
Age-Dependent Microglial Response to Systemic Infection



{kind=link}
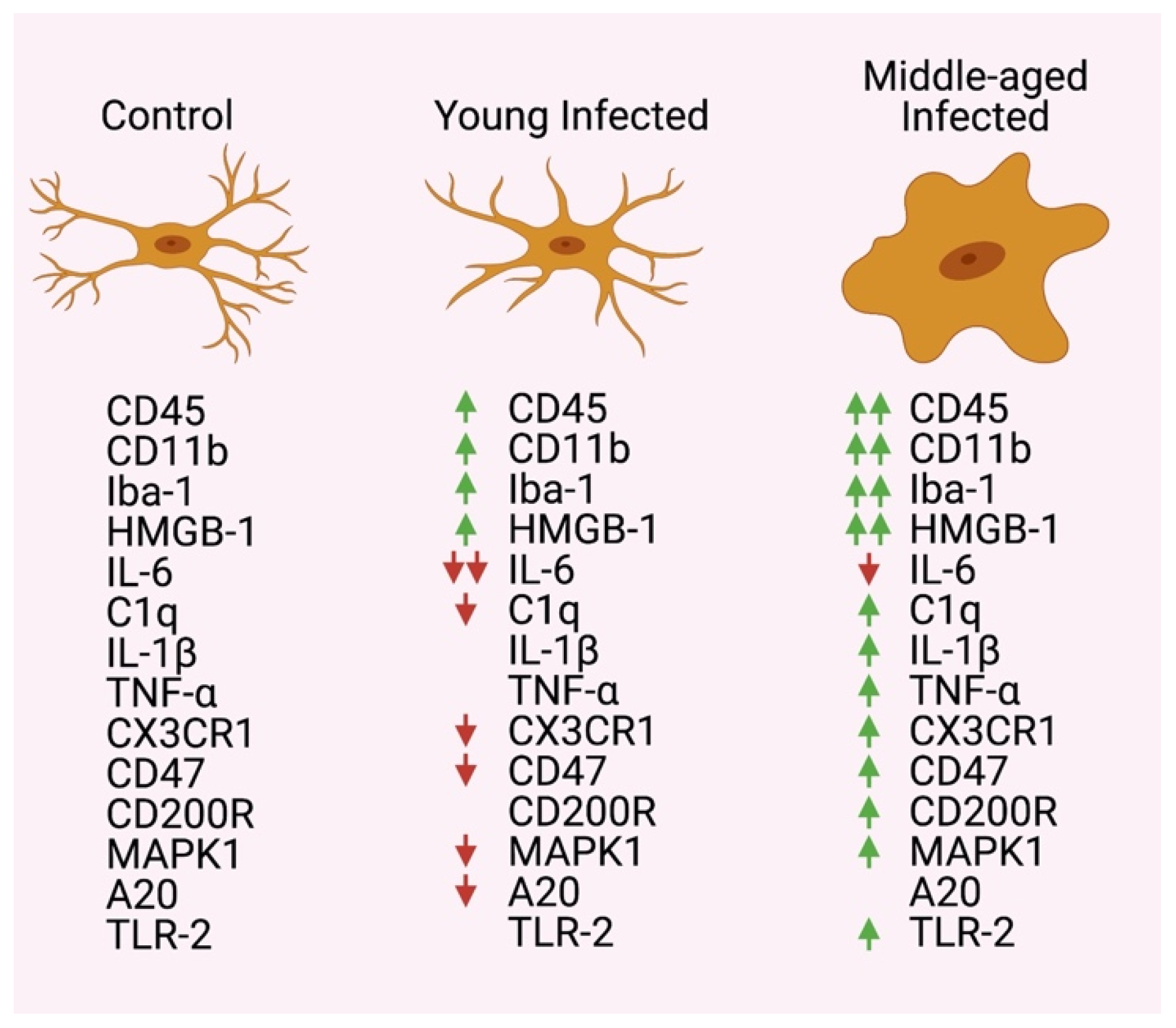
Abstract
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Kipnis, J.; Gadani, S.P.; Derecki, N.C. Pro-cognitive properties of T cells. Nat. Rev. Immunol. 2012, 12, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Dulken, B.W.; Buckley, M.T.; Negredo, P.N.; Saligrama, N.; Cayrol, R.; Leeman, D.S.; George, B.M.; Boutet, S.C.; Hebestreit, K.; Pluvinage, J.V.; et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 2019, 571, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, J.P.D.R.; Dietrich, W.D.; Keane, R.W. Activation and Regulation of Cellular Inflammasomes: Gaps in Our Knowledge for Central Nervous System Injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, J.P.D.R.; Dietrich, W.D.; Keane, R.W. Therapeutics targeting the inflammasome after central nervous system injury. Transl. Res. 2016, 167, 35–45. [Google Scholar] [CrossRef]
- Vaccari, J.P.D.R.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A Molecular Platform in Neurons Regulates Inflammation after Spinal Cord Injury. J. Neurosci. 2008, 28, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, J.P.D.R.; Marcillo, A.; Nonner, D.; Dietrich, W.D.; Keane, R.W. Neuroprotective effects of bone morphogenetic protein 7 (BMP7) treatment after spinal cord injury. Neurosci. Lett. 2009, 465, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, 4. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, G.; Vaccari, J.P.D.R.; Perez, E.R.; Sanchez-Molano, J.; Furones-Alonso, O.; Bramlett, H.M.; Dietrich, W.D. Alterations in Blood-Brain Barrier Permeability to Large and Small Molecules and Leukocyte Accumulation after Traumatic Brain Injury: Effects of Post-Traumatic Hypothermia. J. Neurotrauma 2009, 26, 1123–1134. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Age-Associated Changes in the Immune System and Blood–Brain Barrier Functions. Int. J. Mol. Sci. 2019, 20, 1632. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.; Vaccari, J.P.D.R.; Dietrich, W.D.; Keane, R.W. Neural-respiratory inflammasome axis in traumatic brain injury. Exp. Neurol. 2020, 323, 113080. [Google Scholar] [CrossRef]
- Kerr, M.N.; Vaccari, J.P.D.R.; Weaver, M.C.; Dietrich, W.D.; Ahmed, T.; Keane, R.W. Enoxaparin Attenuates Acute Lung Injury and Inflammasome Activation after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 646–654. [Google Scholar] [CrossRef]
- O’Connor, G.; Jeffrey, E.; Madorma, D.; Marcillo, A.; Abreu, M.T.; Deo, S.K.; Dietrich, W.D.; Daunert, S. Investigation of Microbiota Alterations and Intestinal Inflammation Post-Spinal Cord Injury in Rat Model. J. Neurotrauma 2018, 35, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, J.C.D.R.; Dietrich, W.D.; Keane, R.W. The Inflammasome in Times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Askarova, S.; Umbayev, B.; Masoud, A.-R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The Links Between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell. Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef]
- Baldini, F.; on Behalf of the NCER-PD Consortium; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 1–21. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Rogers, J. Inflammation as a pathogenic mechanism in Alzheimer’s disease. Arzneimittelforschung 1995, 45, 439–442. [Google Scholar]
- Raval, A.P.; Martinez, C.C.; Mejias, N.H.; Vaccari, J.P.D.R. Sexual dimorphism in inflammasome-containing extracellular vesicles and the regulation of innate immunity in the brain of reproductive senescent females. Neurochem. Int. 2019, 127, 29–37. [Google Scholar] [CrossRef]
- Hoogland, I.; Westhoff, D.; Engelen-Lee, J.-Y.; Seron, M.V.; Houben-Weerts, J.; van Westerloo, D.; van der Poll, T.; van Gool, W.; van de Beek, D. Aging and Microglial Response following Systemic Stimulation with Escherichia coli in Mice. Cells 2021, 10, 279. [Google Scholar] [CrossRef]
- D’Avila, J.C.; Siqueira, L.D.; Mazeraud, A.; Azevedo, E.P.; Foguel, D.; Castro-Faria-Neto, H.C.; Sharshar, T.; Chrétien, F.; Bozza, F.A. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflamm. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 2018, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.J.; Nobre, V.; Barichello, T.; Teixeira, A.L. Post-sepsis cognitive impairment and associated risk factors: A systematic review. Aust. Crit. Care 2018, 31, 242–253. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2012, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef]
- Wynne, A.M.; Henry, C.J.; Huang, Y.; Cleland, A.; Godbout, J.P. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 2010, 24, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.M.; Houbolt, C.; Van Westerloo, D.J.; Van Gool, W.A.; Van De Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Westhoff, D.; Engelen-Lee, J.-Y.; Melief, J.; Serón, M.V.; Houben-Weerts, J.H.M.P.; Huitinga, I.; Van Westerloo, D.J.; Van Der Poll, T.; Van Gool, W.A.; et al. Microglial Activation After Systemic Stimulation With Lipopolysaccharide and Escherichia coli. Front. Cell. Neurosci. 2018, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; Vaccari, J.P.D.R. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Spychala, M.S.; Honarpisheh, P.; McCullough, L.D. Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J. Neurosci. Res. 2017, 95, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Skar, G.L.; Synhorst, D.; Beaver, M.; Snowden, J.N. CSF inflammatory markers differ in gram-positive versus gram-negative shunt infections. J. Neuroinflamm. 2019, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Achek, A.; Yesudhas, D.; Choi, S. Toll-like receptors: Promising therapeutic targets for inflammatory diseases. Arch. Pharmacal Res. 2016, 39, 1032–1049. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Sepsis roadmap: What we know, what we learned, and where we are going. Clin. Immunol. 2020, 210, 108264. [Google Scholar] [CrossRef]
- Fu, Q.; Wu, J.; Zhou, X.-Y.; Ji, M.-H.; Mao, Q.-H.; Li, Q.; Zong, M.-M.; Zhou, Z.-Q.; Yang, J.-J. NLRP3/Caspase-1 Pathway-Induced Pyroptosis Mediated Cognitive Deficits in a Mouse Model of Sepsis-Associated Encephalopathy. Inflammation 2019, 42, 306–318. [Google Scholar] [CrossRef]
- Liu, X.; Quan, N. Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front. Neurol. 2018, 9, 8. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cyr, B.; de Rivero Vaccari, J.P. Age-Dependent Microglial Response to Systemic Infection. Cells 2021, 10, 1037. https://doi.org/10.3390/cells10051037
Cyr B, de Rivero Vaccari JP. Age-Dependent Microglial Response to Systemic Infection. Cells. 2021; 10(5):1037. https://doi.org/10.3390/cells10051037
Chicago/Turabian StyleCyr, Brianna, and Juan Pablo de Rivero Vaccari. 2021. "Age-Dependent Microglial Response to Systemic Infection" Cells 10, no. 5: 1037. https://doi.org/10.3390/cells10051037
APA StyleCyr, B., & de Rivero Vaccari, J. P. (2021). Age-Dependent Microglial Response to Systemic Infection. Cells, 10(5), 1037. https://doi.org/10.3390/cells10051037