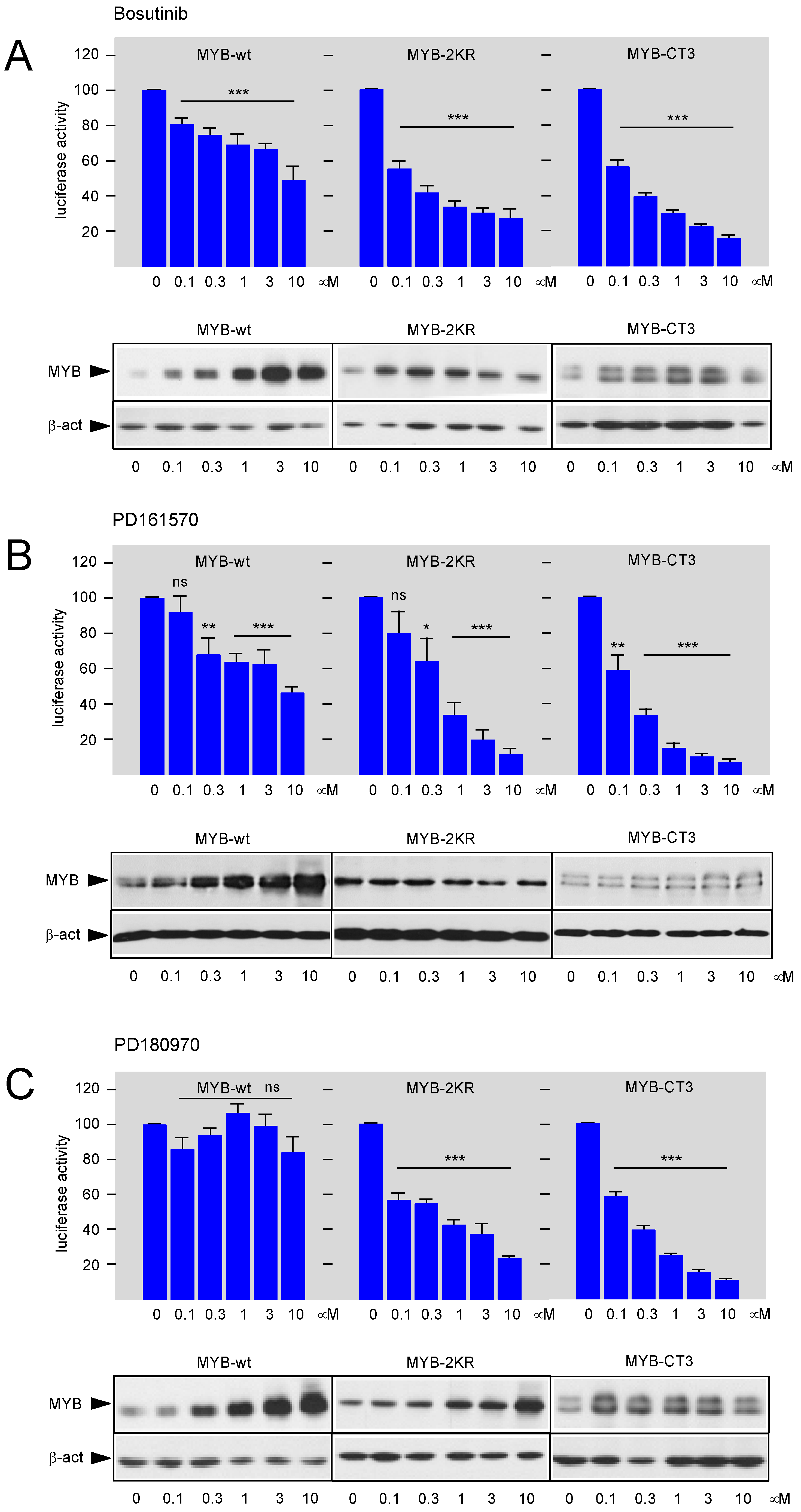
3.2. Bosutinib, PD161570, and PD180970 Affect the Function of the MYB Transactivation Domain
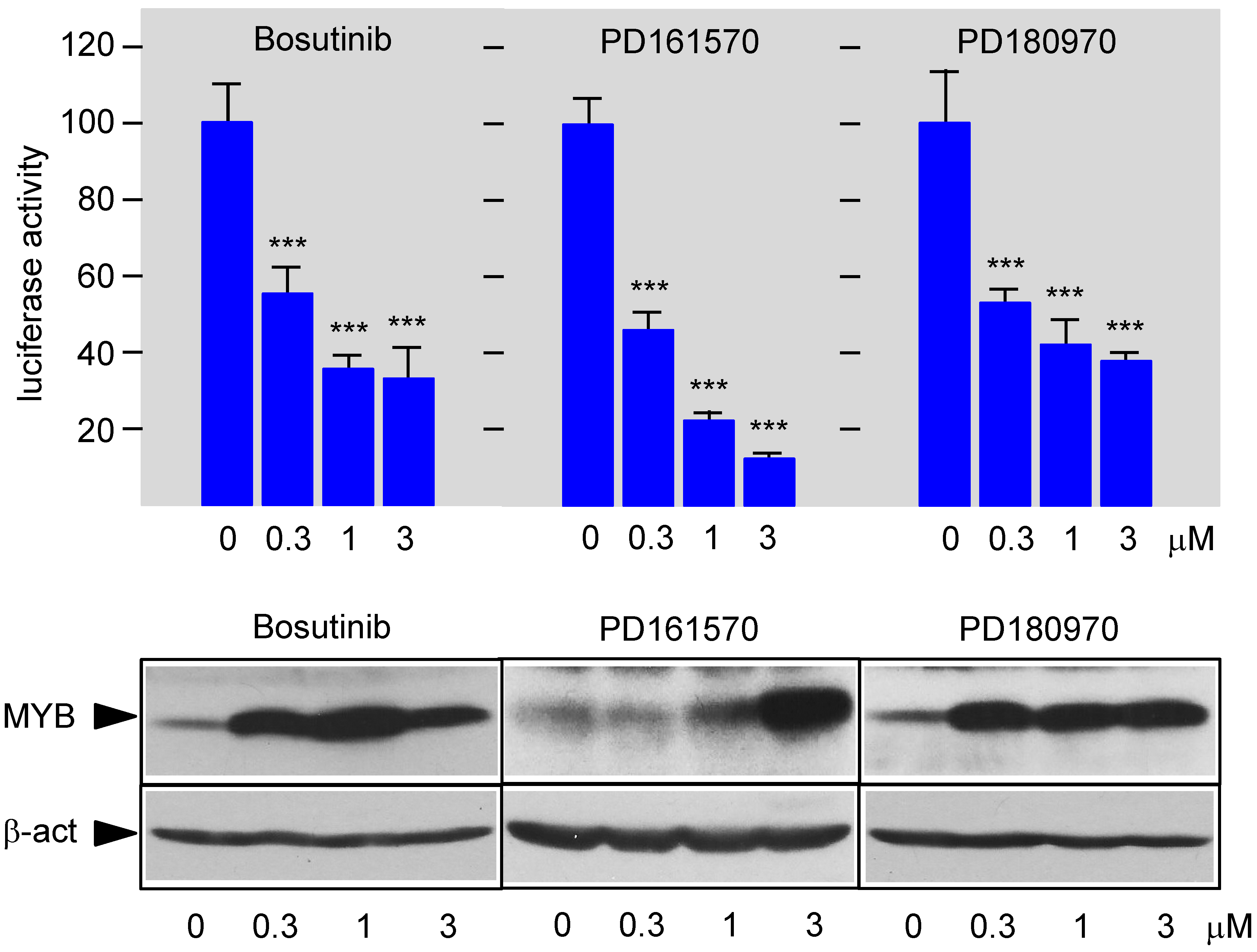
3.3. Bosutinib, PD161570, and PD180970 Disturb the Stimulation of MYB Activity by the Co-Activator p300
3.4. Bosutinib, PD161570, and PD180970 Induce Expression of Myeloid Differentiation Marker CD11b and Cell Death in HL60 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pattabiraman, D.R.; Gonda, T.J. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 2013, 272, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Stenman, G.; Andersson, M.K.; Andrén, Y. New tricks from an old oncogene: Gene fusion and copy number alterations of MYB in human cancer. Cell Cycle 2010, 9, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Uttarkar, S.; Frampton, J.; Klempnauer, K.-H. Targeting the transcription factor Myb by small-molecule inhibitors. Exp. Hematol. 2017, 47, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.K.; Afshari, M.K.; Andrén, Y.; Wick, M.J.; Stenman, G. Targeting the Oncogenic Transcriptional Regulator MYB in Adenoid Cystic Carcinoma by Inhibition of IGF1R/AKT Signaling. J. Natl. Cancer Inst. 2017, 109, djx017. [Google Scholar] [CrossRef]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; et al. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis 2020, 9, 5. [Google Scholar] [CrossRef]
- Zuber, J.; Rappaport, A.R.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.V.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.J.; et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef]
- Takao, S.; Forbes, L.; Uni, M.; Cheng, S.; Pineda, J.M.B.; Tarumoto, Y.; Cifani, P.; Minuesa, G.; Chen, C.; Kharas, M.; et al. Convergent organization of aberrant MYB complex controls oncogenic gene expression in acute myeloid leukemia. eLife 2021, 10, e65905. [Google Scholar] [CrossRef]
- Lahortiga, I.; De Keersmaecker, K.; Van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar] [CrossRef]
- Clappier, E.; Cuccuini, W.; Kalota, A.; Crinquette, A.; Cayuela, J.M.; Dik, W.A.; Langerak, A.W.; Montpellier, B.; Nadel, B.; Walrafen, P.; et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007, 110, 1251–1261. [Google Scholar] [CrossRef]
- O’Neil, J.; Tchinda, J.; Gutierrez, A.; Moreau, L.; Maser, R.S.; Wong, K.K.; Li, W.; McKenna, K.; Liu, X.S.; Feng, B.; et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J. Exp. Med. 2007, 204, 3059–3066. [Google Scholar] [CrossRef]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Magnussen, M.; León, T.E.; Farah, N.; Li, Z.; Abraham, B.J.; Alapi, K.Z.; Mitchell, R.J.; Naughton, T.; Fielding, A.K.; et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 3221–3226. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Andrén, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Klempnauer, K.-H.; Gonda, T.J.; Bishop, J.M. Nucleotide sequence of the retroviral leukemia gene v-myb and its cellular progenitor c-myb: The architecture of a transduced oncogene. Cell 1982, 31, 453–463. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef]
- Zor, T.; De Guzman, R.N.; Dyson, H.J.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J. Mol. Biol. 2004, 337, 521–534. [Google Scholar] [CrossRef]
- Kasper, L.H.; Boussouar, F.; Ney, P.A.; Jackson, C.W.; Rehg, J.; van Deursen, J.M.; Brindle, P.K. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 2002, 419, 738–743. [Google Scholar] [CrossRef]
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev. Cell 2005, 8, 153–166. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef]
- Andersson, M.K.; Åman, P.; Stenman, G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors. Cells 2019, 8, 913. [Google Scholar] [CrossRef]
- Uttarkar, S.; Dukare, S.; Bopp, B.; Goblirsch, M.; Jose, J.; Klempnauer, K.-H. Naphthol AS-E phosphate inhibits the activity of the transcription factor Myb by blocking the interaction with the KIX domain of the coactivator p300. Mol. Cancer Ther. 2015, 14, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Uttarkar, S.; Dassé, E.; Coulibaly, A.; Steinmann, S.; Jakobs, A.; Schomburg, C.; Trentmann, A.; Jose, J.; Schlenke, P.; Berdel, W.E.; et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 Interaction. Blood 2016, 127, 1173–1182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uttarkar, S.; Piontek, T.; Dukare, S.; Schomburg, C.; Schlenke, P.; Berdel, W.E.; Müller-Tidow, C.; Schmidt, T.J.; Klempnauer, K.H. Small-Molecule Disruption of the Myb/p300 Cooperation Targets Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2016, 15, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, K.; Forbes, L.; Minuesa, G.; Gindin, T.; Brown, F.; Kharas, M.G.; Krivtsov, A.V.; Armstrong, S.A.; Still, E.; de Stanchina, E.; et al. Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat. Commun. 2018, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gao, R.; Cao, C.; Forbes, L.; Li, J.; Freeberg, S.; Fredenburg, K.M.; Justice, J.M.; Silver, N.L.; Wu, L.; et al. MYB-activated models for testing therapeutic agents in adenoid cystic carcinoma. Oral Oncol. 2019, 98, 147–155. [Google Scholar] [CrossRef]
- Yusenko, M.V.; Trentmann, A.; Andersson, M.K.; Abdel Ghani, L.; Jakobs, A.; Arteaga Paz, M.F.; Mikesch, J.P.; von Kries, P.J.; Stenman, G.; Klempnauer, K.-H. Monensin, a novel potent MYB inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells. Cancer Lett. 2020, 479, 61–70. [Google Scholar] [CrossRef]
- Yusenko, M.V.; Biyanee, A.; Andersson, M.K.; Radetzki, S.; von Kries, J.P.; Stenman, G.; Klempnauer, K.-H. Proteasome inhibitors suppress MYB oncogenic activity in a p300-dependent manner. Cancer Lett. 2021, 520, 132–142. [Google Scholar] [CrossRef]
- Yusenko, M.; Jakobs, A.; Klempnauer, K.-H. A novel cell-based screening assay for small-molecule MYB inhibitors identifies podophyllotoxins teniposide and etoposide as inhibitors of MYB activity. Sci. Rep. 2018, 8, 13159. [Google Scholar] [CrossRef]
- Dahle, Ø.; Andersen, T.Ø.; Nordgård, O.; Matre, V.; Del Sal, G.; Gabrielsen, O.S. Transactivation properties of c-Myb are critically dependent on two SUMO-1 acceptor sites that are conjugated in a PIASy enhanced manner. Eur. J. Biochem. 2003, 270, 1338–1348. [Google Scholar] [CrossRef]
- Molvaersmyr, A.K.; Saether, T.; Gilfillan, S.; Lorenzo, P.I.; Kvaløy, H.; Matre, V.; Gabrielsen, O.S. A SUMO-regulated activation function controls synergy of c-Myb through a repressor-activator switch leading to differential p300 recruitment. Nucleic Acids Res. 2010, 38, 4970–4984. [Google Scholar] [CrossRef] [PubMed]
- Mink, S.; Haenig, B.; Klempnauer, K.-H. Interaction and functional collaboration of p300 and C/EBPβ. Mol. Cell Biol. 1997, 17, 6609–6617. [Google Scholar] [CrossRef] [PubMed]
- Chayka, O.; Kintscher, J.; Braas, D.; Klempnauer, K.-H. v-Myb mediates cooperation of a cell-specific enhancer with the mim-1 promoter. Mol. Cell Biol. 2005, 25, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Mink, S.; Ringwald, M.; Klempnauer, K.-H. Synergistic activation of the chicken mim-1 gene by v-myb and C/EBP transcription factors. EMBO J. 1993, 12, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P. Xenopus A-myb is expressed during early spermatogenesis. Oncogene 1993, 8, 1931–1941. [Google Scholar] [PubMed]
- Yusenko, M.V.; Klempnauer, K.-H. Characterization of the MYB-inhibitory potential of the Pan-HDAC inhibitor LAQ824. BBA Adv. 2022, 2, 100034. [Google Scholar] [CrossRef]
- Aziz, N.; Miglarese, M.R.; Hendrickson, R.C.; Shabanowitz, J.; Sturgill, T.W.; Hunt, D.F.; Bender, T.P. Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proc. Natl. Acad. Sci. USA 1995, 92, 6429–6433. [Google Scholar] [CrossRef]
- Pani, E.; Ferrari, S. p38MAPKδ controls c-Myb degradation in response to stress. Blood Cells Mol. Dis. 2008, 40, 388–394. [Google Scholar] [CrossRef]
- Pani, E.; Menigatti, M.; Schubert, S.; Hess, D.; Gerrits, B.; Klempnauer, K.-H.; Ferrari, S. Pin1 interacts with c-Myb in a phosphorylation-dependent manner and regulates its transactivation activity. Biochim. Biophys. Acta 2008, 1783, 1121–1128. [Google Scholar] [CrossRef]
- Bies, J.; Sramko, M.; Wolff, L. Stress-induced phosphorylation of Thr486 in c-Myb by p38 mitogen-activated protein kinases attenuates conjugation of SUMO-2/3. J. Biol. Chem. 2013, 288, 36983–36993. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Sun, J.; Dowhan, D.H.; Ishii, S.; Gonda, T.J. Mutations in multiple domains of c-Myb disrupt interaction with CBP/p300 and abrogate myeloid transforming ability. Mol. Cancer Res. 2009, 7, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Ramsay, R.G.; Kanei-Ishii, C.; Ishii, S.; Gonda, T.J. Transformation by carboxyl-deleted Myb reflects increased transactivating capacity and disruption of a negative regulatory domain. Oncogene 1991, 6, 1549–1553. [Google Scholar] [PubMed]
- Walf-Vorderwülbecke, V.; Pearce, K.; Brooks, T.; Hubank, M.; van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Adams, S.; Edwards, D.; Bartram, J.; Samarasinghe, S.; et al. Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia 2018, 32, 882–889. [Google Scholar] [CrossRef]
- Mandelbaum, J.; Shestopalov, I.A.; Henderson, R.E.; Chau, N.G.; Knoechel, B.; Wick, M.J.; Zon, L.I. Zebrafish blastomere screen identifies retinoic acid suppression of MYB in adenoid cystic carcinoma. J. Exp. Med. 2018, 215, 2673–2685. [Google Scholar] [CrossRef] [PubMed]
- Hamby, J.M.; Connolly, C.J.; Schroeder, M.C.; Winters, R.T.; Showalter, H.D.; Panek, R.L.; Major, T.C.; Olsewski, B.; Ryan, M.J.; Dahring, T.; et al. Structure-activity relationships for a novel series of pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. J. Med. Chem. 1997, 40, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhong, L.; Lin, M.; Zhou, X.; Jing, H.; Ying, M.; Luo, P.; Yang, B.; He, Q. MEK/ERK dependent activation of STAT1 mediates dasatinib-induced differentiation of acute myeloid leukemia. PLoS ONE 2013, 8, e66915. [Google Scholar] [CrossRef]
- MacDonald, R.J.; Bunaciu, R.P.; Ip, V.; Dai, D.; Tran, D.; Varner, J.D.; Yen, A. Src family kinase inhibitor bosutinib enhances retinoic acid-induced differentiation of HL-60 leukemia cells. Leuk. Lymphoma 2018, 59, 2941–2951. [Google Scholar] [CrossRef]
- Janknecht, R.; Hunter, T. Transcription. A growing coactivator network. Nature 1996, 383, 22–23. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [CrossRef]
- Oh, A.S.; Lahusen, J.T.; Chien, C.D.; Fereshteh, M.P.; Zhang, X.; Dakshanamurthy, S.; Xu, J.; Kagan, B.L.; Wellstein, A.; Riegel, A.T. Tyrosine phosphorylation of the nuclear receptor coactivator AIB1/SRC-3 is enhanced by Abl kinase and is required for its activity in cancer cells. Mol. Cell. Biol. 2008, 28, 6580–6593. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R.; Nordheim, A. MAP kinase-dependent transcriptional coactivation by Elk-1 and its cofactor CBP. Biochem. Biophys. Res. Commun. 1996, 228, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Chirivia, J.C.; Latchman, D.S. Nerve growth factor up-regulates the transcriptional activity of CBP through activation of the p42/p44 (MAPK) cascade. J. Biol. Chem. 1998, 273, 32400–32407. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.Z.; Thomas, N.S.B.; Latchman, D.S. CBP associates with the p42/p44 MAPK enzymes and is phosphorylated following NGF treatment. NeuroReport 1999, 10, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, P.; Tabilio, A.; Dall’Aglio, P.P.; Ridolo, E.; Carlo-Stella, C.; Pelicci, P.G.; Bonati, A. Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia 2003, 17, 1783–1793. [Google Scholar] [CrossRef]
- Chen, Y.J.; Wang, Y.N.; Chang, W.C. ERK2-mediated C-terminal serine phosphorylation of p300 is vital to the regulation of epidermal growth factor-induced keratin 16 gene expression. J. Biol. Chem. 2007, 282, 27215–27228. [Google Scholar] [CrossRef]
- Paladino, D.; Yue, P.; Furuya, H.; Acoba, J.; Rosser, C.J.; Turkson, J. A novel nuclear Src and p300 signaling axis controls migratory and invasive behavior in pancreatic cancer. Oncotarget 2016, 7, 7253–7267. [Google Scholar] [CrossRef]
- Aoyama, K.; Fukumoto, Y.; Ishibashi, K.; Kubota, S.; Morinaga, T.; Horiike, Y.; Yuki, R.; Takahashi, A.; Nakayama, Y.; Yamaguchi, N. Nuclear c-Abl-mediated tyrosine phosphorylation induces chromatin structural changes through histone modifications that include H4K16 hypoacetylation. Exp. Cell Res. 2011, 317, 2874–2903. [Google Scholar] [CrossRef]
- Veerasubramanian, P.K.; Shao, H.; Meli, V.S.; Phan, T.A.Q.; Luu, T.U.; Liu, W.F.; Downing, T.L. A Src-H3 acetylation signaling axis integrates macrophage mechanosensation with inflammatory response. Biomaterials 2021, 279, 121236. [Google Scholar] [CrossRef]
- Stachowiak, M.K.; Birkaya, B.; Aletta, J.M.; Narla, S.T.; Benson, C.A.; Decker, B.; Stachowiak, E.K. Nuclear FGF receptor-1 and CREB binding protein: An integrative signaling module. J. Cell. Physiol. 2015, 230, 989–1002. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biyanee, A.; Yusenko, M.V.; Klempnauer, K.-H. Src-Family Protein Kinase Inhibitors Suppress MYB Activity in a p300-Dependent Manner. Cells 2022, 11, 1162. https://doi.org/10.3390/cells11071162
Biyanee A, Yusenko MV, Klempnauer K-H. Src-Family Protein Kinase Inhibitors Suppress MYB Activity in a p300-Dependent Manner. Cells. 2022; 11(7):1162. https://doi.org/10.3390/cells11071162
Chicago/Turabian StyleBiyanee, Abhiruchi, Maria V. Yusenko, and Karl-Heinz Klempnauer. 2022. "Src-Family Protein Kinase Inhibitors Suppress MYB Activity in a p300-Dependent Manner" Cells 11, no. 7: 1162. https://doi.org/10.3390/cells11071162
APA StyleBiyanee, A., Yusenko, M. V., & Klempnauer, K.-H. (2022). Src-Family Protein Kinase Inhibitors Suppress MYB Activity in a p300-Dependent Manner. Cells, 11(7), 1162. https://doi.org/10.3390/cells11071162