
In-Depth Annotation of the Drosophila Bithorax-Complex Reveals the Presence of Several Alternative ORFs That Could Encode for Motif-Rich Peptides


Abstract
:1. Introduction
2. Materials and Methods
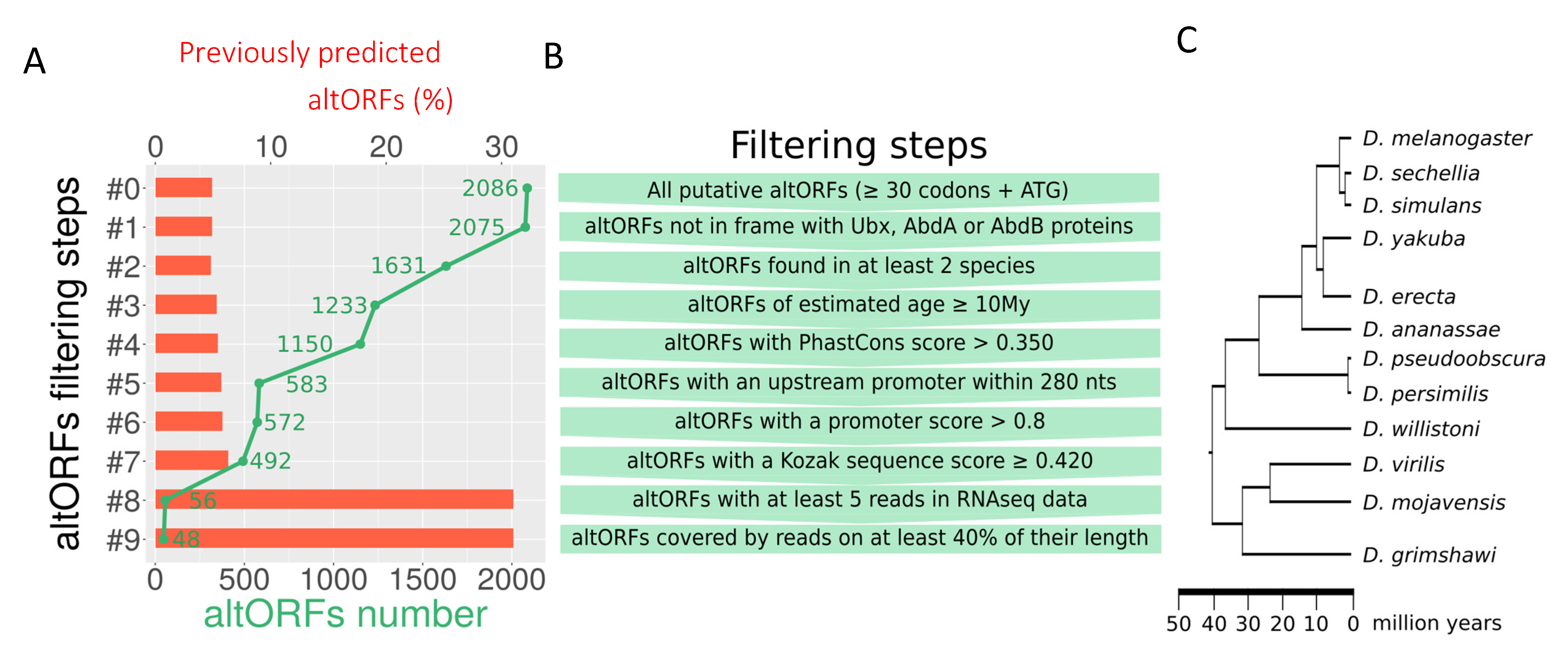
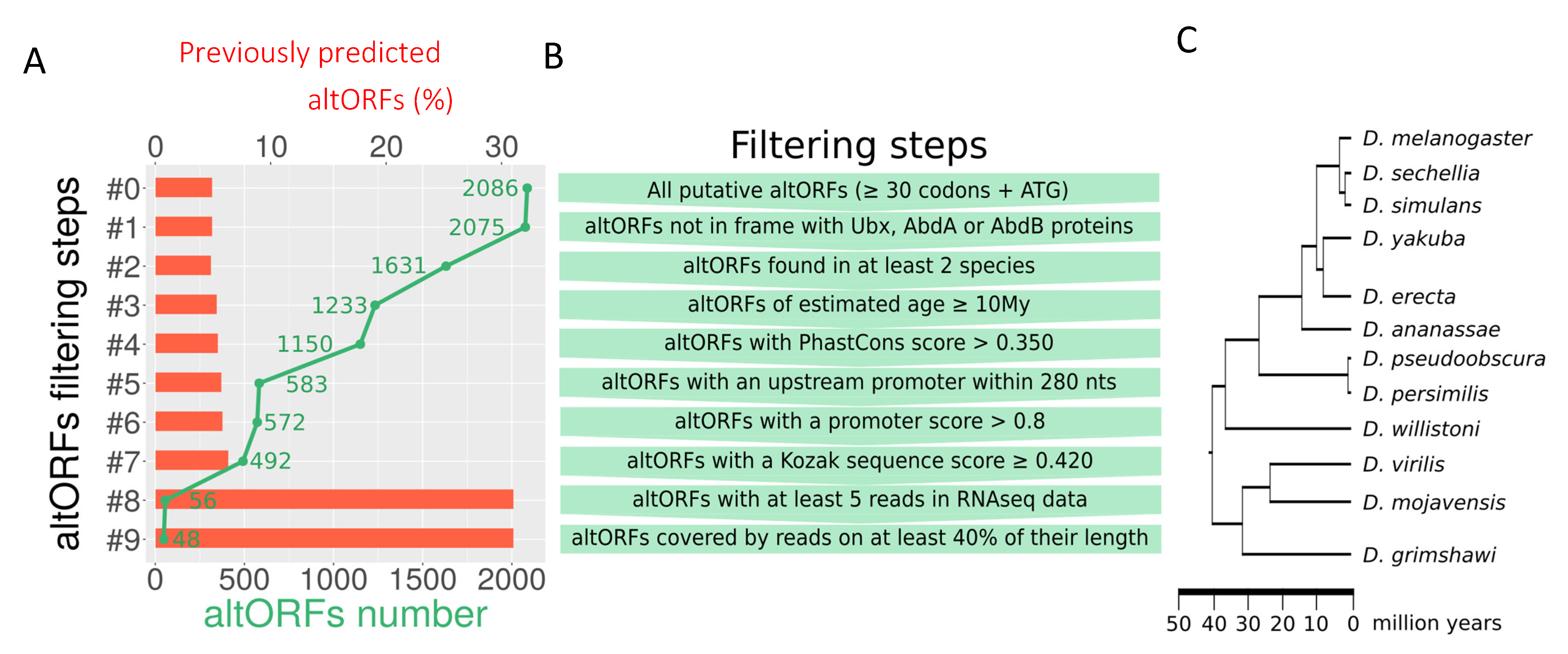
2.1. altORF Prediction
2.2. altORF Characterization
2.2.1. Orthology and Age
2.2.2. Nucleotidic Conservation
2.2.3. Transcription and Translation Features
2.2.4. Overlap of altORFs with Other Genomic Features
2.3. Characterization of the Predicted Peptides
3. Results and Discussion
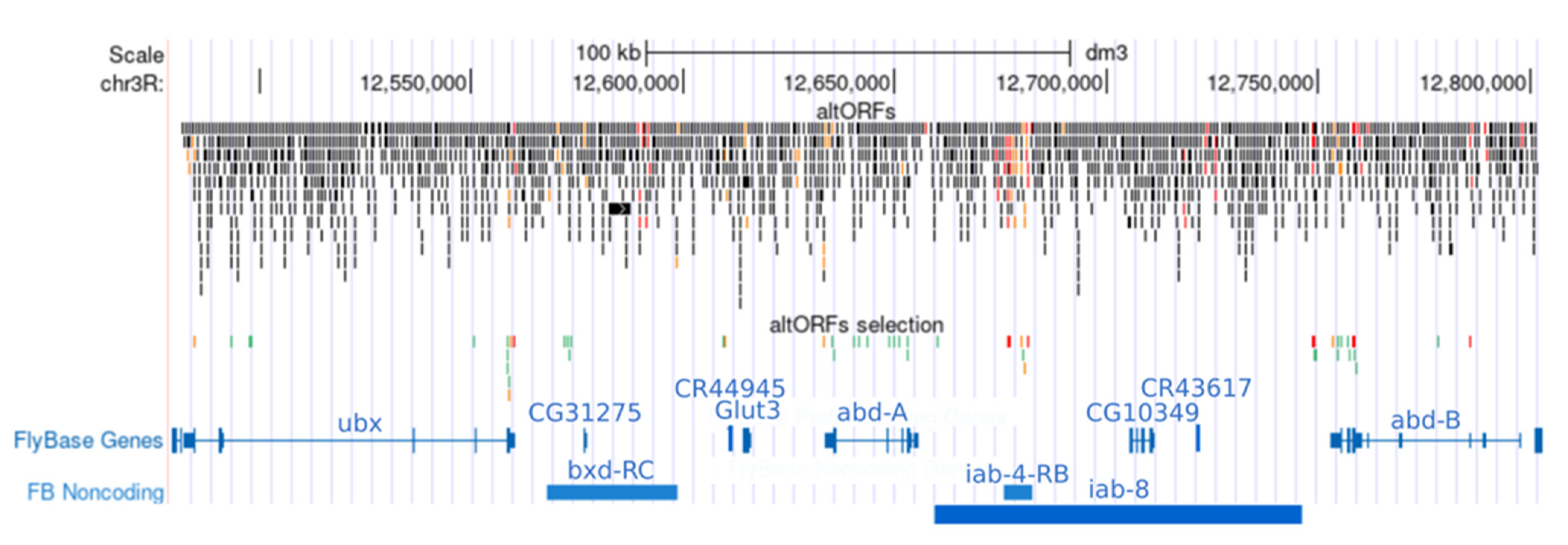
3.1. Annotation of altORFs in BX-C
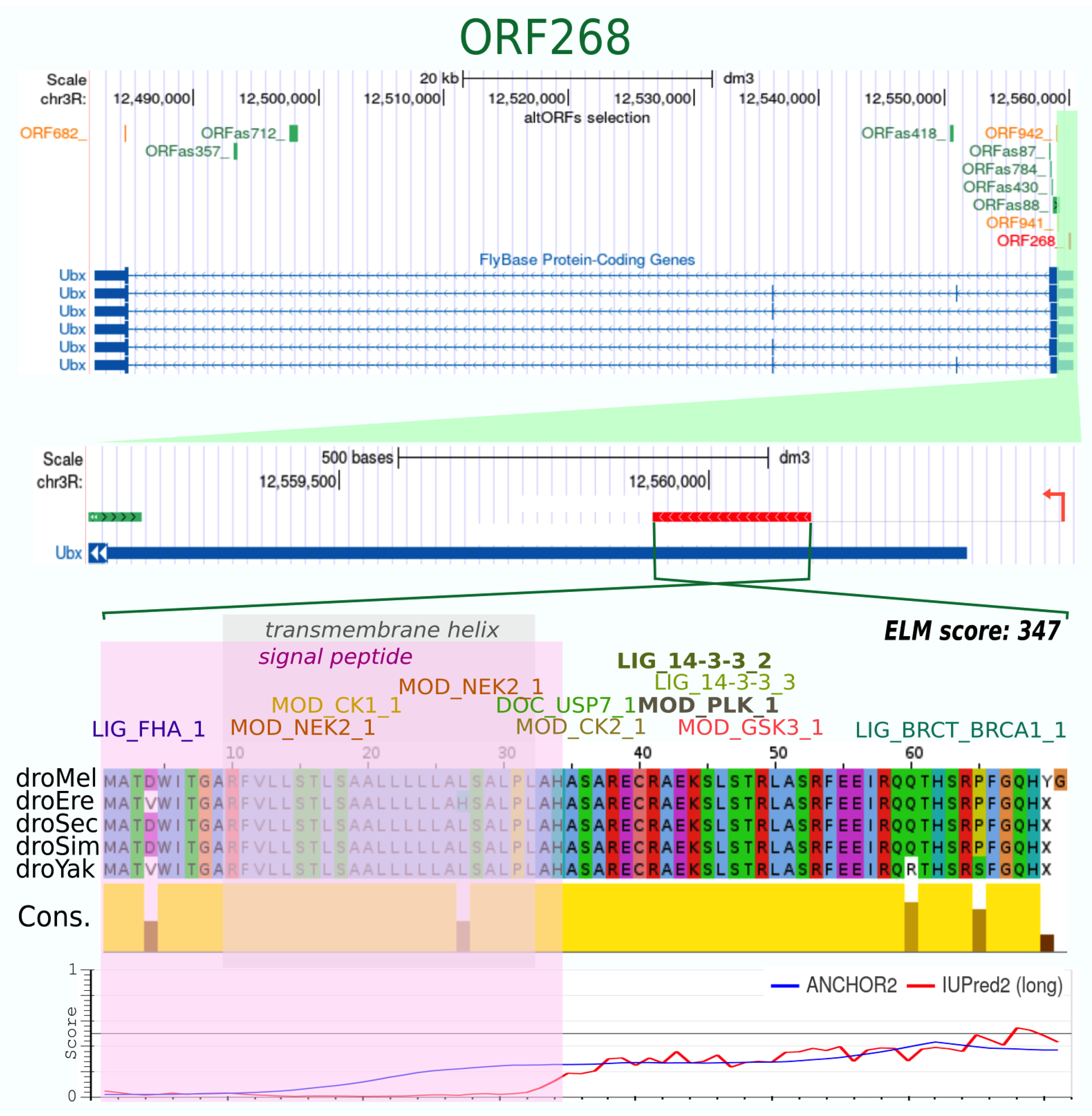
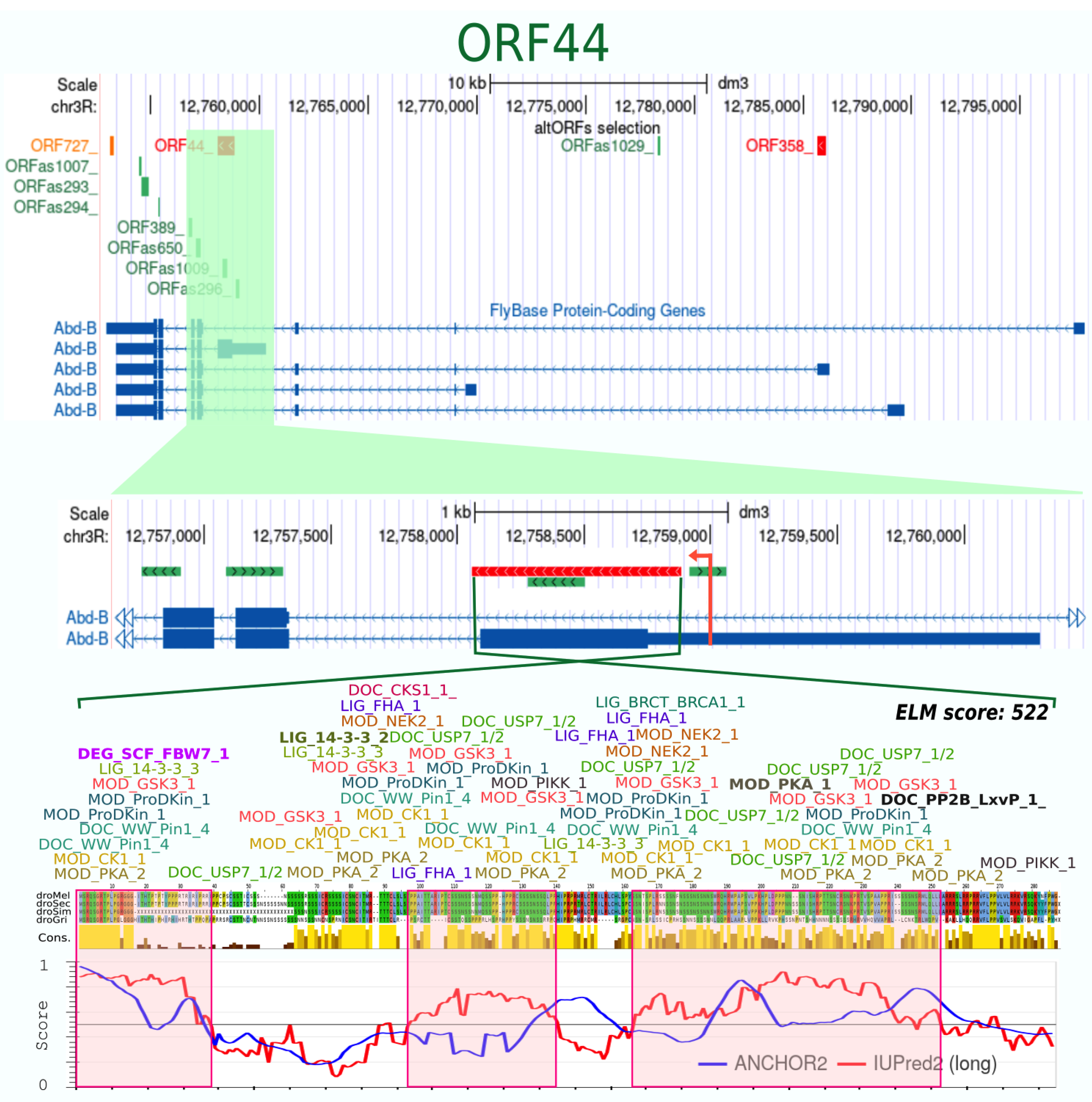
3.2. Structure/Motifs Predictions of Conserved Alternative Peptides of BX-C
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brent, M.R. Genome annotation past, present, and future: How to define an ORF at each locus. Genome Res. 2005, 15, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, M.R. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat. Rev. Genet. 2008, 9, 62–73. [Google Scholar] [CrossRef]
- Windsor, A.J.; Mitchell-Olds, T. Comparative genomics as a tool for gene discovery. Curr. Opin. Biotechnol. 2006, 17, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. An overview of the current status of eukaryote gene prediction strategies. Gene 2010, 461, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays 2007, 29, 288–299. [Google Scholar] [CrossRef]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long Noncoding RNAs: Past, Present, and Future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.W.; Bruford, E.A. Naming “junk”: Human non-protein coding RNA (ncRNA) gene nomenclature. Hum. Genom. 2011, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Ye, R.; Cao, C.; Xue, Y. Enhancer RNA: Biogenesis, function, and regulation. Essays Biochem. 2020, 64, 883–894. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.-B. Alternative ORFs and small ORFs: Shedding light on the dark proteome. Nucleic Acids Res. 2020, 48, 1029–1042. [Google Scholar] [CrossRef]
- Nakamura, A.; Amikura, R.; Mukai, M.; Kobayashi, S.; Lasko, P.F. Requirement for a Noncoding RNA in Drosophila Polar Granules for Germ Cell Establishment. Science 1996, 274, 2075–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, T.; Menze, B.H.; Kirchner, M.; Monigatti, F.; Parker, K.C.; Patterson, T.; Steen, J.J.; Hamprecht, F.A.; Steen, H. Robust Prediction of the MASCOT Score for an Improved Quality Assessment in Mass Spectrometric Proteomics. J. Proteome Res. 2008, 7, 3708–3717. [Google Scholar] [CrossRef]
- Vanderperre, B.; Lucier, J.-F.; Bissonnette, C.; Motard, J.; Tremblay, G.; Vanderperre, S.; Wisztorski, M.; Salzet, M.; Boisvert, F.-M.; Roucou, X. Direct Detection of Alternative Open Reading Frames Translation Products in Human Significantly Expands the Proteome. PLoS ONE 2013, 8, e70698. [Google Scholar] [CrossRef] [Green Version]
- Prabakaran, S.; Hemberg, M.; Chauhan, R.; Winter, D.; Tweedie-Cullen, R.Y.; Dittrich, C.; Hong, E.; Gunawardena, J.; Steen, H.; Kreiman, G.; et al. Quantitative profiling of peptides from RNAs classified as noncoding. Nat. Commun. 2014, 5, 5429. [Google Scholar] [CrossRef] [PubMed]
- Menschaert, G.; Van Criekinge, W.; Notelaers, T.; Koch, A.; Crappé, J.; Gevaert, K.; Van Damme, P. Deep Proteome Coverage Based on Ribosome Profiling Aids Mass Spectrometry-based Protein and Peptide Discovery and Provides Evidence of Alternative Translation Products and Near-cognate Translation Initiation Events*. Mol. Cell. Proteomics 2013, 12, 1780–1790. [Google Scholar] [CrossRef] [Green Version]
- Slavoff, S.A.; Mitchell, A.J.; Schwaid, A.G.; Cabili, M.N.; Ma, J.; Levin, J.Z.; Karger, A.D.; Budnik, B.A.; Rinn, J.L.; Saghatelian, A. Peptidomic discovery of short open reading frame–encoded peptides in human cells. Nat. Chem. Biol. 2013, 9, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Delcourt, V.; Staskevicius, A.; Salzet, M.; Fournier, I.; Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics 2018, 18, 1700058. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Rothnagel, J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef]
- Hayden, C.A.; Bosco, G. Comparative genomic analysis of novel conserved peptide upstream open reading frames in Drosophila melanogaster and other dipteran species. BMC Genom. 2008, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Renz, P.F.; Valdivia-Francia, F.; Sendoel, A. Some like it translated: Small ORFs in the 5′UTR. Exp. Cell Res. 2020, 396, 112229. [Google Scholar] [CrossRef]
- Johnstone, T.G.; Bazzini, A.A.; Giraldez, A.J. Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J. 2016, 35, 706–723. [Google Scholar] [CrossRef] [Green Version]
- Chew, G.-L.; Pauli, A.; Schier, A.F. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun. 2016, 7, 11663. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Higuchi-Takeuchi, M.; Okamoto, M.; Yoshizumi, T.; Shimizu, M.; Nakaminami, K.; Nishi, R.; Ohashi, C.; Iida, K.; Tanaka, M.; et al. Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc. Natl. Acad. Sci. USA 2013, 110, 2395–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenmayer, J.P. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 2006, 16, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Ladoukakis, E.; Pereira, V.; Magny, E.G.; Eyre-Walker, A.; Couso, J. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011, 12, R118. [Google Scholar] [CrossRef] [Green Version]
- Hanyu-Nakamura, K.; Sonobe-Nojima, H.; Tanigawa, A.; Lasko, P.; Nakamura, A. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature 2008, 451, 730–733. [Google Scholar] [CrossRef] [Green Version]
- Zanet, J.; Benrabah, E.; Li, T.; Pelissier-Monier, A.; Chanut-Delalande, H.; Ronsin, B.; Bellen, H.J.; Payre, F.; Plaza, S. Pri sORF peptides induce selective proteasome-mediated protein processing. Science 2015, 349, 1356–1358. [Google Scholar] [CrossRef]
- Galindo, M.I.; Pueyo, J.I.; Fouix, S.; Bishop, S.A.; Couso, J.P. Peptides Encoded by Short ORFs Control Development and Define a New Eukaryotic Gene Family. PLoS Biol. 2007, 5, e106. [Google Scholar] [CrossRef] [Green Version]
- Immarigeon, C.; Frei, Y.; Delbare, S.Y.N.; Gligorov, D.; Machado Almeida, P.; Grey, J.; Fabbro, L.; Nagoshi, E.; Billeter, J.-C.; Wolfner, M.F.; et al. Identification of a micropeptide and multiple secondary cell genes that modulate Drosophila male reproductive success. Proc. Natl. Acad. Sci. USA 2021, 118, e2001897118. [Google Scholar] [CrossRef] [PubMed]
- Aspden, J.L.; Eyre-Walker, Y.C.; Phillips, R.J.; Amin, U.; Mumtaz, M.A.S.; Brocard, M.; Couso, J.-P. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife 2014, 3, E03528. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Orera, J.; Messeguer, X.; Subirana, J.A.; Alba, M.M. Long non-coding RNAs as a source of new peptides. Elife 2014, 3, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Samandi, S.; Roy, A.V.; Delcourt, V.; Lucier, J.-F.; Gagnon, J.; Beaudoin, M.C.; Vanderperre, B.; Breton, M.-A.; Motard, J.; Jacques, J.-F.; et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife 2017, 6, e27860. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.A.; Leblanc, S.; Roucou, X. Reconsidering proteomic diversity with functional investigation of small ORFs and alternative ORFs. Exp. Cell Res. 2020, 393, 112057. [Google Scholar] [CrossRef]
- Granzotto, A.; Lopes, F.R.; Vieira, C.; Carareto, C.M.A. Vertical inheritance and bursts of transposition have shaped the evolution of the BS non-LTR retrotransposon in Drosophila. Mol. Genet. Genom. 2011, 286, 57. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Miura, R.M.; Tian, B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 2006, 22, 2320–2325. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, J.M.; Hoermann, B.; Schlimbach, T.; Teleman, A.A. Changes in global translation elongation or initiation rates shape the proteome via the Kozak sequence. Sci. Rep. 2018, 8, 4018. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Cherbas, L.; Willingham, A.; Zhang, D.; Yang, L.; Zou, Y.; Eads, B.D.; Carlson, J.W.; Landolin, J.M.; Kapranov, P.; Dumais, J.; et al. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 2011, 21, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Saari, G.; Bienz, M. The structure of the Ultrabithorax promoter of Drosophila melanogaster. EMBO J. 1987, 6, 1775–1779. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Čalyševa, J.; et al. ELM—The eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2019, 48, D296–D306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanzoni, A.; Ribeiro, D.M.; Brun, C. Understanding protein multifunctionality: From short linear motifs to cellular functions. Cell. Mol. Life Sci. 2019, 76, 4407–4412. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
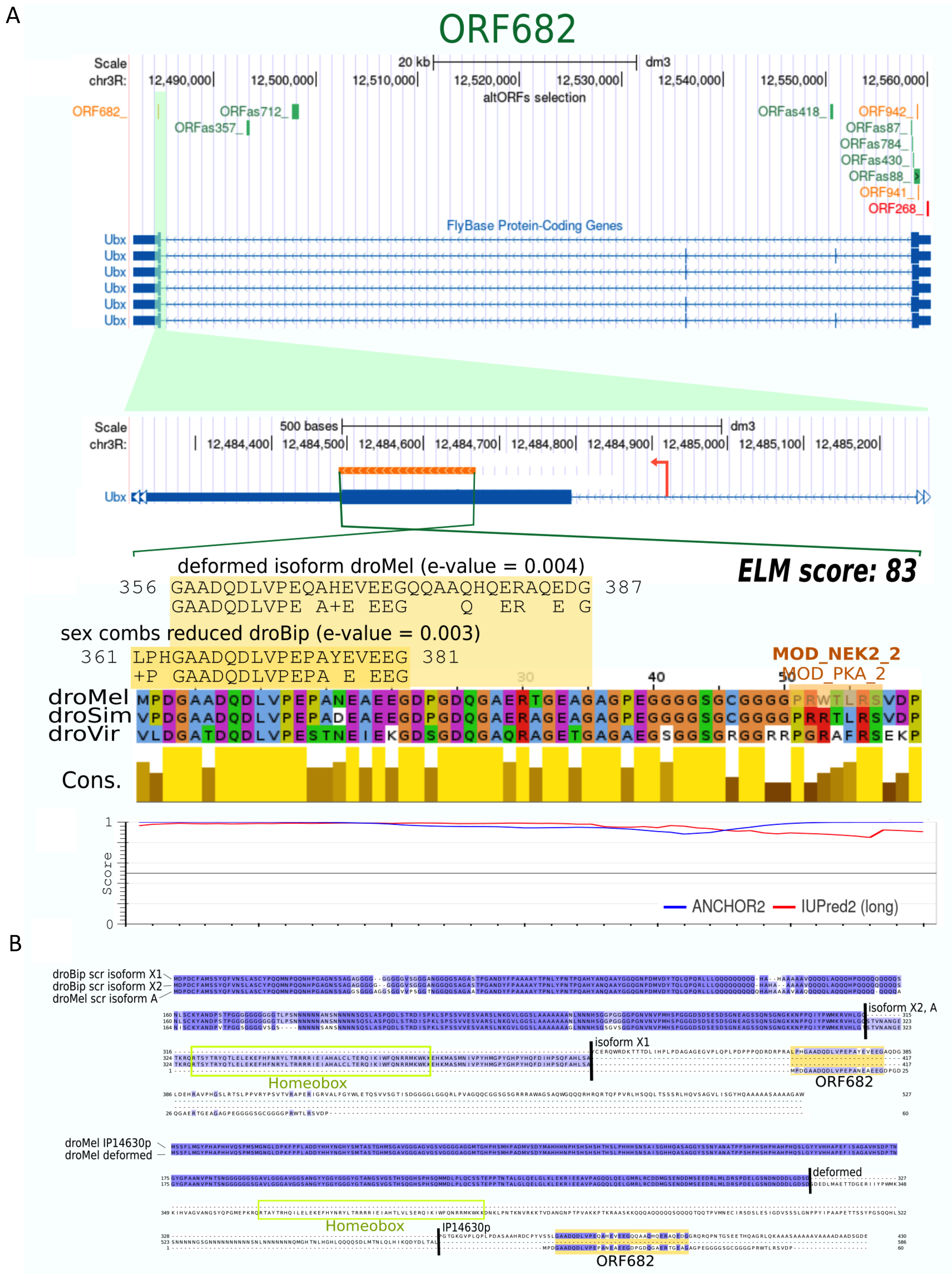{kind=link}
| Name | Distribution and Conservation | Transcription and Translation Features | Peptide Characterization | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length(aa) | Strand | Age(My) | Droso | PhastCons_Score | PromScore | PromDist | KozakScore | Reads | %Reads | Aspden et al. | Samandi et al. | TotalELMs | DifELMs | ELMScore | Signal Peptide, TMhelix | IUPred_structure | BlastP_nr | |
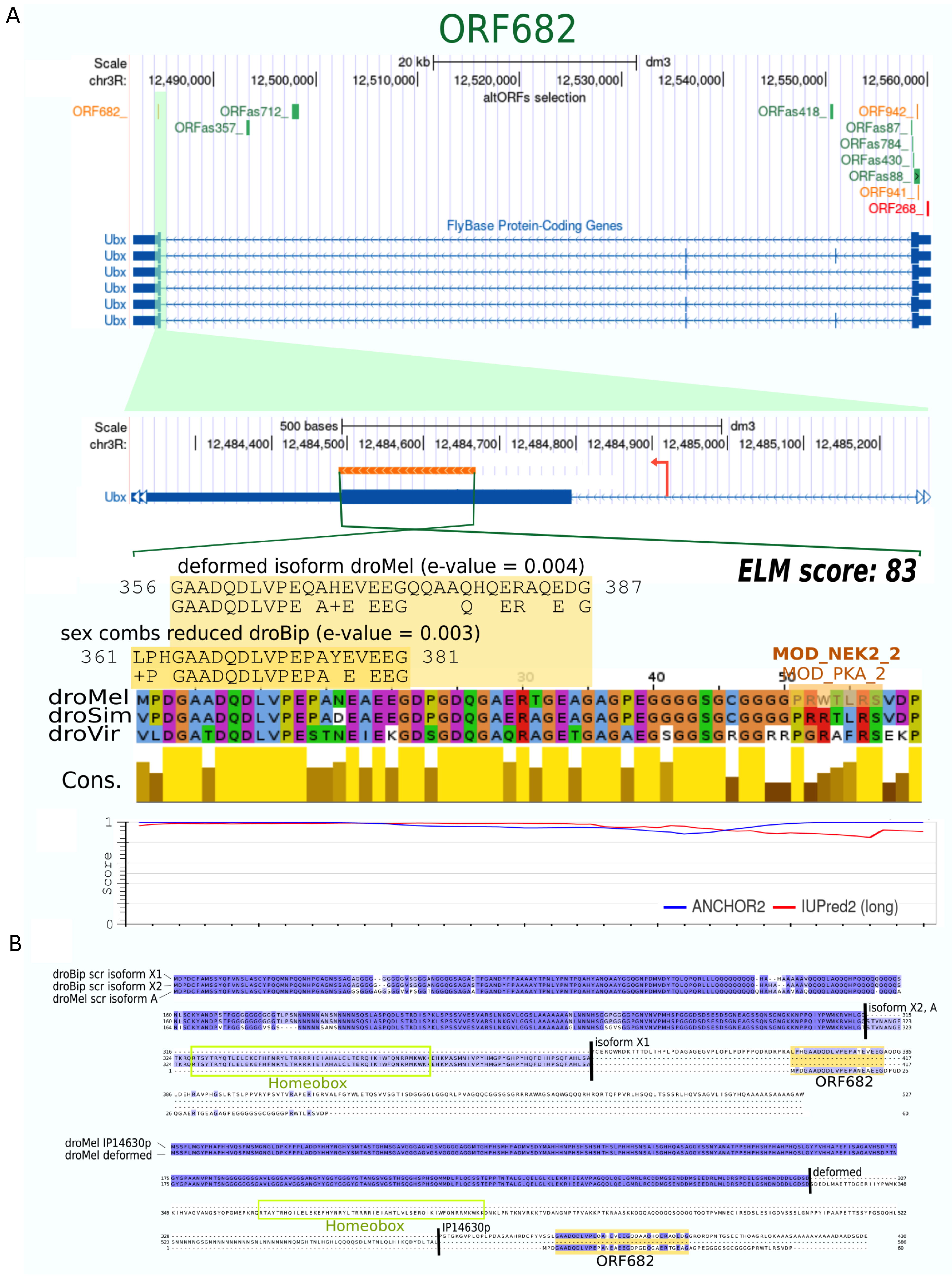
| ORF682_ | 60 | + | 40 | 3 | 0.954 | 0.96 | −246 | 0.643 | 59 | 55 | yes | 2 | 2 | 83 | disordered | Scr, Dfd | ||
| ORFas357_ | 108 | - | 27 | 8 | 0.776 | 0.85 | −191 | 0.874 | 8 | 46 | 14 | 8 | 144 | ordered | ||||
| ORFas712_ | 215 | - | 14 | 5 | 0.718 | 0.98 | −23 | 0.538 | 6 | 45 | 32 | 13 | 184 | ordered | ||||
| ORFas418_ | 88 | - | 10 | 5 | 0.459 | 0.96 | −143 | 0.448 | 5 | 58 | 13 | 8 | 144 | 18% disordered | ||||
| ORFas87_ | 46 | - | 27 | 5 | 0.623 | 0.81 | −47 | 0.755 | 97 | 54 | 5 | 4 | 188 | ordered | ||||
| ORFas784_ | 49 | - | 14 | 6 | 0.920 | 0.93 | −84 | 0.776 | 53 | 76 | 4 | 4 | 220 | ordered | ||||
| ORFas430_ | 51 | - | 10 | 4 | 0.921 | 0.93 | −16 | 0.916 | 90 | 82 | 16 | 10 | 936 | unclear | ||||
| ORFas88_ | 194 | - | 10 | 5 | 0.958 | 1.00 | −232 | 0.881 | 193 | 60 | 18 | 12 | 410 | ordered | ||||
| ORF942_ | 38 | + | 40 | 6 | 0.986 | 0.81 | −48 | 0.434 | 77 | 98 | yes | 5 | 4 | 195 | disordered | |||
| ORF941_ | 37 | + | 10 | 5 | 0.984 | 0.81 | −171 | 0.490 | 55 | 97 | yes | 2 | 2 | 228 | disordered | |||
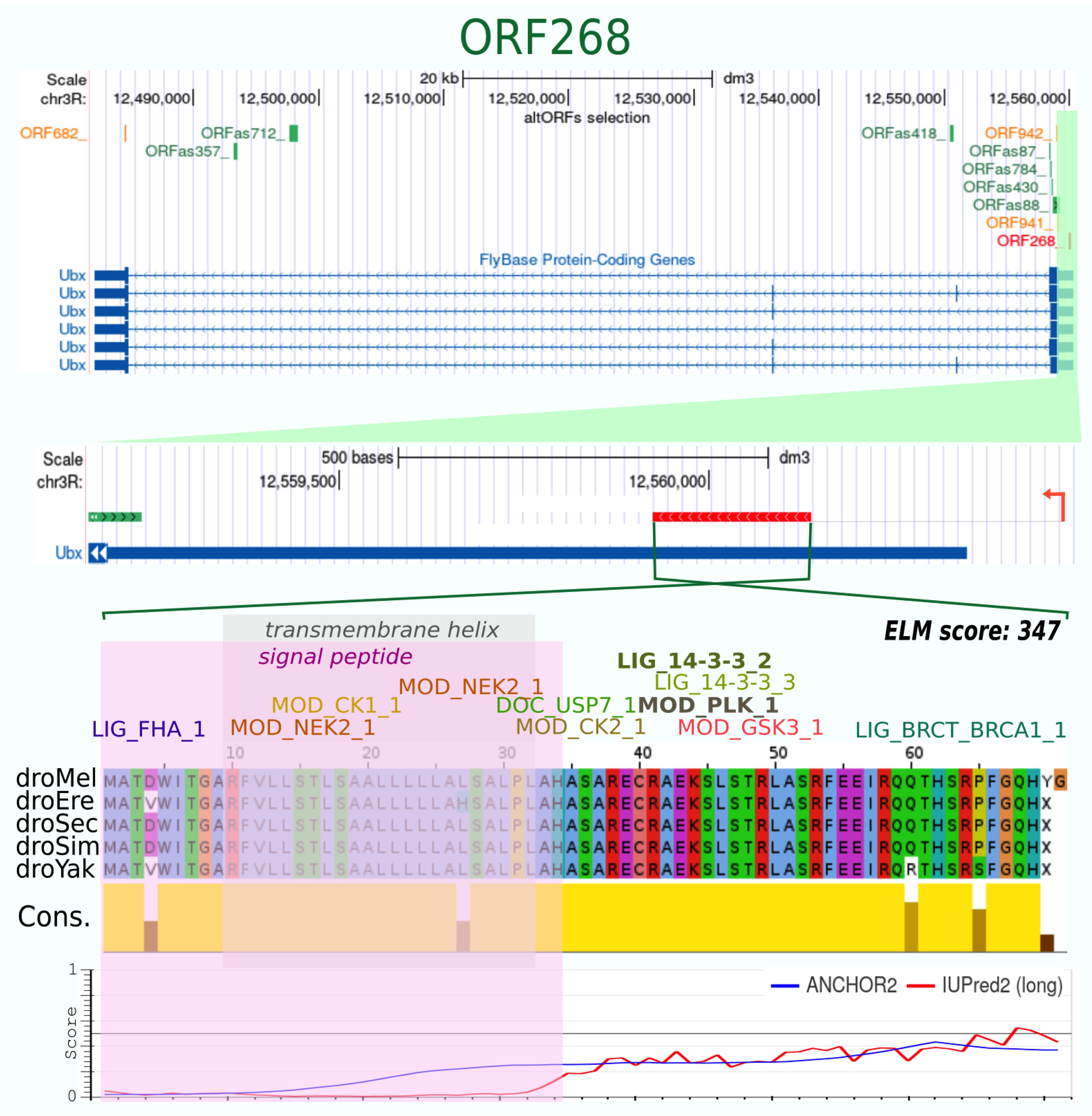
| ORF268_ | 71 | + | 10 | 5 | 0.755 | 0.96 | −278 | 0.594 | 123 | 88 | yes | yes | 15 | 10 | 347 | 1 Signal peptide, 1 TMhelix | ordered | 5′-Ubx |
| ORFas449_ | 61 | - | 40 | 5 | 0.699 | 0.90 | −121 | 0.944 | 9 | 77 | 8 | 6 | 234 | disordered | ||||
| ORFas450_ | 36 | - | 40 | 10 | 0.995 | 0.99 | −9 | 0.986 | 6 | 51 | 1 | 1 | 13 | 32 % disordered | ||||
| ORFas803_ | 116 | - | 10 | 4 | 0.544 | 0.92 | −79 | 0.503 | 10 | 64 | 14 | 9 | 167 | ordered | ||||
| ORFas452_ | 75 | - | 10 | 3 | 0.462 | 0.92 | −167 | 0.825 | 8 | 60 | 15 | 9 | 196 | 1 TMhelix | 7 % disordered | |||
| ORF211_ | 48 | + | 10 | 3 | 0.659 | 0.84 | −8 | 0.643 | 6 | 53 | 10 | 9 | 989 | 36 % disordered | ||||
| ORFas137_ | 113 | - | 10 | 5 | 0.616 | 0.94 | −72 | 0.699 | 9 | 65 | yes | 22 | 10 | 197 | ordered | |||
| ORF861_ | 41 | + | 10 | 5 | 0.765 | 0.88 | −202 | 0.427 | 14 | 89 | yes | 8 | 8 | 432 | ordered | |||
| ORFas875_ | 38 | - | 10 | 5 | 0.906 | 0.95 | −165 | 0.434 | 185 | 98 | 1 | 1 | 38 | ordered | ||||
| ORFas165_ | 62 | - | 14 | 6 | 0.936 | 0.93 | −141 | 0.727 | 82 | 53 | 6 | 3 | 74 | ordered | ||||
| ORF172_ | 80 | + | 10 | 5 | 0.607 | 0.97 | −85 | 0.790 | 12 | 81 | 10 | 8 | 233 | ordered | ||||
| ORF497_ | 109 | + | 40 | 9 | 0.480 | 0.91 | −189 | 0.839 | 7 | 68 | 17 | 9 | 233 | ordered | ||||
| ORFas170_ | 68 | - | 10 | 4 | 0.511 | 1.00 | −96 | 0.434 | 7 | 91 | 12 | 10 | 302 | ordered | ||||
| ORF847_ | 70 | + | 10 | 4 | 0.490 | 0.88 | −130 | 0.427 | 5 | 65 | 28 | 10 | 461 | 14 % disordered | ||||
| ORFas175_ | 114 | - | 40 | 11 | 0.628 | 0.81 | −177 | 0.503 | 16 | 64 | 14 | 9 | 132 | ordered | ||||
| ORF842_ | 34 | + | 10 | 5 | 0.815 | 0.89 | −118 | 0.762 | 5 | 79 | 11 | 8 | 465 | unclear | ||||
| ORFas894_ | 152 | - | 40 | 7 | 0.617 | 0.93 | −42 | 0.497 | 93 | 59 | 13 | 9 | 180 | 6 % disordered | ||||
| ORFas538_ | 57 | - | 10 | 5 | 0.974 | 0.93 | −97 | 0.650 | 79 | 94 | 5 | 5 | 1177 | unclear | ||||
| ORFas545_ | 47 | - | 10 | 5 | 0.600 | 0.84 | −36 | 0.727 | 7 | 42 | 4 | 4 | 247 | ordered | ||||
| ORFas561_ | 72 | - | 10 | 3 | 0.647 | 0.82 | −112 | 0.455 | 8 | 58 | yes | yes | 8 | 6 | 168 | 1 TMhelix | 25 % disordered | |
| ORFas200_ | 54 | - | 10 | 5 | 0.484 | 0.81 | −278 | 0.441 | 6 | 100 | yes | yes | 6 | 5 | 141 | ordered | ||
| ORFas566_ | 80 | - | 10 | 5 | 0.682 | 0.98 | −265 | 0.734 | 13 | 90 | yes | 17 | 11 | 409 | 1 TMhelix | ordered | ||
| ORF459_ | 46 | + | 10 | 4 | 0.571 | 0.85 | −205 | 0.713 | 6 | 100 | 3 | 3 | 131 | 48 % disordered | ||||
| ORFas567_ | 32 | - | 10 | 5 | 0.582 | 0.88 | −47 | 0.580 | 8 | 86 | yes | 7 | 7 | 235 | ordered | |||
| ORF817_ | 49 | + | 10 | 4 | 0.595 | 0.84 | −251 | 0.657 | 8 | 50 | yes | yes | 3 | 3 | 66 | 37 % disordered | ||
| ORF52_ | 242 | + | 10 | 5 | 0.431 | 0.98 | −93 | 0.629 | 51 | 50 | yes | yes | 30 | 12 | 120 | 5 % disordered | ||
| ORF393_ | 241 | + | 10 | 5 | 0.442 | 0.89 | −159 | 0.699 | 27 | 63 | 30 | 13 | 153 | ordered | ||||
| ORF727_ | 53 | + | 10 | 4 | 0.539 | 0.93 | −210 | 0.552 | 11 | 96 | yes | 5 | 4 | 151 | disordered | |||
| ORFas1007_ | 30 | - | 10 | 5 | 0.935 | 0.98 | −105 | 0.832 | 39 | 93 | 5 | 5 | 287 | ordered | ||||
| ORFas293_ | 100 | - | 10 | 4 | 0.523 | 0.95 | −134 | 0.427 | 187 | 84 | 9 | 5 | 65 | 1 TMhelix | ordered | |||
| ORFas294_ | 34 | - | 10 | 2 | 0.691 | 0.95 | −112 | 0.441 | 69 | 66 | 4 | 4 | 161 | ordered | ||||
| ORF389_ | 51 | + | 14 | 3 | 0.499 | 0.99 | −129 | 0.755 | 40 | 46 | 12 | 8 | 352 | 67 % disordered | ||||
| ORFas650_ | 75 | - | 10 | 5 | 0.839 | 1.00 | −234 | 0.538 | 118 | 54 | 6 | 5 | 965 | ordered | ||||
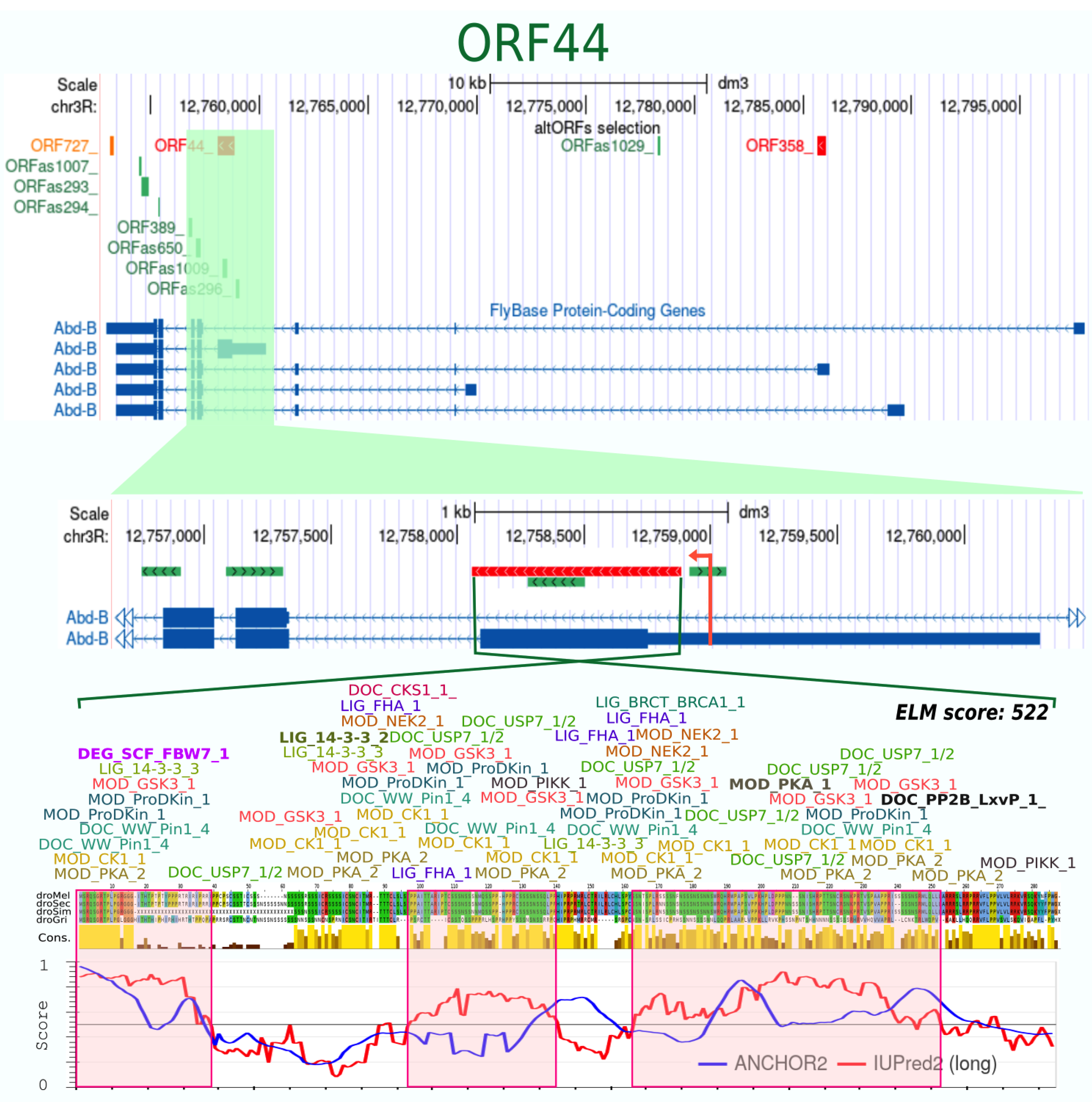
| ORF44_ | 274 | + | 40 | 4 | 0.906 | 0.81 | −100 | 0.818 | 227 | 45 | yes | 125 | 17 | 522 | 61 % disordered | |||
| ORFas1009_ | 75 | - | 10 | 4 | 0.938 | 0.98 | −181 | 0.762 | 79 | 47 | 0 | 0 | 0 | 1 TMhelix | ordered | |||
| ORFas296_ | 49 | - | 10 | 5 | 0.697 | 0.84 | −1 | 0.853 | 17 | 96 | 7 | 6 | 135 | ordered | ||||
| ORFas1029_ | 39 | - | 14 | 5 | 0.632 | 0.90 | −9 | 0.965 | 6 | 58 | 1 | 1 | 31 | ordered | ||||
| ORF358_ | 133 | + | 40 | 8 | 0.484 | 0.86 | −160 | 0.748 | 43 | 47 | yes | 10 | 9 | 100 | ordered | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naville, M.; Merabet, S. In-Depth Annotation of the Drosophila Bithorax-Complex Reveals the Presence of Several Alternative ORFs That Could Encode for Motif-Rich Peptides. Cells 2021, 10, 2983. https://doi.org/10.3390/cells10112983
Naville M, Merabet S. In-Depth Annotation of the Drosophila Bithorax-Complex Reveals the Presence of Several Alternative ORFs That Could Encode for Motif-Rich Peptides. Cells. 2021; 10(11):2983. https://doi.org/10.3390/cells10112983
Chicago/Turabian StyleNaville, Magali, and Samir Merabet. 2021. "In-Depth Annotation of the Drosophila Bithorax-Complex Reveals the Presence of Several Alternative ORFs That Could Encode for Motif-Rich Peptides" Cells 10, no. 11: 2983. https://doi.org/10.3390/cells10112983
APA StyleNaville, M., & Merabet, S. (2021). In-Depth Annotation of the Drosophila Bithorax-Complex Reveals the Presence of Several Alternative ORFs That Could Encode for Motif-Rich Peptides. Cells, 10(11), 2983. https://doi.org/10.3390/cells10112983