
Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. The Human Sec61 Translocon
2.1. Entry of Precursor Polypeptides into the ER
2.2. Targeting of Precursor Polypeptides to the ER
2.3. Structure of the Sec61 Complex

2.4. Dynamics of the Sec61 Complex
2.5. Auxiliary Factors of the Sec61 Complex
2.5.1. Allosteric Effectors of the Sec61 Channel for Channel Opening
2.5.2. Additional Auxiliary Factors of the Sec61 Complex
2.5.3. Structural Considerations
3. Gating of the Sec61 Channel by BiP
3.1. Structure and Dynamics of BiP
3.2. BiP and Sec62 as Allosteric Effectors of the Sec61 Complex for Channel Closing
4. Sec61-Channelopathies
4.1. Bacterial and Fungal Toxins That Target the Sec61 Channel
4.2. Mutated Variants of the Sec61 Channel
4.2.1. Sec61α 1 p.V67G and p.T185A in ADTKD
4.2.2. Sec61α 1 p.Q92R and p.V67G in ADSCN
4.2.3. Sec61α 1 p.V85D and p.E381* in CVID
4.2.4. Sec61α1 p.Y344H in Mice in Diabetes mellitus
5. Diseases That Are Related to Allosteric Effectors of the Sec61 Channel
5.1. Loss of TRAP Function in Congenital Disorders of Glycosylation (CDG)
5.2. Sec63 and Sec61β in Autosomal Dominant Polycystic Liver Disease (ADPCLD)
5.3. BiP and Its Co-Chaperones and NEFs in Diabetes and Neurological Disorders
5.3.1. BiP Deficiency in Hemolytic Uremic Syndrome (HUS)
5.3.2. Marinesco–Sjögren Syndrome (MSS)
5.3.3. ERj3 in Polycystic Kidney Disease (PKD)
6. Tumor Diseases That Are Related to the Sec61 Channel
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Autosomal dominant |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| BiP | Immunoglobulin heavy chain binding protein |
| BS | Binding site |
| CaM | Calmodulin |
| CDG | Congenital disorder of glycosylation |
| CET | Cryo-electron tomography |
| CVID | Common variable immunodeficiency |
| DNAJ | DnaJ homolog |
| EM | Electron microscopy |
| EMC | ER membrane complex |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated (protein) degradation |
| ERj | ER (resident) J-domain (protein) |
| GBM | Glioblastoma multiforme |
| GET | Guided entry of tail-anchored proteins |
| GPI | Glycosylphosphatidylinositol |
| GRP | Glucose-regulated protein |
| GTP | Guanosine triphosphate |
| HSP | Heat shock protein |
| JDP | J-domain protein |
| MSS | Marinesco–Sjögren syndrome |
| NBD | Nucleotide-binding domain |
| NEF | Nucleotide exchange factor |
| OST | Oligosaccharyltransferase |
| PAD | Primary antibody deficiency |
| PC | Polycystin |
| PEX | Peroxisome (protein) |
| PKD | Polycystic kidney disease |
| PLD | Polycystic liver disease |
| RAMP | Ribosome-associated membrane protein |
| RCC | Renal cell carcinoma |
| RNC | Ribosome-nascent chain complex |
| SBD | Substrate-binding domain |
| SCND | Severe congenital neutropenia |
| SEC | (Protein involved in) secretion |
| SERCA | Sarcoplasmic/endoplasmic reticulum ATPase |
| SND | SRP-independent |
| SP | Signal peptide |
| SR | SRP receptor |
| SRP | Signal recognition particle |
| SSR | Signal sequence receptor |
| TA | Tail-anchor(ed) |
| TKD | Tubulointerstitial kidney disease |
| TMEM | Transmembrane (protein) |
| TMH | Transmembrane helix |
| TPR | Tetratricopeptide repeat |
| TRAM | translocating chain-associating membrane (protein) |
| TRAP | Translocon-associated protein |
| TRC | Transmembrane recognition complex |
| TRX | Thioredoxin |
| UPR | Unfolded protein response |
References
- Palade, G. Intracellular aspects of protein synthesis. Science 1975, 189, 347–358. [Google Scholar] [CrossRef]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975, 67, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 1975, 67, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Van, P.N.; Peter, F.; Söling, H.-D. Four intracisternal calcium-binding glycoproteins from rat liver microsomes with high affinity for calcium. J. Biol. Chem. 1989, 264, 17494–17501. [Google Scholar] [CrossRef]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Mogami, H.; Tepikin, A.V.; Petersen, O.H. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J. 1998, 17, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 79–305. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signalling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Sammels, E.; Parys, J.B.; Missiaen, L.; De Smedt, H.; Bultynck, G. Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium 2010, 47, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef]
- Bakowski, D.; Nelson, C.; Parekh, A.B. Endoplasmic reticulum-mitochondria coupling: Local Ca2+ signalling with functional consequences. Eur. J. Physiol. 2012, 464, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channels and disease–From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Kappel, S.; Borgström, A.; Stoklosa, P.; Dörr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough Sheets and Smooth Tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms determining the morphology of the peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef]
- Friedman, J.R.; Voeltz, G.K. The ER in 3D: A multifunctional dynamic membrane network. Trends Cell Biol. 2011, 21, 709–717. [Google Scholar] [CrossRef]
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef] [PubMed]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Pelham, H.R.B. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem. Sci. 1990, 15, 483–486. [Google Scholar] [CrossRef]
- Sambrook, J.F. The involvement of calcium in transport of secretory proteins from the endoplasmic reticulum. Cell 1990, 61, 197–199. [Google Scholar] [CrossRef]
- Schekman, R. Merging cultures in the study of membrane traffic. Nat. Cell Biol. 2004, 6, 483–486. [Google Scholar] [CrossRef]
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.A. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5, a013185. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Noack, J.; Bergmann, T.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Soldá, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M. ER-phagy: Eating the factory. Cell 2020, 78, 811–813. [Google Scholar]
- Madeo, F.; Kroemer, G. Intricate links between ER stress and apoptosis. Mol. Cell 2009, 33, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Ron, D.; Harding, H.P. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb. Perspect. Biol. 2012, 4, a013177. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Vishnu, N.; Jadoon Khan, M.; Karsten, F.; Groschner, L.N.; Waldeck-Weiermair, M.; Rost, R.; Hallström, S.; Imamura, H.; Graier, W.F.; Malli, R. ATP increases within the lumen of the endoplasmic reticulum upon intracellular Ca2+ release. Mol. Biol. Cell 2014, 25, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.-C.; Zimmermann, K.; Schorr, S.; Landini, M.; Klemens, P.; Altensell, J.; Jung, M.; Krause, E.; Nguyen, D.; Helms, V.; et al. AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat. Commun. 2018, 9, 3489. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Bischof, H.; Burgstaller, S.; Siirin, M.; Murphy, A.; Malli, R.; Kaufman, R.J. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca2+. ELife 2019, 8, e49682. [Google Scholar] [CrossRef]
- Zimmermann, R.; Lang, S. A little AXER ABC: ATP, BiP, and Calcium form a triumvirate orchestrating energy homeostasis of the endoplasmic reticulum. Contact 2020. [Google Scholar] [CrossRef]
- Römisch, K. A case for Sec61 channel involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Simon, S.M.; Blobel, G. A protein-conducting channel in the endoplasmic reticulum. Cell 1991, 65, 371–380. [Google Scholar] [CrossRef]
- Görlich, D.; Prehn, S.; Hartmann, E.; Kalies, K.-U.; Rapoport, T.A. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell 1992, 71, 489–503. [Google Scholar] [CrossRef]
- Görlich, D.; Rapoport, T.A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 1993, 75, 615–630. [Google Scholar] [CrossRef]
- Hartmann, E.; Sommer, T.; Prehn, S.; Görlich, D.; Jentsch, S.; Rapoport, T.A. Evolutionary conservation of components of the protein translocation complex. Nature 1994, 367, 654–657. [Google Scholar] [CrossRef]
- Pfeffer, S.; Brandt, F.; Hrabe, T.; Lang, S.; Eibauer, M.; Zimmermann, R.; Förster, F. Structure and 3D arrangement of ER-membrane associated ribosomes. Structure 2012, 20, 1508–1518. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.H.W.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Förster, F. Structure of the mammalian oligosaccharyltransferase in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Förster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal peptide. Science 2016, 351, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Ng, B.; Schaffa, M.; Albert, S.; Plitzko, J.; Baumeister, W.; Zimmermann, R.; Freeze, H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associatecd protein complex. Nat. Commun. 2017, 8, 14516. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel function, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef]
- Lang, S.; Nguyen, D.; Pfeffer, S.; Förster, F.; Helms, V.; Zimmermann, R. Current state of affairs on the eukaryotic ribosome-translocon complex, in Macromolecular Complexes II: Structure and Function. Subcell. Biochem. 2019, 93, 83–141. [Google Scholar] [CrossRef] [PubMed]
- Gemmer, M.; Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340. [Google Scholar] [CrossRef] [PubMed]
- Lomax, R.B.; Camello, C.; Van Coppenolle, F.; Petersen, O.H.; Tepikin, A.V. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 2002, 277, 26479–26485. [Google Scholar] [CrossRef]
- Wirth, A.; Jung, M.; Bies, C.; Frien, M.; Tyedmers, J.; Zimmermann, R.; Wagner, R. The Sec61p complex is a dynamic precursor activated channel. Mol. Cell 2003, 12, 261–268. [Google Scholar] [CrossRef]
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Van Coppenolle, F.; Lehen’kyi, V.; Beck, B.; Skryma, R. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006, 20, 1215–1217. [Google Scholar] [CrossRef]
- Giunti, R.; Gamberucci, A.; Fulceri, R.; Banhegyi, G. Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: A mechanistic study on rat liver microsomes. Arch. Biochem. Biophys. 2007, 462, 115–121. [Google Scholar] [CrossRef]
- Ong, H.L.; Liu, X.; Sharma, A.; Hegde, R.S.; Ambudkar, I.S. Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflug. Arch. 2007, 453, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalié, A.; Zimmermann, R. Sec61 complexes form ubiquitous ER Ca2+ leak channels. Channels 2011, 5, 228–235. [Google Scholar] [CrossRef]
- Erdmann, F.; Schäuble, N.; Lang, S.; Jung, M.; Honigmann, A.; Ahmad, M.; Dudek, J.; Benedix, J.; Harsman, A.; Kopp, A.; et al. Interaction of calmodulin with Sec61α limits Ca2+ leakage from the endoplasmic reticulum. EMBO J. 2011, 30, 17–31. [Google Scholar] [CrossRef]
- Schäuble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, Ö.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Signal sequences. J. Mol. Biol. 1985, 184, 99–105. [Google Scholar] [CrossRef]
- Von Heijne, G. Towards a comparative anatomy of N-terminal topogenic protein sequences. J. Mol. Biol. 1986, 189, 239–242. [Google Scholar] [CrossRef]
- Von Heijne, G.; Gavel, Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988, 174, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.T.; Brown, J.D.; Walter, P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Bernstein, H. The surprising complexity of signal peptides. Trends Biochem. Science 2006, 31, 563–571. [Google Scholar]
- Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targting peptides using deep learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef]
- Wiedmann, M.; Kurzchalia, T.V.; Hartmann, E.; Rapoport, T.A. A signal sequence receptor in the endoplasmic reticulum membrane. Nature 1987, 328, 830–833. [Google Scholar] [CrossRef]
- Menetret, J.F.; Hegde, R.S.; Aguiar, M.; Gygi, S.P.; Park, E.; Rapoport, T.A.; Akey, C.W. Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure 2008, 16, 1126–1137. [Google Scholar] [CrossRef]
- Dierks, T.; Volkmer, J.; Schlenstedt, G.; Jung, C.; Sandholzer, U.; Zachmann, K.; Schlotterhose, P.; Neifer, K.; Schmidt, B.; Zimmermann, R. A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J. 1996, 15, 6931–6942. [Google Scholar] [CrossRef]
- Skowronek, M.H.; Rotter, M.; Haas, I.G. Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biol. Chem. 1999, 380, 1133–1138. [Google Scholar] [CrossRef]
- Mayer, H.-A.; Grau, H.; Kraft, R.; Prehn, S.; Kalies, K.-U.; Hartmann, E. Mammalian Sec61 is associated with Sec62 and Sec63. J. Biol. Chem. 2000, 275, 14550–14557. [Google Scholar] [CrossRef]
- Tyedmers, J.; Lerner, M.; Bies, C.; Dudek, J.; Skowronek, M.H.; Haas, I.G.; Heim, N.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7214–7219. [Google Scholar] [CrossRef]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef]
- Tyedmers, J.; Lerner, M.; Wiedmann, M.; Volkmer, J.; Zimmermann, R. Polypeptide chain binding proteins mediate completion of cotranslational protein translocation into the mammalian endoplasmic reticulum. EMBO Rep. 2005, 4, 505–510. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, evolution and structure of J-domain proteins. Cell Stress Chaperones 2018, 24, 7–15. [Google Scholar] [CrossRef]
- Feige, M.J.; Hendershot, L.M.; Buchner, J. How antibodies fold. Trends Biochem. Sci. 2010, 35, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Haßdenteufel, S.; Klein, M.-C.; Melnyk, A.; Zimmermann, R. Protein transport into the human ER and related diseases: Sec61-channelopathies. Biochem. Cell Biol. 2014, 92, 499–509. [Google Scholar] [CrossRef]
- Jarjanazi, H.; Savas, S.; Pabalan, N.; Dennis, J.W.; Ozcelik, H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins 2008, 70, 394–403. [Google Scholar] [CrossRef]
- Guo, H.; Xiong, Y.; Witkowski, P.; Cui, J.; Wang, L.-J.; Sun, J.; Lara-Lemus, R.; Haataja, L.; Hutchison, K.; Shan, S.O.; et al. Inefficient translocation of preproinsulin contributes to pancreatic ß cell failure and late-onset Diabetes. J. Biol. Chem. 2014, 289, 16290–16302. [Google Scholar] [CrossRef] [PubMed]
- Živná, M.; Hulkova, H.; Matignon, M.; Hodanova, K.; Vylet´al, P.; Kalbacova, M.; Baresova, V.; Sikora, J.; Blazkova, H.; Zivny, J.; et al. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, anch chronic kidney failure. Am. J. Hum. Genet. 2009, 85, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Eisenhaber, F.; Adhikari, S.; Eisenhaber, B.; Gruber, G. Purification and crystallization of yeast glycosylphosphatidylinositol transamidase subunit PIG-S (PIG-S71-467). Acta Cryst. Sect. F 2011, 67, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Kalies, K.-U.; Rapoport, T.A.; Hartmann, E. The beta-subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J. Cell Biol. 1998, 141, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Liaci, A.M.; Steigenberger, B.; Tamara, S.; de Souza, P.C.T.; Gröllers-Mulderji, M.; Ogrissek, P.; Marrink, S.J.; Schletema, R.; Förster, F. Sturcture of the human signal pepidase complex reveals the determinants for signal peptide cleavage. Cell 2021, in press. [Google Scholar] [CrossRef]
- Aviram, N.; Schuldiner, M. Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J. Cell Sci. 2017, 130, 4079–4085. [Google Scholar] [CrossRef]
- Egea, P.F.; Stroud, R.M.; Walter, P. Targeting proteins to membranes: Structure of the signal recognition particle. Curr. Opinion Struct. Biol. 2005, 15, 213–220. [Google Scholar] [CrossRef]
- Halic, M.; Beckmann, R. The signal recognition particle and its interactions during protein targeting. Curr. Opinion Struct. Biol. 2005, 15, 116–125. [Google Scholar] [CrossRef]
- Halic, M.; Blau, M.; Becker, T.; Mielke, T.; Pool, M.R.; Wild, K.; Sinning, I.; Beckmann, R. Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature 2006, 444, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hanebuth, M.A.; Frickey, T.; Deuerling, E. The principle of antagonism ensures protein targeting specificity at the endoplasmic reticulum. Science 2015, 348, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-H.; Lee, J.H.; Chandrasekar, S.; Shan, S.O. A ribosome-associated chaperone enables sustrate triage in a cotranslational protein targeting complex. Nat. Commun. 2020, 11, 5840. [Google Scholar] [CrossRef]
- Aviram, N.; Ast, T.; Costa, E.A.; Arakel, E.; Chuartzman, S.G.; Jan, C.H.; Haßdenteufel, S.; Dudek, J.; Jung, M.; Schorr, S.; et al. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 2016, 540, 134–138. [Google Scholar] [CrossRef]
- Casson, J.; McKenna, M.; Haßdenteufel, S.; Aviram, N.; Zimmermann, R.; High, S. Multiple pathways facilitate the biogenesis of mammalian tail-anchored proteins. J. Cell Sci. 2017, 130, 3851–3861. [Google Scholar] [CrossRef]
- Haßdenteufel, S.; Sicking, M.; Schorr, S.; Aviram, N.; Fecher-Trost, C.; Schuldiner, M.; Jung, M.; Zimmermann, R.; Lang, S. hSnd2 protein represents an alternative targeting factor to the endoplasmic reticulum in human cells. FEBS Lett. 2017, 591, 3211–3224. [Google Scholar] [CrossRef]
- Haßdenteufel, S.; Johnson, N.; Paton, A.W.; Paton, J.C.; High, S.; Zimmermann, R. Chaperone-mediated Sec61 channel gating during ER import of small precursor proteins overcomes Sec61 inhibitor-reinforced energy barrier. Cell Rep. 2018, 23, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Haßdenteufel, S.; Nguyen, D.; Helms, V.; Lang, S.; Zimmermann, R. Components and mechanisms for ER import of small human presecretory proteins. FEBS Lett. 2019, 593, 2506–2524. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.K.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol. Biol. Cell 2012, 23, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Kutay, U.; Hartmann, E.; Rapoport, T.A. A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 1993, 3, 72–75. [Google Scholar] [CrossRef]
- Schuldiner, M.; Metz, J.; Schmid, V.; Denic, V.; Rakwalska, M.; Schmitt, H.D.; Schwappach, B.; Weissman, J.S. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 2008, 134, 634–645. [Google Scholar] [CrossRef]
- Mariappan, M.; Li, X.; Stefanovic, S.; Sharma, A.; Mateja, A.; Keenan, R.J.; Hegde, R.S. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 2010, 466, 1120–1124. [Google Scholar] [CrossRef]
- Borgese, N.; Righi, M. Remote origins of tail-anchored proteins. Traffic 2010, 11, 877–885. [Google Scholar] [CrossRef]
- Borgese, N.; Fasana, E. Targeting pathways of C-tail-anchored proteins. Biochim. Biophys. Acta 2011, 1808, 937–946. [Google Scholar] [CrossRef]
- Vilardi, F.; Lorenz, H.; Dobberstein, B. WRB is the receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins into the ER membrane. J. Cell Sci. 2011, 124, 1301–1307. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Sakisaka, T. Molecular machinery for insertion of tail-anchored membrane proteins into the endoplasmic reticulum membrane in mammalian cells. Mol. Cell 2012, 48, 387–397. [Google Scholar] [CrossRef]
- Wang, F.; Chan, C.; Weir, N.R.; Denic, V. The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 2014, 512, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Borgese, N.; Coy-Vergara, J.; Colombo, S.F.; Schwappach, B. The ways of tails: The GET pathway and more. Proteins 2019, 38, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Pataki, C.I.; Rodrigues, J.; Zhang, L.; Qian, J.; Efron, B.; Hastie, T.; Elias, J.E.; Levitt, M.; Kopito, R.R. Proteomic analysis of monolayer-integrated proteins on lipid droplets identifies amphipathic interfacial α-helical membrane anchors. Proc. Natl. Acad. Sci. USA 2018, 115, E8172–E8180. [Google Scholar] [CrossRef]
- Schrul, B.; Kopito, R.R. Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to ER subdomains. Nat. Cell Biol. 2016, 18, 740. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Sakisaka, T. The peroxisome biogenesis factors posttranslationally target reticulon homology domain-containing proteins to the endoplasmic reticulum. Sci. Rep. 2018, 8, 2322. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, M.J.; Itzhak, D.N.; Hussmann, J.A.; Schirle Oakdale, N.T.; Costa, E.A.; Jonikas, M.; Weibezahn, J.; Popova, K.D.; Jan, C.H.; Sinitcyn, P.; et al. The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. ELife 2018, 7, e37018. [Google Scholar] [CrossRef] [PubMed]
- Chitwood, P.J.; Juszkiewicz, S.; Guna, A.; Shao, S.; Hegde, R.S. EMC is required to initiate accurate membrane protein topogenesis. Cell 2018, 175, 1507–1519. [Google Scholar] [CrossRef]
- Pleiner, T.; Tomaleri, G.P.; Januszyk, K.; Inglis, A.J.; Hazu, M.; Voorhees, R.M. Structural basis for membrane insertion by the human ER membrane protein complex. Science 2020, 369, 433–436. [Google Scholar] [CrossRef]
- Bai, L.; You, Q.; Feng, X.; Kovach, A.; Li, H. Structure of the ER membrane complex, a transmembrane insertase. Nature 2020, 584, 475–478. [Google Scholar] [CrossRef] [PubMed]
- O´Donnel, J.P.; Philips, B.P.; Yagita, Y.; Juszkiewicz, S.; Wagner, A.; Malinverni, D.; Keenan, R.J.; Mille, E.A.; Hegde, R.S. The architecture of EMC reveals a path for membrane protein nsertion. ELife 2020, 9, e57887. [Google Scholar] [CrossRef]
- Ismail, N.; Crawshaw, S.G.; High, S. Active and passive displacement of transmembrane domains both occur during opsin biogenesis at the Sec61 translocon. J. Cell Sci. 2006, 119, 2826–2836. [Google Scholar] [CrossRef]
- Wang, Q.-C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Anghel, S.A.; McGilvray, P.T.; Hegde, R.S.; Keenan, R.J. Identification of Oxa1 homologs operating in the eukaryotic endoplasmic reticulum. Cell Rep. 2017, 21, 3708–3716. [Google Scholar] [CrossRef] [PubMed]
- McGilvray, P.T.; Anghel, S.A.; Sundaram, A.; Zhong, F.; Trnka, M.J.; Fuller, J.R.; Hu, H.; Burlingame, A.L.; Keenan, R.J. An ER translocon for multi-pass mambrane protein biogenesis. ELife 2020, 9, e56889. [Google Scholar] [CrossRef]
- Chitwood, P.J.; Hegde, R.S. An intramembrane chaperone complex facilitates membrane protein biogenesis. Nature 2020, 584, 630–634. [Google Scholar] [CrossRef]
- Van den Berg, B.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.J.; Devaraneni, P.K.; Yang, Z.; David, L.L.; Skach, W.R. Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 2015, 58, 269–283. [Google Scholar] [CrossRef]
- Mahamid, J.; Pfeffer, S.; Schaffer, M.; Villa, E.; Danev, R.; Kuhn Cuellar, L.; Förster, F.; Hyman, A.A.; Plitzko, J.M.; Baumeister, W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 2016, 351, 969–972. [Google Scholar] [CrossRef]
- Tyedmers, J.; Lerner, M.; Nastainczyk, W.; Zimmermann, R. Calumenin and reticulocalbin are associated with the protein translocase of the mammalian endoplasmic reticulum. J. Biol. Sci. 2003, 5, 70–75. [Google Scholar]
- Schorr, S.; Klein, M.-C.; Gamayun, I.; Melnyk, A.; Jung, M.; Schäuble, N.; Wang, Q.; Hemmis, B.; Bochen, F.; Greiner, M.; et al. Co-chaperone specificity in gating of the polypeptide conducting channel in the membrane of the human endoplasmic reticulum. J. Biol. Chem. 2015, 290, 18621–18635. [Google Scholar] [CrossRef]
- Heritage, D.; Wonderlin, W.F. Translocon pores in the endoplasmic reticulum are permeable to a neutral, polar molecule. J. Biol. Chem. 2001, 276, 22655–22662. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Wonderlin, W.F. The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J. Biol. Chem. 2003, 278, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.; Schulten, K. Structural determinants of lateral gate opening in the protein translocon. Biochemistry 2007, 46, 11147–11157. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, R.; Spahn, C.M.; Eswar, N.; Helmers, J.; Penczek, P.A.; Sali, A.; Frank, J.; Blobel, G. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell 2001, 107, 361–372. [Google Scholar] [CrossRef]
- Zhang, B.; Miller, T.F. III Long-timescale dynamics and regulation of Sec-facilitated protein translocation. Cell Rep. 2012, 2, 927–937. [Google Scholar] [CrossRef]
- Trueman, S.F.; Mandon, E.C.; Gilmore, R. A gating motif in the translocation channel sets the hydrophobicity threshold for signal sequence function. J. Cell Biol. 2012, 199, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Goder, V.; Spiess, M. Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J. 2003, 22, 3645–3653. [Google Scholar] [CrossRef]
- Goder, V.; Junne, T.; Spiess, M. Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol. Biol. Cell 2004, 15, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.A.; Wong, W.-C.; Eisenhaber, B.; Warwicker, J.; Eisenhaber, F. Charged residues next to transmembrane regions revisited: “Positive-inside rule” is complemented by the “negative inside depletion/outside enrichment rule”. BMC Biol. 2017, 15, 66. [Google Scholar] [CrossRef]
- Devaraneni, P.K.; Conti, B.; Matsumara, Y.; Yang, Z.; Johnson, A.E.; Skach, W.R. Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell 2011, 146, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Stutz, R.; Schorr, S.; Lang, S.; Pfeffer, S.; Freeze, H.F.; Förster, F.; Helms, V.; Dudek, J.; Zimmermann, R. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 2018, 9, 37639. [Google Scholar] [CrossRef]
- Schorr, S.; Nguyen, D.; Haßdenteufel, S.; Nagaraj, N.; Cavalié, A.; Greiner, M.; Weissgerber, P.; Loi, M.; Paton, A.W.; Paton, J.C.; et al. Proteomics identifies signal peptide features determining the substrate specificity in human Sec62/Sec63-dependent ER protein import. FEBS J. 2020, 287, 4612–4640. [Google Scholar] [CrossRef]
- Jung, S.J.; Kim, J.E.H.; Reithinger, J.H.; Kim, H. The Sec62–Sec63 translocon facilitates translocation of the C-terminus of membrane proteins. J. Cell Sci. 2014, 127, 4270–4278. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Greiner, M.; Müller, A.; Hendershot, L.M.; Kopsch, K.; Nastainczyk, W.; Zimmermann, R. ERj1p plays a basic role in protein biogenesis at the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2005, 12, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Blau, M.; Mullapudi, S.; Becker, T.; Dudek, J.; Zimmermann, R.; Penczek, P.A.; Beckmann, R. ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat. Struct. Mol. Biol. 2005, 12, 1015–1016. [Google Scholar] [CrossRef]
- Benedix, J.; Lajoie, P.; Jaiswal, H.; Burgard, C.; Greiner, M.; Zimmermann, R.; Rospert, S.; Snapp, E.L.; Dudek, J. BiP modulates the affinity of its co-chaperone ERj1 to ribosomes. J. Biol. Chem. 2010, 285, 36427–36433. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Diaz de Escauriaza, M.; Lajoie, P.; Theis, M.; Jung, M.; Müller, A.; Burgard, C.; Greiner, M.; Snapp, E.L.; Dudek, J.; et al. Evolutionary gain of function of the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 2010, 21, 691–703. [Google Scholar] [CrossRef]
- Snapp, E.L.; Reinhart, G.A.; Bogert, B.A.; Lippincott-Schwartz, J.; Hegde, R.S. The organization of engaged and quiescent translocons in the endoplasmic reticulum of mammalian cells. J. Cell Biol. 2004, 164, 997–1007. [Google Scholar] [CrossRef]
- Jadhav, B.; McKenna, M.; Johnson, N.; High, S.; Sinning, I.; Pool, M.R. Mammalian SRP receptor switches the Sec61 translocase from Sec62 to SRP-dependent translocation. Nat. Commun. 2015, 6, 10133. [Google Scholar] [CrossRef]
- Lambert, R.; Prange, R. Chaperone action in the posttranslational topological reorientation of the hepatitis B virus large envelope protein: Implications for translocational regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 5199–5204. [Google Scholar] [CrossRef] [PubMed]
- Reithinger, J.H.; Kim, J.E.H.; Kim, H. Sec62 protein mediates membrane insertion and orientation of moderately hydrophobic signal anchor proteins in the endoplasmic reticulum (ER). J. Biol. Chem. 2013, 288, 18058–18067. [Google Scholar] [CrossRef]
- Sommer, N.; Junne, T.; Kalies, K.-U.; Spiess, M.; Hartmann, E. TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim. Biophys. Acta 2013, 1833, 3104–3111. [Google Scholar] [CrossRef] [PubMed]
- Fons, R.D.; Bogert, B.A.; Hegde, R.S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 2003, 160, 529–539. [Google Scholar] [CrossRef]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schäuble, N.; Jalal, C.; Greiner, M.; Haßdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61α-, Sec62 and Sec63-depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef]
- Ziska, A.; Tatzelt, J.; Dudek, J.; Paton, A.W.; Paton, J.C.; Zimmermann, R.; Haßdenteufel, S. The signal peptide plus a cluster of positive charges in prion protein dictate chaperone-mediated Sec61-channel gating. Biol. Open 2019, 8, bio040691. [Google Scholar] [CrossRef] [PubMed]
- Nicchitta, C.V.; Blobel, G. Lumenal proteins of the mammalian endoplasmic reticulum are required to complete protein translocation. Cell 1993, 73, 989–998. [Google Scholar] [CrossRef]
- Wada, I.; Rindress, D.; Cameron, P.H.; Ou, W.-J.; Doherty, J.J.; Louvard, D.; Bell, A.W.; Dignard, D.; Thomas, D.Y.; Bergeron, J.J.M. SSRα and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J. Biol. Chem. 1991, 266, 19599–19610. [Google Scholar] [CrossRef]
- Shaffer, K.L.; Sharma, A.; Snapp, E.L.; Hegde, R.S. Regulation of protein compartmentalization expands the diversity of protein function. Dev. Cell 2005, 9, 545–554. [Google Scholar] [CrossRef]
- Dejgaard, K.; Theberge, J.-F.; Heath-Engel, H.; Chevet, E.; Tremblay, M.L.; Thomas, D.Y. Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 2010, 9, 1763–1771. [Google Scholar] [CrossRef]
- Bano-Polo, M.; Martinez-Garay, C.A.; Grau, B.; Martinez-Gil, L.; Mingarro, I. Membrane insertion and topology of the translocon-associated protein (TRAP) gamma subunit. Biochem. Biophys. Acta 2017, 1859, 903–909. [Google Scholar] [CrossRef]
- Görlich, D.; Hartmann, E.; Prehn, S.; Rapoport, T.A. A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature 1992, 357, 47–52. [Google Scholar] [CrossRef]
- High, S.; Martoglio, B.; Görlich, D.; Andersen, S.S.L.; Ashford, A.A.; Giner, A.; Hartmann, E.; Prehn, S.; Rapoport, T.A.; Dobberstein, B.; et al. Site-specific photocross-linking reveals that Sec61p and TRAM contact different regions of a membrane-inserted signal sequence. J. Biol. Chem. 1993, 268, 26745–26751. [Google Scholar] [CrossRef]
- Mothes, W.; Prehn, S.; Rapoport, T.A. Systematic probing of the environment of a translocating secretory protein translocation through the ER membrane. EMBO J. 1994, 13, 3973–3982. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Falcone, D.; Lin, J.; Andrews, D.W.; Johnson, A.E. The cotranslational integration of membrane proteins into the phospholipid bilayer is a multistep process. Cell 1996, 85, 369–378. [Google Scholar] [CrossRef]
- Voigt, S.; Jungnickel, B.; Hartmann, E.; Rapoport, T.A. Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Voigt, S.; Rapoport, T.A.; Lingappa, V.R. TRAM regulates the exposure of nascent secretory proteins to the cytosol during translocation into the endoplasmic reticulum. Cell 1998, 92, 621–631. [Google Scholar] [CrossRef]
- McCormick, P.J.; Miao, Y.; Shao, Y.; Lin, J.; Johnson, A.E. Cotranslational protein integration into the ER membrane is mediated by the binding of nascent chains to translocon proteins. Mol. Cell 2003, 12, 329–341. [Google Scholar] [CrossRef]
- Sadlish, H.; Pitonzo, D.; Johnson, A.E.; Skach, W.R. Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat. Struct. Mol. Biol. 2005, 12, 870–878. [Google Scholar] [CrossRef]
- Sauri, A.; McCormick, P.J.; Johnson, A.E.; Mingarro, I. Sec61alpha and TRAM are sequentially adjacent to a nascent viral membrane protein during its ER integration. J. Mol. Biol. 2007, 366, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.-C.; Lerner, M.; Nguyen, D.; Pfeffer, S.; Dudek, J.; Förster, F.; Helms, V.; Lang, S.; Zimmermann, R. TRAM1 protein may support ER protein import by modulating the phospholipid bilayer near the lateral gate of the Sec61 channel. Channels 2020, 14, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Stefanonvic, B.; Stefanovic, L.; Schnabl, B.; Bataller, R.; Brenner, D.A. TRAM2 protein interacts with endoplasmic reticulum Ca2+ pump SERCA2b and is necessary for collagen type I synthesis. Mol. Cell. Biol. 2004, 24, 1758–1768. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Denard, B.; Lee, C.E.; Han, S.; Ye, J.S.; Ye, J. Inverting the topology of a transmembrane protein by regulating the translocation of the first transmembrane helix. Mol. Cell 2016, 63, 567–578. [Google Scholar] [CrossRef]
- Yau, W.-M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef]
- Chen, Y.; Capponi, S.; Zu, L.; Gallenbeck, P.; Freites, J.A.; White, S.H.; Dalbey, R.E. YidC insertase of Escherichia coli: Water accessibility and membrane shaping. Structure 2017, 25, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Itskanov, S.; Park, E. Structure of the posttranslational Sec protein-translocation channel complex from yeast. Science 2019, 363, 84–87. [Google Scholar] [CrossRef]
- Wu, X.; Cabanos, C.; Rapoport, T.A. Structure of the post-translational protein translocation machinery of the ER membrane. Nature 2019, 566, 136–139. [Google Scholar] [CrossRef]
- Weng, T.-H.; Steinchen, W.; Beatrix, B.; Berninghausen, O.; Becker, T.; Bange, G.; Cheng, J.; Beckmann, R. Architecture of the active post-translational SEC translocon. EMBO J. 2021, 40, e105643. [Google Scholar] [CrossRef]
- Itskanov, S.; Kuo, K.M.; Gumbart, J.C.; Park, E. Stepwise gating of the Sec61 protein-conducting channel by Sec62 and Sec63. Nat. Struct. Mol. Biol. 2021, 28, 162–172. [Google Scholar] [CrossRef]
- Trueman, S.F.; Mandon, E.C.; Gilmore, R. Translocation channel gating kinetics balances protein translocation efficiency with signal sequence recognition fidelity. Mol. Biol. Cell 2011, 22, 2983–2993. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Scheckman, R. A Sec63-BiP complex is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 1993, 123, 1355–1363. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Goeckeler, J.; Schekman, R. BiP and Sec63p are required for both co- and posttranslational protein translocation into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1995, 92, 9643–9646. [Google Scholar] [CrossRef]
- Lyman, S.K.; Schekman, R. Interaction between BiP and Sec63p is required for the completion of protein translocation into the ER of Saccharomyces cerevisiae. J. Cell Biol. 1995, 131, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Lyman, S.K.; Schekman, R. Binding of secretory precursor polypeptides to a translocon subcomplex is regulated by BiP. Cell 1997, 88, 85–96. [Google Scholar] [CrossRef]
- Bole, D.G.; Hendershot, L.M.; Kearney, J.F. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 1986, 102, 1558–1566. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R.B. An Hsp70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986, 46, 291–300. [Google Scholar] [CrossRef]
- Lievremont, J.P.; Rizzuto, R.; Hendershot, L.M.; Meldolesi, J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879. [Google Scholar] [CrossRef]
- Tatu, U.; Helenius, A. Interactions between newly synthesized glycoproteins, calnexin and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 1997, 136, 555–565. [Google Scholar] [CrossRef]
- Meunier, L.; Usherwood, Y.-K.; Chung, K.T.; Hendershot, L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 2002, 13, 4456–4469. [Google Scholar] [CrossRef]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell Biol. 2006, 15, 5688–5697. [Google Scholar] [CrossRef]
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485. [Google Scholar] [CrossRef]
- Awad, W.; Estrada, I.; Shen, Y.; Hendershot, L.M. BiP mutants that are unable to interact with endoplasmic reticulum DnaJ proteins provide insights into interdomain interactions in BiP. Proc. Natl. Acad. Sci. USA 2008, 105, 1164–1169. [Google Scholar] [CrossRef]
- Zhuravieva, A.; Gierasch, L. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. USA 2015, 112, E2865–E2873. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef]
- Xu, M.; Marsh, H.M.; Sevier, C.S. A conserved cysteine within the ATPase domain of the endoplasmic reticulum chaperone BiP is necessary for a complete complement of BiP activities. J. Mol. Biol. 2016, 428, 4168–4184. [Google Scholar] [CrossRef]
- Kopp, M.C.; Larburo, N.; Duraiaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Stuct. Mol. Biol. 2020, 26, 1053–1062. [Google Scholar] [CrossRef]
- Hamman, B.D.; Hendershot, L.M.; Johnson, A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 1998, 92, 747–758. [Google Scholar] [CrossRef]
- Alder, N.A.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanism underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef]
- Hennessy, F.; Nicoll, W.S.; Zimmermann, R.; Cheetham, M.E.; Blatch, G.L. Not all J domains are created equal: Implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 2005, 14, 1697–1709. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Masso-Welch, P.; Di, Y.-P.; Cai, J.-W.; Shen, J.-W.; Subjeck, J.R. The 170-kDa glucose-regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol. Biol. Cell 1993, 4, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Kitao, Y.; Hashimoto, K.; Matsuyama, T.; Iso, H.; Tamatani, T.; Hori, O.; Stern, D.M.; Kano, M.; Ozawa, K.; Ogawa, S. ORP150/HSP12A regulates purkinje cell survival: A role for endoplasmic reticulum stress in cerebellar development. J. Neurosci. 2004, 24, 1486–1496. [Google Scholar] [CrossRef]
- Weitzmann, A.; Volkmer, J.; Zimmermann, R. The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 2006, 580, 5237–5240. [Google Scholar] [CrossRef]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef]
- Shomura, Y.; Dragovic, Z.; Chang, H.C.; Tzvetkov, N.; Young, J.C.; Brodsky, J.L.; Guerriero, V.; Hartl, F.U.; Bracher, A. Regulation of Hsp70 function by HspBP1: Structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol. Cell 2005, 17, 367–379. [Google Scholar]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef]
- Haigh, N.G.; Johnson, A.E. A new role for BiP: Closing the aqueous translocon pore during protein integration into the ER membrane. J. Cell Biol. 2002, 156, 261–270. [Google Scholar] [CrossRef]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 2017, 171, 1625–1637. [Google Scholar] [CrossRef]
- Brightman, S.E.; Blatch, G.L.; Zetter, B.R. Isolation of a mouse cDNA encoding MTJ1, a new murine member of the DnaJ family of proteins. Gene 1995, 153, 249–254. [Google Scholar] [CrossRef]
- Bies, C.; Guth, S.; Janoschek, K.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. A Scj1p homolog and folding catalysts present in dog pancreas microsomes. Biol. Chem. 1999, 380, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, R.H.A.; Haslam, D.B. HEDJ, an Hsp40 Co-chaperone localized to the endoplasmic reticulum of human cells. J. Biol. Chem. 2000, 275, 24984–24992. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Volkmer, J.; Bies, C.; Guth, S.; Müller, A.; Lerner, M.; Feick, P.; Schäfer, K.H.; Morgenstern, E.; Hennessy, F.; et al. A novel type of cochaperone mediates transmembrane recruitment of DnaK-like chaperones to ribosomes. EMBO J. 2002, 21, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Meunier, L.; Hendershot, L.M. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 2002, 277, 15947–15956. [Google Scholar] [CrossRef]
- Hosoda, A.; Kimata, Y.; Tsuru, A.; Kohno, K. JPDI, a novel endoplasmic reticulum-resident protein containing both a BiP-interacting J-domain and thioredoxin-like motifs. J. Biol. Chem. 2003, 278, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, J.; Honma, A.; Miyajima, H.; Kondo, S.; Okumura, M.; Imaizumi, K. MDG1/ERdj4, an ER-resident DnaJ family member, suppresses cell death induced by ER stress. Genes Cells 2003, 8, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Cunnea, P.M.; Miranda-Vizuete, A.; Bertoli, G.; Simmen, T.; Damdimopoulos, A.E.; Hermann, S.; Leinonen, S.; Huikko, M.P.; Gustafsson, J.-A.; Sitia, R.; et al. ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J. Biol. Chem. 2003, 278, 1059–1066. [Google Scholar] [CrossRef]
- Bies, C.; Blum, R.; Dudek, J.; Nastainczyk, W.; Oberhauser, S.; Jung, M.; Zimmermann, R. Characterization of pancreatic ERj3p, a homolog of yeast DnaJ-like protein Scj1p. Biol. Chem. 2004, 385, 389–395. [Google Scholar] [CrossRef]
- Shen, Y.; Hendershot, L.M. ERdj3p, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP´s interactions with unfolded substrates. Mol. Biol. Cell 2004, 16, 40–50. [Google Scholar] [CrossRef]
- Kroczynska, B.; Evangelista, C.M.; Samant, S.S.; Elguindi, E.C.; Blond, S.Y. The SANT2 domain of murine tumor cell DnaJ-like protein 1 human homologue interacts with alpha1-antichymotrypsin and kinetically interferes with its serpin inhibitory activity. J. Biol. Chem. 2004, 279, 11432–11443. [Google Scholar] [CrossRef]
- Ladiges, W.C.; Knoblaugh, S.E.; Morton, J.F.; Korth, M.J.; Sopher, B.L.; Baskin, C.R.; MacAuley, A.; Goodman, A.G.; LeBoeuf, R.C.; Katze, M.G. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005, 54, 1074–1081. [Google Scholar] [CrossRef]
- Weitzmann, A.; Baldes, C.; Dudek, J.; Zimmermann, R. The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum-a quantitative approach. FEBS J. 2007, 274, 5175–5187. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kang, S.W.; Goodman, A.G.; Garrison, J.L.; Taunton, J.; Katze, M.G.; Kaufman, R.J.; Hedge, R.S. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Petrova, K.; Oyadomari, S.; Hendershot, L.M.; Ron, D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008, 27, 2862–2872. [Google Scholar] [CrossRef]
- Dong, M.; Bridges, J.P.; Apsley, K.; Xu, Y.; Weaveret, T.E. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 2008, 19, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Awad, W.; Petrova, K.; Hendershot, L.M. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J. 2008, 27, 2873–2882. [Google Scholar] [CrossRef]
- Jin, Y.; Zhuang, M.; Hendershot, L.M. ERdj3, a luminal ER DnaJ homologue, binds directly to unfolded proteins in the mammalian ER: Identification of critical residues. Biochemistry 2009, 48, 41–49. [Google Scholar] [CrossRef]
- Zahedi, R.P.; Völzing, C.; Schmitt, A.; Frien, M.; Jung, M.; Dudek, J.; Wortelkamp, S.; Sickmann, A.; Zimmermann, R. Analysis of the membrane proteome of canine pancreatic rough microsomes identifies a novel Hsp40, termed ERj7. Proteomics 2009, 9, 3463–3473. [Google Scholar] [CrossRef]
- Svärd, M.; Biterova, E.I.; Bourhis, J.-M.; Guy, J.E. The crystal structure of the human co-chaperone P58IPK. PLoS ONE 2011, 6, e22337. [Google Scholar] [CrossRef]
- Hagiwara, M.; Maegawa, K.-I.; Suzuki, M.; Ushioda, R.; Araki, K.; Matsumoto, J.H.; Nagata, K.; Inaba, K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 2011, 41, 432–444. [Google Scholar] [CrossRef]
- Lai, C.W.; Otero, J.H.; Hendershot, L.M.; Snapp, E. ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J. Biol. Chem. 2012, 287, 7969–7978. [Google Scholar] [CrossRef]
- Oka, O.B.V.; Pringle, M.A.; Schopp, I.M.; Braakman, I.; Bulleid, N.J. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol. Cell 2013, 50, 793–804. [Google Scholar] [CrossRef]
- Fritz, J.M.; Dong, M.; Apsley, K.S.; Martin, E.P.; Na, C.-L.; Sitaraman, S.; Weaver, T.E. Deficiency of the BiP cochaperone ERdj4 causes constitutive endoplasmic reticulum stress and metabolic defects. Mol. Biol. Cell 2014, 25, 431–440. [Google Scholar] [CrossRef]
- Yamamoto, Y.H.; Kasai, A.; Omori, H.; Takino, T.; Sugihara, M.; Umemoto, T.; Hamasaki, M.; Hatta, T.; Natsume, T.; Morimoto, R.I.; et al. ERdj8 governs the size of autophagosomes during the formation process. J. Cell Biol. 2020, 219, e201903127. [Google Scholar]
- Harsman, A.; Kopp, A.; Wagner, R.; Zimmermann, R.; Jung, M. Calmodulin regulation of the calcium-leak channel Sec61 is unique to vertebrates. Channels 2011, 5, 293–298. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schorr, S.; Jung, M.; Schäuble, N.; Linxweiler, J.; Langer, F.; Schäfers, H.-J.; Cavalié, A.; Zimmermann, R.; Greiner, M. Targeting cell migration and the ER stress response with calmodulin antagonists: A clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer 2013, 13, 574. [Google Scholar] [CrossRef]
- Luesch, H.; Paavalainen, V.O. Natural products as modulators of eukayotic secretion. Nat. Prod. Rep. 2020, 37, 717. [Google Scholar] [CrossRef]
- Zehner, M.; Marschall, A.L.; Bos, E.; Schloetel, J.-G.; Kreer, C.; Fehrenschild, D.; Limmer, A.; Ossendorp, F.; Lang, T.; Koster, A.J.; et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8+ T cells. Immunity 2015, 42, 850–863. [Google Scholar] [CrossRef]
- Garrison, J.L.; Kunkel, E.J.; Hegde, R.S.J.; Taunton, J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 2005, 436, 285–289. [Google Scholar] [CrossRef]
- Besemer, J.; Harent, H.; Wang, S.; Oberhauser, B.; Marquardt, K.; Foster, C.A.; Schreiner, E.P.; de Vries, J.E.; Dascher-Nadel, C.; Lindley, I.J.D. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 2005, 436, 290–293. [Google Scholar] [CrossRef]
- Cross, B.C.S.; McKibbin, C.; Callan, A.C.; Roboti, P.; Piacenti, M.; Rabu, C.; Wilson, C.M.; Whitehead, R.; Flitsch, S.L.; Pool, M.R.; et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 2009, 122, 4393–4400. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Hill, K.; McKenna, M.; Ogbechi, J.; High, S.; Willis, A.E.; Simmonds, R.E. The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, Mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathol. 2014, 10, e1004061. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.L.; Paavilainen, V.O.; Sharma, A.; Hegde, R.S.; Taunton, J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. ELife 2014, 3, e01483. [Google Scholar] [CrossRef] [PubMed]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin kills cells by direct blockade of the Sec61 protein translocation channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef]
- Baron, L.; Paatero, A.O.; Morel, J.-D.; Impens, F.; Guenin-Macé, L.; Saint-Auret, S.; Blanchard, N.; Dillmann, R.; Niang, F.; Pellegrini, S.; et al. Maycolactone subervts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 2016, 213, 2885–2896. [Google Scholar] [CrossRef]
- Grotzke, J.E.; Kozik, P.; Morel, J.-D.; Impens, F.; Pietrosemoli, N.; Cresswell, P.; Amigorena, S.; Demangel, C. Sec61 blockade by mycolactone inhibits antigen cross-presentation indepently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. USA 2017, 114, E5910–E5919. [Google Scholar] [CrossRef]
- McKenna, M.; Simmonds, R.E.; High, S. Mycolactone reveals the substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J. Cell Sci. 2017, 130, 1307–1320. [Google Scholar] [CrossRef]
- Morel, J.-D.; Paatero, A.O.; Wei, J.; Yewdell, J.W.; Guenin-Macé, L.; Van Haver, D.; Impens, F.; Pietrosemoli, N.; Paavilainen, V.O.; Demangel, C. Proteomics reveals scope of Mycolactone-mediated Sec61 blockade and distinctive stress signature. Mol. Cell. Prot. 2018, 17, 1750–1765. [Google Scholar] [CrossRef]
- Zong, G.; Hu, Z.; O´Keefe, S.; Tranter, D.; Lanotti, M.J.; Baron, L.; Hall, B.S.; Corfield, K.; Paatero, A.O.; Henderson, M.J.; et al. Ipomoeassin F binds Sec61α to inhibit protein translocation. J. Am. Chem. Soc. 2019, 141, 8450–8461. [Google Scholar] [CrossRef]
- Gamayun, I.; O’Keefe, S.; Pick, T.; Klein, M.-C.; Nguyen, D.; McKibbin, C.; Piacenti, M.; Williams, H.M.; Flitch, S.L.; Whitehead, R.C.; et al. Eeyarestatin compounds selectively enhance Sec61-mediated Ca2+ leakage from the endoplasmic reticulum. Cell Chem. Biol. 2019, 26, 571–583. [Google Scholar] [CrossRef]
- Tranter, D.; Paatero, A.O.; Kawaguchi, S.; Kazemi, S.; Serrill, J.D.; Kellosalo, J.; Vogel, W.K.; Richter, U.; Mattos, D.R.; Wan, X.; et al. Coibamide A targets Sec61 to prevent biogenesis of secretory and membrane proteins. ACS Chem. Biol. 2020, 15, 2125–2136. [Google Scholar] [CrossRef]
- Gérard, S.F.; Hall, B.S.; Zaki, A.M.; Corfield, K.A.; Mayerhofer, P.U.; Costa, C.; Wheligan, D.K.; Biggin, P.C.; Simmonds, R.E.; Higgins, M.K. Structure of the inhibites state of the Sec translocon. Mol. Cell 2020, 79, 406–415. [Google Scholar] [CrossRef] [PubMed]
- O´Keefe, S.; Roboti, P.; Duah, K.B.; Zong, G.; Schneider, H.; Shi, W.Q.; High, S. Ipomoeassin-F inhibits the in vitro biogenesis oft he SARS-CoV-2 spike protein and ist host cell membrane receptor. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Koopmann, J.-O.; Albring, J.; Hüter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hämmerling, G.J.; Momburg, F. Export of antigenis peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 2000, 13, 117–127. [Google Scholar] [CrossRef]
- Schäuble, N.; Cavalié, A.; Zimmermann, R.; Jung, M. Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels 2014, 8, 76–83. [Google Scholar] [CrossRef]
- Bolar, N.A.; Golzio, C.; Živná, M.; Hayot, G.; Van Hemelrijk, C.; Schepers, D.; Vandeweyer, G.; Hoischen, A.; Huyghe, J.R.; Raes, A.; et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am. J. Hum. Genet. 2016, 299, 174–187. [Google Scholar] [CrossRef]
- Espino-Hernández, M.; Milla, C.P.; Vara-Martin, J.; González-Granado, L.I. De novo SEC61A1 mutation in autosomal dominant tubule-interstitial kidney disease: Phenotype expansion and review of the literature. J. Pedr. Child. Health 2021. [Google Scholar] [CrossRef]
- Schubert, D.; Klein, M.-C.; Haßdenteufel, S.; Caballero-Oteyza, A.; Yang, L.; Proietti, M.; Bulashevska, A.; Kemming, J.; Kühn, J.; Winzer, S.; et al. Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 (SEC61A1). J. Allergy Clin. Immunol. 2018, 141, 1427–1438. [Google Scholar] [CrossRef]
- Van Nieuwenhove, E.; Barber, J.; Smeets, E.; Neumann, J.; Willemsen, M.; Pasciuto, E.; Prezzemolo, T.; Lagou, V.; Seldeslachts, L.; Malengier-Devlies, B.; et al. Defective Sec61α1 underlies a novel cause of autosomal dominant severe congenital neutropenia. J. Allergy Clin. Immunol. 2020, 146, 1180–1192. [Google Scholar] [CrossRef]
- Lloyd, D.J.; Wheeler, M.C.; Gekakis, N. A point mutation in Sec61α leads to Diabetes and hepatosteatisis in mice. Diabetes 2010, 59, 460–470. [Google Scholar] [CrossRef]
- Synofzik, M.; Haack, T.B.; Kopajtich, R.; Gorza, M.; Rapoport, D.; Greiner, M.; Schönfeld, C.; Freiberg, C.; Schorr, S.; Holl, R.W.; et al. Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Gen. 2014, 95, 689–697. [Google Scholar] [CrossRef]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.-U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. 2019, 5, 60. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in Perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Losfeld, M.E.; Ng, B.G.; Kircher, M.; Buckingham, K.J.; Turner, E.H.; Eroshkin, A.; Smith, J.D.; Shendure, J.; Nickerson, D.A.; Bamshag, M.J.; et al. A new congenital disorder of glycosylation caused by a mutation in SSR4, the signal sequence receptor 4 protein of the TRAP-complex. Hum. Mol. Genet. 2014; 23, 1602–1605. [Google Scholar] [CrossRef]
- Ng, B.G.; Raymond, K.; Kircher, M.; Buckingham, K.J.; Wood, T.; Shendure, J.; Nickerson, D.A.; Bamshag, M.J.; University of Washington Center for Mendelian Genomics; Wong, J.T.S.; et al. Expanding the Molecular and Clinical Phenotype of SSR4-CDG. Hum. Mutat. 2015, 36, 1048–1051. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, A.R.; Germino, G.G.; Somlo, S. Molecular advances in autosomal dominant polycystic kidney disease. Adv. Chr. Kid Dis. 2010, 17, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P.H.; Martina, J.A.; van de Kerkhof, R.; Bonifacio, J.S.; Jansen, J.B.M.J. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol. Med. 2005, 11, 37–42. [Google Scholar] [CrossRef]
- Drenth, J.P.H.; te Morsche, R.H.M.; Smink, R.; Bonifacio, J.S.; Jansen, J.B.M.J. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003, 33, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Davila, S.; Furu, L.; Gharavi, A.G.; Tian, X.; Onoe, T.; Qian, Q.; Li, A.; Cai, Y.; Kamath, P.S.; King, B.F.; et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 2004, 36, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Waanders, E.; van Krieken, J.H.J.M.; Lameris, A.L.L.; Drenth, J.P.H. Disrupted cell adhesison but not proliferation mediates cyst formation in polycystic liver disease. Mod. Pathol. 2008, 21, 1293–1302. [Google Scholar] [CrossRef]
- Fedeles, S.V.; Tian, X.; Gallagher, A.-R.; Mitobe, M.; Nishio, S.; Lee, S.H.; Cai, Y.; Geng, L.; Crews, C.M.; Somlo, S. A genetic interaction network of five genes for human polycystic kidney and liver disease defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 2011, 43, 639–647. [Google Scholar] [CrossRef]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.-R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effector s of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilche, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar] [CrossRef]
- Villa, A.; Podini, P.; Panzeri, M.C.; Söling, H.D.; Volpe, P.; Meldolesi, J. The endoplasmic-sarcoplasmic reticulum of smooth muscle: Immunocytochemistry of vas deferens fibers reveals specialized subcompartments differently equipped for the control of Ca2+ homeostasis. J. Cell Biol. 1993, 121, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Krieger, M.; Stendel, C.; Bergmann, C.; Moser, M.; Breitbach-Faller, N.; Rudinik-Schoneborn, S.; Blaschek, A.; Wolf, N.; Harting, I.; et al. Mutations in Sil1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat. Genet. 2005, 37, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, A.-K.; Mahjneh, I.; Hämäläinen, R.H.; Lagier-Tourenne, C.; Kopra, O.; Waris, L.; Anttonen, M.; Joensuu, T.; Kalimo, H.; Paetau, A.; et al. The gene disrupted in Marinesco-Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat. Genet. 2005, 37, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Longo-Guess, C.; Harris, B.S.; Lee, J.W.; Ackerman, S.L. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat. Genet. 2005, 37, 974–979. [Google Scholar] [CrossRef]
- Zhao, L.; Rosales, C.; Seburn, K.; Ron, D.; Ackerman, S.L. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjögren syndrome. Human Mol. Gen. 2009, 19, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M.; Roos, A.; Stendel, C.; Caeys, K.G.; Sonmez, F.M.; Baudis, M.; Bauer, P.; Bornemann, A.; de Goede, C.; Dufke, A.; et al. SIL1 mutations and clinical spectrum in patients with Marinesco-Sjögren syndrome. Brain 2013, 136, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Buchkremer, S.; Kollipara, L.; Labisch, T.; Gatz, C.; Zitzelsberger, M.; Brauers, E.; Nolte, K.; Schröder, J.M.; Kirschner, J.; et al. Myopathy in Marinesco-Sjögren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. 2014, 127, 761–777. [Google Scholar] [CrossRef]
- De L´Etang, A.F.; Maharjan, N.; Brana, M.C.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J.; et al. Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress. Nat. Neurosci. 2015, 18, 227–238. [Google Scholar] [CrossRef]
- Liu, Z.-C.; Chu, J.; Lin, L.; Song, J.; Ning, L.-N.; Luo, H.-B.; Yang, S.-S.; Shi, S.; Wang, Q.; Qu, N.; et al. SIL1 rescued BiP elevation-related Tau hyperphosphorylation in ER stress. Mol. Neurobiol. 2015, 53, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Buchkremer, S.; Corasp, G.; Weis, J.; Roos, A. SIL1-mutant mice elucidate chaperone function in neurological disorders. J. Neuromus. Dis. 2016, 3, 169–181. [Google Scholar] [CrossRef]
- Roos, A.; Kollipara, L.; Buchkremer, S.; Labisch, T.; Brauers, E.; Gatz, C.; Gerardo-Nava, J.; Weis, J.; Zahedi, R.P. Cellular signature of SIL1 depletion: Disease pathogenesis due to alterations in protein composition beyond the ER machinery. Mol. Neurobiol. 2016, 53, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Kollipara, L.; Buchkremer, S.; Weis, J.; Brauers, E.; Hoss, M.; Rütten, S.; Caviedes, P.; Zahedi, R.P.; Roos, A. Proteome profiling and ultrastructural characterization of the human RCMH cell Line: Myoblastic properties and suitability for myopathological studies. J. Proteome Res. 2016, 15, 945–955. [Google Scholar] [CrossRef]
- Labisch, T.; Buchkremer, S.; Phan, V.; Kollipara, L.; Gatz, C.; Lentz, C.; Nolte, K.; Vervoorts, J.; Coraspe, J.A.G.; Sickmann, A.; et al. Trafficking effects of SIL1 increase: Taking a closer look beyond the consequences of elevated expression level. Mol. Neurobiol. 2018, 55, 2524–2546. [Google Scholar] [CrossRef]
- Phan, V.; Cox, D.; Cipriani, S.; Spendiff, S.; Buchkremer, S.; O‘Connor, E.; Horvath, R.; Goebel, H.H.; Hathazi, D.; Lochmüller, H.; et al. SIL1 deficiency causes degenerative changes of peripheral nerves and neuromuscular junctions in fish, mice and human. Neurobiol. Dis. 2019, 124, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Gatz, C.; Hthazi, D.; Münchberg, U.; Buchkremer, S.; Munro, B.; Horvath, R.; Töpf, A.; Weis, J.; Roos, A. Identification of cellular pathogenicity markers for SIL1 mutations linked to Marinesco-Sjögren syndrome. Front. Neurol. 2019, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ohta, Y. Enforced expression of oxygen-regulated protein, ORP150, induces vacuolar degeneration in mouse myocardium. Transgen. Res. 2003, 12, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Takita, Y.; Suzuki, A.; Katsu, Y.; Iguchi, T.; Ohta, Y. Vacuolar degeneration of skeletal muscle in transgenic mice overexpressing ORP150. J. Vet. Med. Sci. 2008, 70, 115–118. [Google Scholar] [CrossRef][Green Version]
- Ozon, Z.A.; Alikasifoglu, A.; Kandemir, N.; Aydin, B.; Gonc, E.N.; Karaosmamoglu, B.; Celik, N.B.; Eroglu-Ertugrul, N.G.; Taskiran, E.Z.; Haliloglu, G.; et al. Novel insights into diabetes mellitus due to DNAJC3-defect: Evolution of neurological and endocrine phenotype in the pediatric age group. Pediatr. Diabetes 2020, 21, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Senée, V.; Salpea, P.; Fantuzzi, F.; Philippi, A.; Abdulkarim, B.; Sawatini, T.; Marin-Cnas, S.; Pachera, N.; Degavre, A.; et al. DNAJC3 deficency induces ß-cell mitochondrial apoptosis and causes syndromic young-onset diabetes. Eur. J. Endocrin. 2021, 184, 459–472. [Google Scholar] [CrossRef]
- Osman, A.M.; van Loveren, H. Matrin 3 co-immunoprecipitates with the heat shock proteins glucose-regulated protein 78 (GRP78), GRP75 and glutathione S-transferase 𝛑 isoform 2 (GST𝛑2) in thymoma cells. Biochimie 2014, 101, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Garvey, S.M.; Krieger, M.; Guergueltcheva, V.; Urtizberea, A.; Roos, A.; Elbracht, M.; Stendel, C.; Tournev, I.; Mihailova, I.; et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am. J. Hum. Genet. 2009, 84, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Gan-Or, A.; Spiegelman, D.; Laurent, S.B.; Szuto, A.; Hodgkinson, A.; Dionne-Laporte, A.; Provencher, P.; de Carvalho, M.; Orrú, S.; et al. Replication study of MATR3 in familial sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 37, e17–e209. [Google Scholar] [CrossRef]
- Zhang, X.; Yamashita, S.; Hara, K.; Doki, T.; Tawara, N.; Ikeda, T.; Misumi, Y.; Zhang, Z.; Matsuo, Y.; Nagai, M.; et al. A mutant MATR3 mouse model to explain multisystsm proteinopathy. J. Pathol. 2019, 249, 182–192. [Google Scholar] [CrossRef]
- Ijuin, T.; Mochizuki, Y.; Fukami, K.; Funaki, M.; Asano, T.; Takenawa, T. Identification and characterization of a novel inositol polyphosphate 5-phosphatase. J. Biol. Chem. 2000, 275, 10870–10875. [Google Scholar] [CrossRef]
- Ijuin, T.; Hatano, N.; Takenawa, T. Glucose-regulated protein 78 (GRP78) binds directly to PIP3 phosphatase SKIP and determines its localization. Genes Cells 2016, 21, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhu, T.; Benedetti, L.; Gowrishankar, S.; Deng, H.; Cai, Y.; Wang, X.; Shen, K.; De Camilli, P. The inositol 5-phosphatase INPP5K participates in the control of ER organization. J. Cell Biol. 2018, 217, 3577–3592. [Google Scholar] [CrossRef]
- D´Amico, A.; Fattori, F.; Nicita, F.; Barresi, S.; Tasca, G.; Verardo, M.; Pizzi, S.; Moroni, I.; De Mitri, F.; Frongia, A.; et al. A recurrent pathogenic variant of INPP5K underlies autosomal recessive congenital muscular dystrophy with cataracts and intellectual disability: Evidence for a founder effect in Southern Italy. Front. Genet. 2020, 10, 3389. [Google Scholar] [CrossRef]
- Wiessner, M.; Roos, A.; Munn, C.J.; Vishwanathan, R.; Whyte, T.; Cox, D.; Schoser, B.; Sewry, C.; Roper, H.; Phadke, R.; et al. Mutations in INPP5K, encoding a phosphoinositide 5-phosphatase. Cause congenital muscular dystrophy with cataracts and mild cognitive impairment. Am. J. Hum. Gen. 2017, 102, 832–844. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.J.; Eramo, M.J.; Gurung, R.; Sriratana, A.; Gehrig, S.M.; Lynch, G.S.; Lourdes, S.R.; Koentgen, F.; Feeney, S.F.; Lazarou, M.; et al. Defectice lysosome formation during autophagy causes skeletal muscle disease. J. Clin. Investig. 2021, 131, e135124. [Google Scholar] [CrossRef]
- Kollipara, L.; Buchkremer, S.; Coraspe, J.A.G.; Hathazi, D.; Senderek, J.; Weis, J.; Zahedi, R.P.; Ross, A. In-depth phenotyping of lymphoblastoid cells suggests selective cellular vulnerability in Marinesco-Sjögren syndrome. Oncotarget 2017, 8, 68493–68516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hathazi, D.; Cox, D.; D‘Amico, A.; Tasca, G.; Charlton, R.; Carlier, R.-Y.; Baumann, J.; Kollipara, L.; Zahedi, R.P.; Feldmann, I.; et al. INPP5K and SIL1 associated pathologies with overlapping clinical phenotypes converge through dysregulation of PHGDH. Brain 2021, in press. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrezet, M.-P.; Hopp, K.; Porath, B.; Shi, B.; et al. Genkyst Study Group, the Halt Progression of Polycystic Kidney Disease Group, the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease, Harris, P.C. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Gen. 2018, 102, 832–844. [Google Scholar] [CrossRef]
- Macario, A.J.; Conway de Macario, E. Molecular chaperones: Multiple functions, pathologies, and potential applications. Front. Biosci. 2007, 12, 2588–2600. [Google Scholar] [CrossRef] [PubMed]
- Aridor, M. Visiting the ER: The endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv. Drug Deliv. Rev. 2007, 59, 759–781. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Sato, F.; Selaru, F.M.; Olaru, A.; Perry, K.; Kimos, M.C.; Tamura, G.; Matsubara, N.; Wang, S.; Xu, Y.; et al. Instabilotyping reveals unique mutational spectra in microsatellite-unstable gastric cancers. Cancer Res. 2002, 62, 3641–3645. [Google Scholar]
- Schulmann, K.; Brasch, F.E.; Kunstmann, E.; Engel, C.; Pagenstecher, C.; Vogelsang, H.; Krüger, S.; Vogel, T.; Knaebel, H.-P.; Rüschoff, J.; et al. For the German HNPCC consortium. HNPCC-associated small bowel cancer: Clinical and molecular characteristics. Gastroenterology 2005, 128, 590–599. [Google Scholar] [CrossRef]
- Eschrich, S.; Yang, I.; Bloom, G.; Kwong, K.Y.; Boulware, D.; Cantor, A.; Coppola, D.; Kruhoffer, M.; Aaltonen, L.; Orntoft, T.F.; et al. Molecular staging or surival prediction of colorectal cancer patients. J. Clin. Oncol. 2005, 23, 3526–3535. [Google Scholar] [CrossRef]
- Jung, V.; Kindich, R.; Kamradt, J.; Jung, M.; Mueller, M.; Schulz, W.A.; Engers, R.; Unteregger, G.; Stoeckle, M.; Zimmermann, R.; et al. Genomic and expression analysis of the 3q25-q26 amplicon reveals TLOC1/SEC62 as a probable target gene in prostate cancer. Mol. Cancer Res. 2006, 4, 169–176. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pinheiro, C.; Lambros, M.B.K.; Milanezi, F.; Carvalho, S.; Savage, K.; Simpson, P.T.; Jones, C.; Swift, S.; Mackay, A.; et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J. Pathol. 2006, 209, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Uchida, T.; Karnan, S.; Noguchi, T.; Nguyen, L.T.; Tanigawa, M.; Takeuchi, I.; Matsuura, K.; Hijiya, N.; Nakada, C.; et al. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. J. Pathol. 2008, 216, 471–482. [Google Scholar] [CrossRef]
- Lu, Z.; Zhou, L.; Killela, P.; Rasheed, A.B.; Di, C.; Poe, W.E.; McLendon, R.E.; Bigner, D.D.; Nicchitta, C.; Yan, H. Glioblastoma protooncogene SEC61γ is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009, 69, 9105–9111. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Chen, K.-T.; Fan, C.-W.; Han, C.-L.; Chen, Y.-J.; Yu, J.-S.; Chang, Y.-S.; Chien, C.-W.; Wu, C.-P.; Hung, R.-P.; et al. Comparison of membrane fraction proteomic profiles of normal and cancerous human colorectal tissues with gel-assisted digestion and iTRAQ labeling mass spectrometry. FEBS J. 2010, 277, 3028–3038. [Google Scholar] [CrossRef]
- Greiner, M.; Kreutzer, B.; Jung, V.; Grobholz, R.; Hasenfus, A.; Stöhr, R.; Franz, R.; Tornillo, L.; Dudek, J.; Stöckle, M.; et al. Silencing of the SEC62 gene inhibits migratory and invasive potential of various tumor cells. Int. J. Cancer 2011, 128, 2284–2295. [Google Scholar] [CrossRef]
- Greiner, M.; Kreutzer, B.; Lang, S.; Jung, V.; Cavalié, A.; Unteregger, G.; Zimmermann, R.; Wullich, B. Sec62 protein level is crucial for ER-stress tolerance of prostate cancer. Prostate 2011, 71, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, M.; Linxweiler, J.; Barth, M.; Benedix, J.; Jung, V.; Kim, Y.-J.; Bohle, R.; Zimmermann, R.; Greiner, M. Sec62 bridges the gap from 3q amplification to molecular cell biology in Non-Small Cell Lung Cancer. Am. J. Pathol. 2012, 180, 473–483. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring ultidimensional cancer genomics data. Cancer Discover. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Weng, L.; Du, J.; Zhou, Q.; Cheng, B.; Li, J.; Zhang, D.; Ling, C. Identification of cyclin B1 and Sec62 as biomarkers for recurrence in patients with HBV-related hepatocellular carcinoma after surgical resection. Mol. Cancer 2012, 11, 39. [Google Scholar] [CrossRef]
- Fan, C.-W.; Chan, C.-C.; Chen, K.-T.; Twu, J.; Huang, Y.-S.; Han, C.L.; Chen, Y.-J.; Yu, J.-S.; Kuo, Y.-B.; Chan, E.-C. Identification of SEC61β and its autoantibody as biomarkers for colorectal cancer. Clin. Chim. Acta 2011, 412, 887–893. [Google Scholar] [CrossRef]
- Casper, M.; Weber, S.N.; Kloor, M.; Müllenbach, R.; Grobholz, R.; Lammert, F. Hepatocellular carcinoma as extracolonic manifestation of Lynch syndrome indicates SEG63 as potential target gene in hepatocarcinogenesis. Sca. J. Gastroenterol. 2013, 48, 344–351. [Google Scholar] [CrossRef]
- Hagerstrand, D.; Tong, A.; Schumacher, S.E.; Ilic, N.; Shen, R.R.; Cheung, H.W.; Vazquez, F.; Shrestha, Y.; Kim, S.Y.; Giacomelli, A.O.; et al. Systematic interrogation of 3q26 identifies TLOC1 and SKL as cancer drivers. Cancer Discov. 2013, 3, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, J.; Kollipara, L.; Zahedi, R.; Lampel, P.; Zimmermann, R.; Greiner, M. Proteomic insights into non-small cell lung cancer: New ideas for cancer diagnosis and therapy from a functional viewpoint. EuPA Open Proteom. 2014. [Google Scholar] [CrossRef]
- Linxweiler, M.; Bochen, F.; Schick, B.; Wemmert, S.; Al Kadah, B.; Greiner, M.; Hasenfus, A.; Bohle, R.-M.; Juhasz-Böss, I.; Solomayer, E.-F.; et al. Identification of SEC62 as a potential marker for 3q amplification and cellular migration in dysplastic cervical lesions. BMC Cancer 2016, 16, 676. [Google Scholar] [CrossRef]
- Wemmert, S.; Lindner, Y.; Linxweiler, J.; Wagenpfeil, S.; Bohle, R.; Niewald, M.; Schick, B. Initial evidence for Sec62 as a prognostic marker in advanced head and neck squamous cell carcinoma. Oncol. Lett. 2016, 11, 1661–1670. [Google Scholar] [CrossRef]
- Bochen, F.; Adisurya, H.; Wemmert, S.; Lerner, C.; Greiner, M.; Zimmermann, R.; Hasenfus, A.; Wagner, M.; Smola, S.; Pfuhl, T.; et al. Effect of 3q oncogenes SEC62 and SOX2 on lymphatic metastasis and clinical outcome of head and neck squamous cell carcinomas. Oncotarget 2017, 8, 4922–4934. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Lets talk about Secs: Sec61, Sec62, Sec63 in signal transduction, oncology and personalized medicine. Signal. Transduct. Target. Ther. 2017, 2, e17002. [Google Scholar] [CrossRef]
- Bergmann, T.J.; Fumagalli, F.; Loi, M.; Molinari, M. Role of SEC62 in ER maintenance: A link with ER stress tolerance in SEC62-overexpressing tumors? Mol. Cell. Oncol. 2017, 4, e1264351. [Google Scholar] [CrossRef] [PubMed]
- Körbel, C.; Linxweiler, M.; Wemmert, S.; Bochen, F.; Schick, B.; Meyer, M.; Maurer, H.; Menger, M.D.; Zimmermann, R.; Greiner, M. Treatment of SEC62 over-expressing tumors by thapsigargin and trifluoperazine. BioMol Conc. 2018, 9, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Takacs, F.Z.; Radosa, J.C.; Linxweiler, M.; Kasohah, M.; Bohle, R.M.; Bochen, F.; Unger, C.; Solomayer, E.-F.; Schick, B.; Juhasz-Böss, I. Identification of 3q oncogene SEC62 as a marker for distant metastasis and poor clinical outcome in invasive ductal breast cancer. Arch. Gynecol. Obstet. 2019, 299, 1405–1413. [Google Scholar] [CrossRef]
- Takacs, F.Z.; Radosa, J.C.; Bochen, F.; Juhasz-Böss, I.; Solomayer, E.-F.; Bohle, R.M.; Breitbach, R.M.; Schick, B.; Linxweiler, M. Sec62/Ki67 and p16/Ki67 dual-staining immunocytochemistry in vulvar cytology for the identification of vulvar intraepithelial neoplasia and vulvar cancer: A pilot study. Arch. Gynecol. Obstet. 2019, 299, 825–833. [Google Scholar] [CrossRef]
- Takacs, F.Z.; Radosa, J.C.; Bohle, R.M.; Bochen, F.; Juhasz-Böss, I.; Solomayer, E.-F.; Schick, B.; Linxweiler, M. Sec62/Ki67 dual staining in cervical cytology specimens: A new marker for high-grade dysplasia. Arch. Gynecol. Obstet. 2019, 299, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, J.; Liao, Y.; Jin, C.; Zhang, Z.; Zhao, J.; Liu, K.; Huang, H.; Cao, H.; Cheng, Q. Identification of SEC61G as a novel prognostic marker for predicting survival and response to therapies in patients with glioblastoma. Med. Sci. Monit. 2019, 25, 3624. [Google Scholar] [CrossRef]
- Li, W.T.; Zou, A.E.; Honda, C.O.; Zheng, H.; Wang, X.Q.; Kisseleva, T.; Chang, E.Y.; Ongkeko, W.M. Etiology-specific analysis of hepatocellular carcinoma transcriptome reveals genetic dysregulation in pathways implicated in immunotherapy efficacy. Cancers 2019, 11, 1273. [Google Scholar] [CrossRef]
- Müller, C.S.; Kreie, L.; Bochen, F.; Pfuhl, T.; Smola, S.; Gräber, S.; Vogt, T.; Schick, B.; Linxweiler, M. Expression of 3q oncogene SEC62 in atypical fibroxanthoma immunohistochemical analysis of 41 cases and correlation with clinical, viral and histopathologic features. Oncol. Lett. 2019, 17, 1768–1776. [Google Scholar] [CrossRef]
- Du, J.; Zhao, Z.; Zhao, H.; Liu, D.; Chen, J.; Cheng, B.; Zhai, X.; Yin, Z.; Zhang, Y.; Ling, C. Sec62 promotes early recurrence of hepatocellular carcinoma through activating integrinα/CAV1 signalling. Oncogenesis 2019, 8, 74. [Google Scholar] [CrossRef]
- Casper, M.; Linxweiler, M.; Linxweiler, J.; Zimmermann, R.; Glanemann, M.; Lammert, F.; Weber, S.N. SEC62 and SEC63 expression in hepatocellular carcinoma (HCC) and tumor-surrounding liver tissue. Visc. Med. 2021, 37, 110–115. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://ars.els-cdn.com/content/image/1-s2.0-S221112471200280X-mmc2.mp4 (accessed on 21 April 2021).
- Dudek, J.; Pfeffer, S.; Lee, P.-H.; Jung, M.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. Protein transport into the human endoplasmic reticulum. J. Mol. Biol. 2015, 427, 1159–1175. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Schäuble, N.; Cavalié, A.; Zimmermann, R. Live cell calcium imaging in combination with siRNA mediated gene silencing identifies Ca2+ leak channels in the ER membrane and their regulatory mechanisms. J. Vis. Exp. 2011, 53, e2730. [Google Scholar] [CrossRef]
- cBioPortal. Available online: https://www.cbioportal.org (accessed on 21 April 2021).










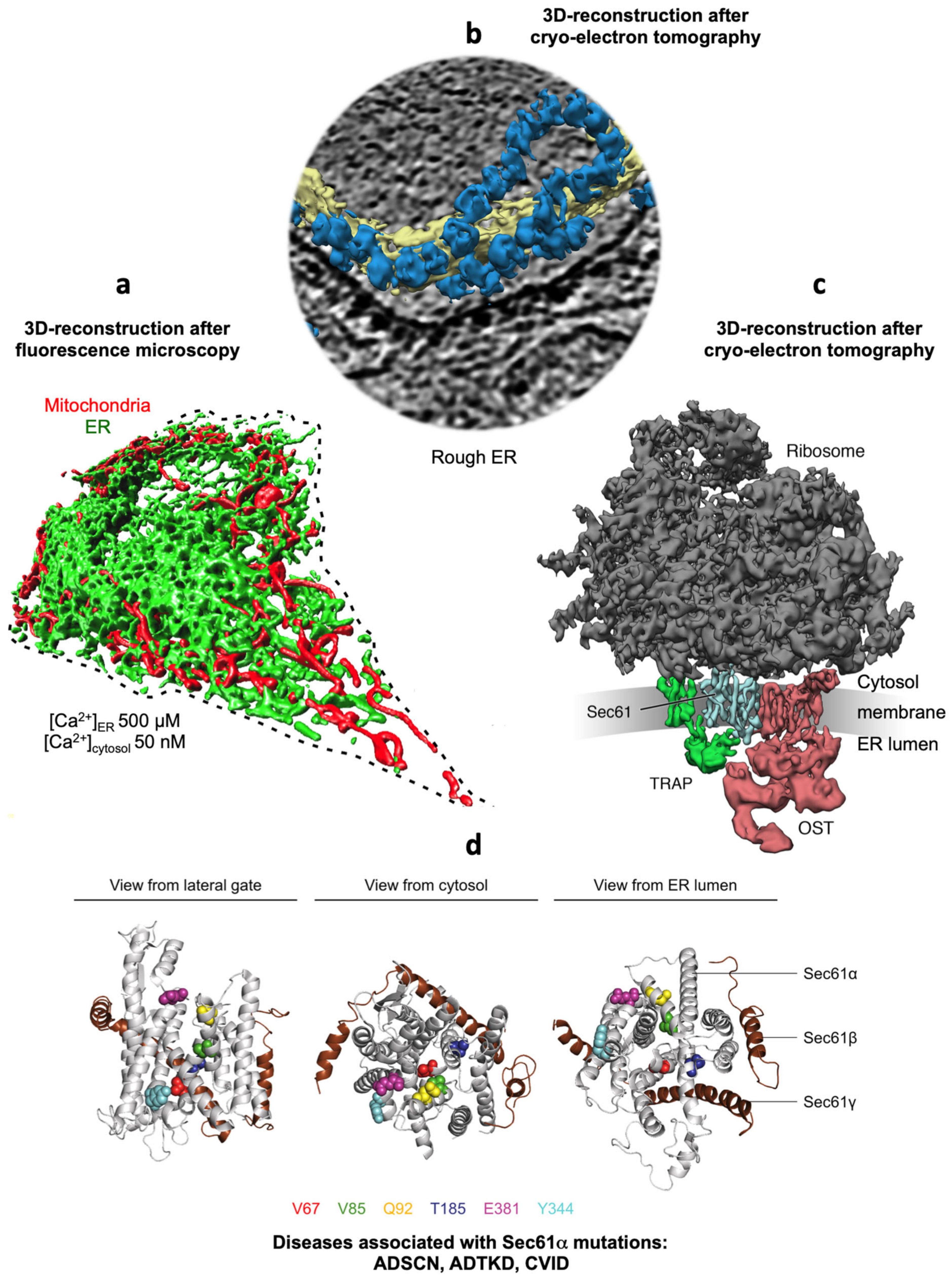{kind=link}
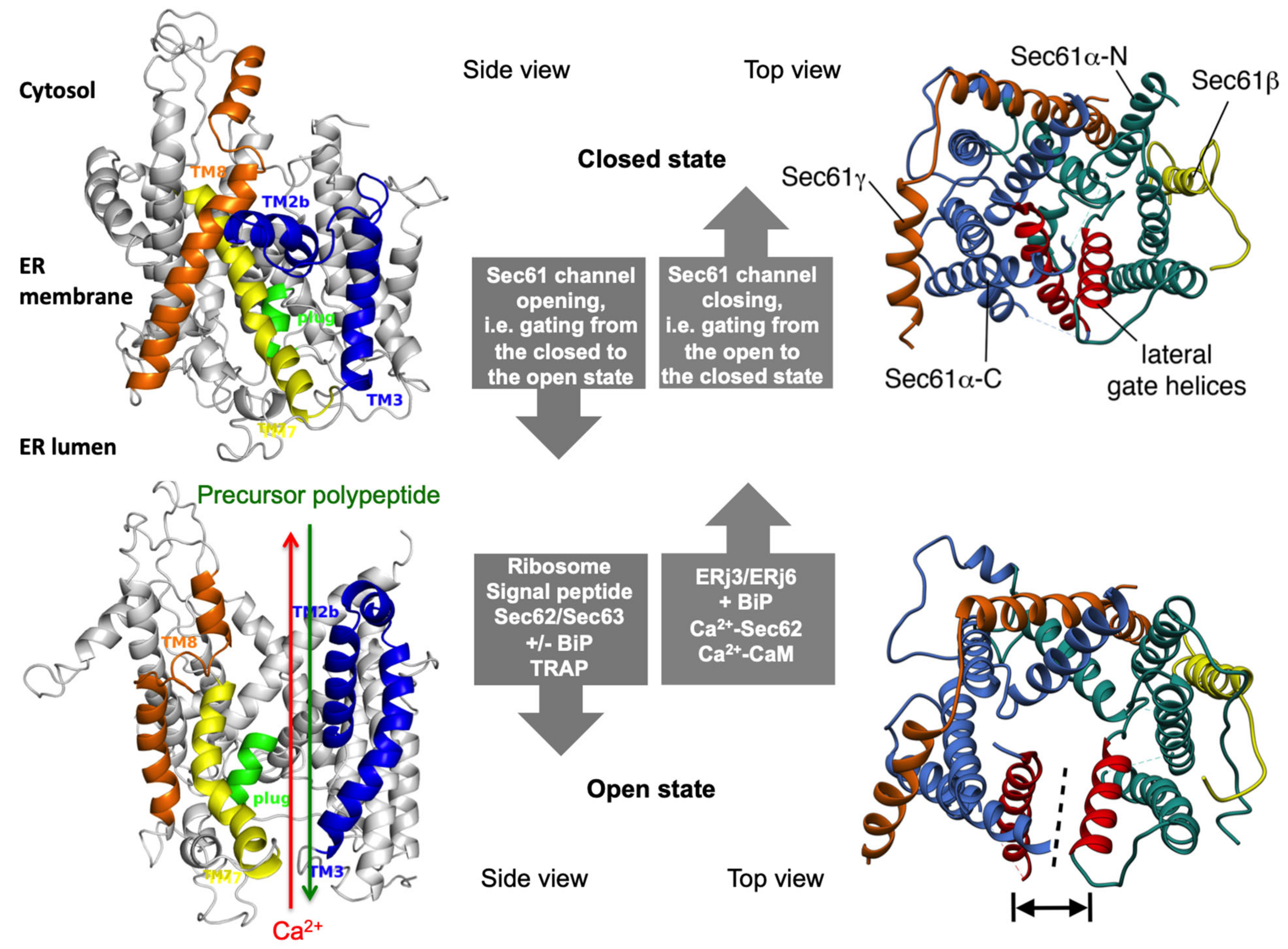{kind=link}
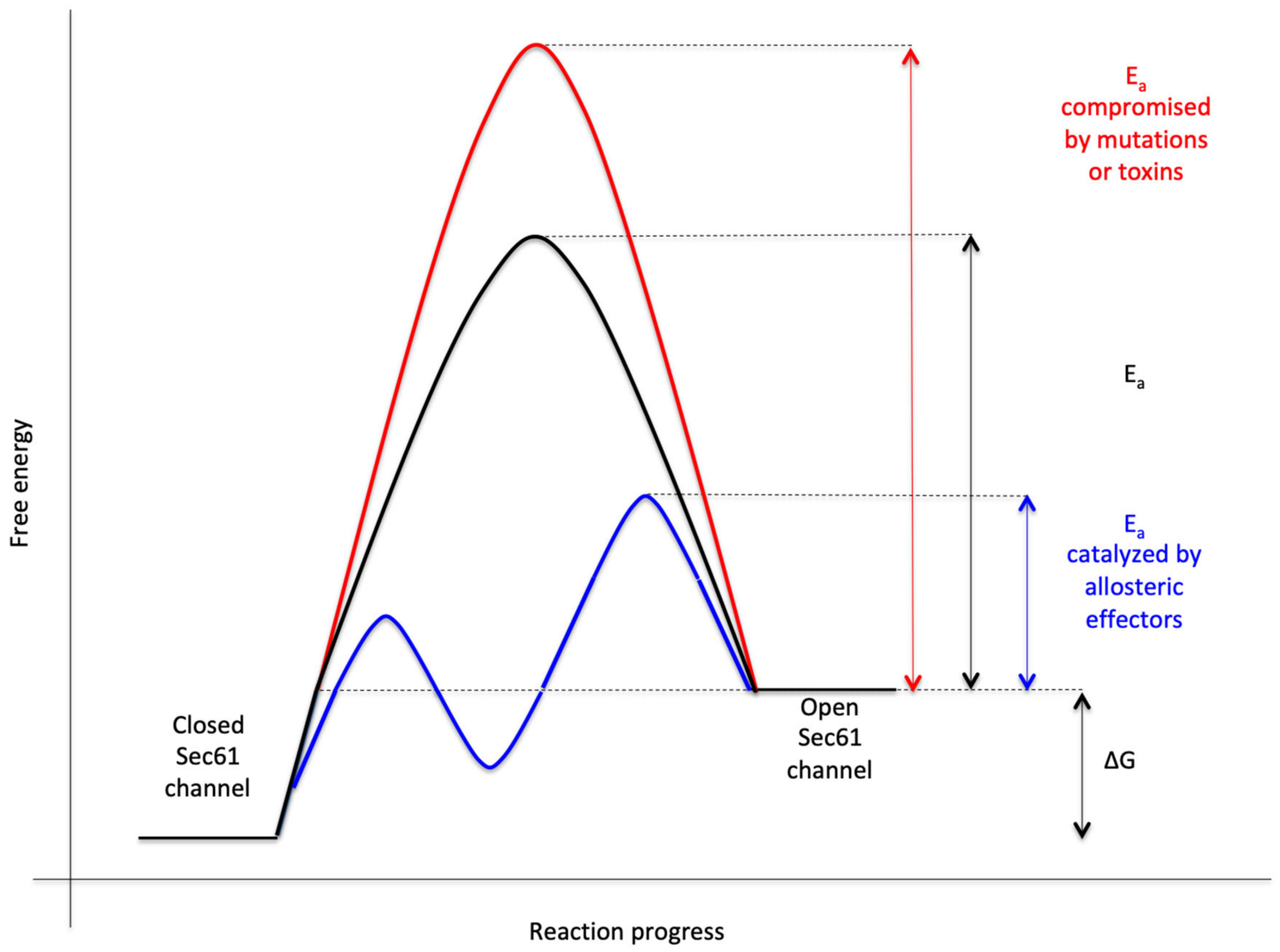{kind=link}
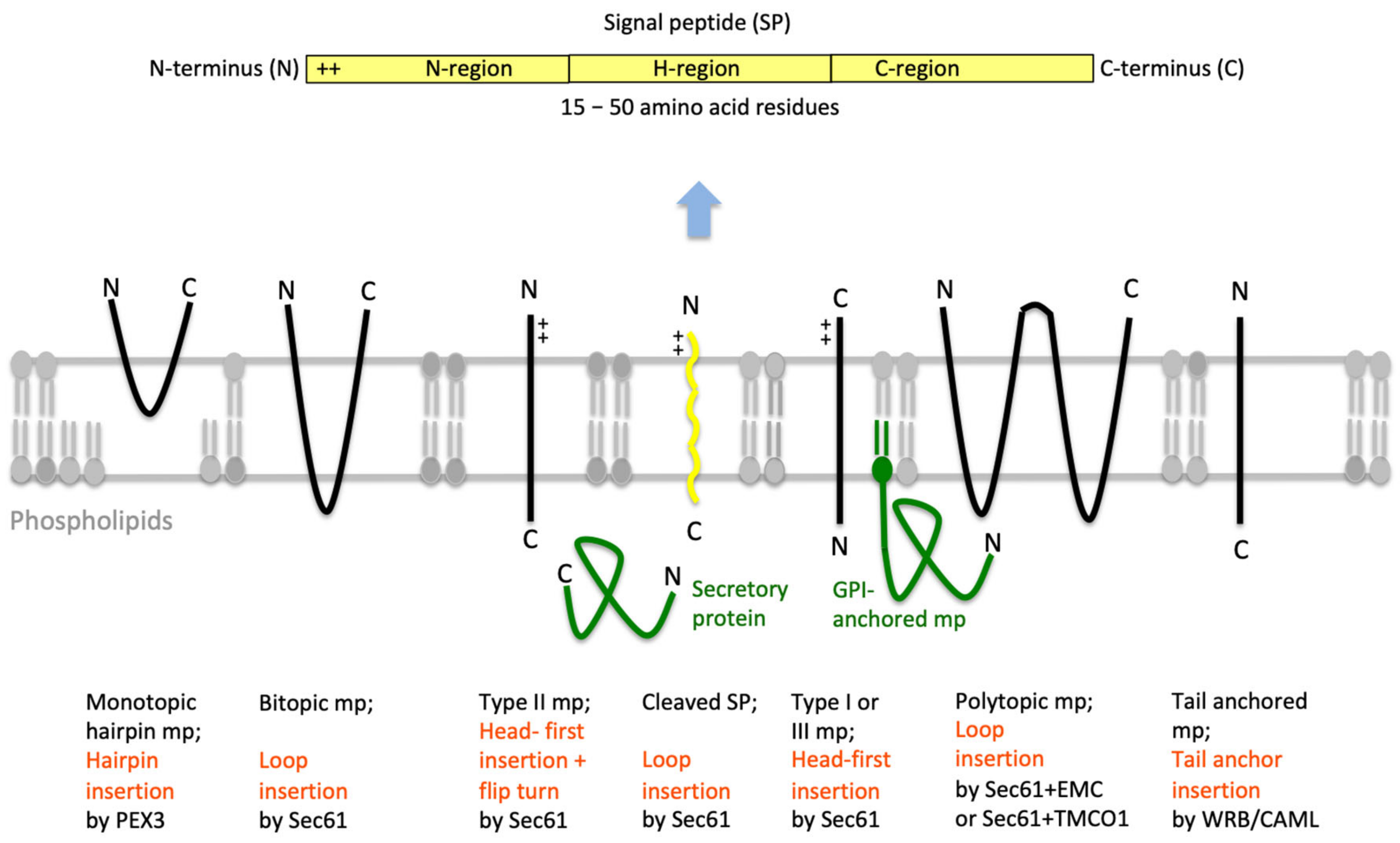{kind=link}
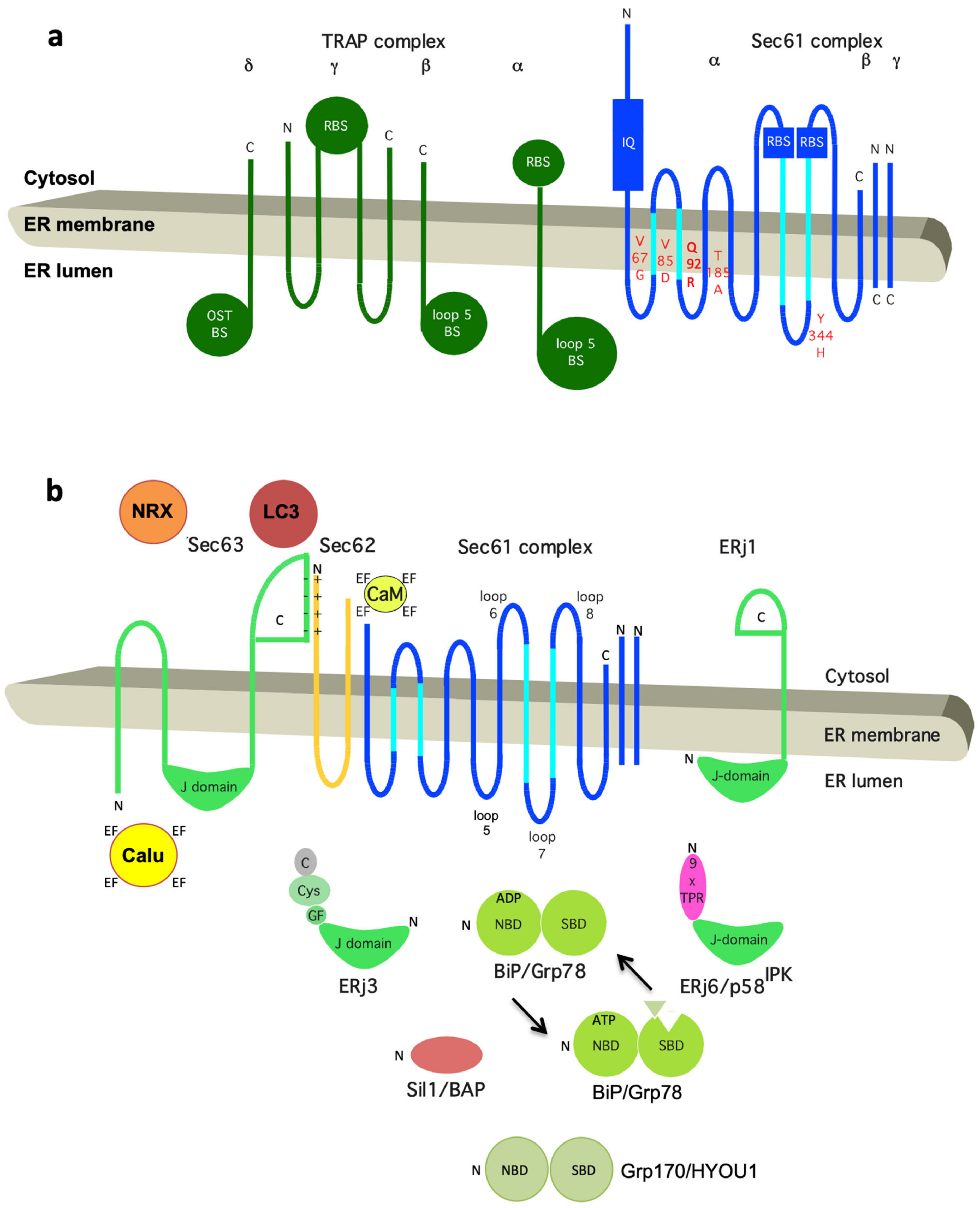{kind=link}
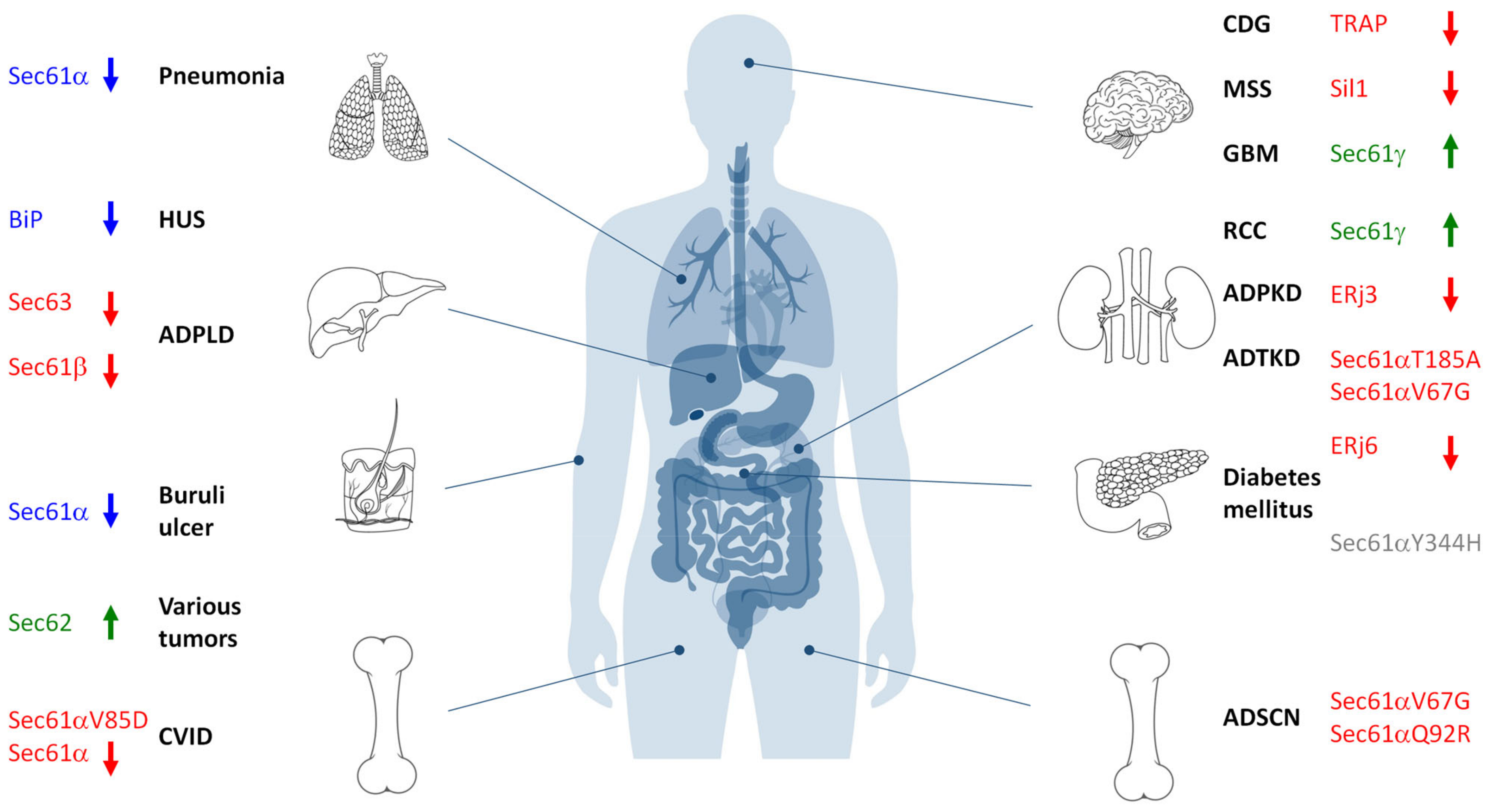{kind=link}
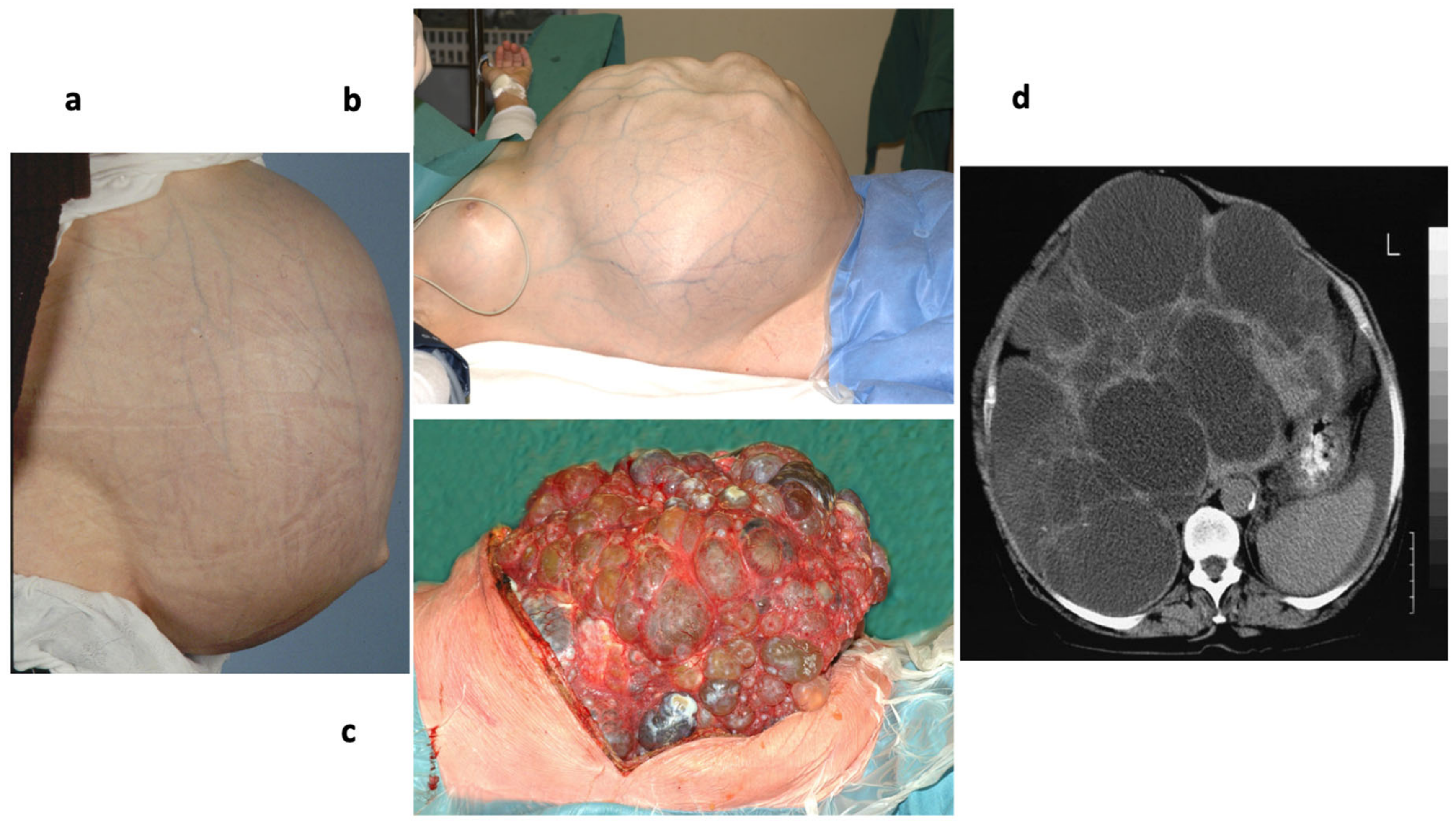{kind=link}
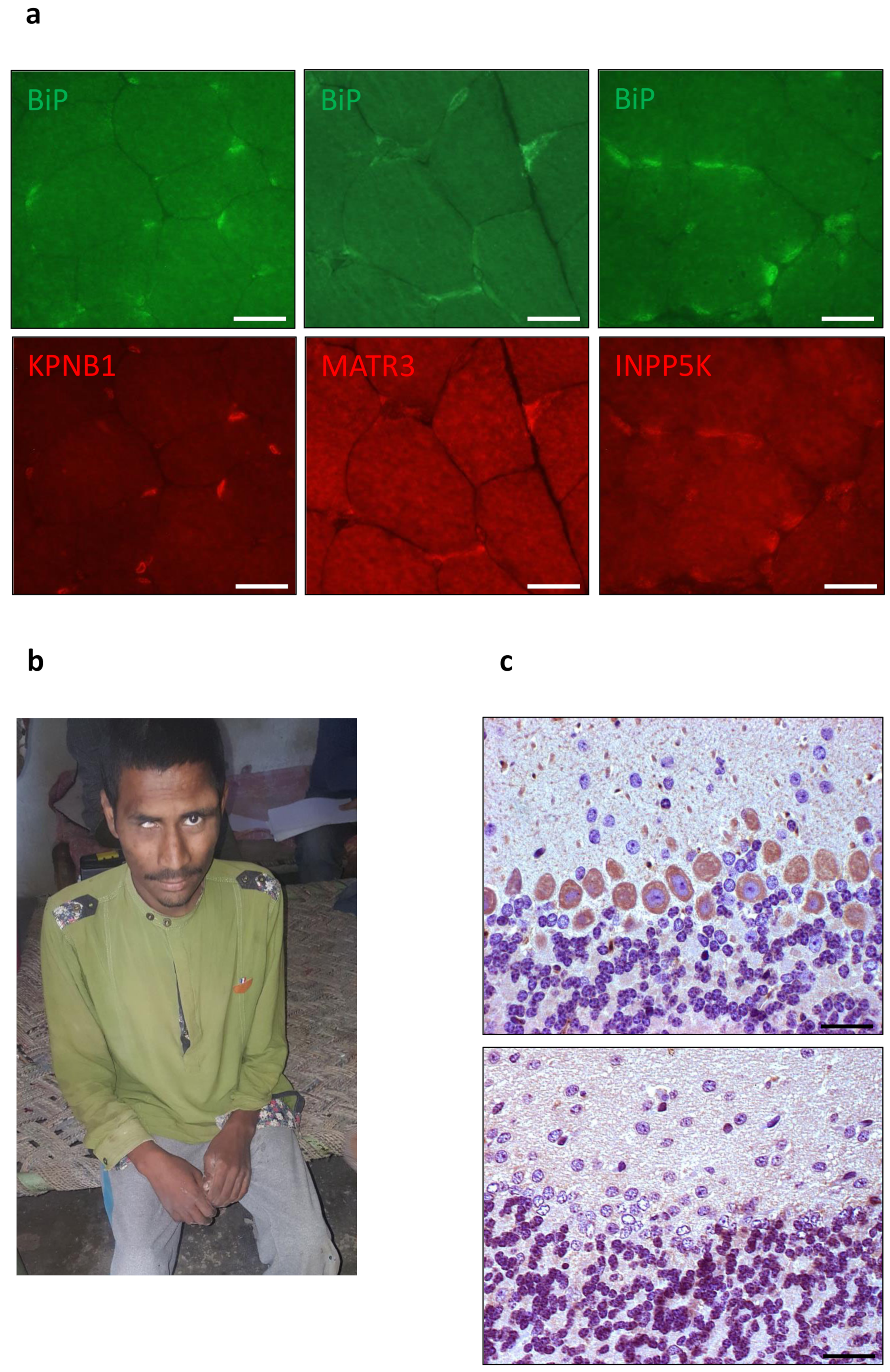{kind=link}
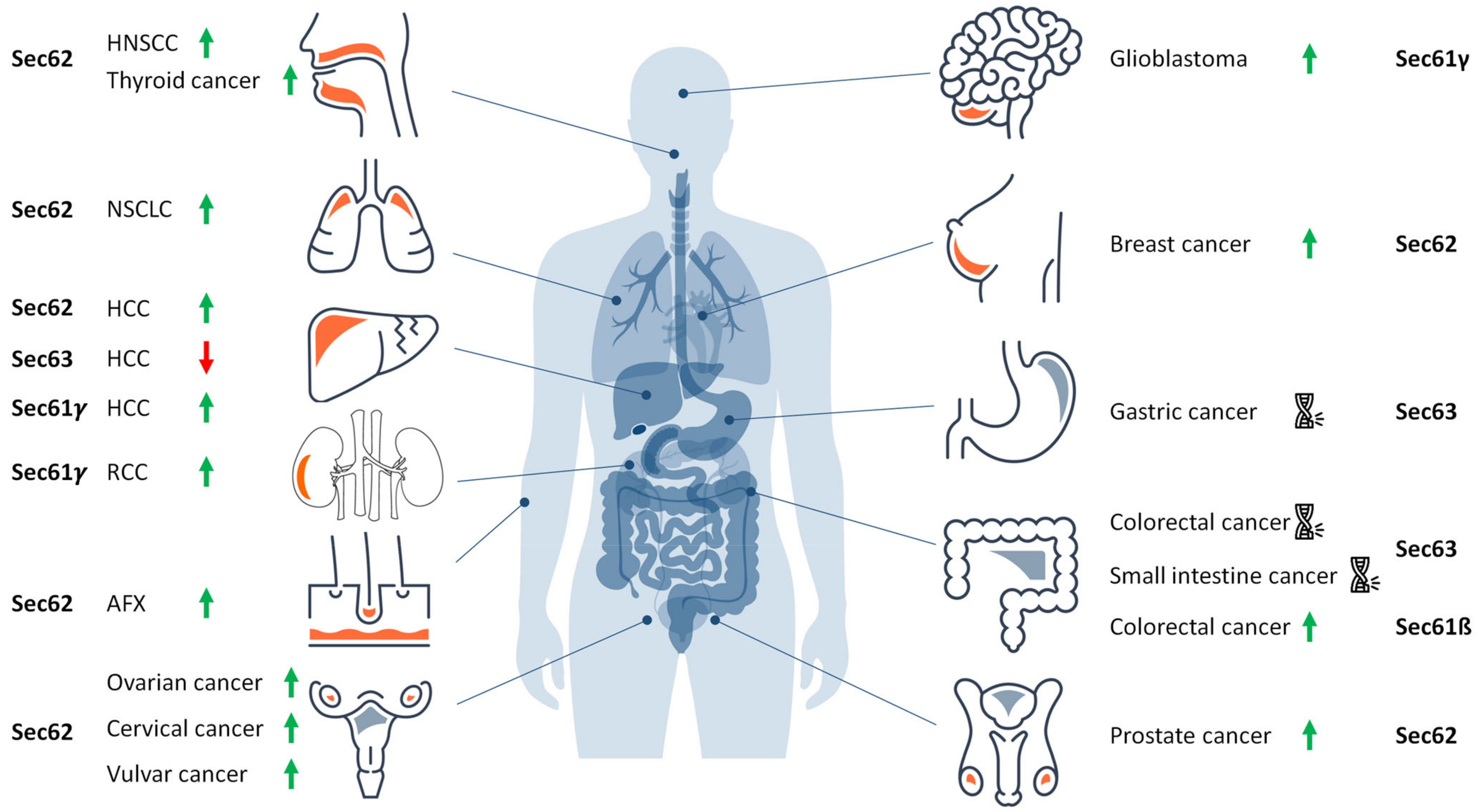{kind=link}
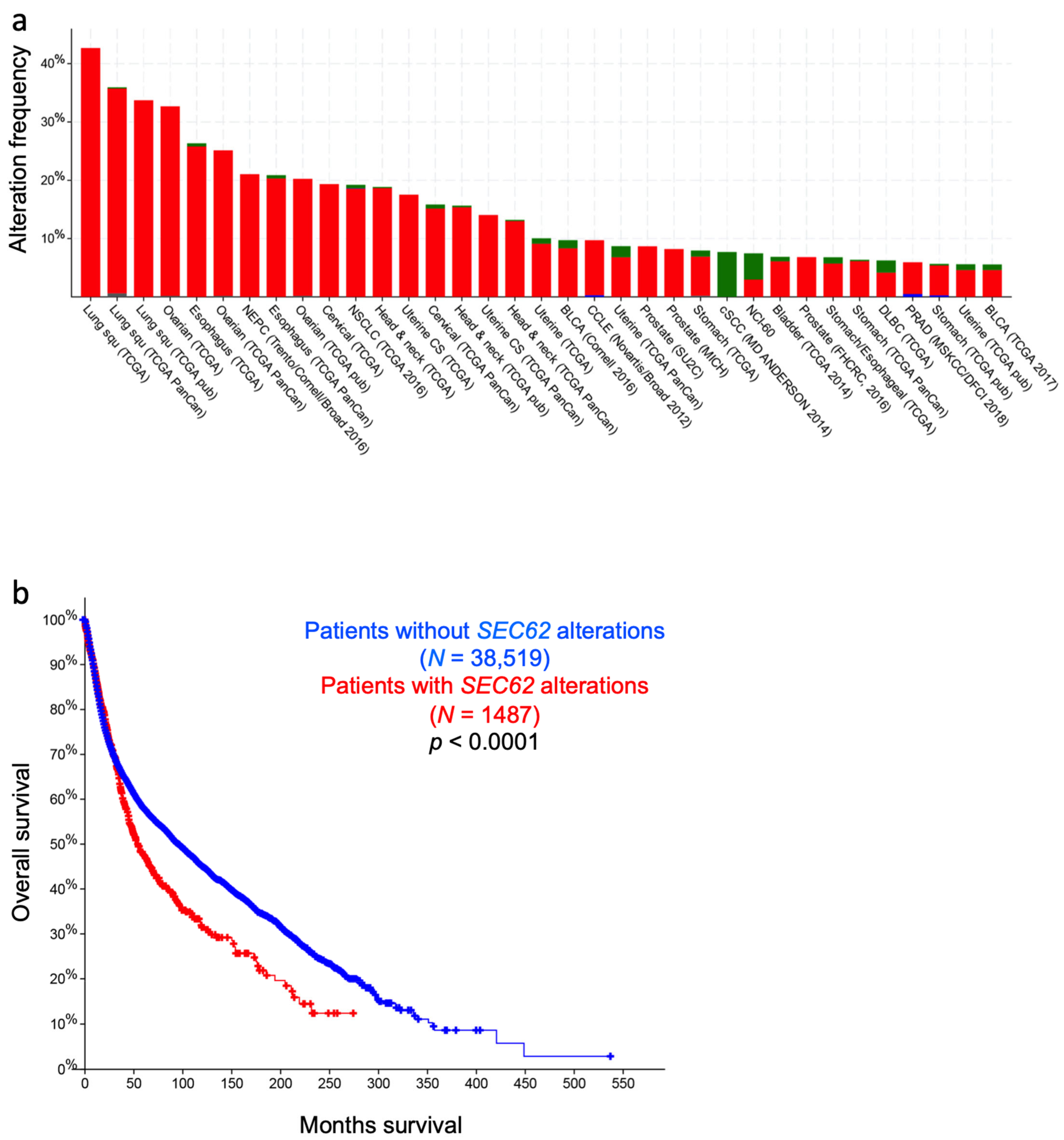{kind=link}
| Component/subunit | Abundance | Location | Linked Diseases |
|---|---|---|---|
| Calmodulin | 9428 | C | |
| Cytosolic Chaperones | C | ||
| Hsc70 (HSPA8) | 3559 | ||
| Hdj2 (DNAJA1) | 660 | ||
| Bag1 (HAP, RAP46) | 46 | ||
| #NAC | C | ||
| - NACα | 1412 | ||
| - NACβ | |||
| #SRP | C | ||
| - SRP72 | 355 | Aplasia, Myelodysplasia | |
| - SRP68 | 197 | ||
| - SRP54 | 228 | Neutropenia, Pancreas Insufficiency | |
| - SRP19 | 33 | ||
| - SRP14 | 4295 | ||
| - SRP9 | 3436 | ||
| - 7SL RNA | |||
| SRP receptor | ERM | ||
| - SRα (docking protein) | 249 | ||
| - SRβ | 173 | ||
| hSnd1 | unknown | ||
| Snd receptor | ERM | ||
| - hSnd2 (TMEM208) | 81 | ||
| - hSnd3 | 49 | ||
| #Bag6 complex | C | ||
| - TRC35 (Get4) | 171 | ||
| - Ubl4A | 177 | ||
| - Bag6 (Bat3) | 133 | ||
| SGTA | 549 | C | |
| TRC40 (Asna1, Get3) | 381 | C | |
| TA receptor | ERM | ||
| - CAML (CAMLG, Get2) | 5 | ||
| - WRB (CHD5, Get1) | 4 | Congenital Heart Disease | |
| ERM protein complex | ERM | ||
| - EMC1 | 124 | ||
| - EMC2 | 300 | ||
| - EMC3 | 270 | ||
| - EMC4 | 70 | ||
| - EMC5 (MMGT1) | 35 | ||
| - EMC6 (TMEM93) | 5 | ||
| - EMC7 | 247 | ||
| - EMC8 | 209 | ||
| - EMC9 | 1 | ||
| - EMC10 | 3 | ||
| #TMCO1 complex | ERM | ||
| - TMCO1 ## | 2013 | Glaucoma, Cerebrofaciothoracic Dysplasia | |
| - Nicalin | 99 | ||
| - TMEM147 | 21 | ||
| - CCDC47 (Calumin) | 193 | ||
| - NOMO | 267 | ||
| PAT complex | ERM | ||
| - PAT10 (Asterix) | |||
| - CCDC47 (Calumin) | 193 | ||
| PEX19 | 80 | C | Zellweger Syndrome |
| PEX3 | 103 | ERM | Zellweger Syndrome |
| #Sec61 complex ## | ERM | ||
| - Sec61α1 | 139 | Diabetes **, CVID, TKD, Neutropenia | |
| - Sec61β | 456 | PLD, Colorectal Cancer | |
| - Sec61γ | 400 | GBM, Hepatocellular Carcinoma, RCC | |
| #Sec62 (TLOC1) | 26 | ERM | Breast-, Prostate-, Cervix-, Lung-Cancer et al. |
| ER Chaperones | |||
| Sec63 (ERj2) | 168 | ERM | PLD, Colorectal Cancer et al. |
| #ERj1 (DNAJC1) | 8 | ERM | |
| ERj3 (DNAJB11) | 1001 | ERL | Polycystic Kidney Disease (PKD) |
| ERj4 (DNAJB9) | 12 | ERL | |
| ERj5 (DNAJC10) | 43 | ERL | |
| ERj6 (DNAJC3, p58IPK) | 237 | ERL | Diabetes, Neurodegeneration |
| ERj7 (DNAJC25) | 10 | ERM | Hyperinsulinismus, Allergic Asthma |
| ERj8 (DNAJC16) | 24 | ERM | |
| ERj9 (DNAJC22) | ERM | ||
| BiP (Grp78, HSPA5) | 8253 | ERL | Hemolytic Uremic Syndrome (HUS) |
| Grp170 (HYOU1) | 923 | ERL | Immunodeficincy & Hypoglycemia |
| Sil1 (BAP) | 149 | ERL | Marinesco-Sjögren-Syndrome (MSS) |
| Grp94 (CaBP4, Hsp90B1) | 4141 | ERL | |
| PPIB (Cyclophilin B) | 1289 | ERL | |
| FKBP2 (FKBP13) | 894 | ERL | |
| PDIA1 (PDI, ERp59) | 3624 | ERL | Cole-Carpenter Syndrome |
| PDIA2 (PDIp) | ERL | ||
| PDIA3 (ERp61, Grp57) | 3730 | ERL | |
| PDIA4 (ERp72, CaBP2) | 2173 | ERL | |
| PDIA5 (PDIR) | 37 | ERL | |
| PDIA6 (P5, CaBP1) | 3001 | ERL | |
| PDIA9 (ERp29) | ERL | ||
| Calreticulin (CaBP3, ERp60) | 14521 | ERL | |
| #Calnexinpalmitoylated | 7278 | ERM | |
| #TRAM1 | 26 | ERM | |
| TRAM2 | 40 | ERM | |
| #TRAP complex | ERM | ||
| - TRAPα (SSR1) | 568 | ||
| - TRAPβ (SSR2) | |||
| - TRAPγ (SSR3) | 1701 | CDG, Hepatocellular Carcinoma | |
| - TRAPδ (SSR4) | 3212 | CDG | |
| #RAMP4 (SERP1) | ERM | ||
| #Oligosaccharyltransferase (OST-A) | ERM | ||
| - RibophorinI (Rpn1) | 1956 | ||
| - RibophorinII (Rpn2) | 527 | ||
| - OST48 | 273 | CDG | |
| - Dad1 | 464 | ||
| - OST4 | |||
| - TMEM258 | |||
| - Stt3A * | 430 | CDG | |
| - DC2 | |||
| - Kcp2 | |||
| Oligosaccharyltransferase (OST-B) | ERM | ||
| - RibophorinI (Rpn1) | 1956 | ||
| - RibophorinII (Rpn2) | 527 | ||
| - OST48 | 273 | CDG | |
| - Dad1 | 464 | ||
| - OST4 | |||
| - TMEM258 | |||
| - Stt3B* | 150 | CDG | |
| - TUSC3 | CDG | ||
| - MagT1 | 33 | ||
| Signal peptidase (SPC-A) | ERM | ||
| - SPC12 | 2733 | ||
| - SPC18 * (SEC11A) | |||
| - SPC22/23 | 334 | ||
| - SPC25 | 94 | ||
| Signal peptidase (SPC-C) | ERM | ||
| - SPC12 | 2733 | ||
| - SPC21 * (SEC11C) | |||
| - SPC22/23 | 334 | ||
| - SPC25 | 94 | ||
| GPI transamidase (GPI-T) | ERM | ||
| - GPAA1 | 9 | ||
| - PIG-K | 38 | ||
| - PIG-S | 86 | ||
| - PIG-T | 20 | ||
| - PIG-U | 42 | ||
| Additional modifying enzymes | |||
| ALG8 | 10 | ERM | CDG, PLD |
| UGGT | 232 | ERL | |
| Glucosidase IIα (GIIα) | ERL | PLD | |
| Glucosidase IIβ (PRKCSH, GIIβ) | ERL | PLD | |
| Proly-4-hydroxylase α (4-PH) | ERL | ||
| Proly-4-hydroxylase β (PDI) | 3624 | ERL | Cole-Carpenter Syndrome |
| SUMF1 | 23 | ERL | Multiple Sulfatase Deficiency |
| SUMF2 | 386 | ERL | Multiple Sulfatase Deficiency |
| #p34 (LRC59, LRRC59) | 2480 | ERM | |
| #p180 (RRBP1) | 135 | ERM | Hepatocellular Carcinoma, Colorectal Cancer |
| Kinectin 1 (KTN1) | 263 | ERM |
| Inhibitor | Binding Site in Sec61α | Effect | Linked Disease(s) |
|---|---|---|---|
| Apratoxin A | T86 (TMH2), Y131 (TMH5) | non-selective | |
| CAM741 | selective inhibition | ||
| Coibamide A | non-selective | ||
| Cotransin CT8 | R66 (loop 1), G80 (TMH2), S82 (loop 1), M136 (TMH3) | selective inhibition | |
| Eeyarestatin ES24 | non-selective Ca2+ leak inducing | ||
| Exotoxin A | N-terminus | non-selective Ca2+ leak blocking | Pneumonia, Sepsis |
| Ipomoeassin F | non-selective | ||
| Mycolactone | R66 (loop 1), S82 (loop 1) | selective inhibition Ca2+ leak inducing | Buruli ulcer |
| Disease | Linked Gene(s) | Sec61 Effect | MIM |
|---|---|---|---|
| ADPKD | DNAJB11 | Ca2+ leak induction * | |
| PKD1, PKD2, UMOD | |||
| ADPLD | ALG8, GANAB, PRKCSH, | ||
| SEC61B, SEC63 | selective transport inhibition | 617004 | |
| ADSCN | ELANE, JAGN1, SEC61A1 | transport inhibition | |
| Ca2+ leak induction | |||
| ADTKD | HNF1B, MUC1, REN, | selective transport inhibition | |
| SEC61A1, UMOD | Ca2+ leak induction | 617056 | |
| CDG | ALG8, SSR3, SSR4, OST48, | selective transport inhibition | |
| STT3A, STT3B, TUSC3 | |||
| CVID | CD19, CD20, CD21, | transport inhibition | |
| CD80, SEC61A1 | Ca2+ leak induction | ||
| Diabetes | DNAJC3, INS | Ca2+ leak induction * | |
| MSS | SIL1 | 248800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sicking, M.; Lang, S.; Bochen, F.; Roos, A.; Drenth, J.P.H.; Zakaria, M.; Zimmermann, R.; Linxweiler, M. Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells 2021, 10, 1036. https://doi.org/10.3390/cells10051036
Sicking M, Lang S, Bochen F, Roos A, Drenth JPH, Zakaria M, Zimmermann R, Linxweiler M. Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells. 2021; 10(5):1036. https://doi.org/10.3390/cells10051036
Chicago/Turabian StyleSicking, Mark, Sven Lang, Florian Bochen, Andreas Roos, Joost P. H. Drenth, Muhammad Zakaria, Richard Zimmermann, and Maximilian Linxweiler. 2021. "Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex" Cells 10, no. 5: 1036. https://doi.org/10.3390/cells10051036
APA StyleSicking, M., Lang, S., Bochen, F., Roos, A., Drenth, J. P. H., Zakaria, M., Zimmermann, R., & Linxweiler, M. (2021). Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells, 10(5), 1036. https://doi.org/10.3390/cells10051036