
The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease


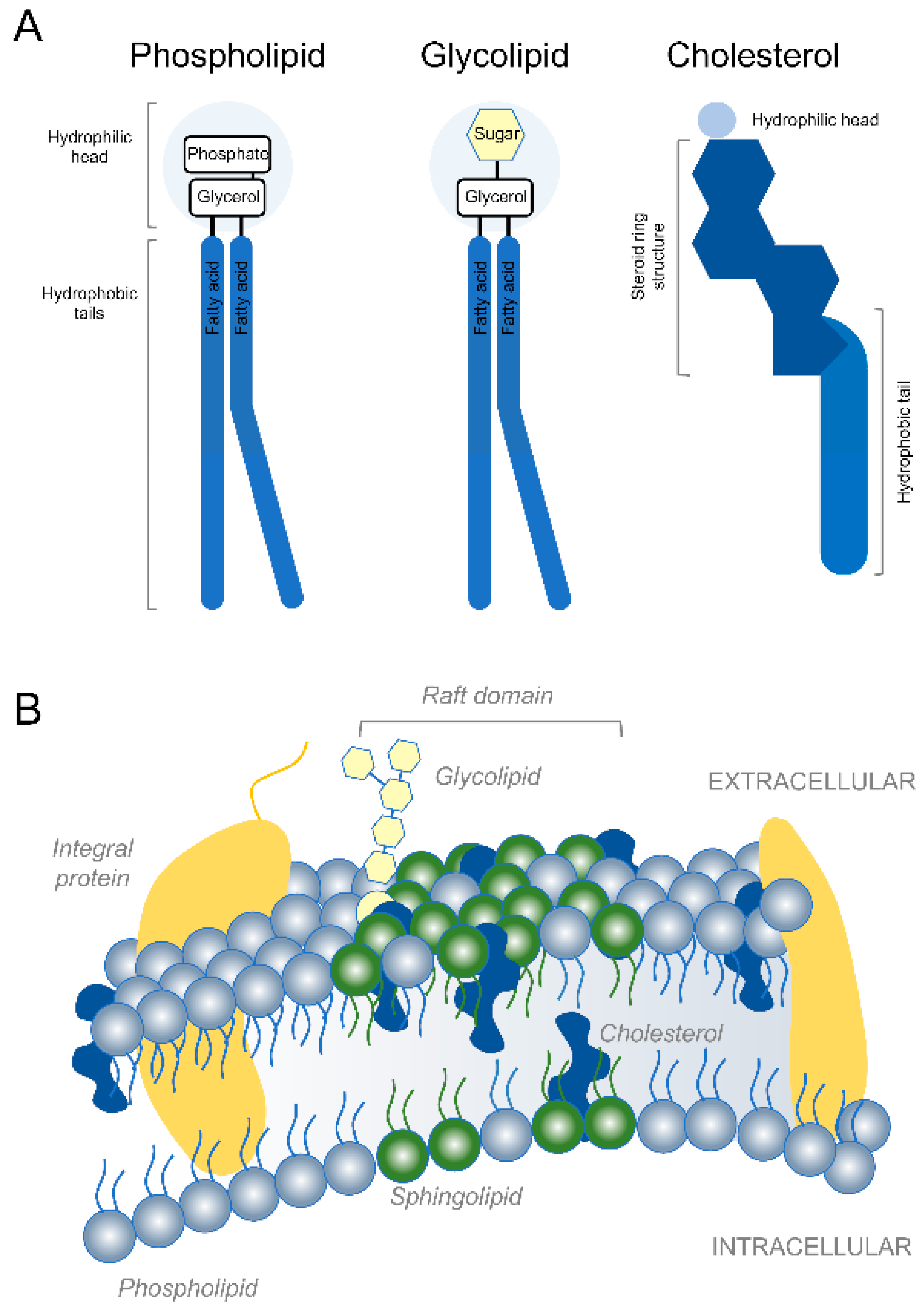{kind=link}
{kind=link}
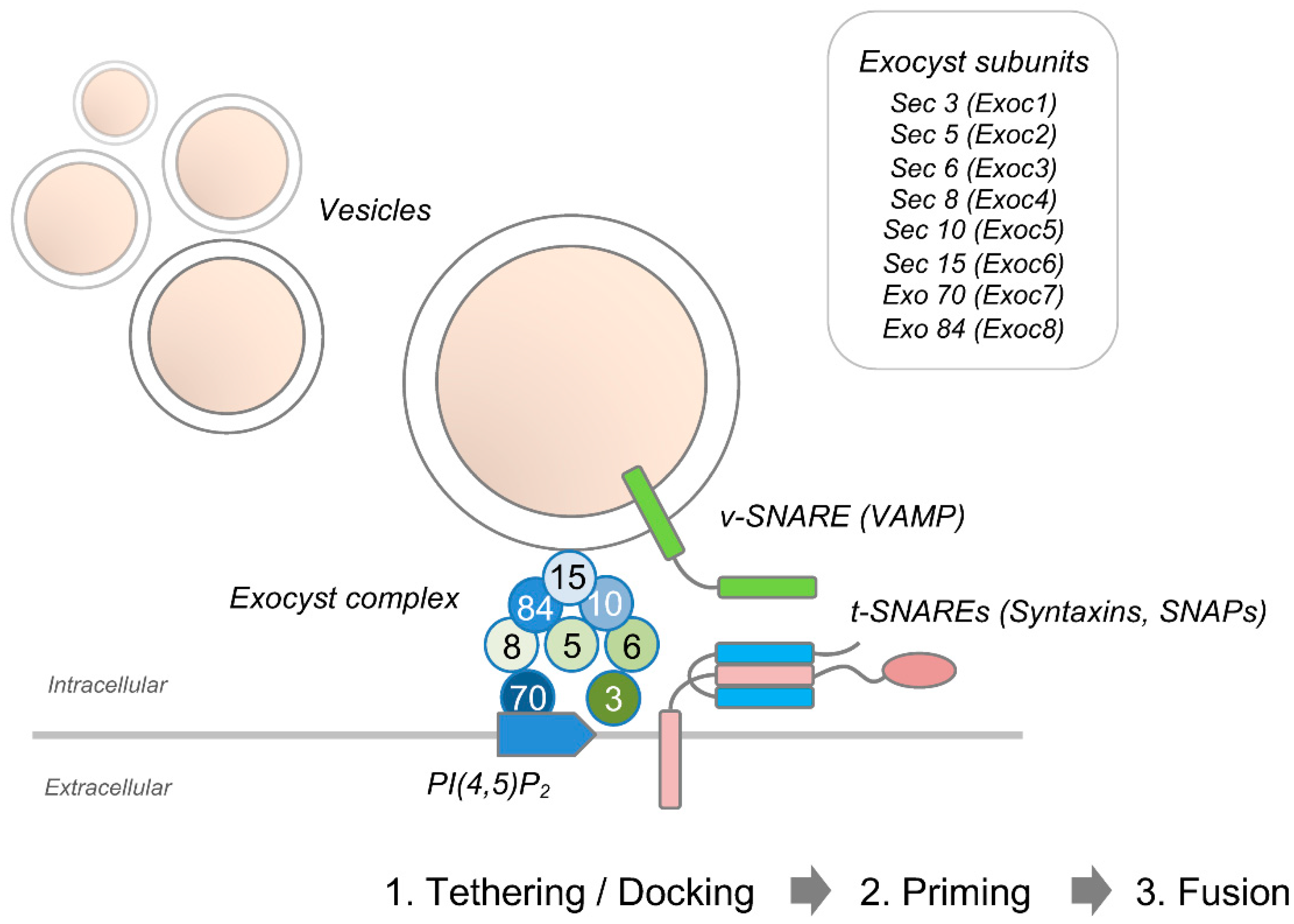{kind=link}
{kind=link}
Abstract
1. Introduction
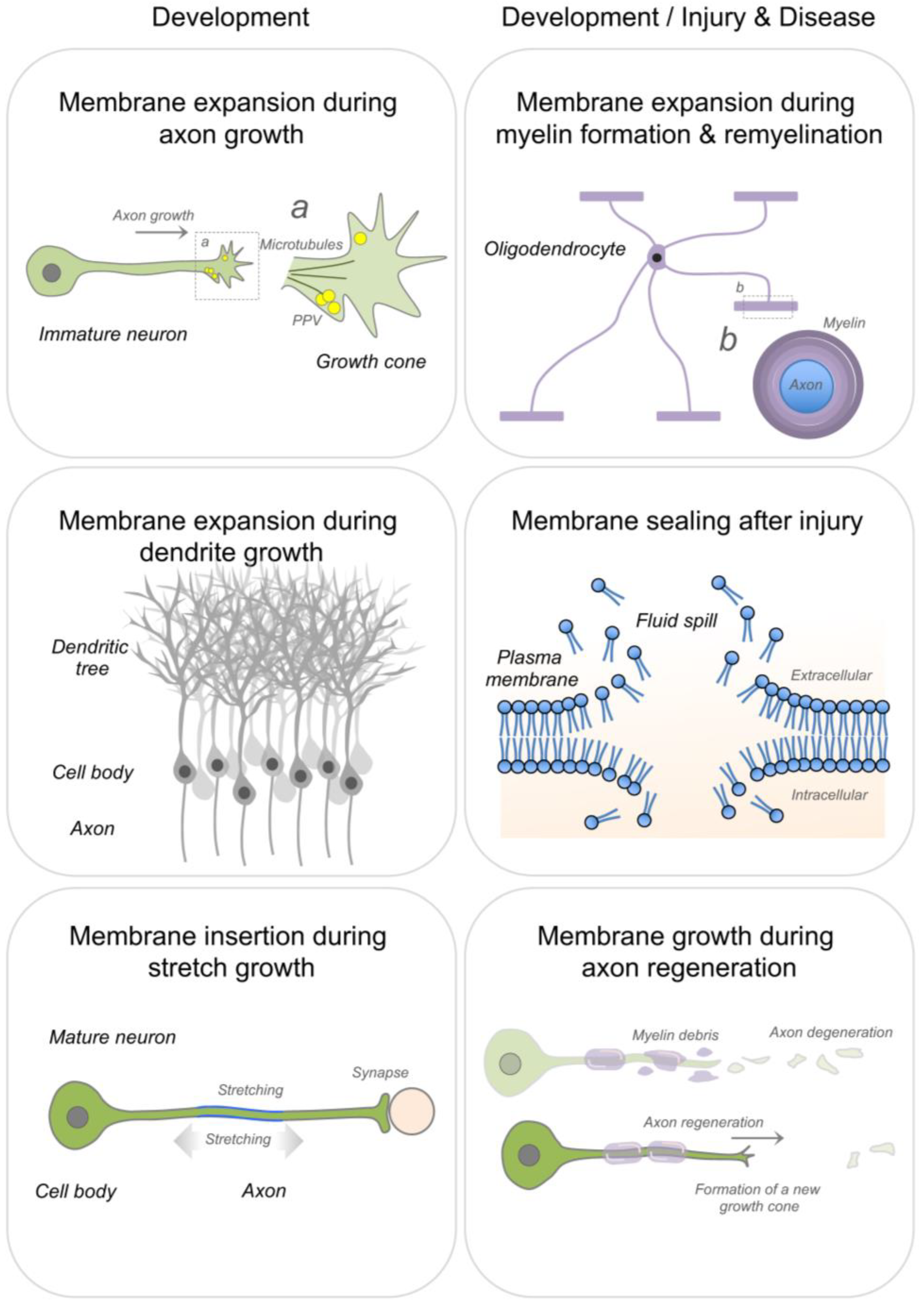
2. Membrane Expansion
2.1. Axon Growth & Regeneration
2.2. Dendritic Growth
2.3. Myelin Formation and Repair
3. Energy Metabolism and Mitochondrial Transport
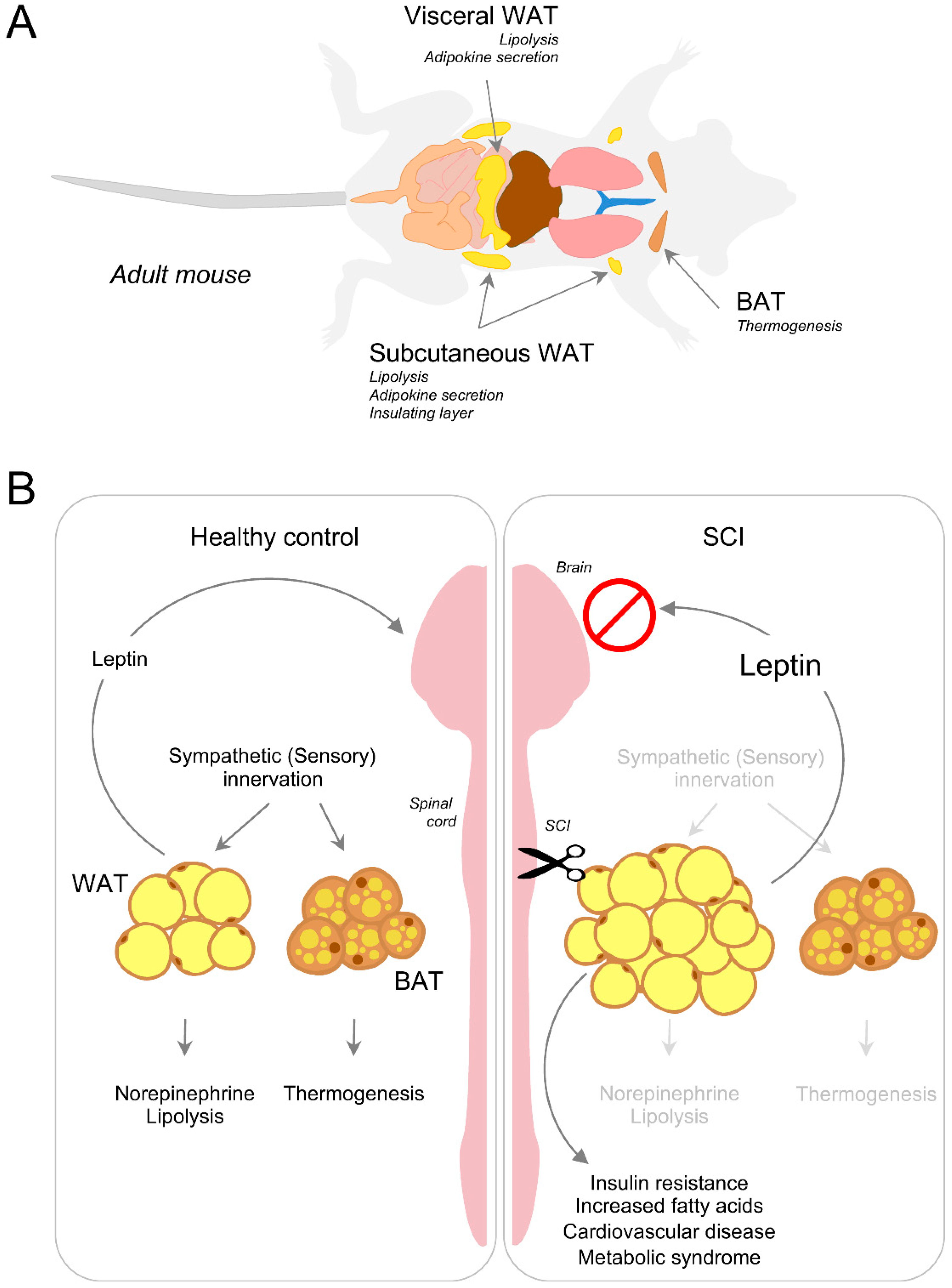
4. Energy Balance, Lipid Accumulation and Adipose Tissue
4.1. Lipolysis, Adaptive Thermogenesis and Innervation of Adipose Tissue
4.2. Disruption of Adipose Tissue Innervation and Energy Metabolism after Injury
4.3. Adiposity and CNS Injury
4.4. Ectopic Liver Fat Accumulation after SCI
4.5. Glucose Intolerance, Insulin Resistance and Cardiovascular Complications
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Polleux, F.; Snider, W. Initiating and growing an axon. Cold Spring Harb. Perspect Biol. 2010, 2, a001925. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Jin, Y. Intrinsic Control of Axon Regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef]
- Pfister, B.J.; Iwata, A.; Meaney, D.F.; Smith, D.H. Extreme stretch growth of integrated axons. J. Neurosci. 2004, 24, 7978–7983. [Google Scholar] [CrossRef]
- Lu, P.; Wang, Y.; Graham, L.; McHale, K.; Gao, M.; Wu, D.; Brock, J.; Blesch, A.; Rosenzweig, E.S.; Havton, L.A.; et al. Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell 2012, 150, 1264–1273. [Google Scholar] [CrossRef]
- Pfenninger, K.H. Plasma membrane expansion: A neuron’s Herculean task. Nat. Rev. Neurosci. 2009, 10, 251–261. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Wang, J.; Wang, X.; Chen, W.; Lu, N.; Siniossoglou, S.; Yao, Z.; Liu, K. Rewiring Neuronal Glycerolipid Metabolism Determines the Extent of Axon Regeneration. Neuron 2020, 105, 276–292.e5. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Dun, X.P.; Parkinson, D.B. Visualizing peripheral nerve regeneration by whole mount staining. PLoS ONE 2015, 10, e0119168. [Google Scholar] [CrossRef]
- Scheib, J.; Hoke, A. Advances in peripheral nerve regeneration. Nat. Rev. Neurol. 2013, 9, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 2010, 13, 1075–1081. [Google Scholar] [CrossRef]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Bradke, F. Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr. Opin. Neurobiol. 2017, 42, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Dissecting spinal cord regeneration. Nature 2018, 557, 343–350. [Google Scholar] [CrossRef]
- Sun, W.; Larson, M.J.; Kiyoshi, C.M.; Annett, A.J.; Stalker, W.A.; Peng, J.; Tedeschi, A. Gabapentinoid treatment promotes corticospinal plasticity and regeneration following murine spinal cord injury. J. Clin. Investig. 2020, 130, 345–358. [Google Scholar] [CrossRef]
- Buchovecky, C.M.; Turley, S.D.; Brown, H.M.; Kyle, S.M.; McDonald, J.G.; Liu, B.; Pieper, A.A.; Huang, W.; Katz, D.M.; Russell, D.W.; et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013, 45, 1013–1020. [Google Scholar] [CrossRef]
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ewan, E.E.; Avraham, O.; Carlin, D.; Goncalves, T.M.; Zhao, G.; Cavalli, V. Ascending dorsal column sensory neurons respond to spinal cord injury and downregulate genes related to lipid metabolism. Sci. Rep. 2021, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef]
- Chrast, R.; Saher, G.; Nave, K.A.; Verheijen, M.H. Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. J. Lipid. Res. 2011, 52, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, S.; Bisbal, M.; Caceres, A. Regulation of plasma membrane expansion during axon formation. Dev. Neurobiol. 2018, 78, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.B.; Thiele, C.; Tenedini, F.; Richard, M.; Leyendecker, P.; Hoermann, A.; Soba, P.; Tavosanis, G. Cell-autonomous control of neuronal dendrite expansion via the fatty acid synthesis regulator SREBP. Cell Rep. 2017, 21, 3346–3353. [Google Scholar] [CrossRef] [PubMed]
- Avraham, O.; Deng, P.Y.; Jones, S.; Kuruvilla, R.; Semenkovich, C.F.; Klyachko, V.A.; Cavalli, V. Satellite glial cells promote regenerative growth in sensory neurons. Nat. Commun. 2020, 11, 4891. [Google Scholar] [CrossRef] [PubMed]
- de Chaves, E.I.; Rusinol, A.E.; Vance, D.E.; Campenot, R.B.; Vance, J.E. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J. Biol. Chem. 1997, 272, 30766–30773. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E.; Campenot, R.B.; Vance, D.E. The synthesis and transport of lipids for axonal growth and nerve regeneration. Biochim. Biophys. Acta 2000, 1486, 84–96. [Google Scholar] [CrossRef]
- Lockerbie, R.O.; Miller, V.E.; Pfenninger, K.H. Regulated plasmalemmal expansion in nerve growth cones. J. Cell Biol. 1991, 112, 1215–1227. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef]
- Boyles, J.K.; Zoellner, C.D.; Anderson, L.J.; Kosik, L.M.; Pitas, R.E.; Weisgraber, K.H.; Hui, D.Y.; Mahley, R.W.; Gebicke-Haerter, P.J.; Ignatius, M.J.; et al. A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J. Clin. Investig. 1989, 83, 1015–1031. [Google Scholar] [CrossRef]
- Muse, E.D.; Jurevics, H.; Toews, A.D.; Matsushima, G.K.; Morell, P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J. Neurochem. 2001, 76, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Brugger, B.; Lappe-Siefke, C.; Mobius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.L.; Brophy, P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005, 6, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Trotter, J. Wrapping it up: The cell biology of myelination. Curr. Opin. Neurobiol. 2007, 17, 533–540. [Google Scholar] [CrossRef]
- Craig, A.M.; Wyborski, R.J.; Banker, G. Preferential addition of newly synthesized membrane protein at axonal growth cones. Nature 1995, 375, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Erez, H.; Malkinson, G.; Prager-Khoutorsky, M.; De Zeeuw, C.I.; Hoogenraad, C.C.; Spira, M.E. Formation of microtubule-based traps controls the sorting and concentration of vesicles to restricted sites of regenerating neurons after axotomy. J. Cell Biol. 2007, 176, 497–507. [Google Scholar] [CrossRef]
- Popov, S.; Brown, A.; Poo, M.M. Forward plasma membrane flow in growing nerve processes. Science 1993, 259, 244–246. [Google Scholar] [CrossRef]
- He, B.; Guo, W. The exocyst complex in polarized exocytosis. Curr. Opin. Cell Biol. 2009, 21, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Ting, A.E.; Hazuka, C.D.; Davanger, S.; Kenny, J.W.; Kee, Y.; Scheller, R.H. The mammalian brain rsec6/8 complex. Neuron 1996, 17, 1209–1219. [Google Scholar] [CrossRef]
- TerBush, D.R.; Maurice, T.; Roth, D.; Novick, P. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 1996, 15, 6483–6494. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Xi, F.; Zhang, X.; Zhang, J.; Guo, W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007, 26, 4053–4065. [Google Scholar] [CrossRef]
- Boyd, C.; Hughes, T.; Pypaert, M.; Novick, P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J. Cell Biol. 2004, 167, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zajac, A.; Zhang, J.; Wang, P.; Li, M.; Murray, J.; TerBush, D.; Guo, W. The critical role of Exo84p in the organization and polarized localization of the exocyst complex. J. Biol. Chem. 2005, 280, 20356–20364. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Chang, L.; Hwang, J.; Chiang, S.H.; Saltiel, A.R. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature 2003, 422, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Chiang, S.H.; Chang, L.; Chen, X.W.; Saltiel, A.R. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol. Biol. Cell. 2006, 17, 2303–2311. [Google Scholar] [CrossRef]
- Gracias, N.G.; Shirkey-Son, N.J.; Hengst, U. Local translation of TC10 is required for membrane expansion during axon outgrowth. Nat. Commun. 2014, 5, 3506. [Google Scholar] [CrossRef]
- Dupraz, S.; Grassi, D.; Bernis, M.E.; Sosa, L.; Bisbal, M.; Gastaldi, L.; Jausoro, I.; Caceres, A.; Pfenninger, K.H.; Quiroga, S. The TC10-Exo70 complex is essential for membrane expansion and axonal specification in developing neurons. J. Neurosci. 2009, 29, 13292–13301. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 1998, 395, 347–353. [Google Scholar] [CrossRef]
- Grote, E.; Carr, C.M.; Novick, P.J. Ordering the final events in yeast exocytosis. J. Cell Biol. 2000, 151, 439–452. [Google Scholar] [CrossRef]
- Bennett, M.K.; Calakos, N.; Scheller, R.H. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 1992, 257, 255–259. [Google Scholar] [CrossRef]
- Novick, P.; Schekman, R. Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1979, 76, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; De Craene, J.O.; Ferro-Novick, S.; Novick, P. Functional specialization within a vesicle tethering complex: Bypass of a subset of exocyst deletion mutants by Sec1p or Sec4p. J. Cell Biol. 2004, 167, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 1995, 131 Pt 2, 1747–1758. [Google Scholar] [CrossRef]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef]
- Bradke, F.; Fawcett, J.W.; Spira, M.E. Assembly of a new growth cone after axotomy: The precursor to axon regeneration. Nat. Rev. Neurosci. 2012, 13, 183–193. [Google Scholar] [CrossRef]
- Hur, E.M.; Saijilafu; Zhou, F.Q. Growing the growth cone: Remodeling the cytoskeleton to promote axon regeneration. Trends. Neurosci. 2012, 35, 164–174. [Google Scholar] [CrossRef]
- Chauhan, M.Z.; Arcuri, J.; Park, K.K.; Zafar, M.K.; Fatmi, R.; Hackam, A.S.; Yin, Y.; Benowitz, L.; Goldberg, J.L.; Samarah, M.; et al. Multi-omic analyses of growth cones at different developmental stages provides insight into pathways in adult Neuroregeneration. iScience 2020, 23, 100836. [Google Scholar] [CrossRef]
- Carman, G.M.; Han, G.S. Fat-regulating phosphatidic acid phosphatase: A review of its roles and regulation in lipid homeostasis. J. Lipid. Res. 2019, 60, 2–6. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Kawaguchi, R.; Zhang, Y.; Wang, Q.; Monavarfeshani, A.; Yang, Z.; Chen, B.; Shi, Z.; Meng, H.; et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature 2020, 587, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Poplawski, G.H.D.; Kawaguchi, R.; Van Niekerk, E.; Lu, P.; Mehta, N.; Canete, P.; Lie, R.; Dragatsis, I.; Meves, J.M.; Zheng, B.; et al. Injured adult neurons regress to an embryonic transcriptional growth state. Nature 2020, 581, 77–82. [Google Scholar] [CrossRef]
- Tedeschi, A.; Dupraz, S.; Laskowski, C.J.; Xue, J.; Ulas, T.; Beyer, M.; Schultze, J.L.; Bradke, F. The calcium channel subunit alpha2delta2 suppresses axon regeneration in the adult CNS. Neuron 2016, 92, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, I.; Mehra, V.; Wang, Z.; Califf, B.; Blackmore, M.G. Developmental chromatin restriction of pro-growth gene networks acts as an epigenetic barrier to axon regeneration in cortical neurons. Dev. Neurobiol. 2018, 78, 960–977. [Google Scholar] [CrossRef]
- Chang, K.C.; Bian, M.; Xia, X.; Madaan, A.; Sun, C.; Wang, Q.; Li, L.; Nahmou, M.; Noro, T.; Yokota, S.; et al. Posttranslational modification of Sox11 regulates RGC survival and axon regeneration. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Lindborg, J.A.; Tran, N.M.; Chenette, D.M.; DeLuca, K.; Foli, Y.; Kannan, R.; Sekine, Y.; Wang, X.; Wollan, M.; Kim, I.J.; et al. Optic nerve regeneration screen identifies multiple genes restricting adult neural repair. Cell Rep. 2021, 34, 108777. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Omura, K.; Tedeschi, A.; Riva, P.; Painter, M.W.; Rojas, L.; Martin, J.; Lisi, V.; Huebner, E.A.; Latremoliere, A.; et al. Robust axonal regeneration occurs in the injured CAST/Ei mouse CNS. Neuron 2015, 86, 1215–1227. [Google Scholar] [CrossRef]
- Puttagunta, R.; Tedeschi, A.; Soria, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat. Commun. 2014, 5, 3527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Reynolds, A.; Kirry, A.; Nienhaus, C.; Blackmore, M.G. Overexpression of Sox11 promotes corticospinal tract regeneration after spinal injury while interfering with functional recovery. J. Neurosci. 2015, 35, 3139–3145. [Google Scholar] [CrossRef]
- Lee, J.; Shin, J.E.; Lee, B.; Kim, H.; Jeon, Y.; Ahn, S.H.; Chi, S.W.; Cho, Y. The stem cell marker Prom1 promotes axon regeneration by down-regulating cholesterol synthesis via Smad signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 15955–15966. [Google Scholar] [CrossRef]
- Rosello-Busquets, C.; de la Oliva, N.; Martinez-Marmol, R.; Hernaiz-Llorens, M.; Pascual, M.; Muhaisen, A.; Navarro, X.; Del Valle, J.; Soriano, E. Cholesterol depletion regulates axonal growth and enhances central and peripheral nerve regeneration. Front. Cell Neurosci. 2019, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Shabanzadeh, A.P.; Charish, J.; Tassew, N.G.; Farhani, N.; Feng, J.; Qin, X.; Sugita, S.; Mothe, A.J.; Walchli, T.; Koeberle, P.D.; et al. Cholesterol synthesis inhibition promotes axonal regeneration in the injured central nervous system. Neurobiol. Dis. 2021, 150, 105259. [Google Scholar] [CrossRef]
- Chernoff, G.F. Shiverer: An autosomal recessive mutant mouse with myelin deficiency. J. Hered. 1981, 72, 128. [Google Scholar] [CrossRef] [PubMed]
- Mar, F.M.; da Silva, T.F.; Morgado, M.M.; Rodrigues, L.G.; Rodrigues, D.; Pereira, M.I.L.; Marques, A.; Sousa, V.F.; Coentro, J.; Sa-Miranda, C.; et al. Myelin lipids inhibit axon regeneration following spinal cord injury: A novel perspective for therapy. Mol. Neurobiol. 2016, 53, 1052–1064. [Google Scholar] [CrossRef]
- Hanani, M. Satellite glial cells in sensory ganglia: From form to function. Brain. Res. Brain. Res. Rev. 2005, 48, 457–476. [Google Scholar] [CrossRef]
- Hanani, M.; Spray, D.C. Emerging importance of satellite glia in nervous system function and dysfunction. Nat. Rev. Neurosci. 2020, 21, 485–498. [Google Scholar] [CrossRef]
- Meltzer, S.; Bagley, J.A.; Perez, G.L.; O’Brien, C.E.; DeVault, L.; Guo, Y.; Jan, L.Y.; Jan, Y.N. Phospholipid homeostasis regulates dendrite morphogenesis in drosophila sensory neurons. Cell Rep. 2017, 21, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Kim, J.B.; Graves, R.A.; Spiegelman, B.M. ADD1: A novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol. Cell Biol. 1993, 13, 4753–4759. [Google Scholar] [CrossRef]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Pavlidis, P.; Ramaswami, M.; Tanouye, M.A. The Drosophila easily shocked gene: A mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell 1994, 79, 23–33. [Google Scholar] [CrossRef]
- Rawson, R.B. The SREBP pathway--insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 2003, 4, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Dobrosotskaya, I.Y.; Seegmiller, A.C.; Brown, M.S.; Goldstein, J.L.; Rawson, R.B. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 2002, 296, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Wang, W.; Wessells, R.J.; Ocorr, K.; Bodmer, R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes. Dev. 2011, 25, 189–200. [Google Scholar] [CrossRef]
- Aggarwal, S.; Yurlova, L.; Simons, M. Central nervous system myelin: Structure, synthesis and assembly. Trends. Cell Biol. 2011, 21, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar] [CrossRef]
- Hille, B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978, 22, 283–294. [Google Scholar] [CrossRef]
- Waxman, S.G. Determinants of conduction velocity in myelinated nerve fibers. Muscle Nerve 1980, 3, 141–150. [Google Scholar] [CrossRef]
- Bei, F.; Lee, H.H.C.; Liu, X.; Gunner, G.; Jin, H.; Ma, L.; Wang, C.; Hou, L.; Hensch, T.K.; Frank, E.; et al. Restoration of visual function by enhancing conduction in regenerated axons. Cell 2016, 164, 219–232. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Ishiyama, N.; Hill, C.M.; Bates, I.R.; Libich, D.S.; Fares, C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 2004, 35, 503–542. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Stoffel, W. Proteolipid protein (PLP) of CNS myelin: Positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry 1992, 31, 12289–12296. [Google Scholar] [CrossRef] [PubMed]
- Jahn, O.; Siems, S.B.; Kusch, K.; Hesse, D.; Jung, R.B.; Liepold, T.; Uecker, M.; Sun, T.; Werner, H.B. The CNS Myelin Proteome: Deep Profile and Persistence After Post-mortem Delay. Front. Cell Neurosci. 2020, 14, 239. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.T.; Poduslo, S.E. Myelination in rat brain: Method of myelin isolation. J. Neurochem. 1973, 21, 749–757. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid. Res. 1965, 6, 537–544. [Google Scholar] [CrossRef]
- Norton, W.T.; Poduslo, S.E. Myelination in rat brain: Changes in myelin composition during brain maturation. J. Neurochem. 1973, 21, 759–773. [Google Scholar] [CrossRef]
- Saher, G.; Simons, M. Cholesterol and myelin biogenesis. Subcell. Biochem. 2010, 51, 489–508. [Google Scholar]
- Malheiro, A.R.; Correia, B.; Ferreira da Silva, T.; Bessa-Neto, D.; Van Veldhoven, P.P.; Brites, P. Leukodystrophy caused by plasmalogen deficiency rescued by glyceryl 1-myristyl ether treatment. Brain. Pathol. 2019, 29, 622–639. [Google Scholar] [CrossRef] [PubMed]
- Huttner, W.B.; Zimmerberg, J. Implications of lipid microdomains for membrane curvature, budding and fission. Curr. Opin. Cell Biol. 2001, 13, 478–484. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef]
- Smith, M.E. The turnover of myelin in the adult rat. Biochim. Biophys. Acta 1968, 164, 285–293. [Google Scholar] [CrossRef]
- Espenshade, P.J.; Hughes, A.L. Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 2007, 41, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef]
- Miller, L.G., Jr.; Young, J.A.; Ray, S.K.; Wang, G.; Purohit, S.; Banik, N.L.; Dasgupta, S. Sphingosine Toxicity in EAE and MS: Evidence for Ceramide Generation via Serine-Palmitoyltransferase Activation. Neurochem. Res. 2017, 42, 2755–2768. [Google Scholar] [CrossRef]
- Dimas, P.; Montani, L.; Pereira, J.A.; Moreno, D.; Trotzmuller, M.; Gerber, J.; Semenkovich, C.F.; Kofeler, H.C.; Suter, U. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Klopfleisch, S.; Merkler, D.; Schmitz, M.; Kloppner, S.; Schedensack, M.; Jeserich, G.; Althaus, H.H.; Bruck, W. Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J. Neurosci. 2008, 28, 13609–13614. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Zehntner, S.P.; Kuhlmann, T.; Ludwin, S.K.; Owens, T.; Kennedy, T.E.; Bedell, B.J.; Antel, J.P. Statin therapy inhibits remyelination in the central nervous system. Am. J. Pathol. 2009, 174, 1880–1890. [Google Scholar] [CrossRef]
- Triplet, E.M.; Scarisbrick, I.A. Statin use is associated with reduced motor recovery after spinal cord injury. Spinal Cord Ser. Cases 2021, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.G.; Hilton, B.J.; Tetzlaff, W.; Zheng, B. Evidence for an age-dependent decline in axon regeneration in the adult mammalian central nervous system. Cell Rep. 2016, 15, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.C.; Geoffroy, C.G. The influence of neuron-extrinsic factors and aging on injury progression and axonal repair in the central nervous system. Front. Cell Dev. Biol. 2020, 8, 190. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lutjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Cartoni, R.; Norsworthy, M.W.; Bei, F.; Wang, C.; Li, S.; Zhang, Y.; Gabel, C.V.; Schwarz, T.L.; He, Z. The mammalian-specific protein armcx1 regulates mitochondrial transport during axon regeneration. Neuron 2016, 92, 1294–1307. [Google Scholar] [CrossRef]
- Gallo, G. The bioenergetics of neuronal morphogenesis and regeneration: Frontiers beyond the mitochondrion. Dev. Neurobiol. 2020, 80, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Xie, Y.; Ordaz, J.D.; Huh, A.J.; Huang, N.; Wu, W.; Liu, N.; Chamberlain, K.A.; Sheng, Z.H.; Xu, X.M. Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab. 2020, 31, 623–641.e8. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Baig, H.S.; Hammarlund, M. Mitochondria localize to injured axons to support regeneration. Neuron 2016, 92, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yu, P.; Lin, M.Y.; Sun, T.; Chen, Y.; Sheng, Z.H. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol. 2016, 214, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Wojtczak, L.; Schonfeld, P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta 1993, 1183, 41–57. [Google Scholar] [CrossRef]
- Yang, S.Y.; He, X.Y.; Schulz, H. Fatty acid oxidation in rat brain is limited by the low activity of 3-ketoacyl-coenzyme A thiolase. J. Biol. Chem. 1987, 262, 13027–13032. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Jimenez-Riani, M.; Diaz-Amarilla, P.; Isasi, E.; Casanova, G.; Barbeito, L.; Olivera-Bravo, S. Ultrastructural features of aberrant glial cells isolated from the spinal cord of paralytic rats expressing the amyotrophic lateral sclerosis-linked SOD1G93A mutation. Cell Tissue Res. 2017, 370, 391–401. [Google Scholar] [CrossRef]
- Gallo, G. The cytoskeletal and signaling mechanisms of axon collateral branching. Dev. Neurobiol. 2011, 71, 201–220. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Deinhardt, K.; Miyoshi, G.; Bennett, A.M.; Chao, M.V. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat. Neurosci. 2010, 13, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Kiyoshi, C.; Tedeschi, A. Axon growth and synaptic function: A balancing act for axonal regeneration and neuronal circuit formation in CNS trauma and disease. Dev. Neurobiol. 2020, 80, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.P.; Lilley, B.N.; Pan, Y.A.; Plummer, L.J.; Powell, A.W.; Raines, A.N.; Sanes, J.R.; Polleux, F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 2007, 129, 549–563. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Shelly, M.; Cancedda, L.; Heilshorn, S.; Sumbre, G.; Poo, M.M. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 2007, 129, 565–577. [Google Scholar] [CrossRef]
- Courchet, J.; Lewis, T.L., Jr.; Lee, S.; Courchet, V.; Liou, D.Y.; Aizawa, S.; Polleux, F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 2013, 153, 1510–1525. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Kong, G.; Zhou, L.; Serger, E.; Palmisano, I.; De Virgiliis, F.; Hutson, T.H.; McLachlan, E.; Freiwald, A.; La Montanara, P.; Shkura, K.; et al. AMPK controls the axonal regenerative ability of dorsal root ganglia sensory neurons after spinal cord injury. Nat. Metab. 2020, 2, 918–933. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Ohtake, Y.; Sami, A.; Jiang, X.; Horiuchi, M.; Slattery, K.; Ma, L.; Smith, G.M.; Selzer, M.E.; Muramatsu, S.I.; Li, S. Promoting axon regeneration in adult cns by targeting liver kinase B1. Mol. Ther. 2019, 27, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Sheng, Z.H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118 Pt 23, 5411–5419. [Google Scholar] [CrossRef]
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125 Pt 9, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold. Spring Harb. Perspect Biol. 2013, 5. [Google Scholar] [CrossRef]
- Baas, P.W. Microtubule transport in the axon. Int. Rev. Cytol. 2002, 212, 41–62. [Google Scholar]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104 Pt 3, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.G.; Awal, M.R.; Gabel, C.V. O-GlcNAc signaling orchestrates the regenerative response to neuronal injury in caenorhabditis elegans. Cell Rep. 2018, 24, 1931–1938.e3. [Google Scholar] [CrossRef]
- Beirowski, B.; Babetto, E.; Golden, J.P.; Chen, Y.J.; Yang, K.; Gross, R.W.; Patti, G.J.; Milbrandt, J. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat. Neurosci. 2014, 17, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 2020, 23, 1215–1228. [Google Scholar] [CrossRef]
- Li, F.; Sami, A.; Noristani, H.N.; Slattery, K.; Qiu, J.; Groves, T.; Wang, S.; Veerasammy, K.; Chen, Y.X.; Morales, J.; et al. Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. 2020, 32, 767–785.e7. [Google Scholar] [CrossRef]
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.P.; Forss, I.; Mandrup, S.; MacDougald, O.A. SnapShot: Niche determines adipocyte character I. Cell Metab. 2018, 27, 266. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. The adipose organ: Morphological perspectives of adipose tissues. Proc. Nutr. Soc. 2001, 60, 319–328. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid. Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.S.; Jessen, N.; Jorgensen, J.O.; Moller, N.; Lund, S. Dissecting adipose tissue lipolysis: Molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 2014, 52, R199–R222. [Google Scholar] [CrossRef]
- Yu, Y.H.; Ginsberg, H.N. Adipocyte signaling and lipid homeostasis: Sequelae of insulin-resistant adipose tissue. Circ. Res. 2005, 96, 1042–1052. [Google Scholar] [CrossRef]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. 1), S130–S135. [Google Scholar] [CrossRef]
- Chondronikola, M.; Volpi, E.; Borsheim, E.; Porter, C.; Saraf, M.K.; Annamalai, P.; Yfanti, C.; Chao, T.; Wong, D.; Shinoda, K.; et al. Brown adipose tissue activation is linked to distinct systemic effects on lipid metabolism in humans. Cell Metab. 2016, 23, 1200–1206. [Google Scholar] [CrossRef]
- Francois, M.; Qualls-Creekmore, E.; Berthoud, H.R.; Munzberg, H.; Yu, S. Genetics-based manipulation of adipose tissue sympathetic innervation. Physiol. Behav. 2018, 190, 21–27. [Google Scholar] [CrossRef]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef]
- Wolf, Y.; Boura-Halfon, S.; Cortese, N.; Haimon, Z.; Sar Shalom, H.; Kuperman, Y.; Kalchenko, V.; Brandis, A.; David, E.; Segal-Hayoun, Y.; et al. Brown-adipose-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat. Immunol. 2017, 18, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Eric Nyam, T.T.; Ho, C.H.; Chio, C.C.; Lim, S.W.; Wang, J.J.; Chang, C.H.; Kuo, J.R.; Wang, C.C. Traumatic brain injury increases the risk of major adverse cardiovascular and cerebrovascular events: A 13-year, population-based study. World Neurosurg. 2019, 122, e740–e753. [Google Scholar] [CrossRef]
- Karelina, K.; Sarac, B.; Freeman, L.M.; Gaier, K.R.; Weil, Z.M. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 2016, 43, 1034–1043. [Google Scholar] [CrossRef]
- Silveira, S.L.; Winter, L.L.; Clark, R.; Ledoux, T.; Robinson-Whelen, S. Baseline dietary intake of individuals with spinal cord injury who are overweight or obese. J. Acad. Nutr. Diet. 2019, 119, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Bartness, T.J.; Liu, Y.; Shrestha, Y.B.; Ryu, V. Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocrinol. 2014, 35, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Dogiel, A. Die sensiblen nervenendigungen im herzen und in den blutgefässen der säugethiere. Archiv. Für Mikroskopische Anatomie 1898, 52, 44–70. [Google Scholar] [CrossRef]
- Correll, J.W. Adipose tissue: Ability to respond to nerve stimulation in vitro. Science 1963, 140, 387–388. [Google Scholar] [CrossRef]
- Blaszkiewicz, M.; Willows, J.W.; Dubois, A.L.; Waible, S.; DiBello, K.; Lyons, L.L.; Johnson, C.P.; Paradie, E.; Banks, N.; Motyl, K.; et al. Neuropathy and neural plasticity in the subcutaneous white adipose depot. PLoS ONE 2019, 14, e0221766. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.T.; Bartness, T.J. Sympathetic but not sensory denervation stimulates white adipocyte proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1630–R1637. [Google Scholar] [CrossRef] [PubMed]
- Slavin, B.G.; Ballard, K.W. Morphological studies on the adrenergic innervation of white adipose tissue. Anat. Rec. 1978, 191, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Huesing, C.; Qualls-Creekmore, E.; Lee, N.; Francois, M.; Torres, H.; Zhang, R.; Burk, D.H.; Yu, S.; Morrison, C.D.; Berthoud, H.R.; et al. Sympathetic innervation of inguinal white adipose tissue in the mouse. J. Comp. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, N.M.; Stefanidis, A.; Oldfield, B.J. Characterization of the central neural projections to brown, white, and beige adipose tissue. FASEB J. 2017, 31, 4879–4890. [Google Scholar] [CrossRef] [PubMed]
- Song, C.K.; Bartness, T.J. CNS sympathetic outflow neurons to white fat that express MEL receptors may mediate seasonal adiposity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R666–R672. [Google Scholar] [CrossRef] [PubMed]
- Fishman, R.B.; Dark, J. Sensory innervation of white adipose tissue. Am. J. Physiol. 1987, 253 Pt 2, R942–R944. [Google Scholar] [CrossRef]
- Song, C.K.; Schwartz, G.J.; Bartness, T.J. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R501–R511. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Lee, S.; Elmquist, J.K.; Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 2018, 19, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Schwartz, G.J.; Nguyen, N.L.; Mendez, J.M.; Ryu, V.; Bartness, T.J. Leptin-sensitive sensory nerves innervate white fat. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1338–E1347. [Google Scholar] [CrossRef]
- Bagchi, D.P.; MacDougald, O.A. Identification and dissection of diverse mouse adipose depots. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Foster, D.O.; Depocas, F.; Zaror-Behrens, G. Unilaterality of the sympathetic innervation of each pad of rat interscapular brown adipose tissue. Can. J. Physiol. Pharmacol. 1982, 60, 107–113. [Google Scholar] [CrossRef]
- Francois, M.; Torres, H.; Huesing, C.; Zhang, R.; Saurage, C.; Lee, N.; Qualls-Creekmore, E.; Yu, S.; Morrison, C.D.; Burk, D.; et al. Sympathetic innervation of the interscapular brown adipose tissue in mouse. Ann. N. Y. Acad. Sci. 2019, 1454, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Nogueiras, R.; Dieguez, C.; Rahmouni, K.; Lopez, M. Traveling from the hypothalamus to the adipose tissue: The thermogenic pathway. Redox Biol. 2017, 12, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Harlan, S.M.; Morgan, D.A.; Agassandian, K.; Guo, D.F.; Cassell, M.D.; Sigmund, C.D.; Mark, A.L.; Rahmouni, K. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res. 2011, 108, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Henriques, F.; Bedard, A.H.; Czech, M.P. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Bauman, W.A.; Spungen, A.M.; Zhong, Y.G.; Mobbs, C.V. Plasma leptin is directly related to body adiposity in subjects with spinal cord injury. Horm. Metab. Res. 1996, 28, 732–736. [Google Scholar] [CrossRef]
- Latifi, S.; Koushki, D.; Norouzi Javidan, A.; Matin, M.; Sabour, H. Changes of leptin concentration in plasma in patients with spinal cord injury: A meta-analysis. Spinal Cord 2013, 51, 728–731. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Pan, Z.; Zeng, Y. Serum leptin, bone mineral density and the healing of long bone fractures in men with spinal cord injury. Bosn. J. Basic Med. Sci. 2015, 15, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, X.; Zhang, H.; Yuan, J.; Ding, H.; Wei, Y. Elevated leptin expression in rat model of traumatic spinal cord injury and femoral fracture. J. Spinal Cord Med. 2011, 34, 501–509. [Google Scholar] [CrossRef]
- Buchholz, A.C.; Pencharz, P.B. Energy expenditure in chronic spinal cord injury. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 635–639. [Google Scholar] [CrossRef]
- Commins, S.P.; Watson, P.M.; Levin, N.; Beiler, R.J.; Gettys, T.W. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different beta-adrenoceptor subtypes. J. Biol. Chem. 2000, 275, 33059–33067. [Google Scholar] [CrossRef] [PubMed]
- Siegrist-Kaiser, C.A.; Pauli, V.; Juge-Aubry, C.E.; Boss, O.; Pernin, A.; Chin, W.W.; Cusin, I.; Rohner-Jeanrenaud, F.; Burger, A.G.; Zapf, J.; et al. Direct effects of leptin on brown and white adipose tissue. J. Clin. Investig. 1997, 100, 2858–2864. [Google Scholar] [CrossRef]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Westgren, N.; Levi, R. Quality of life and traumatic spinal cord injury. Arch. Phys. Med. Rehabil. 1998, 79, 1433–1439. [Google Scholar] [CrossRef]
- Claydon, V.E.; Steeves, J.D.; Krassioukov, A. Orthostatic hypotension following spinal cord injury: Understanding clinical pathophysiology. Spinal Cord 2006, 44, 341–351. [Google Scholar] [CrossRef]
- Furlan, J.C.; Fehlings, M.G. Cardiovascular complications after acute spinal cord injury: Pathophysiology, diagnosis, and management. Neurosurg. Focus 2008, 25, E13. [Google Scholar] [CrossRef]
- Krassioukov, A.V.; Bunge, R.P.; Pucket, W.R.; Bygrave, M.A. The changes in human spinal sympathetic preganglionic neurons after spinal cord injury. Spinal Cord 1999, 37, 6–13. [Google Scholar] [CrossRef]
- Gorgey, A.S.; Gater, D.R., Jr. Prevalence of obesity after spinal cord injury. Top. Spinal Cord Inj. Rehabil. 2007, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spungen, A.M.; Adkins, R.H.; Stewart, C.A.; Wang, J.; Pierson, R.N., Jr.; Waters, R.L.; Bauman, W.A. Factors influencing body composition in persons with spinal cord injury: A cross-sectional study. J. Appl. Physiol. 2003, 95, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Gorgey, A.S.; Wells, K.M.; Austin, T.L. Adiposity and spinal cord injury. World J. Orthop. 2015, 6, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Legge, M.; Goulding, A. Healthy body mass index values often underestimate body fat in men with spinal cord injury. Arch. Phys. Med. Rehabil. 2003, 84, 1068–1071. [Google Scholar] [CrossRef]
- Laughton, G.E.; Buchholz, A.C.; Martin Ginis, K.A.; Goy, R.E.; Group, S.S.R. Lowering body mass index cutoffs better identifies obese persons with spinal cord injury. Spinal Cord 2009, 47, 757–762. [Google Scholar] [CrossRef]
- Yarar-Fisher, C.; Chen, Y.; Jackson, A.B.; Hunter, G.R. Body mass index underestimates adiposity in women with spinal cord injury. Obesity 2013, 21, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Gorgey, A.S.; Mather, K.J.; Gater, D.R. Central adiposity associations to carbohydrate and lipid metabolism in individuals with complete motor spinal cord injury. Metabolism 2011, 60, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Modlesky, C.M.; Bickel, C.S.; Slade, J.M.; Meyer, R.A.; Cureton, K.J.; Dudley, G.A. Assessment of skeletal muscle mass in men with spinal cord injury using dual-energy X-ray absorptiometry and magnetic resonance imaging. J. Appl. Physiol. 2004, 96, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Gorgey, A.S.; Mather, K.J.; Poarch, H.J.; Gater, D.R. Influence of motor complete spinal cord injury on visceral and subcutaneous adipose tissue measured by multi-axial magnetic resonance imaging. J. Spinal Cord Med. 2011, 34, 99–109. [Google Scholar] [CrossRef]
- Benova, A.; Tencerova, M. Obesity-induced changes in bone marrow homeostasis. Front. Endocrinol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Pierce, J.L.; Begun, D.L.; Westendorf, J.J.; McGee-Lawrence, M.E. Defining osteoblast and adipocyte lineages in the bone marrow. Bone 2019, 118, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Boroumand, P.; Klip, A. Bone marrow adipose cells—Cellular interactions and changes with obesity. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Fink, L.N.; Costford, S.R.; Lee, Y.S.; Jensen, T.E.; Bilan, P.J.; Oberbach, A.; Bluher, M.; Olefsky, J.M.; Sams, A.; Klip, A. Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity 2014, 22, 747–757. [Google Scholar] [CrossRef]
- Singer, K.; DelProposto, J.; Morris, D.L.; Zamarron, B.; Mergian, T.; Maley, N.; Cho, K.W.; Geletka, L.; Subbaiah, P.; Muir, L.; et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 2014, 3, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Gorgey, A.S.; Poarch, H.J.; Adler, R.A.; Khalil, R.E.; Gater, D.R. Femoral bone marrow adiposity and cortical bone cross-sectional areas in men with motor complete spinal cord injury. PM R 2013, 5, 939–948. [Google Scholar] [CrossRef] [PubMed]
- de Groot, S.; Adriaansen, J.J.; Tepper, M.; Snoek, G.J.; van der Woude, L.H.; Post, M.W. Metabolic syndrome in people with a long-standing spinal cord injury: Associations with physical activity and capacity. Appl. Physiol. Nutr. Metab. 2016, 41, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Sauerbeck, A.D.; Laws, J.L.; Bandaru, V.V.; Popovich, P.G.; Haughey, N.J.; McTigue, D.M. Spinal cord injury causes chronic liver pathology in rats. J. Neurotrauma 2015, 32, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [PubMed]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ojha, S.; Rai, P.; Joshi, A.; Kamat, S.S.; Mallik, R. Insulin activates intracellular transport of lipid droplets to release triglycerides from the liver. J. Cell Biol. 2019, 218, 3697–3713. [Google Scholar] [CrossRef]
- La Fountaine, M.F.; Cirnigliaro, C.M.; Emmons, R.R.; Kirshblum, S.C.; Galea, M.; Spungen, A.M.; Bauman, W.A. Lipoprotein heterogeneity in persons with Spinal Cord Injury: A model of prolonged sitting and restricted physical activity. Lipids Health Dis. 2015, 14, 81. [Google Scholar] [CrossRef][Green Version]
- Rankin, K.C.; O’Brien, L.C.; Segal, L.; Khan, M.R.; Gorgey, A.S. Liver Adiposity and Metabolic Profile in Individuals with Chronic Spinal Cord Injury. Biomed. Res. Int. 2017, 2017, 1364818. [Google Scholar] [CrossRef]
- van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Goodus, M.T.; McTigue, D.M. Hepatic dysfunction after spinal cord injury: A vicious cycle of central and peripheral pathology? Exp. Neurol. 2020, 325, 113160. [Google Scholar] [CrossRef]
- Popovich, P.G.; Wei, P.; Stokes, B.T. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J. Comp. Neurol. 1997, 377, 443–464. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martin-Romero, C.; Perez-Perez, A.; Gonzalez-Yanes, C.; Sanchez-Margalet, V. Role of leptin in the activation of immune cells. Mediators. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Lago, R.; Gomez, R.; Dieguez, C.; Lago, F.; Gomez-Reino, J.; Gualillo, O. Towards a pro-inflammatory and immunomodulatory emerging role of leptin. Rheumatology 2006, 45, 944–950. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef]
- Kim, J.K.; Wi, J.K.; Youn, J.H. Plasma free fatty acids decrease insulin-stimulated skeletal muscle glucose uptake by suppressing glycolysis in conscious rats. Diabetes 1996, 45, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Solomon, S.S.; Jallepalli, P.; Heckemeyer, C.; Finnern, J.; Powers, A. Glucose intolerance due to insulin resistance in patients with spinal cord injuries. Diabetes 1980, 29, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Bauman, W.A.; Spungen, A.M. Disorders of carbohydrate and lipid metabolism in veterans with paraplegia or quadriplegia: A model of premature aging. Metabolism 1994, 43, 749–756. [Google Scholar] [CrossRef]
- Libin, A.; Tinsley, E.A.; Nash, M.S.; Mendez, A.J.; Burns, P.; Elrod, M.; Hamm, L.F.; Groah, S.L. Cardiometabolic risk clustering in spinal cord injury: Results of exploratory factor analysis. Top. Spinal Cord Inj. Rehabil. 2013, 19, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Selassie, A.; Snipe, L.; Focht, K.L.; Welldaregay, W. Baseline prevalence of heart diseases, hypertension, diabetes, and obesity in persons with acute traumatic spinal cord injury: Potential threats in the recovery trajectory. Top. Spinal Cord Inj. Rehabil. 2013, 19, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Soden, R.J.; Walsh, J.; Middleton, J.W.; Craven, M.L.; Rutkowski, S.B.; Yeo, J.D. Causes of death after spinal cord injury. Spinal Cord 2000, 38, 604–610. [Google Scholar] [CrossRef]
- Flank, P.; Fahlstrom, M.; Bostrom, C.; Lewis, J.E.; Levi, R.; Wahman, K. Self-reported physical activity and risk markers for cardiovascular disease after spinal cord injury. J. Rehabil. Med. 2014, 46, 886–890. [Google Scholar] [CrossRef]
- Angin, Y.; Steinbusch, L.K.; Simons, P.J.; Greulich, S.; Hoebers, N.T.; Douma, K.; van Zandvoort, M.A.; Coumans, W.A.; Wijnen, W.; Diamant, M.; et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem. J. 2012, 448, 43–53. [Google Scholar] [CrossRef]
- Greenwalt, D.E.; Scheck, S.H.; Rhinehart-Jones, T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J. Clin. Investig. 1995, 96, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef]
- Petrova, V.; Nieuwenhuis, B.; Fawcett, J.W.; Eva, R. Axonal organelles as molecular platforms for axon growth and regeneration after injury. Int. J. Mol. Sci. 2021, 22, 1798. [Google Scholar] [CrossRef] [PubMed]
- Passmore, J.B.; Nijenhuis, W.; Kapitein, L.C. From observing to controlling: Inducible control of organelle dynamics and interactions. Curr. Opin. Cell Biol. 2021, 71, 69–76. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, D.; Tedeschi, A. The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease. Cells 2021, 10, 1078. https://doi.org/10.3390/cells10051078
Roy D, Tedeschi A. The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease. Cells. 2021; 10(5):1078. https://doi.org/10.3390/cells10051078
Chicago/Turabian StyleRoy, Debasish, and Andrea Tedeschi. 2021. "The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease" Cells 10, no. 5: 1078. https://doi.org/10.3390/cells10051078
APA StyleRoy, D., & Tedeschi, A. (2021). The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease. Cells, 10(5), 1078. https://doi.org/10.3390/cells10051078