
Rho GTPases in Skeletal Muscle Development and Homeostasis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
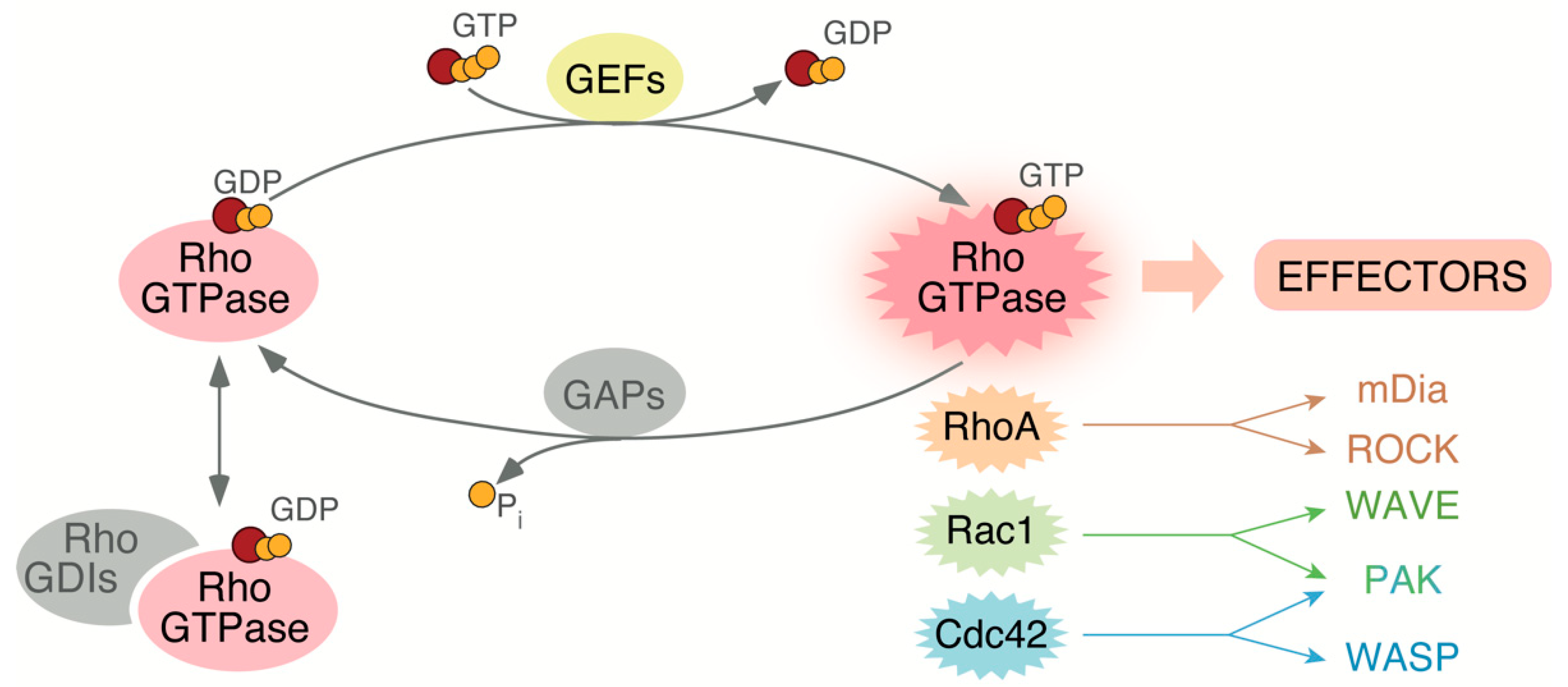
2. Rho GTPases Regulation, Family Members, and Roles
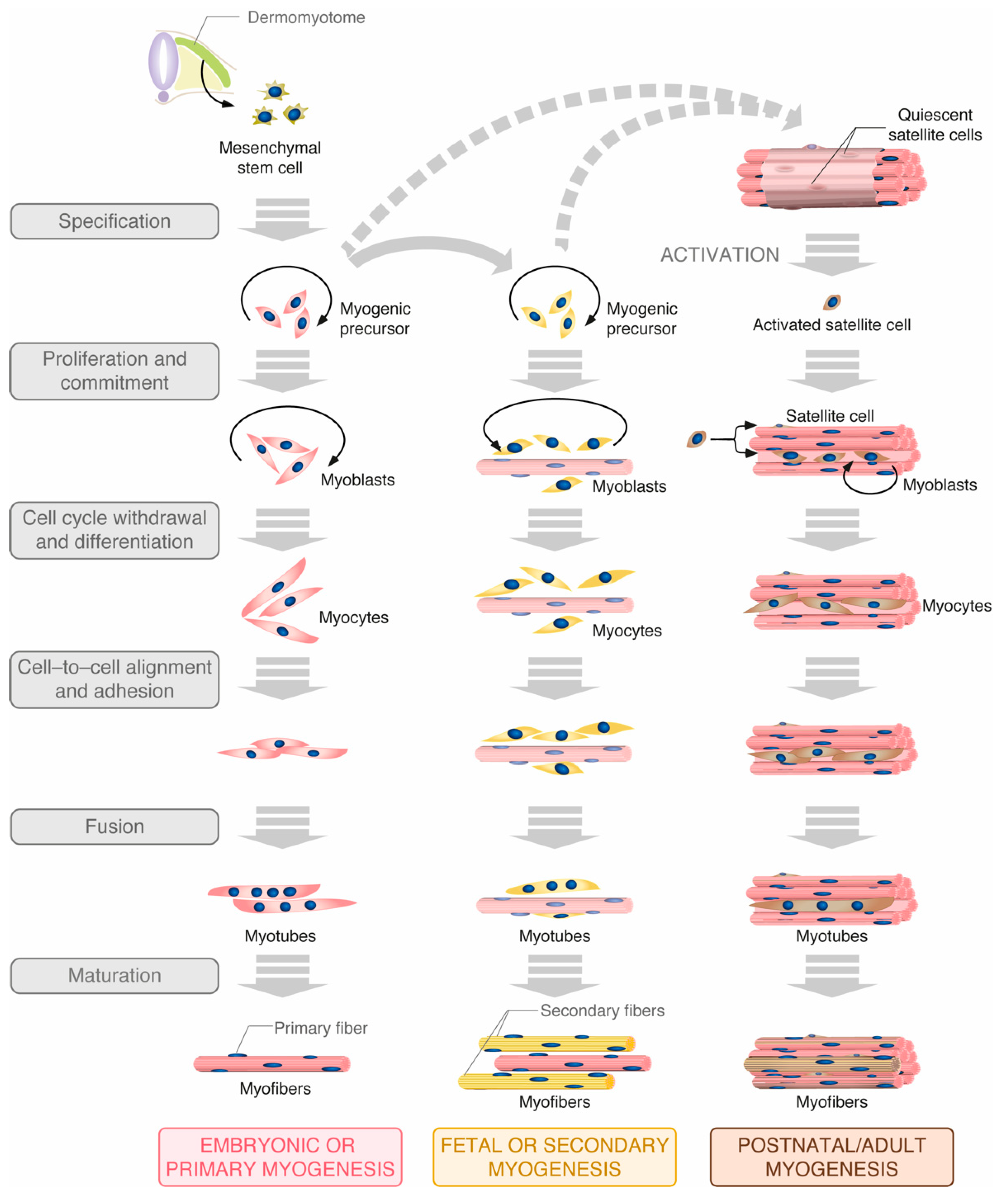
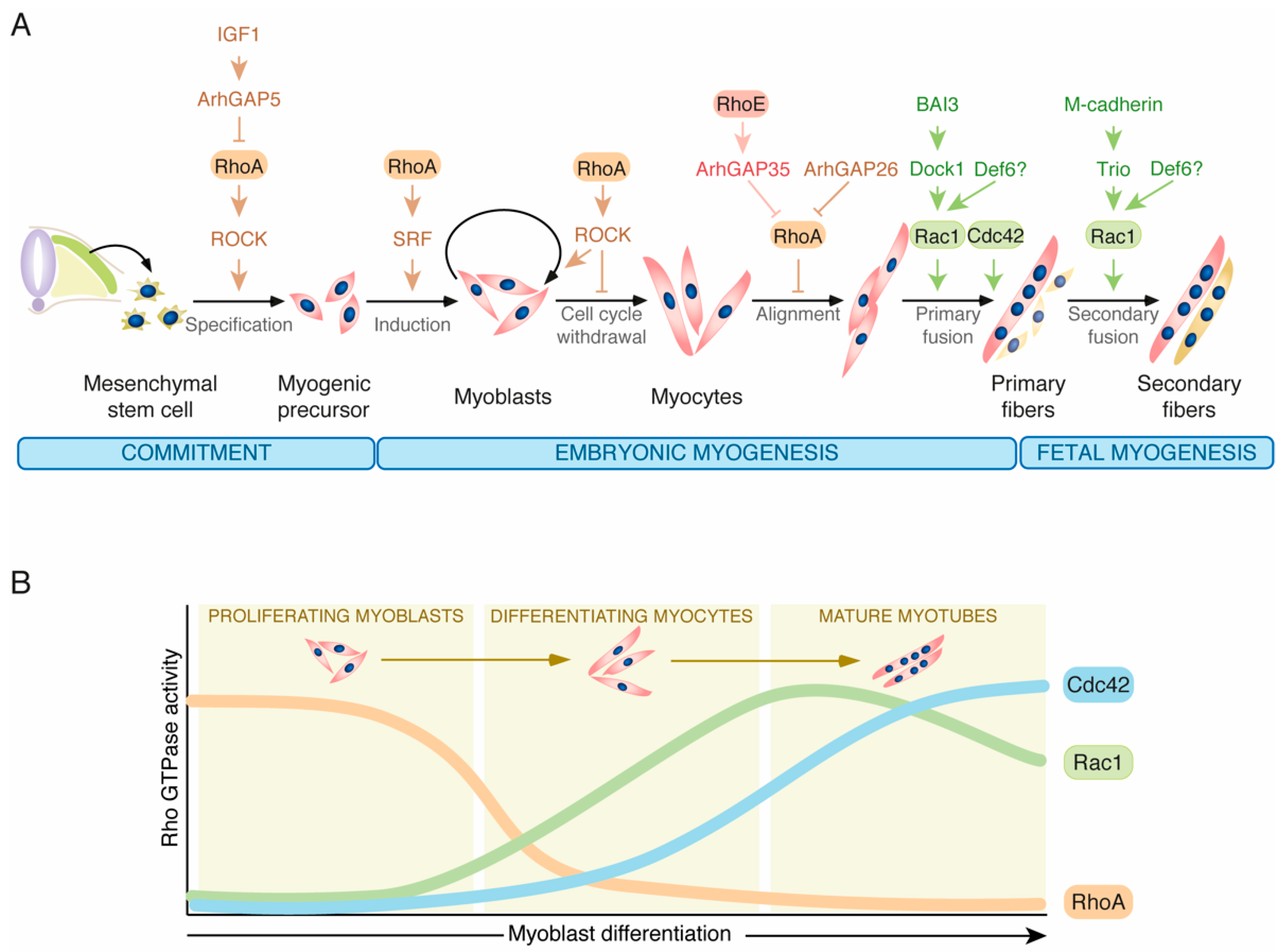
2.1. Rho GTPases in Muscle Differentiation
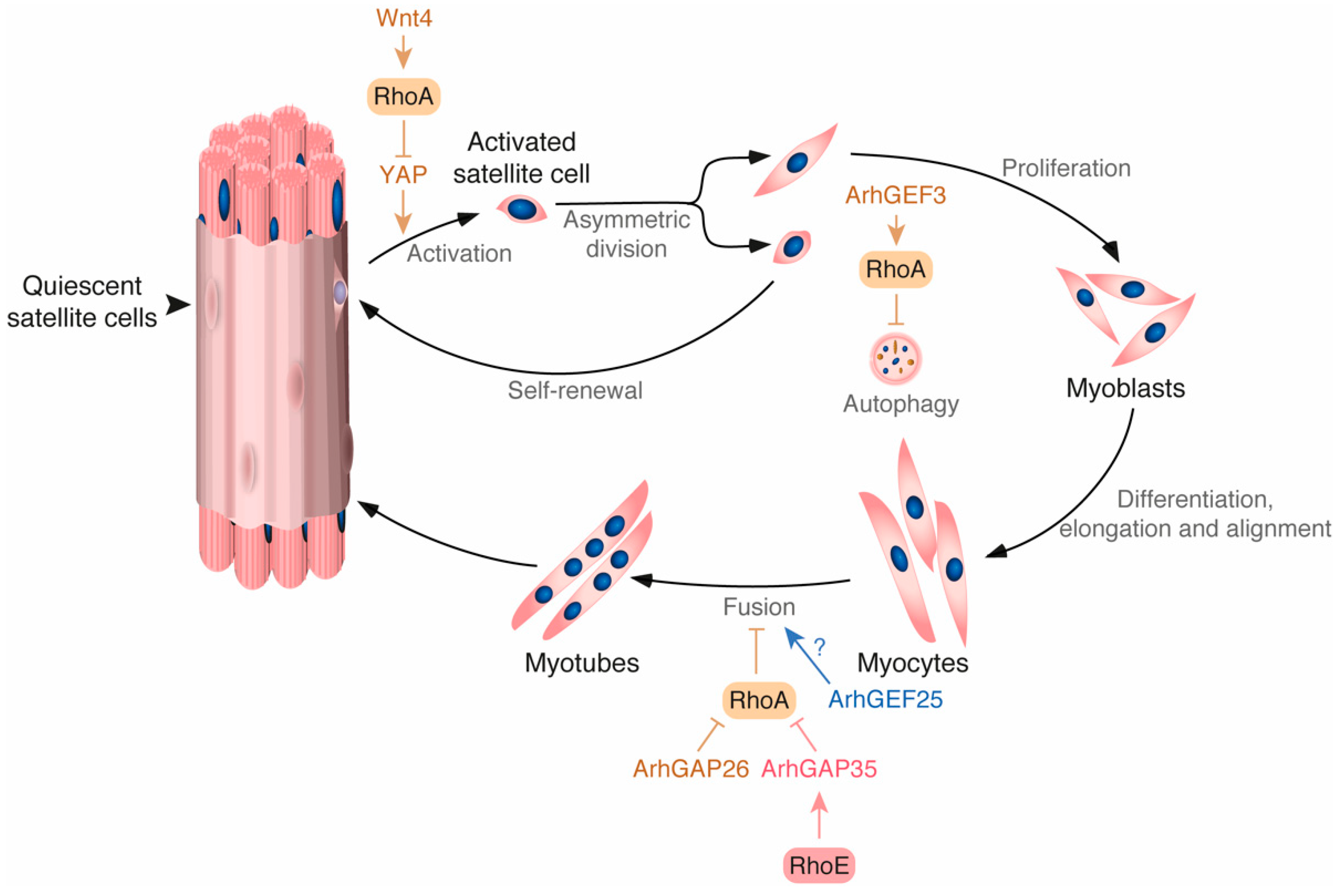
2.2. Rho GTPases in Muscle Regeneration
2.3. Rho GTPases in Muscle Mass Regulation
2.4. Rho GTPases Signalling in Sarcomere Banding Patterning
2.5. Rho GTPases in Skeletal Muscle-Related Diseases
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Petersen, K.F.; Shulman, G.I. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 11G–18G. [Google Scholar] [CrossRef]
- Betz, M.J.; Enerback, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef]
- Dennison, E.M.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Messina, G.; Cossu, G. The origin of embryonic and fetal myoblasts: A role of Pax3 and Pax7. Genes Dev. 2009, 23, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Hindi, S.M.; Tajrishi, M.M.; Kumar, A. Signaling mechanisms in mammalian myoblast fusion. Sci. Signal. 2013, 6, re2. [Google Scholar] [CrossRef] [PubMed]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Goncalves, D.A.; Silveira, W.A.; Manfredi, L.H.; Graca, F.A.; Armani, A.; Bertaggia, E.; O’Neill, B.T.; Lautherbach, N.; Machado, J.; Nogara, L.; et al. Insulin/IGF1 signalling mediates the effects of beta2 -adrenergic agonist on muscle proteostasis and growth. J. Cachexia Sarcopenia Muscle 2019, 10, 455–475. [Google Scholar] [CrossRef]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef] [PubMed]
- Velloso, C.P. Regulation of muscle mass by growth hormone and IGF-I. Br. J. Pharmacol. 2008, 154, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras superfamily of small GTPases: The unlocked secrets. In Ras Signaling; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1120, pp. 1–18. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. Bioessays 2007, 29, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Touaitahuata, H.; Blangy, A.; Vives, V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases 2014, 5, e28119. [Google Scholar] [CrossRef] [PubMed]
- Otsu, K.; Harada, H. Rho GTPases in ameloblast differentiation. Jpn. Dent. Sci. Rev. 2016, 52, 32–40. [Google Scholar] [CrossRef]
- Bryan, B.A.; Li, D.; Wu, X.; Liu, M. The Rho family of small GTPases: Crucial regulators of skeletal myogenesis. Cell. Mol. Life Sci. 2005, 62, 1547–1555. [Google Scholar] [CrossRef]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Machin, P.A.; Tsonou, E.; Hornigold, D.C.; Welch, H.C.E. Rho Family GTPases and Rho GEFs in Glucose Homeostasis. Cells 2021, 10, 915. [Google Scholar] [CrossRef]
- Moller, L.L.V.; Klip, A.; Sylow, L. Rho GTPases-Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells 2019, 8, 434. [Google Scholar] [CrossRef]
- El Masri, R.; Delon, J. RHO GTPases: From new partners to complex immune syndromes. Nat. Rev. Immunol. 2021, 21, 499–513. [Google Scholar] [CrossRef]
- Fabbiano, S.; Menacho-Marquez, M.; Sevilla, M.A.; Albarran-Juarez, J.; Zheng, Y.; Offermanns, S.; Montero, M.J.; Bustelo, X.R. Genetic dissection of the vav2-rac1 signaling axis in vascular smooth muscle cells. Mol. Cell. Biol. 2014, 34, 4404–4419. [Google Scholar] [CrossRef]
- Sauzeau, V.; Jerkic, M.; Lopez-Novoa, J.M.; Bustelo, X.R. Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol. Biol. Cell 2007, 18, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Sevilla, M.A.; Montero, M.J.; Bustelo, X.R. The Rho/Rac exchange factor Vav2 controls nitric oxide-dependent responses in mouse vascular smooth muscle cells. J. Clin. Investig. 2010, 120, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fdez, S.; Lorenzo-Martín, L.F.; Fabbiano, S.; Menacho-Márquez, M.; Sauzeau, V.; Dosil, M.; Bustelo, X.R. New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System. Biology 2021, 10, 857. [Google Scholar] [CrossRef]
- Boettner, B.; Van Aelst, L. The role of Rho GTPases in disease development. Gene 2002, 286, 155–174. [Google Scholar] [CrossRef]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Charrasse, S.; Causeret, M.; Comunale, F.; Bonet-Kerrache, A.; Gauthier-Rouviere, C. Rho GTPases and cadherin-based cell adhesion in skeletal muscle development. J. Muscle Res. Cell Motil. 2003, 24, 309–313. [Google Scholar] [CrossRef]
- Chiu, T.T.; Jensen, T.E.; Sylow, L.; Richter, E.A.; Klip, A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell. Signal. 2011, 23, 1546–1554. [Google Scholar] [CrossRef]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef]
- Aspenstrom, P.; Ruusala, A.; Pacholsky, D. Taking Rho GTPases to the next level: The cellular functions of atypical Rho GTPases. Exp. Cell Res. 2007, 313, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on Rho GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- You, J.S.; Singh, N.; Reyes-Ordonez, A.; Khanna, N.; Bao, Z.; Zhao, H.; Chen, J. ARHGEF3 Regulates Skeletal Muscle Regeneration and Strength through Autophagy. Cell Rep. 2021, 34, 108594. [Google Scholar] [CrossRef] [PubMed]
- Laurin, M.; Fradet, N.; Blangy, A.; Hall, A.; Vuori, K.; Cote, J.F. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 15446–15451. [Google Scholar] [CrossRef]
- Moore, C.A.; Parkin, C.A.; Bidet, Y.; Ingham, P.W. A role for the myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development 2007, 134, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Charrasse, S.; Comunale, F.; Fortier, M.; Portales-Casamar, E.; Debant, A.; Gauthier-Rouviere, C. M-cadherin activates Rac1 GTPase through the Rho-GEF trio during myoblast fusion. Mol. Biol. Cell 2007, 18, 1734–1743. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef]
- Rodriguez-Fdez, S.; Lorenzo-Martin, L.F.; Fernandez-Pisonero, I.; Porteiro, B.; Veyrat-Durebex, C.; Beiroa, D.; Al-Massadi, O.; Abad, A.; Dieguez, C.; Coppari, R.; et al. Vav2 catalysis-dependent pathways contribute to skeletal muscle growth and metabolic homeostasis. Nat. Commun. 2020, 11, 5808. [Google Scholar] [CrossRef] [PubMed]
- Young, P.; Ehler, E.; Gautel, M. Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J. Cell Biol. 2001, 154, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Ford-Speelman, D.L.; Roche, J.A.; Bowman, A.L.; Bloch, R.J. The rho-guanine nucleotide exchange factor domain of obscurin activates rhoA signaling in skeletal muscle. Mol. Biol. Cell 2009, 20, 3905–3917. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bi, F.; Zhou, X.; Zheng, Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012, 22, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef] [PubMed]
- Lenhart, K.C.; Becherer, A.L.; Li, J.; Xiao, X.; McNally, E.M.; Mack, C.P.; Taylor, J.M. GRAF1 promotes ferlin-dependent myoblast fusion. Dev. Biol. 2014, 393, 298–311. [Google Scholar] [CrossRef][Green Version]
- Doherty, J.T.; Lenhart, K.C.; Cameron, M.V.; Mack, C.P.; Conlon, F.L.; Taylor, J.M. Skeletal Muscle Differentiation and Fusion Are Regulated by the BAR-containing Rho-GTPase-activating Protein (Rho-GAP), GRAF1. J. Biol. Chem. 2011, 286, 25903–25921. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Jiang, W.; Chen, G.C.; Curto, M.; Settleman, J. Modulation of rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell 2003, 113, 147–158. [Google Scholar] [CrossRef]
- Fortier, M.; Comunale, F.; Kucharczak, J.; Blangy, A.; Charrasse, S.; Gauthier-Rouviere, C. RhoE controls myoblast alignment prior fusion through RhoA and ROCK. Cell Death Differ. 2008, 15, 1221–1231. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ‘invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Rocklin, G.; Seo, J.Y.; Bokoch, G.M. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell 2006, 17, 4760–4768. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Schnelzer, A.; Bokoch, G.M. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol. Cell 2004, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Navarro-Lerida, I.; Muriel, O.; Couceiro, J.R.; Pimentel-Muinos, F.X.; Del Pozo, M.A.; Bustelo, X.R. Coronin 1A promotes a cytoskeletal-based feedback loop that facilitates Rac1 translocation and activation. EMBO J. 2011, 30, 3913–3927. [Google Scholar] [CrossRef]
- Fort, P.; Blangy, A. The Evolutionary Landscape of Dbl-Like RhoGEF Families: Adapting Eukaryotic Cells to Environmental Signals. Genome Biol. Evol. 2017, 9, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Rac-ing to the plasma membrane: The long and complex work commute of Rac1 during cell signaling. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Bagci, H.; Sriskandarajah, N.; Robert, A.; Boulais, J.; Elkholi, I.E.; Tran, V.; Lin, Z.Y.; Thibault, M.P.; Dube, N.; Faubert, D.; et al. Mapping the proximity interaction network of the Rho-family GTPases reveals signalling pathways and regulatory mechanisms. Nat. Cell Biol. 2020, 22, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Kato, T.; Fujita, A.; Ishizaki, T.; Narumiya, S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1999, 1, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Eden, S.; Rohatgi, R.; Podtelejnikov, A.V.; Mann, M.; Kirschner, M.W. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 2002, 418, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef]
- Hill, C.S.; Wynne, J.; Treisman, R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995, 81, 1159–1170. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sanchez-Perez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Teramoto, H.; Coso, O.A.; Miyata, H.; Igishi, T.; Miki, T.; Gutkind, J.S. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 1996, 271, 27225–27228. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Carnac, G.; Primig, M.; Kitzmann, M.; Chafey, P.; Tuil, D.; Lamb, N.; Fernandez, A. RhoA GTPase and serum response factor control selectively the expression of MyoD without affecting Myf5 in mouse myoblasts. Mol. Biol. Cell 1998, 9, 1891–1902. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, W.; Croissant, J.D.; Johansen, F.E.; Prywes, R.; Balasubramanyam, A.; Schwartz, R.J. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 1998, 273, 30287–30294. [Google Scholar] [CrossRef]
- Castellani, L.; Salvati, E.; Alema, S.; Falcone, G. Fine regulation of RhoA and rock is required for skeletal muscle differentiation. J. Biol. Chem. 2006, 281, 15249–15257. [Google Scholar] [CrossRef]
- Iwasaki, K.; Hayashi, K.; Fujioka, T.; Sobue, K. Rho/Rho-associated kinase signal regulates myogenic differentiation via myocardin-related transcription factor-A/Smad-dependent transcription of the Id3 gene. J. Biol. Chem. 2008, 283, 21230–21241. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Kii, I.; Kudo, A. Inactivation of Rho/ROCK signaling is crucial for the nuclear accumulation of FKHR and myoblast fusion. J. Biol. Chem. 2004, 279, 47311–47319. [Google Scholar] [CrossRef]
- Travaglione, S.; Messina, G.; Fabbri, A.; Falzano, L.; Giammarioli, A.M.; Grossi, M.; Rufini, S.; Fiorentini, C. Cytotoxic necrotizing factor 1 hinders skeletal muscle differentiation in vitro by perturbing the activation/deactivation balance of Rho GTPases. Cell Death Differ. 2005, 12, 78–86. [Google Scholar] [CrossRef]
- Taglietti, V.; Angelini, G.; Mura, G.; Bonfanti, C.; Caruso, E.; Monteverde, S.; Le Carrou, G.; Tajbakhsh, S.; Relaix, F.; Messina, G. RhoA and ERK signalling regulate the expression of the transcription factor Nfix in myogenic cells. Development 2018, 145, dev163956. [Google Scholar] [CrossRef]
- Messina, G.; Biressi, S.; Monteverde, S.; Magli, A.; Cassano, M.; Perani, L.; Roncaglia, E.; Tagliafico, E.; Starnes, L.; Campbell, C.E.; et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 2010, 140, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Maroli, G.; Cermenati, S.; Monteverde, S.; Ferrante, A.; Rossi, G.; Cossu, G.; Beltrame, M.; Messina, G. Nfix Induces a Switch in Sox6 Transcriptional Activity to Regulate MyHC-I Expression in Fetal Muscle. Cell Rep. 2016, 17, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Antonini, S.; Bonfanti, C.; Monteverde, S.; Vezzali, C.; Tajbakhsh, S.; Cossu, G.; Messina, G. Nfix Regulates Temporal Progression of Muscle Regeneration through Modulation of Myostatin Expression. Cell Rep. 2016, 14, 2238–2249. [Google Scholar] [CrossRef]
- Vasyutina, E.; Martarelli, B.; Brakebusch, C.; Wende, H.; Birchmeier, C. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 8935–8940. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Jin, P.; Luo, F.B.; Zhang, G.F.; Anderson, N.; Chen, E.H. Group I PAKs function downstream of Rac to promote podosome invasion during myoblast fusion in vivo. J. Cell Biol. 2012, 199, 169–185. [Google Scholar] [CrossRef]
- Joseph, G.A.; Lu, M.; Radu, M.; Lee, J.K.; Burden, S.J.; Chernoff, J.; Krauss, R.S. Group I Paks Promote Skeletal Myoblast Differentiation In Vivo and In Vitro. Mol. Cell. Biol. 2017, 37, e00222-16. [Google Scholar] [CrossRef]
- Haralalka, S.; Shelton, C.; Cartwright, H.N.; Katzfey, E.; Janzen, E.; Abmayr, S.M. Asymmetric Mbc, active Rac1 and F-actin foci in the fusion-competent myoblasts during myoblast fusion in Drosophila. Development 2011, 138, 1551, correction in Development 2013, 140, 1370–1371. [Google Scholar] [CrossRef] [PubMed]
- Pajcini, K.V.; Pomerantz, J.H.; Alkan, O.; Doyonnas, R.; Blau, H.M. Myoblasts and macrophages share molecular components that contribute to cell-cell fusion. J. Cell Biol. 2008, 180, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Hamoud, N.; Tran, V.; Aimi, T.; Kakegawa, W.; Lahaie, S.; Thibault, M.P.; Pelletier, A.; Wong, G.W.; Kim, I.S.; Kania, A.; et al. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin-2 controls myoblast fusion. Nat. Commun. 2018, 9, 4470. [Google Scholar] [CrossRef]
- Hamoud, N.; Tran, V.; Croteau, L.P.; Kania, A.; Cote, J.F. G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. Proc. Natl. Acad. Sci. USA 2014, 111, 3745–3750. [Google Scholar] [CrossRef]
- Samson, T.; Will, C.; Knoblauch, A.; Sharek, L.; von der Mark, K.; Burridge, K.; Wixler, V. Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain alpha7A and influences myoblast differentiation. J. Biol. Chem. 2007, 282, 15730–15742. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef]
- Eliazer, S.; Muncie, J.M.; Christensen, J.; Sun, X.; D’Urso, R.S.; Weaver, V.M.; Brack, A.S. Wnt4 from the Niche Controls the Mechano-Properties and Quiescent State of Muscle Stem Cells. Cell Stem Cell 2019, 25, 654–665. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef]
- Zhang, L.; Noguchi, Y.T.; Nakayama, H.; Kaji, T.; Tsujikawa, K.; Ikemoto-Uezumi, M.; Uezumi, A.; Okada, Y.; Doi, T.; Watanabe, S.; et al. The CalcR-PKA-Yap1 Axis Is Critical for Maintaining Quiescence in Muscle Stem Cells. Cell Rep. 2019, 29, 2154–2163. [Google Scholar] [CrossRef]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.B.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125, 6009–6019. [Google Scholar] [CrossRef]
- Call, J.A.; Nichenko, A.S. Autophagy: An essential but limited cellular process for timely skeletal muscle recovery from injury. Autophagy 2020, 16, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; Mitchell, D.C.; Zhao, L.; Ma, W.; Stafford, L.J.; Teng, B.B.; Liu, M. Modulation of muscle regeneration, myogenesis, and adipogenesis by the Rho family guanine nucleotide exchange factor GEFT. Mol. Cell. Biol. 2005, 25, 11089–11101. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.A.; Hung, M.; Goel, A.J.; Hong, M.; Rieder, M.K.; Beckmann, N.D.; Serasinghe, M.N.; Chipuk, J.E.; Devarakonda, P.M.; Goldhamer, D.J.; et al. Late-onset megaconial myopathy in mice lacking group I Paks. Skelet. Muscle 2019, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017, 17, 165–178. [Google Scholar] [CrossRef]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The Transcription Factor Nfix Requires RhoA-ROCK1 Dependent Phagocytosis to Mediate Macrophage Skewing during Skeletal Muscle Regeneration. Cells 2020, 9, 708. [Google Scholar] [CrossRef]
- Cerquone Perpetuini, A.; Re Cecconi, A.D.; Chiappa, M.; Martinelli, G.B.; Fuoco, C.; Desiderio, G.; Castagnoli, L.; Gargioli, C.; Piccirillo, R.; Cesareni, G. Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy. J. Cachexia Sarcopenia Muscle 2018, 9, 727–746. [Google Scholar] [CrossRef]
- Sparrow, J.C.; Schock, F. The initial steps of myofibril assembly: Integrins pave the way. Nat. Rev. Mol. Cell Biol. 2009, 10, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Coisy-Quivy, M.; Touzet, O.; Bourret, A.; Hipskind, R.A.; Mercier, J.; Fort, P.; Philips, A. TC10 controls human myofibril organization and is activated by the sarcomeric RhoGEF obscurin. J. Cell Sci. 2009, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Coisy-Quivy, M.; Sanguesa-Ferrer, J.; Weill, M.; Johnson, D.S.; Donnay, J.M.; Hipskind, R.; Fort, P.; Philips, A. Identification of Rho GTPases implicated in terminal differentiation of muscle cells in ascidia. Biol. Cell 2006, 98, 577–588. [Google Scholar] [CrossRef]
- Perry, N.A.; Ackermann, M.A.; Shriver, M.; Hu, L.Y.; Kontrogianni-Konstantopoulos, A. Obscurins: Unassuming giants enter the spotlight. IUBMB Life 2013, 65, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, D.; Pierantozzi, E.; Rossi, D.; Sorrentino, V. The potential of obscurin as a therapeutic target in muscle disorders. Expert Opin. Ther. Targets 2017, 21, 897–910. [Google Scholar] [CrossRef]
- Camporez, J.P.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc. Natl. Acad. Sci. USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef]
- Hamrick, M.W.; Pennington, C.; Webb, C.N.; Isales, C.M. Resistance to body fat gain in ‘double-muscled’ mice fed a high-fat diet. Int. J. Obes. 2006, 30, 868–870. [Google Scholar] [CrossRef]
- Christoffolete, M.A.; Silva, W.J.; Ramos, G.V.; Bento, M.R.; Costa, M.O.; Ribeiro, M.O.; Okamoto, M.M.; Lohmann, T.H.; Machado, U.F.; Musaro, A.; et al. Muscle IGF-1-induced skeletal muscle hypertrophy evokes higher insulin sensitivity and carbohydrate use as preferential energy substrate. BioMed Res. Int. 2015, 2015, 282984. [Google Scholar] [CrossRef]
- Yeung, C.H.C.; Au Yeung, S.L.; Fong, S.S.M.; Schooling, C.M. Lean mass, grip strength and risk of type 2 diabetes: A bi-directional Mendelian randomisation study. Diabetologia 2019, 62, 789–799. [Google Scholar] [CrossRef]
- Yasuoka, M.; Muraki, I.; Imano, H.; Jinnouchi, H.; Kubota, Y.; Hayama-Terada, M.; Umesawa, M.; Yamagishi, K.; Ohira, T.; Kitamura, A.; et al. Joint impact of muscle mass and waist circumference on type 2 diabetes in Japanese middle-aged adults: The Circulatory Risk in Communities Study (CIRCS). J. Diabetes 2020, 12, 677–685. [Google Scholar] [CrossRef]
- Son, J.W.; Lee, S.S.; Kim, S.R.; Yoo, S.J.; Cha, B.Y.; Son, H.Y.; Cho, N.H. Low muscle mass and risk of type 2 diabetes in middle-aged and older adults: Findings from the KoGES. Diabetologia 2017, 60, 865–872. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Pehmoller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2014, 26, 323–331. [Google Scholar] [CrossRef]
- Ueda, S.; Kitazawa, S.; Ishida, K.; Nishikawa, Y.; Matsui, M.; Matsumoto, H.; Aoki, T.; Nozaki, S.; Takeda, T.; Tamori, Y.; et al. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 2010, 24, 2254–2261. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Nielsen, I.L.; Kleinert, M.; Moller, L.L.; Ploug, T.; Schjerling, P.; Bilan, P.J.; Klip, A.; Jensen, T.E.; Richter, E.A. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 2016, 594, 4997–5008. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Hojlund, K.; Kiens, B.; Wojtaszewski, J.; Prats, C.; Schjerling, P.; Richter, E.A. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 2013, 62, 1865–1875. [Google Scholar] [CrossRef]
- Ueda, S.; Kataoka, T.; Satoh, T. Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 2008, 100, 645–657. [Google Scholar] [CrossRef]
- Sylow, L.; Moller, L.L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Rac1—A novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Exp. Physiol. 2014, 99, 1574–1580. [Google Scholar] [CrossRef]
- Sylow, L.; Moller, L.L.V.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Stretch-stimulated glucose transport in skeletal muscle is regulated by Rac1. J. Physiol. 2015, 593, 645–656. [Google Scholar] [CrossRef] [PubMed]
- You, G.Y.; Lee, J.O.; Kim, J.H.; Kim, N.; Lee, S.K.; Moon, J.W.; Jie, S.; Lee, H.J.; Kim, S.J.; Park, S.H.; et al. Tiam-1, a GEF for Rac1, plays a critical role in metformin-mediated glucose uptake in C2C12 cells. Cell. Signal. 2013, 25, 2558–2565. [Google Scholar] [CrossRef]
- Yue, Y.; Zhang, C.; Zhao, X.; Liu, S.; Lv, X.; Zhang, S.; Yang, J.; Chen, L.; Duan, H.; Zhang, Y.; et al. Tiam1 mediates Rac1 activation and contraction-induced glucose uptake in skeletal muscle cells. FASEB J. 2021, 35, e21210. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.O.; Lee, Y.W.; Kim, S.A.; Park, S.H.; Kim, H.S. Kalirin, a GEF for Rac1, plays an important role in FSTL-1-mediated glucose uptake in skeletal muscle cells. Cell. Signal. 2017, 29, 150–157. [Google Scholar] [CrossRef]
- Furukawa, N.; Ongusaha, P.; Jahng, W.J.; Araki, K.; Choi, C.S.; Kim, H.J.; Lee, Y.H.; Kaibuchi, K.; Kahn, B.B.; Masuzaki, H.; et al. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005, 2, 119–129. [Google Scholar] [CrossRef]
- Lee, S.H.; Huang, H.; Choi, K.; Lee, D.H.; Shi, J.; Liu, T.; Chun, K.H.; Seo, J.A.; Lima, I.S.; Zabolotny, J.M.; et al. ROCK1 isoform-specific deletion reveals a role for diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E332–E343. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.H.; Choi, K.D.; Lee, D.H.; Jung, Y.; Henry, R.R.; Ciaraldi, T.P.; Kim, Y.B. In vivo activation of ROCK1 by insulin is impaired in skeletal muscle of humans with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E536–E542. [Google Scholar] [CrossRef]
- Zhou, X.; Li, R.; Liu, X.; Wang, L.; Hui, P.; Chan, L.; Saha, P.K.; Hu, Z. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci. Rep. 2016, 6, 29669. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Chockalingam, P.S.; Cholera, R.; Oak, S.A.; Zheng, Y.; Jarrett, H.W.; Thomason, D.B. Dystrophin-glycoprotein complex and Ras and Rho GTPase signaling are altered in muscle atrophy. Am. J. Physiol. Cell Physiol. 2002, 283, C500–C511. [Google Scholar] [CrossRef] [PubMed]
- Oak, S.A.; Zhou, Y.W.; Jarrett, H.W. Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J. Biol. Chem. 2003, 278, 39287–39295. [Google Scholar] [CrossRef]
- Lenhart, K.C.; O’Neill, T.J.; Cheng, Z.; Dee, R.; Demonbreun, A.R.; Li, J.; Xiao, X.; McNally, E.M.; Mack, C.P.; Taylor, J.M. GRAF1 deficiency blunts sarcolemmal injury repair and exacerbates cardiac and skeletal muscle pathology in dystrophin-deficient mice. Skelet. Muscle 2015, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B.; Vieland, V.J.; Dunn, D.M.; Kaminoh, Y.; Flanigan, K.M.; United Dystrophinopathy, P. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in duchenne muscular dystrophy. Ann. Neurol. 2018, 84, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Tang, Y.; Takayama, K.; Chen, W.; Lu, A.; Wang, B.; Weiss, K.; Huard, J. RhoA/ROCK inhibition improves the beneficial effects of glucocorticoid treatment in dystrophic muscle: Implications for stem cell depletion. Hum. Mol. Genet. 2017, 26, 2813–2824. [Google Scholar] [CrossRef]
- Mu, X.; Usas, A.; Tang, Y.; Lu, A.; Wang, B.; Weiss, K.; Huard, J. RhoA mediates defective stem cell function and heterotopic ossification in dystrophic muscle of mice. FASEB J. 2013, 27, 3619–3631. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Fdez, S.; Bustelo, X.R. Rho GTPases in Skeletal Muscle Development and Homeostasis. Cells 2021, 10, 2984. https://doi.org/10.3390/cells10112984
Rodríguez-Fdez S, Bustelo XR. Rho GTPases in Skeletal Muscle Development and Homeostasis. Cells. 2021; 10(11):2984. https://doi.org/10.3390/cells10112984
Chicago/Turabian StyleRodríguez-Fdez, Sonia, and Xosé R. Bustelo. 2021. "Rho GTPases in Skeletal Muscle Development and Homeostasis" Cells 10, no. 11: 2984. https://doi.org/10.3390/cells10112984
APA StyleRodríguez-Fdez, S., & Bustelo, X. R. (2021). Rho GTPases in Skeletal Muscle Development and Homeostasis. Cells, 10(11), 2984. https://doi.org/10.3390/cells10112984