
A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteria Strains
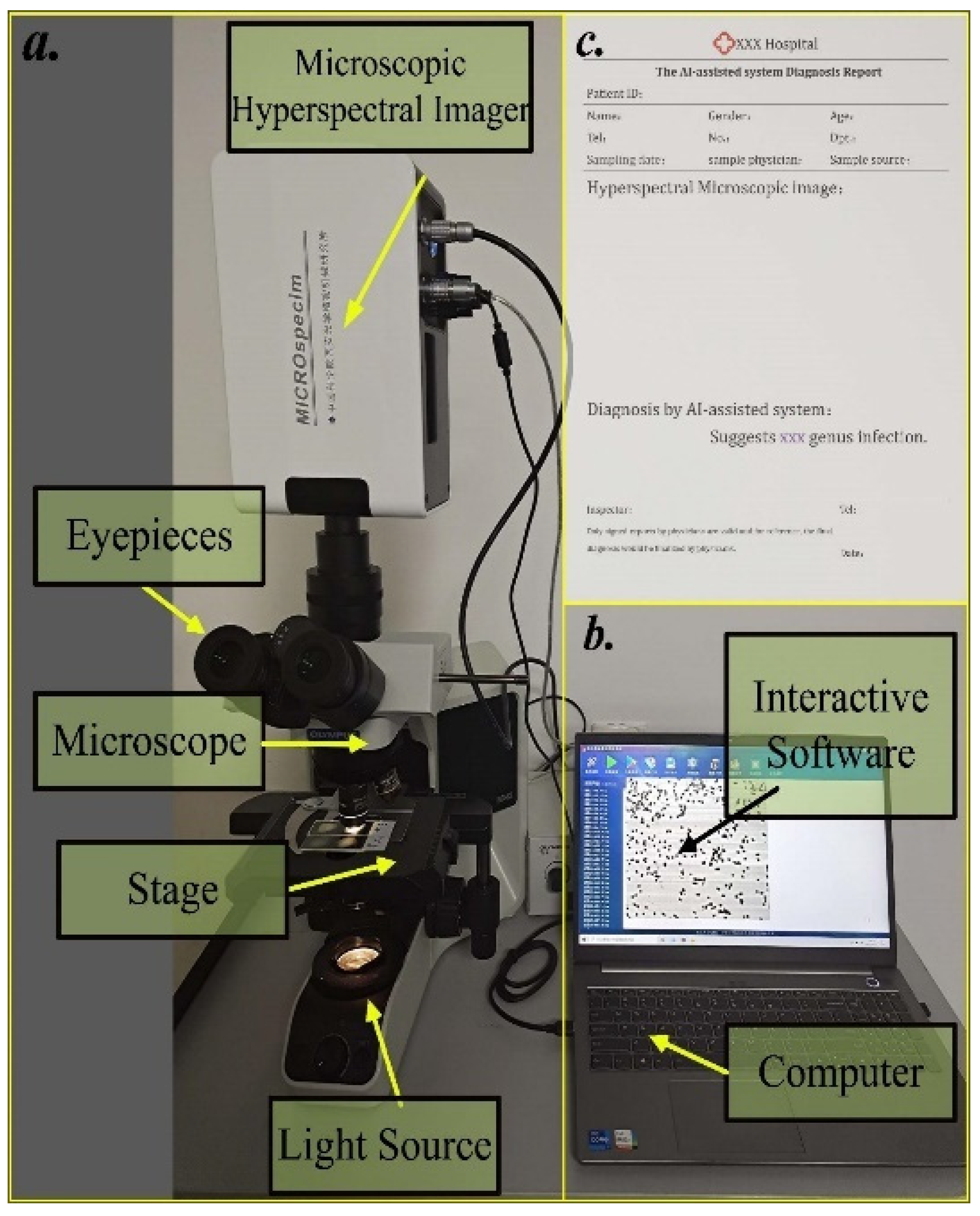
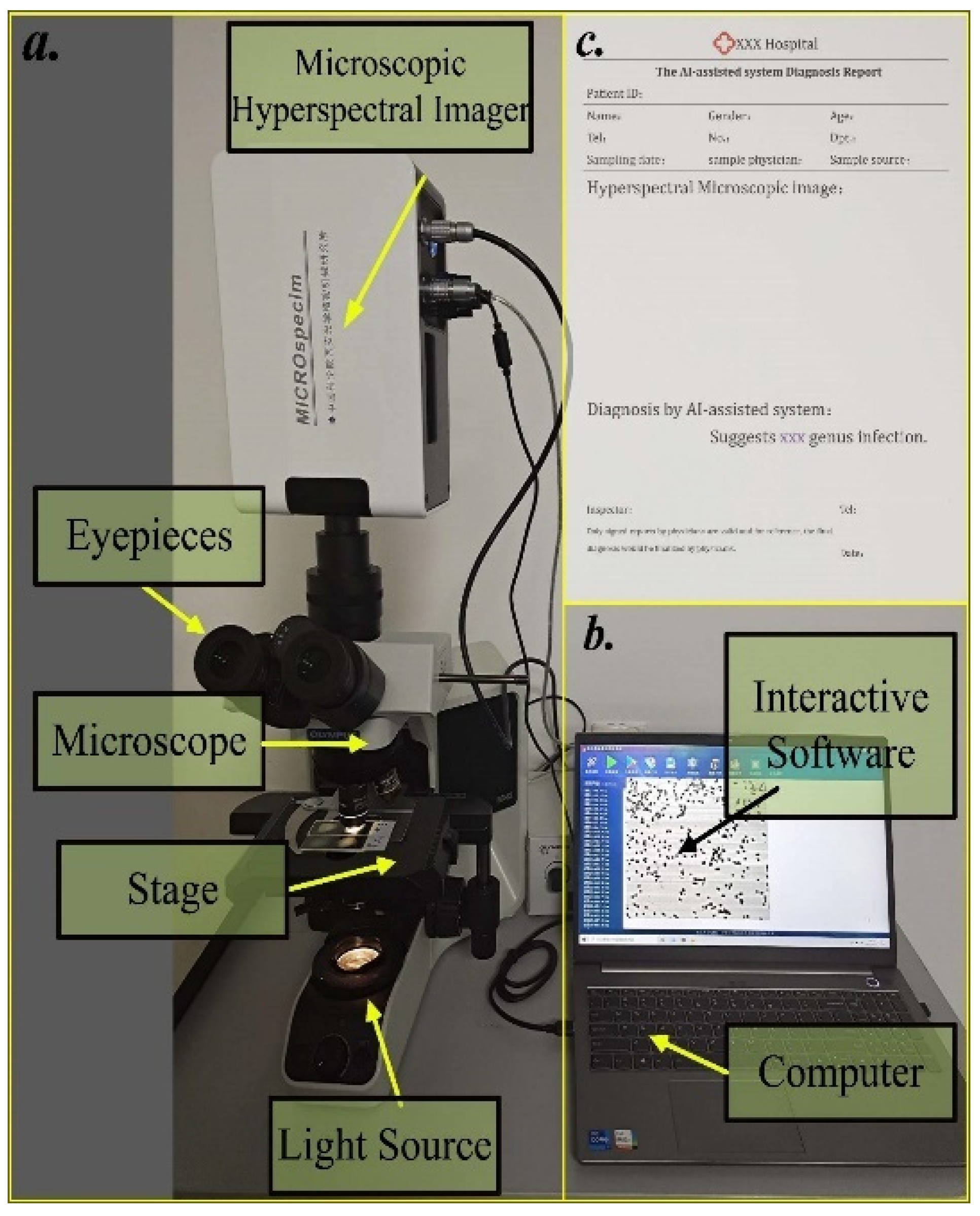
2.2. Hyperspectral Microscopic Imaging (HMI) System
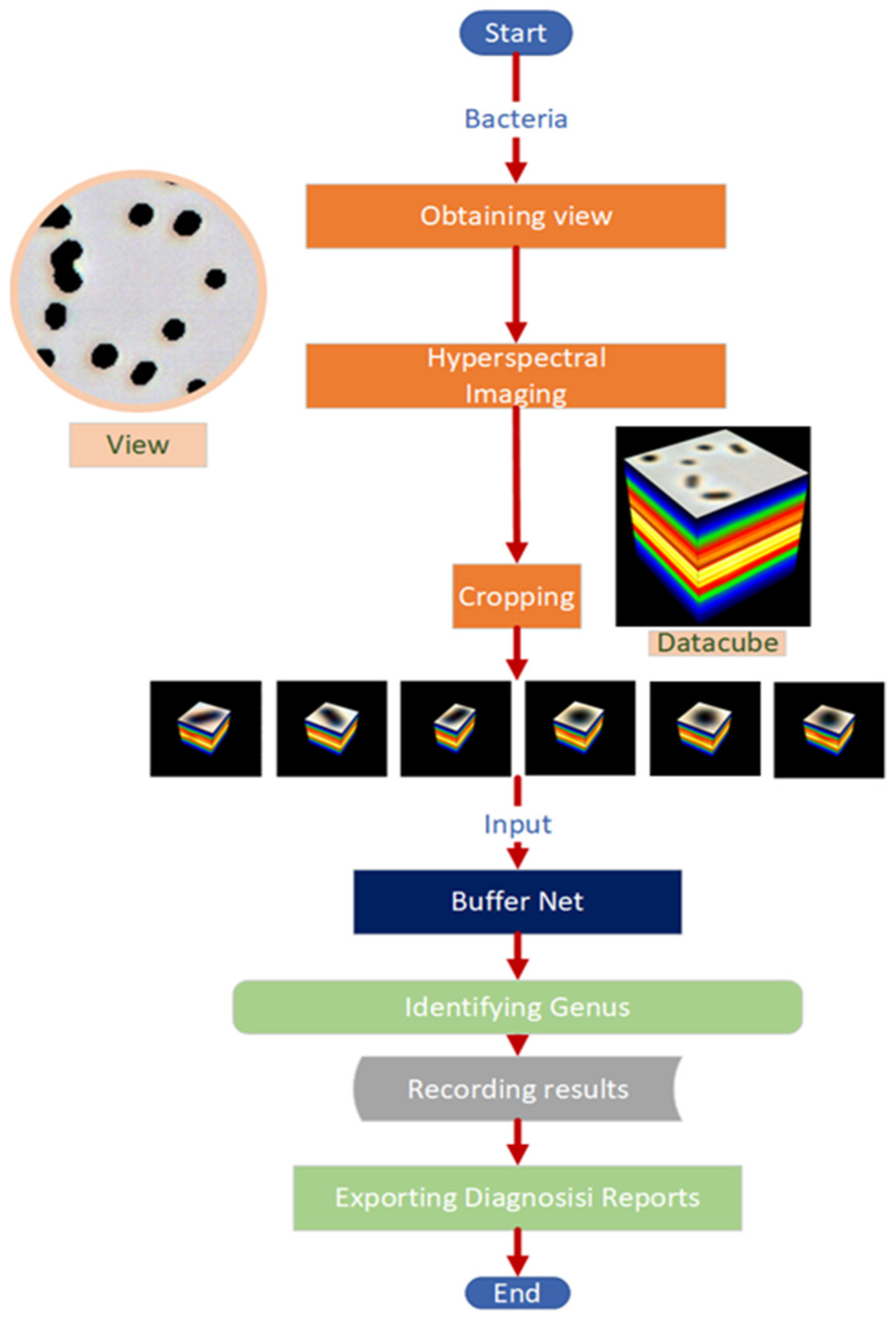
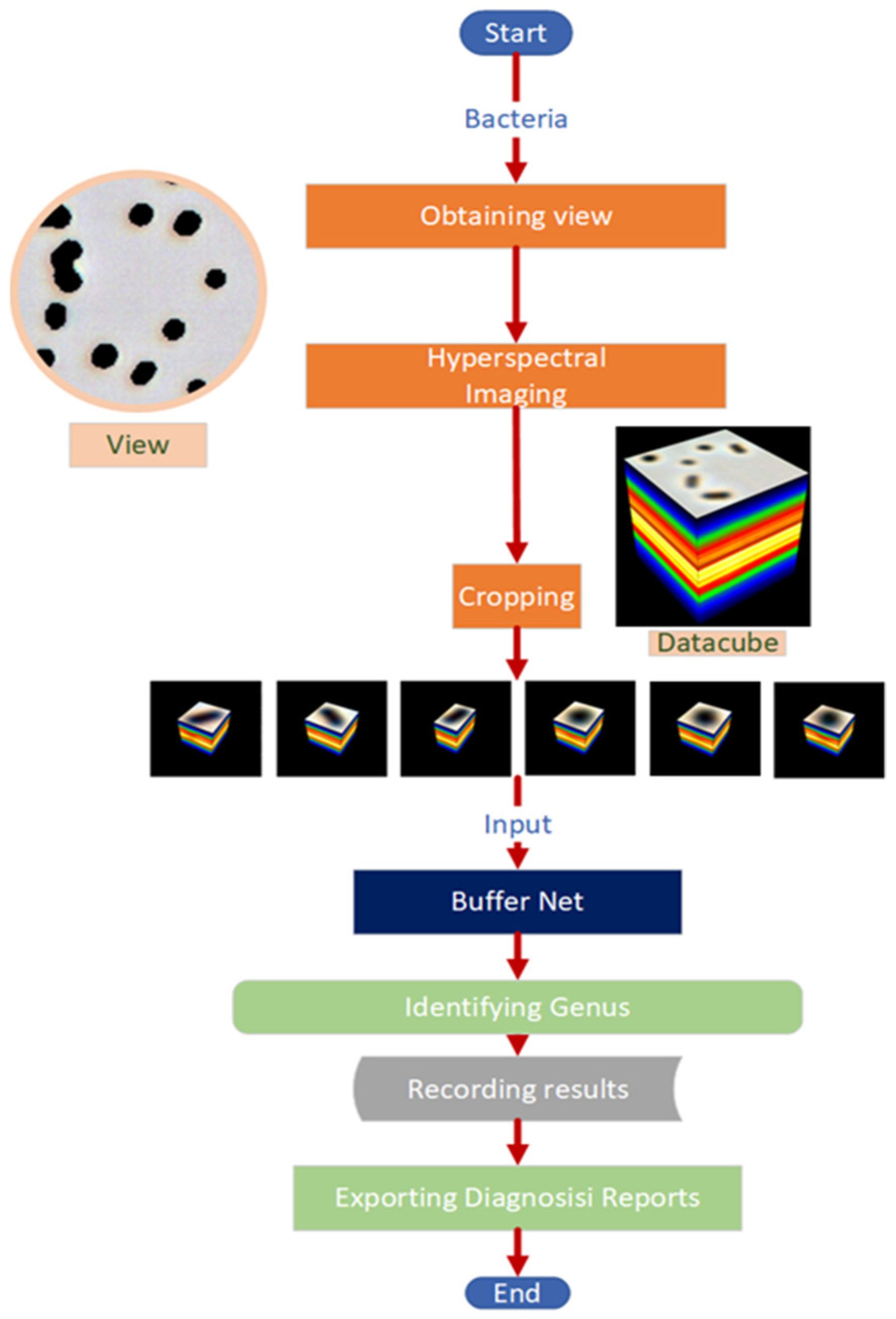
2.3. Data Collection and Preprocessing
2.4. Model Design
2.4.1. Deep Learning
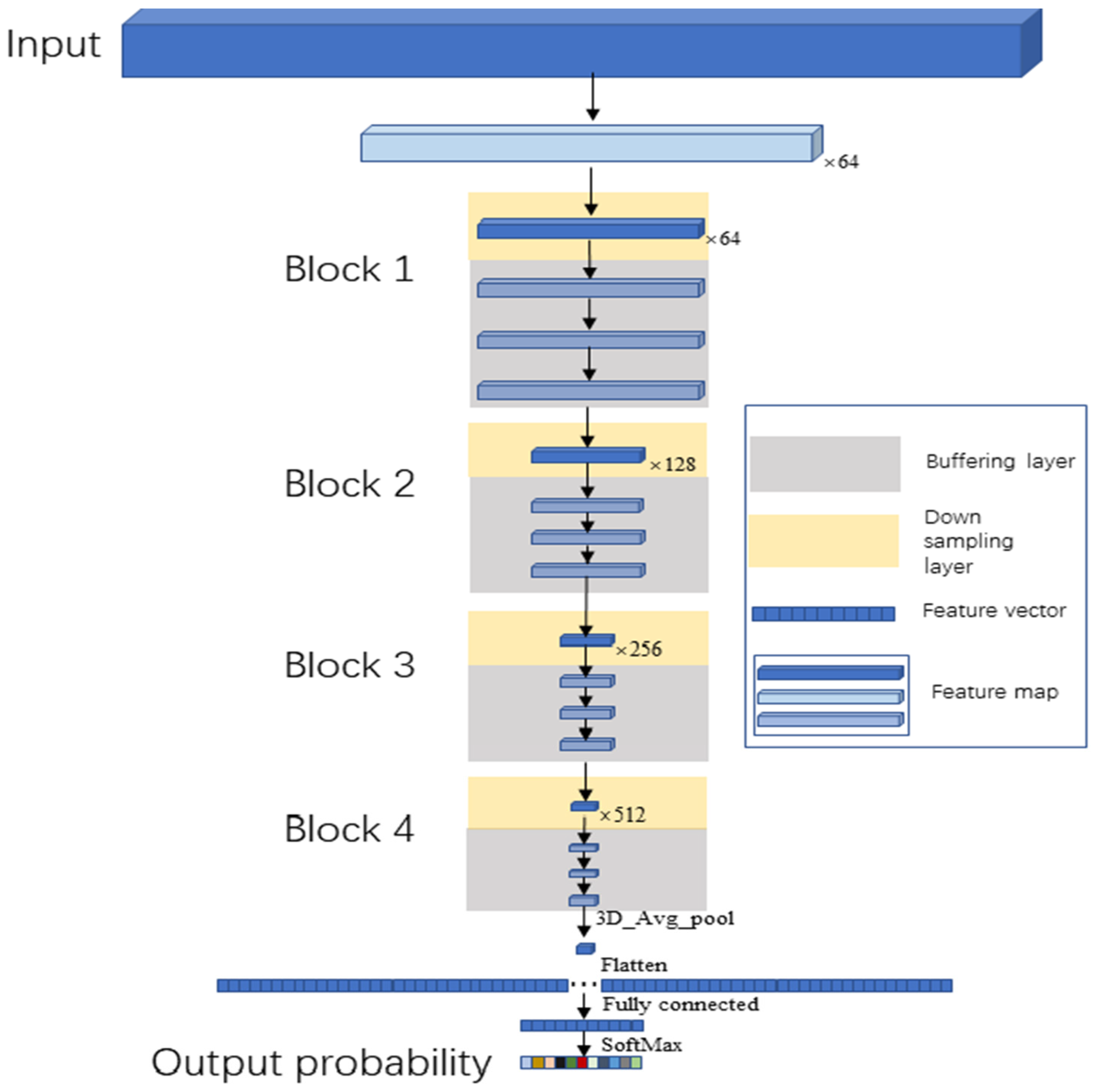
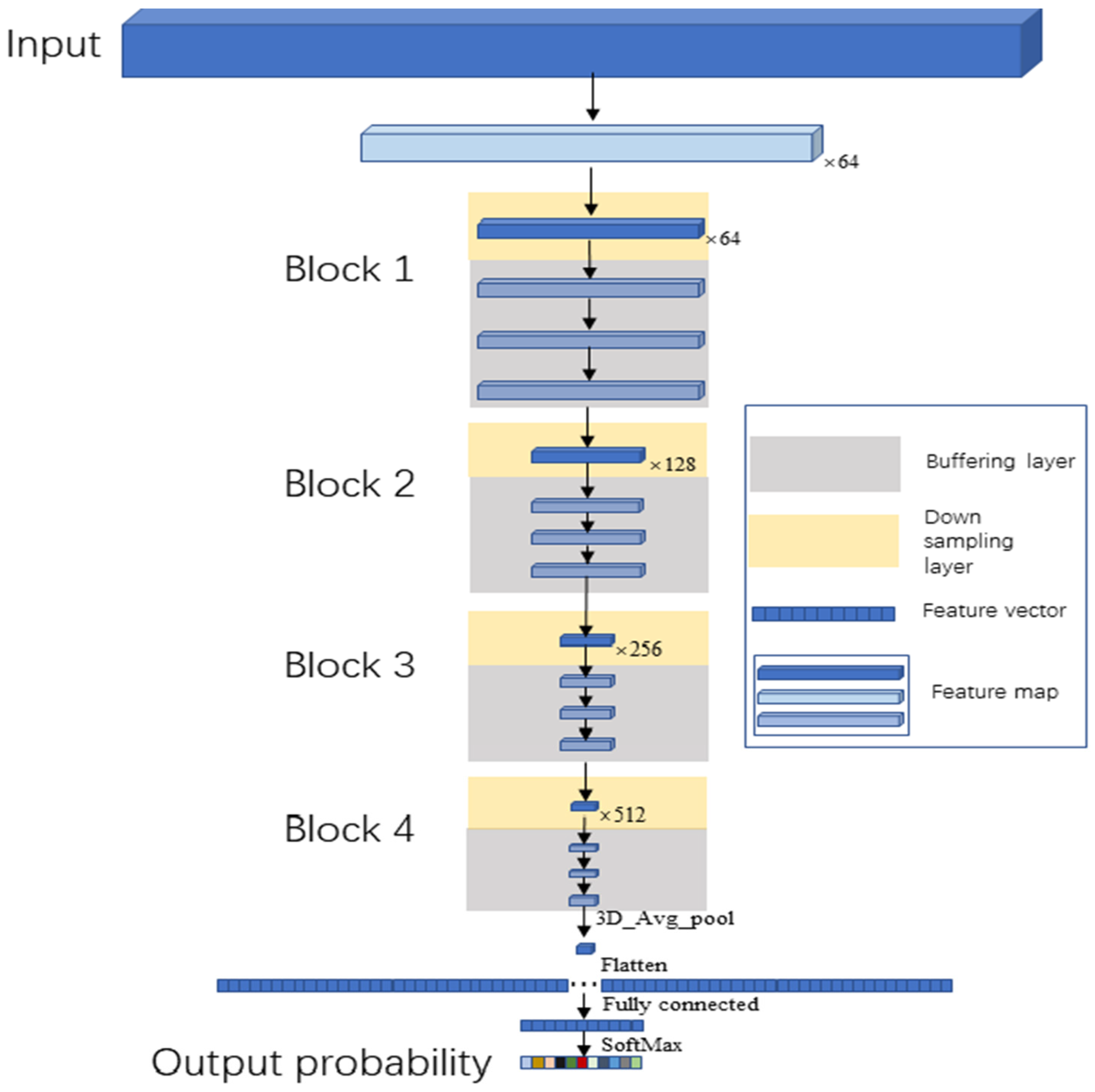
2.4.2. Buffer Net
2.5. Development Language and Training Details
2.6. System Integration
2.7. Evaluation Metrics
3. Results
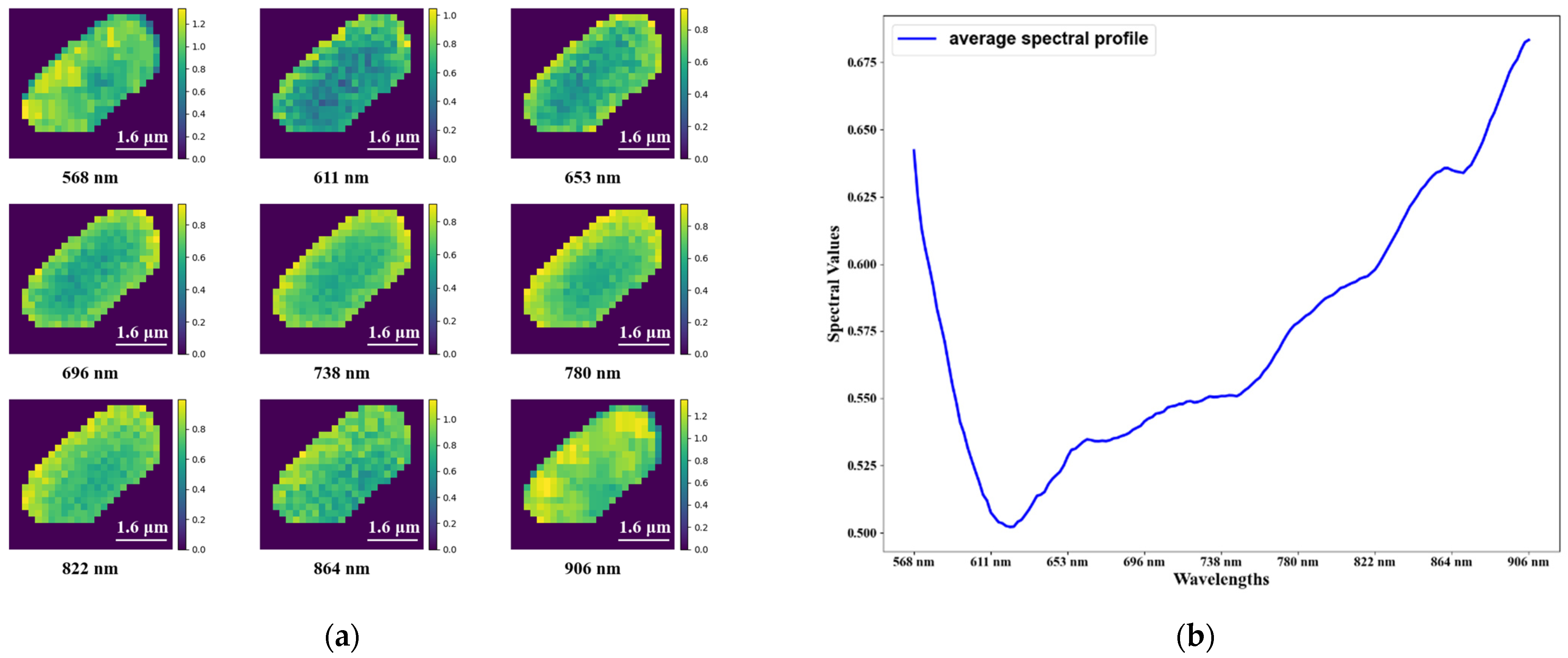
3.1. Hyperspectral Microscopic Images
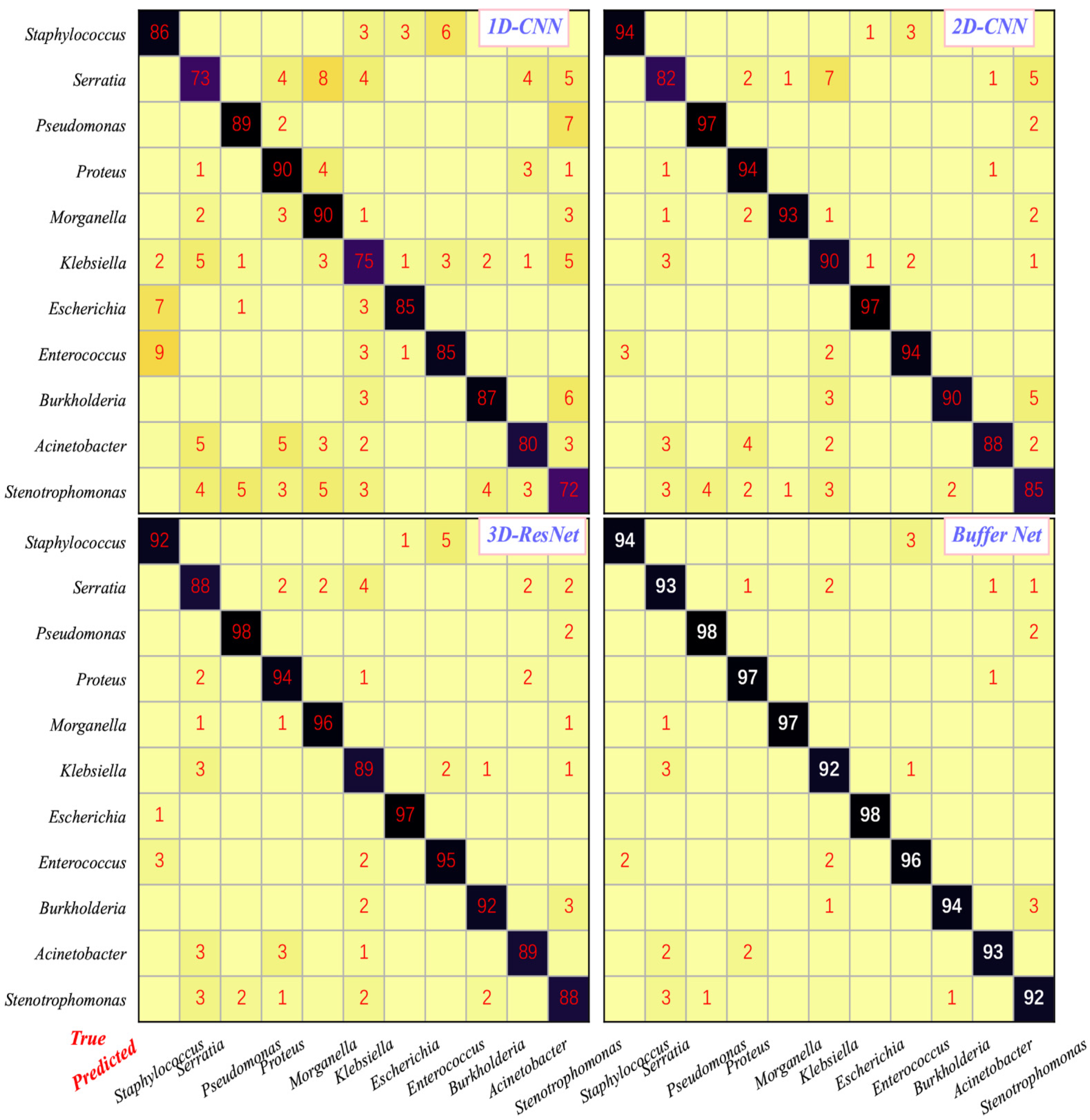
3.2. Classification Performance of the AI-Assisted System
3.3. The Differentiation Speed of Our AI-Assisted HMI System
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gan, Y.; Li, C.; Peng, X.; Wu, S.; Li, Y.; Tan, J.P.; Yang, Y.Y.; Yuan, P.; Ding, X. Fight bacteria with bacteria: Bacterial membrane vesicles as vaccines and delivery nanocarriers against bacterial infections. Nanomed. Nanotechnol. Biol. Med. 2021, 35, 102398. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano Tinoco, E.-A.; García-Contreras, S.-J.; García-Contreras, R. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front. Microbiol. 2015, 6, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadarangani, M. Protection against invasive infections in children caused by encapsulated bacteria. Front. Immunol. 2018, 9, 2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Versalovic, J. Manual of Clinical Microbiology; American Society for Microbiology Press: Washington, DC, USA, 2011; Volume 1. [Google Scholar]
- Engelmann, I.; Alidjinou, E.K.; Ogiez, J.; Pagneux, Q.; Miloudi, S.; Benhalima, I.; Ouafi, M.; Sane, F.; Hober, D.; Roussel, A. Preanalytical issues and cycle threshold values in SARS-CoV-2 real-time RT-PCR testing: Should test results include these? ACS Omega 2021, 6, 6528–6536. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Rave, A.; Kuss, A.; Peil, G.; Ladeira, S.; Villarreal, J.; Nascente, P. Biochemical identification techniques and antibiotic susceptibility profile of lipolytic ambiental bacteria from effluents. Braz. J. Biol. 2018, 79, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-T.; Ha, S.; Park, H.-E.; Shim, S.; Hur, T.Y.; Yoo, H.S. Comparative analysis of serological tests and fecal detection in the diagnosis of Mycobacterium avium subspecies paratuberculosis infection. Korean J. Vet. Res. 2020, 60, 117–122. [Google Scholar] [CrossRef]
- Roux-Dalvai, F.; Gotti, C.; Leclercq, M.; Hélie, M.-C.; Boissinot, M.; Arrey, T.N.; Dauly, C.; Fournier, F.; Kelly, I.; Marcoux, J. Fast and Accurate Bacterial Species Identification in Urine Specimens Using LC-MS/MS Mass Spectrometry and Machine Learning. Mol. Cell. Proteom. 2019, 18, 2492–2505. [Google Scholar] [CrossRef] [Green Version]
- Leekha, S.; Terrell, C.L.; Edson, R.S. General principles of antimicrobial therapy. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2011; pp. 156–167. [Google Scholar]
- Alexandrakis, D.; Downey, G.; Scannell, A.G. Detection and identification of bacteria in an isolated system with near-infrared spectroscopy and multivariate analysis. J. Agric. Food Chem. 2008, 56, 3431–3437. [Google Scholar] [CrossRef]
- Yoon, S.-C.; Lawrence, K.; Siragusa, G.; Line, J.; Park, B.; Feldner, P. Hyperspectral reflectance imaging for detecting a foodborne pathogen: Campylobacter. Trans. ASABE 2009, 52, 651–662. [Google Scholar] [CrossRef]
- Windham, W.R.; Yoon, S.-C.; Ladely, S.R.; Heitschmidt, J.W.; Lawrence, K.C.; Park, B.; Narrang, N.; Cray, W.C. The effect of regions of interest and spectral pre-processing on the detection of non-0157 Shiga-toxin producing Escherichia coli serogroups on agar media by hyperspectral imaging. J. Near Infrared Spectrosc. 2012, 20, 547–558. [Google Scholar] [CrossRef]
- Yoon, S.-C.; Windham, W.R.; Ladely, S.R.; Heitschmidt, J.W.; Lawrence, K.C.; Park, B.; Narang, N.; Cray, W.C. Hyperspectral imaging for differentiating colonies of non-0157 Shiga-toxin producing Escherichia coli (STEC) serogroups on spread plates of pure cultures. J. Near Infrared Spectrosc. 2013, 21, 81–95. [Google Scholar] [CrossRef]
- Kammies, T.-L.; Manley, M.; Gouws, P.A.; Williams, P.J. Differentiation of foodborne bacteria using NIR hyperspectral imaging and multivariate data analysis. Appl. Microbiol. Biotechnol. 2016, 100, 9305–9320. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Park, B.; Hinton, A.; Yoon, S.-C.; Lawrence, K.C. Identification of Staphylococcus species with hyperspectral microscope imaging and classification algorithms. J. Food Meas. Charact. 2016, 10, 253–263. [Google Scholar] [CrossRef]
- Kang, R.; Park, B.; Eady, M.; Ouyang, Q.; Chen, K. Single-cell classification of foodborne pathogens using hyperspectral microscope imaging coupled with deep learning frameworks. Sens. Actuators B Chem. 2020, 309, 127789. [Google Scholar] [CrossRef]
- Kang, R.; Park, B.; Eady, M.; Ouyang, Q.; Chen, K. Classification of foodborne bacteria using hyperspectral microscope imaging technology coupled with convolutional neural networks. Appl. Microbiol. Biotechnol. 2020, 104, 3157–3166. [Google Scholar] [CrossRef]
- Kang, R.; Park, B.; Ouyang, Q.; Ren, N. Rapid identification of foodborne bacteria with hyperspectral microscopic imaging and artificial intelligence classification algorithms. Food Control 2021, 130, 108379. [Google Scholar] [CrossRef]
- Seibert, J.A.; Boone, J.M.; Lindfors, K.K. Flat-field correction technique for digital detectors. In Medical Imaging 1998: Physics of Medical Imaging; SPIE: Bellingham, WA, USA, 1998; pp. 348–354. [Google Scholar]
- Likas, A.; Vlassis, N.; Verbeek, J.J. The global k-means clustering algorithm. Pattern Recognit. 2003, 36, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Processing Syst. 2012, 25, 1097–1105. [Google Scholar] [CrossRef]
- Tian, S.; Wang, S.; Xu, H. Early detection of freezing damage in oranges by online Vis/NIR transmission coupled with diameter correction method and deep 1D-CNN. Comput. Electron. Agric. 2022, 193, 106638. [Google Scholar] [CrossRef]
- Hsieh, T.-H.; Kiang, J.-F. Comparison of CNN algorithms on hyperspectral image classification in agricultural lands. Sensors 2020, 20, 1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, S.; Xu, W.; Yang, M.; Yu, K. 3D convolutional neural networks for human action recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 35, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.-Y.; Maire, M.; Belongie, S.; Hays, J.; Perona, P.; Ramanan, D.; Dollár, P.; Zitnick, C.L. Microsoft coco: Common objects in context. In European Conference on Computer Vision; Springer: Berlin/Heidelberg, Germany, 2014; pp. 740–755. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Wang, H.; Ceylan Koydemir, H.; Qiu, Y.; Bai, B.; Zhang, Y.; Jin, Y.; Tok, S.; Yilmaz, E.C.; Gumustekin, E.; Rivenson, Y. Early detection and classification of live bacteria using time-lapse coherent imaging and deep learning. Light Sci. Appl. 2020, 9, 118. [Google Scholar] [CrossRef]
- Kim, G.; Ahn, D.; Kang, M.; Jo, Y.; Ryu, D.; Kim, H.; Song, J.; Ryu, J.S.; Choi, G.; Chung, H.J. Rapid and label-free identification of individual bacterial pathogens exploiting three-dimensional quantitative phase imaging and deep learning. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.; Park, S.; Jung, J.; Yoon, J.; Joo, H.; Kim, M.-H.; Kang, S.-J.; Choi, M.C.; Lee, S.Y.; Park, Y. Holographic deep learning for rapid optical screening of anthrax spores. Sci. Adv. 2017, 3, e1700606. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
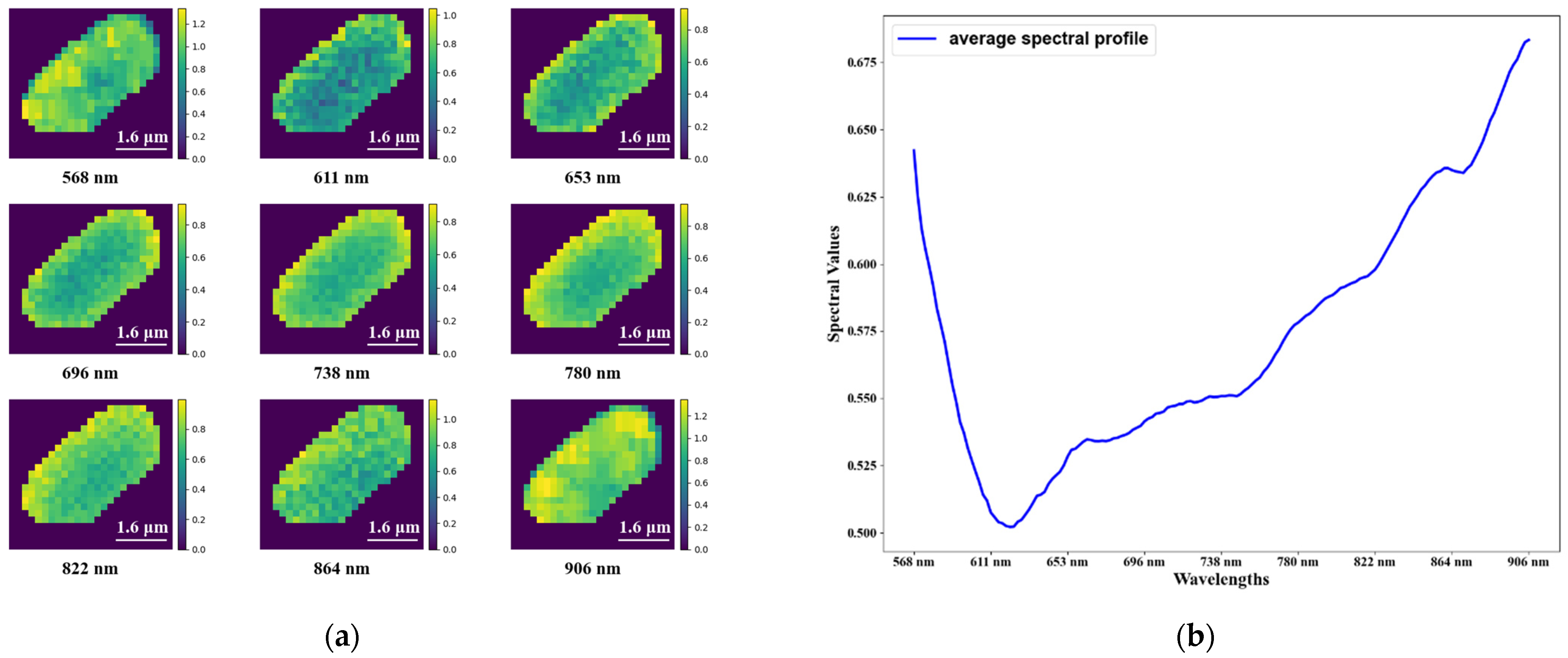{kind=link}
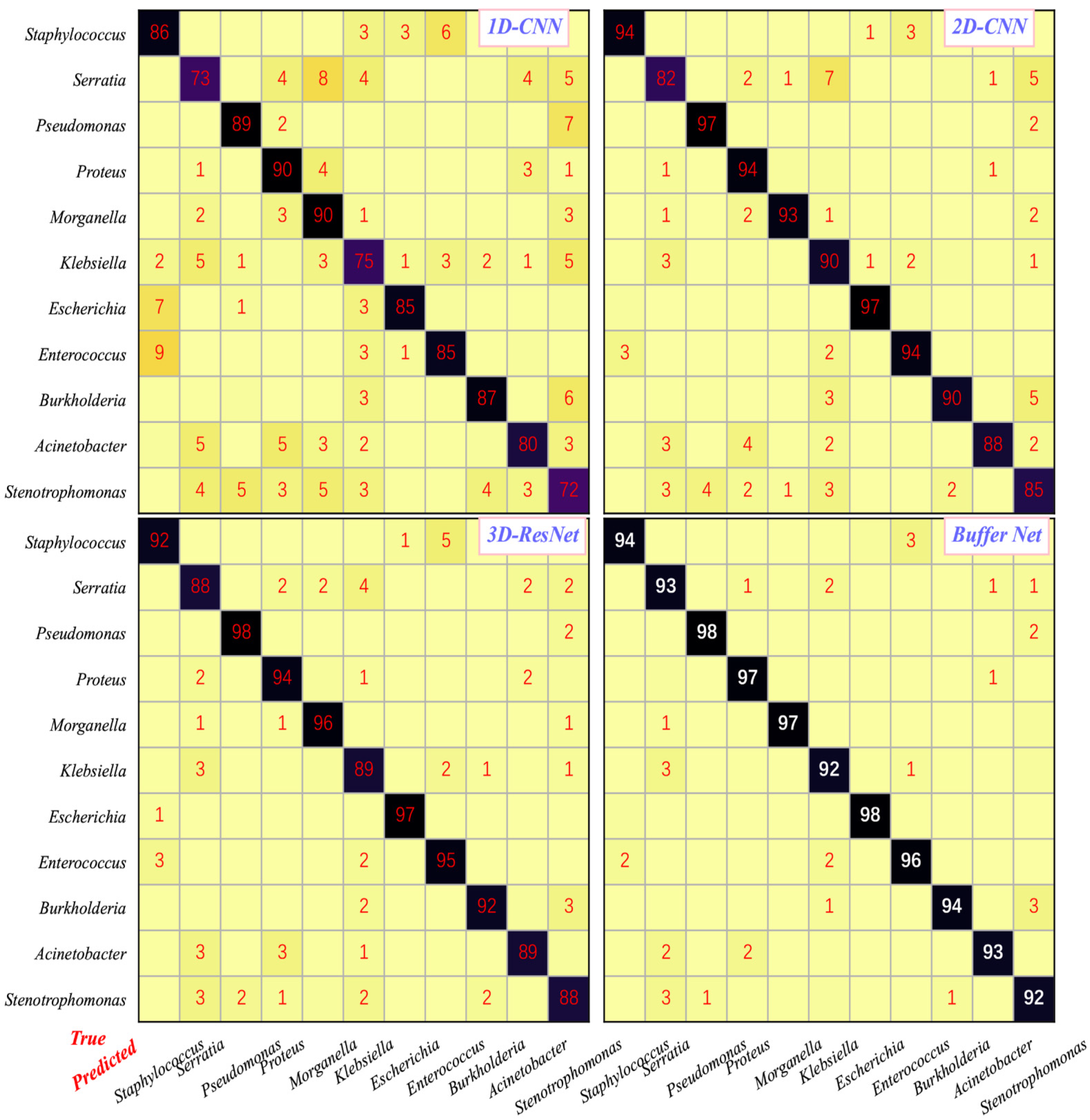{kind=link}
| Genera | Numbers for Training | Numbers for Testing | Total |
|---|---|---|---|
| Stenotrophomonas | 9070 | 3888 | 12,958 |
| Escherichia | 7135 | 3058 | 10,193 |
| Morganella | 7384 | 3165 | 10,549 |
| Burkholderia | 6337 | 2717 | 9054 |
| Serratia | 8126 | 3483 | 11,609 |
| Pseudomonas | 8954 | 3838 | 12,792 |
| Acinetobacter | 7185 | 3080 | 10,265 |
| Klebsiella | 11,255 | 4825 | 16,080 |
| Proteus | 11,435 | 4902 | 16,337 |
| Staphylococcus | 8182 | 3508 | 11,690 |
| Enterococcus | 8887 | 3810 | 12,697 |
| total | 93,950 | 40,274 | 134,224 |
| Algorithm | 1D-CNN | 2D-CNN | 3D-ResNet | Buffer Net (Without) | Buffer Net (With) |
|---|---|---|---|---|---|
| Accuracy | 82.6 | 91.3 | 92.3 | 88.4 | 94.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, C.; Du, J.; Tang, Y.; Wang, J.; Dong, K.; Yang, M.; Hu, B.; Zhang, Z. A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images. Cells 2022, 11, 2237. https://doi.org/10.3390/cells11142237
Tao C, Du J, Tang Y, Wang J, Dong K, Yang M, Hu B, Zhang Z. A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images. Cells. 2022; 11(14):2237. https://doi.org/10.3390/cells11142237
Chicago/Turabian StyleTao, Chenglong, Jian Du, Yingxin Tang, Junjie Wang, Ke Dong, Ming Yang, Bingliang Hu, and Zhoufeng Zhang. 2022. "A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images" Cells 11, no. 14: 2237. https://doi.org/10.3390/cells11142237
APA StyleTao, C., Du, J., Tang, Y., Wang, J., Dong, K., Yang, M., Hu, B., & Zhang, Z. (2022). A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images. Cells, 11(14), 2237. https://doi.org/10.3390/cells11142237