
Sarcoma Metabolomics: Current Horizons and Future Perspectives

 , , ,
, , ,
Abstract
1. Introduction
2. Cancer Metabolic Fingerprints
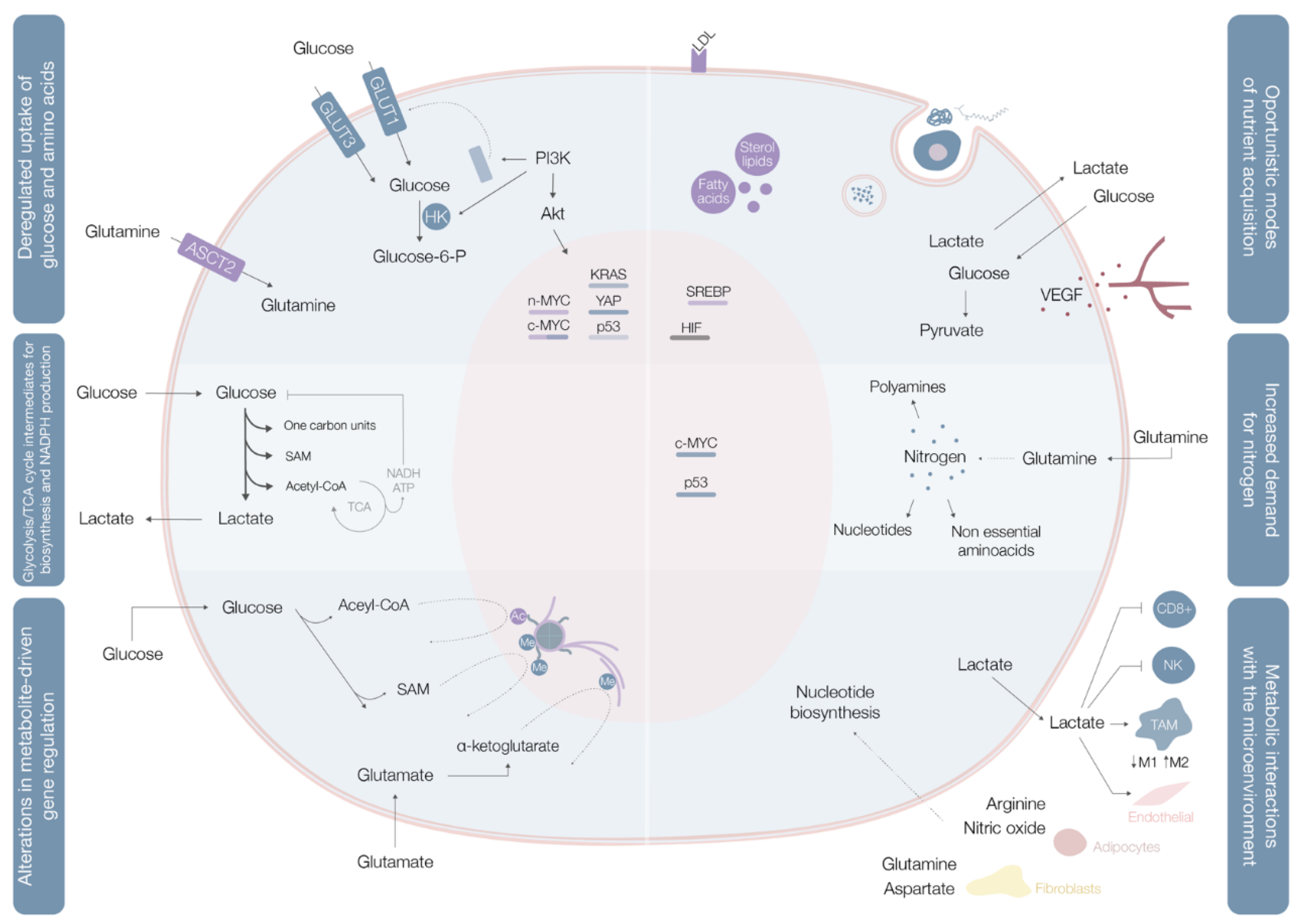
2.1. Deregulated Glucose and Amino Acid Uptake
2.2. Use of Opportunistic Modes of Nutrient Acquisition
2.3. Use of Glycolysis/TCA Cycle Intermediates for Biosynthesis and NADPH Production
2.4. Increased Demand for Nitrogen
2.5. Alterations in Metabolite-Driven Gene Regulation
2.6. Metabolic Interactions with the Microenvironment
3. Sarcoma Metabolomics
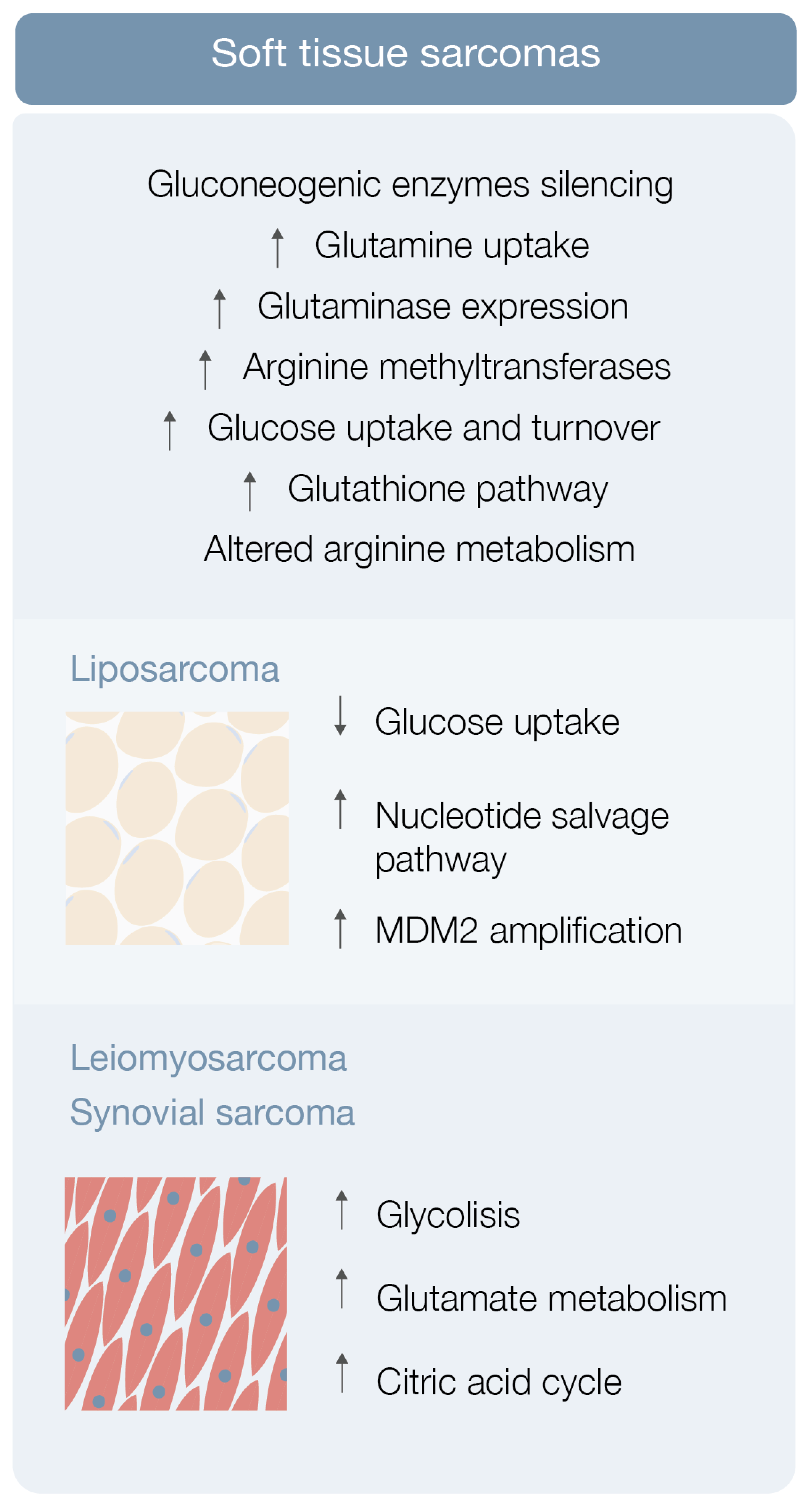
3.1. Soft Tissue Sarcoma Metabolomics
3.1.1. Liposarcoma
3.1.2. Leiomyosarcoma, Synovial Sarcoma and Others STS
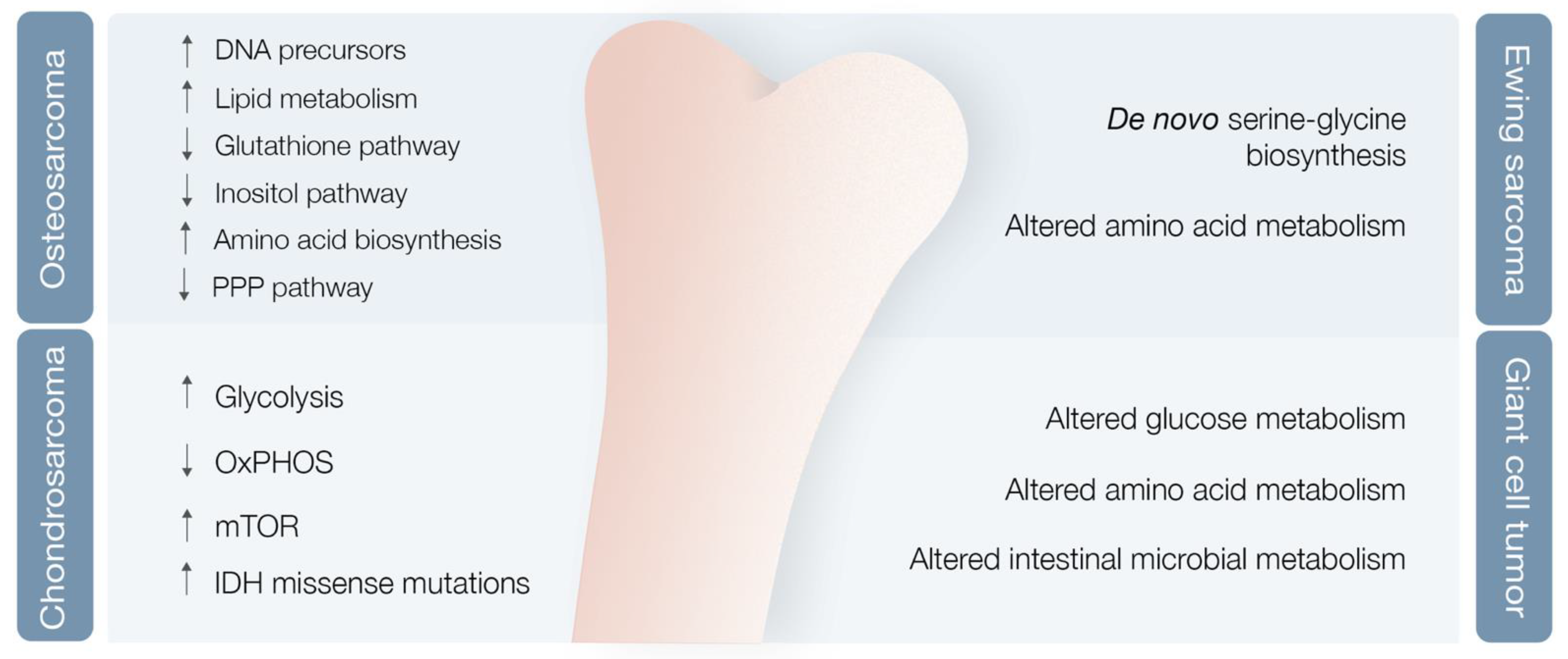
3.2. Bone Sarcoma Metabolomics
3.2.1. Osteosarcoma
3.2.2. Chondrosarcoma
3.2.3. Ewing Sarcoma
3.2.4. Giant Cell Tumor of the Bone
3.3. The Special Case of Gastrointestinal Stromal Tumors
4. From Biomarkers to Therapeutic Targets
4.1. mTOR Signaling Pathway Inhibition
4.2. β-Catenin Gene Mutation Modulation
4.3. BCR-ABL and Src Signaling Inhibition
4.4. PARP and Nicotinamide Phosphoribosyltransferase Activity Inhibition
4.5. MicroRNAs (miRNA) Inhibition
4.6. Isolated Proteasome Inhibition and Combined Proteasome and Histone Deacetylases (HDAC) Inhibition
4.7. Immune Checkpoint Inhibition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.H. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlem; Arnold Constable: London, UK, 1930. [Google Scholar]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBernardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Torrence, M.E.; Manning, B.D. Nutrient sensing in cancer. Annu. Rev. Cancer Biol. 2018, 2, 251–269. [Google Scholar] [CrossRef]
- Mele, L.; del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Mele, L.; La Noce, M.; Paino, F.; Regad, T.; Wagner, S.; Liccardo, D.; Papaccio, G.; Lombardi, A.; Caraglia, M.; Tirino, V.; et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Haigis, M.C. Metabolites and the tumor microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Florou, V.; Nascimento, A.G.; Giulia, A. Global health perspective in sarcomas and other rare cancers. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Abecassis, N.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; Broto, J.; et al. Soft tissue and visceral sarcomas: ESMO–EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv51–iv67. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. cancer statistics. CA Cancer. J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic phenotyping in health and disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef]
- Lou, S.; Balluff, B.; Cleven, A.H.G.; Bovée, J.V.M.G.; McDonnell, L.A. Prognostic Metabolite Biomarkers for Soft Tissue Sarcomas Discovered by Mass Spectrometry Imaging. J. Am. Soc. Mass Spectrom. 2017, 28, 376–383. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; DeCarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef]
- Sadri, N.; Zhang, P.J. Hypoxia-inducible factors: Mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers 2013, 5, 320–333. [Google Scholar] [CrossRef]
- Miolo, G.; Di Gregorio, E.; Saorin, A.; Lombardi, D.; Scalone, S.; Buonadonna, A.; Steffan, A.; Corona, G. Integration of serum metabolomics into clinical assessment to improve outcome prediction of metastatic soft tissue sarcoma patients treated with trabectedin. Cancers 2020, 12, 1983. [Google Scholar] [CrossRef]
- Huangyang, P.; Li, F.; Lee, P.; Nissim, I.; Weljie, A.M.; Mancuso, A.; Li, B.; Keith, B.; Yoon, S.S.; Simon, M.C. Fructose-1,6-biphosphatase 2 inhibits sarcoma progression by restraining mitochondrial genesis. Cell Metab. 2020, 31, 174–188. [Google Scholar] [CrossRef]
- Lee, P.; Malik, D.; Perkons, N.; Huangyang, P.; Khare, S.; Rhoades, S.; Gong, Y.-Y.; Burrows, M.; Finan, J.M.; Nissim, I.; et al. Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Ge, S. Arginine methyltransferase inhibitor-1 inhibits sarcoma viability in vitro and in vivo. Oncol. Lett. 2018, 16, 2161–2166. [Google Scholar] [CrossRef]
- Min, L.; Choy, E.; Tu, C.; Hornicek, F.; Duan, Z. Application of metabolomics in sarcoma: From biomarkers to therapeutic targets. Crit. Rev. Oncol. 2017, 116, 1–10. [Google Scholar] [CrossRef]
- Krikelis, D.; Judson, I. Role of chemotherapy in the management of soft tissue sarcomas. Expert Rev. Anticancer Ther. 2010, 10, 249–260. [Google Scholar] [CrossRef]
- Crago, A.M.; Singer, S. Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr. Opin. Oncol. 2011, 23, 373–378. [Google Scholar] [CrossRef]
- Braas, D.; Ahler, E.; Tam, B.; Nathanson, D.; Riedinger, M.; Benz, M.R.; Smith, K.B.; Eilber, F.C.; Witte, O.N.; Tap, W.D.; et al. Metabolomics Strategy Reveals Subpopulation of Liposarcomas Sensitive to Gemcitabine Treatment. Cancer Discov. 2012, 2, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Patt, A.; Demoret, B.; Stets, C.; Bill, K.-L.; Smith, P.; Vijay, A.; Patterson, A.; Hays, J.; Hoang, M.; Chen, J.L.; et al. MDM2-dependent rewiring of metabolomic and lipidomic profiles in dedifferentiated liposarcoma models. Cancers 2020, 12, 2157. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.D.; Breitkopf, S.B.; Yuan, M.; Goldsmith, J.; Spentzos, D.; Asara, J.M. Metabolomic profiling from formalin-fixed, paraffin-embedded tumor tissue using targeted LC/MS/MS: Application in sarcoma. PLoS ONE 2011, 6, e25357. [Google Scholar] [CrossRef] [PubMed]
- Monti, E.; Fanzani, A. Uncovering metabolism in rhabdomyosarcoma. Cell Cycle 2016, 15, 184–195. [Google Scholar] [CrossRef]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current treatment and a collaborative pathway to success. J. Clin. Oncol. 2015, 33, 3028. [Google Scholar] [CrossRef]
- Lv, D.; Zou, Y.; Zeng, Z.; Yao, H.; Ding, S.; Bian, Y.; Xie, X. Comprehensive metabolomic profiling of osteosarcoma based on UHPLC-HRMS. Metabolomics 2020, 16, 120. [Google Scholar] [CrossRef]
- Hua, Y.; Qiu, Y.; Zhao, A.; Wang, X.; Chen, T.; Zhang, Z.; Chi, Y.; Li, Q.; Sun, W.; Li, G.; et al. Dynamic Metabolic Transformation in Tumor Invasion and Metastasis in Mice with LM-8 Osteosarcoma Cell Transplantation. J. Proteome Res. 2011, 10, 3513–3521. [Google Scholar] [CrossRef] [PubMed]
- Kort, W.J.; Hulsmann, W.C.; Stehman, T.E. Modulation of metastatic ability by inhibition of cholesterol synthesis. Clin. Exp. Metastasis 1989, 7, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.C.; Shen, S.; Hornicek, F.J.; Duan, Z. From genomics to metabolomics: Emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev. 2018, 37, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Hong, E.S.; Mendoza, A.; Issaq, S.; Hoang, C.T.; Lizardo, M.; LeBlanc, A.; Khanna, C. Metabolomics uncovers a link between inositol metabolism and osteosarcoma metastasis. Oncotarget 2017, 8, 38541. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.P.; Singh, T.; Kumar, P. Metabolic adaptations in cancer stem cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef] [PubMed]
- La Noce, M.; Paino, F.; Mele, L.; Papaccio, G.; Regad, T.; Lombardi, A.; Papaccio, F.; Desiderio, V.; Tirino, V. HDAC2 depletion promotes osteosarcoma’s stemness both in vitro and in vivo: A study on a putative new target for CSCs directed therapy. J. Exp. Clin. Cancer Res. 2018, 37, 296. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy Metabolism Characterization of a Novel Cancer Stem Cell-Like Line 3AB-OS. J. Cell. Biochem. 2013, 115, 368–379. [Google Scholar] [CrossRef]
- Escobar, M.Q.; Costa, T.B.B.C.; Martins, L.G.; Costa, S.S.; Lengert, A.V.; Boldrini, E.; Da Silva, S.R.M.; Lopes, L.F.; Vidal, D.O.; Krepischi, A.C.V.; et al. Insights in Osteosarcoma by Proton Nuclear Magnetic Resonance Serum Metabonomics. Front. Oncol. 2020, 10, 506959. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.A. Chondrosarcoma: Biology, genetics and epigenetics. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, L.B.; Hameetman, L.; van Wezel, T.; Taminiau, A.H.; Cleton-Jansen, A.M.; Hogendoorn, P.C.; Bovée, J.V. cDNA expression profiling of chondrosarcomas: Ollier disease resembles solitary tumours and alteration in genes coding for components of energy metabolism occurs with increasing grade. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2005, 207, 61–71. [Google Scholar] [CrossRef]
- Zhang, Y.X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; Van Wezel, T.; Bühnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovée, J.V.M.G. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Addie, R.D.; de Jong, Y.; Alberti, G.; Kruisselbrink, A.B.; Que, I.; Baelde, H.; Bovée, J.V. Exploration of the chondrosarcoma metabolome; the mTOR pathway as an important pro-survival pathway. J. Bone Oncol. 2019, 15, 100222. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Paz, A.C.; Wilky, B.A.; Johnson, B.; Galoian, K.; Rosenberg, A.; Hu, G.; Tinoco, G.; Bodamer, O.; Trent, J.C. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PLoS ONE 2015, 10, e0133813. [Google Scholar] [CrossRef]
- Eder, J.P.; Doroshow, D.B.; Do, K.T. Clinical efficacy of olaparib in IDH1/IDH2-mutant mesenchymal sarcomas. JCO Precis. Oncol. 2021, 5, 466–472. [Google Scholar] [CrossRef]
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Tanner, J.M.; Bensard, C.; Wei, P.; Krah, N.M.; Schell, J.C.; Gardiner, J.D.; Schiffman, J.D.; Lessnick, S.L.; Rutter, J. EWS/FLI is a Master Regulator of Metabolic Reprogramming in Ewing Sarcoma. Mol. Cancer Res. 2017, 15, 1517–1530. [Google Scholar] [CrossRef]
- Sen, N.; Cross, A.M.; Lorenzi, P.L.; Khan, J.; Gryder, B.E.; Kim, S.; Caplen, N.J. EWS-FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine-glycine biosynthesis. Mol. Carcinog. 2018, 57, 1342–1357. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T. Matrix screen identifies synergistic combination of parp inhibitors and nicotinamide phosphoribosyltransferase (NAMPT) inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef]
- Mavrogenis, A.F.; Igoumenou, V.G.; Megaloikonomos, P.D.; Panagopoulos, G.N.; Papagelopoulos, P.J.; Soucacos, P.N. Giant cell tumor of bone revisited. SICOT-J 2017, 3, 54. [Google Scholar] [CrossRef]
- Alberghini, M.; Kliskey, K.; Krenacs, T.; Picci, P.; Kindblom, L.; Forsyth, R.; Athanasou, N.A. Morphological and immunophenotypic features of primary and metastatic giant cell tumour of bone. Virchows Archiv. 2009, 456, 97–103. [Google Scholar] [CrossRef]
- Whelan, J.S.; Davis, L.E. Osteosarcoma, chondrosarcoma, and chordoma. J. Clin. Oncol. 2018, 36, 188–193. [Google Scholar] [CrossRef]
- Wang, W.; Liu, X.; Wu, J.; Kang, X.; Xie, Q.; Sheng, J.; Xu, W.; Liu, D.; Zheng, W. Plasma metabolite profiling reveals potential biomarkers of giant cell tumor of bone by using NMR-based metabolic profiles: A cross-sectional study. Medicine 2019, 98, e17445. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.K.; Gao, J.; Chen, Z.; Shi, H.; Yuan, J.; Cui, H.L.; Yeh, C.-N.; Bränström, R.; Larsson, C.; Li, S.; et al. Heterogeneity of metabolic vulnerability in imatinib-resistant gastrointestinal stromal tumor. Cells 2020, 9, 1333. [Google Scholar] [CrossRef] [PubMed]
- Van den Abbeele, A.D. The lessons of GIST-PET and PET/CT: A new paradigm for imaging. Oncologist 2008, 13, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Tang, M.D.; Lin, D.Y.; Liu, D.J.; Lyu, Q.H.; Zhang, J.P.; Cai, Z.H. The value of (18)F-FDG PET-CT imaging in predicting the malignant potential of GIST. Zhonghua Zhong Liu Za Zhi Chin. J. Oncol. 2017, 39, 821–827. [Google Scholar]
- Albano, D.; Bosio, G.; Tomasini, D.; Bonù, M.; Giubbini, R.; Bertagna, F. Metabolic behavior and prognostic role of pretreatment 18F-FDG PET/CT in gist. Asia-Pac. J. Clin. Oncol. 2020, 16, 207. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signaling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.-Y. Updating progress in sarcoma therapy with mTOR inhibitors. Ann. Oncol. 2011, 22, 280–287. [Google Scholar] [CrossRef]
- Yoo, C.; Lee, J.; Rha, S.Y.; Park, K.H.; Kim, T.M.; Kim, Y.J.; Lee, H.J.; Lee, K.H.; Ahn, J.-H. Multicenter phase II study of everolimus in patients with metastatic or recurrent bone and soft-tissue sarcomas after failure of anthracycline and ifosfamide. Investig. New Drugs 2013, 31, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Pink, D.; Hohenberger, P.; Schuette, H.; Casali, P.G.; Pustowka, A.; Reichardt, P. Multicenter, triple-arm, single-stage, phase II trial to determine the efficacy and safety of everolimus (RAD001) in patients with refractory bone or soft tissue sarcomas including GIST. J. Clin. Oncol. 2010, 28, 10038. [Google Scholar] [CrossRef]
- van der Graaf, W.T.A.; Gelderblom, H. New systemic therapy options for advanced sarcomas. Curr. Treat Options Oncol. 2012, 13, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.-O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 2008, 7, 1237–1245. [Google Scholar] [CrossRef]
- Di Gialleonardo, V.; Aldeborgh, H.N.; Miloushev, V.; Folkers, K.M.; Granlund, K.; Tap, W.D.; Lewis, J.S.; Weber, W.A.; Keshari, K.R. Multinuclear NMR and MRI Reveal an Early Metabolic Response to mTOR Inhibition in Sarcoma. Cancer Res. 2017, 77, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Mercier, K.A.; Al-Jazrawe, M.; Poon, R.; Acuff, Z.; Alman, B. A Metabolomics Pilot Study on Desmoid Tumors and Novel Drug Candidates. Sci. Rep. 2018, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Kruh, G.D. Introduction to resistance to anticancer agents. Oncogene 2003, 22, 7262–7264. [Google Scholar] [CrossRef]
- Cubitt, C.L.; Menth, J.; Dawson, J.; Martinez, G.V.; Foroutan, P.; Morse, D.L.; Bui, M.M.; Letson, G.D.; Sullivan, D.M.; Reed, D.R. Rapid Screening of Novel Agents for Combination Therapy in Sarcomas. Sarcoma 2013, 2013, 365723. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Rixe, U.; Superti-Furga, G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk. Lymphoma 2008, 49, 615–619. [Google Scholar] [CrossRef]
- Hingorani, P.; Zhang, W.; Gorlick, R.; Kolb, E.A. Inhibition of Src Phosphorylation Alters Metastatic Potential of Osteosarcoma In vitro but not In vivo. Clin. Cancer Res. 2009, 15, 3416–3422. [Google Scholar] [CrossRef]
- Shor, A.C.; Keschman, E.A.; Lee, F.Y. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007, 67, 2800–2808. [Google Scholar] [CrossRef]
- Shor, A.C. Src kinase inhibitors for the treatment of sarcomas: Cellular and molecular mechanisms of action. Graduate Theses and Dissertations 2007. Available online: https://scholarcommons.usf.edu/etd/2364 (accessed on 25 February 2021).
- van der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Mangoni, M.; Sottili, M.; Salvatore, G.; Campanacci, D.; Scoccianti, G.; Beltrami, G.; Paoli, C.D.; Dominici, L.; Maragna, V.; Olmetto, E.; et al. Soft tissue sarcomas: New opportunity of treatment with PARP inhibitors? La Radiol. Med. 2019, 124, 282–289. [Google Scholar] [CrossRef]
- Montecucco, B.F.; Cea, M.; Bauer, I.; Soncini, D.; Caffa, I.; Lasigliè, D.; Aimable, N.; Antonio, U.; Santina, B.; Alessio, N. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors as therapeutics: Rationales, controversies, clinical experience. Curr. Drug Targets 2013, 14, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Leithner, A.; Posch, F.; Szkandera, J.; Liegl-Atzwanger, B.; Pichler, M. MicroRNAs in different histologies of soft tissue sarcoma: A comprehensive review. Int. J. Mol. Sci. 2017, 18, 1960. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Calin, G.A. MicroRNAs in cancer: From developmental genes in worms to their clinical application in patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Lui, W.O.; Lee, C.H.; Espinosa, I.; Nielsen, T.O.; Heinrich, M.C.; Corless, C.L.; Fire, A.Z.; Van De Rijn, M. MicroRNA expression signature of human sarcomas. Oncogene 2007, 27, 2015–2026. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Shen, J.; Wu, W.K.K.; Chan, M.T. MicroRNA expression and its clinical implications in Ewing’s sarcoma. Cell Prolif. 2015, 48, 1–6. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef]
- Nunes, A.T.; Annunziata, C.M. Proteasome Inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef]
- Shukla, N.; Somwar, R.; Smith, R.S.; Ambati, S.; Munoz, S.; Merchant, M.; D’Arcy, P.; Wang, X.; Kobos, R.; Antczak, C.; et al. Proteasome Addiction Defined in Ewing Sarcoma Is Effectively Targeted by a Novel Class of 19S Proteasome Inhibitors. Cancer Res. 2016, 76, 4525–4534. [Google Scholar] [CrossRef]
- Lu, G.; Punj, V.; Chaudhary, P.M. Proteasome inhibitor Bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing’s sarcoma family of tumors and synergizes with TRAIL. Cancer Biol. Ther. 2008, 7, 603–608. [Google Scholar] [CrossRef]
- Laporte, A.N.; Barrott, J.J.; Yao, R.J.; Poulin, N.M.; Brodin, B.A.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. HDAC and Proteasome Inhibitors Synergize to Activate Pro-Apoptotic Factors in Synovial Sarcoma. PLoS ONE 2017, 12, e0169407. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar]
- Lim, J.; Poulin, N.M.; Nielsen, T.O. New strategies in sarcoma: Linking genomic and immunotherapy approaches to molecular subtype. Clin. Cancer Res. 2015, 21, 4753–4759. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; Ingham, M.; Spraker, M.B.; Schwartz, G.K. Emerging targeted and imune-based therapies in sarcoma. J. Clin. Oncol. 2017, 36, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Beird, H.; Livingston, J. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Metabolic Hallmark | Alterations and Adaptations in Cancer | Outcome/Acquired Phenotype |
|---|---|---|
| Deregulated uptake of glucose and amino acids [5] | (1) Mutations of the oncogenes c-MYC, KRAS and YAP [8] (2) Overexpression of YAP and loss-of-function mutations in p53 [8] (3) Phosphoinositide 3-kinase (PI3K)/Akt pathway hyperactivation [5,8] (4) C-MYC, n-MYC, mTORC1, IL-4 and lactate modulation [8] (5) RAS mutations [8] | (1) Upregulate glucose transporter (GLUT) 1 expression [8] (2) Augments GLUT3 expression [8] (1) and (2) Increase entrance of glucose into the cell [8] (3) Promotes GLUT1 mRNA expression and GLUT1 protein translocation from the inner membranes to the cell surface [5] and hexokinase (HK)2 activity upregulation, trapping glucose inside the cell [8] (4) Upregulates ASCT2 glutamine transporter expression increasing entrance of glutamine into the cell [8] (5) Increases glutamine uptake by micropinocytosis [8] |
| Use of opportunistic modes of nutrient acquisition [5] | (1) Hypoxia triggers the expression of transcription factors called hypoxia-inducible factors (HIF) [9] (2) Cholesterol depletion induces activation of sterol regulatory element-binding proteins [9] (3) Amino acid deprivation leads to activation of the GCN2 kinase [9] (4) Ras or c-Src mutations [5] (5) Prolonged periods of extracellular nutrients absence lead to macroautophagy [9] | (1) Stimulates glucose uptake, lactate export, glycolysis and angiogenesis (by induction of VEGF expression) [9] (2) Stimulates the expression of enzymes required for de novo synthesis of fatty acid and sterol lipids, increases LDL receptors expression and enhances NADPH production [9] (3) Promotes selective translation of mRNAs like ATF4, promoting the transcription of amino acids transporters and enzymes involved in the generation of non-essential amino acids [9] (4) Enhances the recovery of free amino acids by lysosomal digestion of extracellular proteins by several processes including micropinocytosis, degradation of entire living cells (entosis) and digestion of apoptotic cellular corpses [5] (5) Sequestrates and promotes lysosomal digestion of cytosolic macromolecules and organelles, allowing the recycling of these cellular components into nutrients [9] |
| Use of glycolysis/TCA cycle intermediates for biosynthesis and NADPH production [5] | (1) C-MYC and β-catenin/TCF signaling hyperactivation [5] | (1) Leads to overexpression of multiple key enzymes for generation of diverse glycolytic and TCA cycle intermediates that are biosynthetic precursors [5] |
| Increased demand for nitrogen [5] | (1) C-MYC signaling hyperactivation [5] (2) Asparagine synthetase upregulation [5] (3) Glutamine synthetase upregulation [5] | (1) Promotes celular glutamine uptake, upregulates the expression of different enzymes with roles in nucleotide biosynthesis and upregulates glutaminase [5] (2) Increases asparagine synthesis (crucial in glutamine deprived conditions) [5] (3) Augments intracelular de novo glutamine production (fundamental in glutamine deprived conditions) [5] |
| Alterations in metabolite-driven gene regulation [5] | (1) Diverse oncogenic pathways hyperactivation [10] (2) Loss-of-function SDH and FH mutations [10] (3) Gain-of-function IDH1 and IDH2 mutations [10] | (1) Enhances total histone acetylation, leading to increased and broader oncogene expression [10] (2) Succinate and fumarate accumulation leads to inhibition of demethylases (JmJC and TET), increase of genome wide DNA and histone hypermethylation, enabling oncogenic promoter-enhancer interactions, inducing epithelial-to-mesenchymal transition, and disrupting DNA repair mechanisms [10] (3) Catalyzes the conversion of α-ketoglutarate to 2-HG, leading to 2-HG accumulation, DNA and histone hypermethylation with downregulation of genes associated with tumor-suppression and cellular differentiation blockade [10] |
| Metabolic interactions with the microenvironment [5] | (1) Low glucose and aminoacids (glutamine, L-arginine, methionine) extracellular availability and extracellular lactate accumulation [11] (2) Increased CAF glycolytic and glutamine anabolic metabolism [11] (3) CAF-derived exosomes proliferation [11] (4) Metabolic plasticity (glycolysis vs. mitochondrial metabolism) relative to local oxygen availability [11] | (1) Decreases mTOR activity leading to an impairment of T cell (CD8+) and NK cell function and proliferation and promotes a macrophage M2 polarization [11] (2) Leads to use of resultant metabolites from CAF glycolysis and glutamine metabolism to fuel cancer cells [11] (3) Supplies cancer cells with amino acids, lipids and TCA intermediates [11] (4) Sustains glucose consumption, glycolysis and OXPHOS in cancer cells located in well perfused areas, while cells in poorly perfused areas depend on other carbon sources [11] |
| Therapeutic Target | Alterations and Adaptations in Cancer |
|---|---|
| mTOR signalling pathway inhibition | Rapamycin, Temsirolimus, Everolimus and Ridaforolimus |
| Beta-catenin gene mutations modulation | Dasatinib and FAK inhibitor 14 |
| BCR-ABL and Src signalling inhibition | Imatinib, Dasatinib, Pazopanib and Olaratumab |
| PARP and NAMPT activity inhibition | PARP inhibitors and NAMPT inhibitors |
| miRNAs inhibition | miRNAs |
| Proteosome and HDAC inhibition | Bortezomib and Quinostat |
| Immune checkpoint inhibition | Pembrolizumab (monotherapy or combined with Axitinib or Gemcitabine) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esperança-Martins, M.; Fernandes, I.; Soares do Brito, J.; Macedo, D.; Vasques, H.; Serafim, T.; Costa, L.; Dias, S. Sarcoma Metabolomics: Current Horizons and Future Perspectives. Cells 2021, 10, 1432. https://doi.org/10.3390/cells10061432
Esperança-Martins M, Fernandes I, Soares do Brito J, Macedo D, Vasques H, Serafim T, Costa L, Dias S. Sarcoma Metabolomics: Current Horizons and Future Perspectives. Cells. 2021; 10(6):1432. https://doi.org/10.3390/cells10061432
Chicago/Turabian StyleEsperança-Martins, Miguel, Isabel Fernandes, Joaquim Soares do Brito, Daniela Macedo, Hugo Vasques, Teresa Serafim, Luís Costa, and Sérgio Dias. 2021. "Sarcoma Metabolomics: Current Horizons and Future Perspectives" Cells 10, no. 6: 1432. https://doi.org/10.3390/cells10061432
APA StyleEsperança-Martins, M., Fernandes, I., Soares do Brito, J., Macedo, D., Vasques, H., Serafim, T., Costa, L., & Dias, S. (2021). Sarcoma Metabolomics: Current Horizons and Future Perspectives. Cells, 10(6), 1432. https://doi.org/10.3390/cells10061432