Regulation of Osteoclast Differentiation and Activity by Lipid Metabolism




Abstract
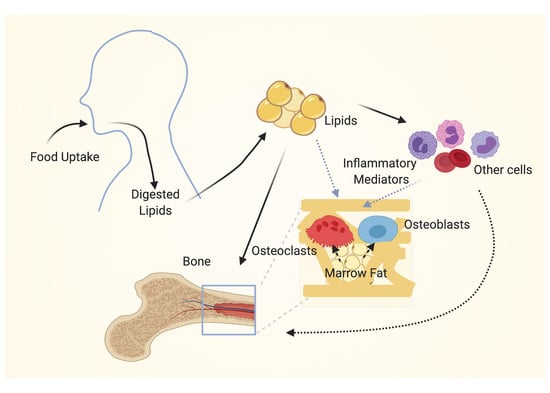
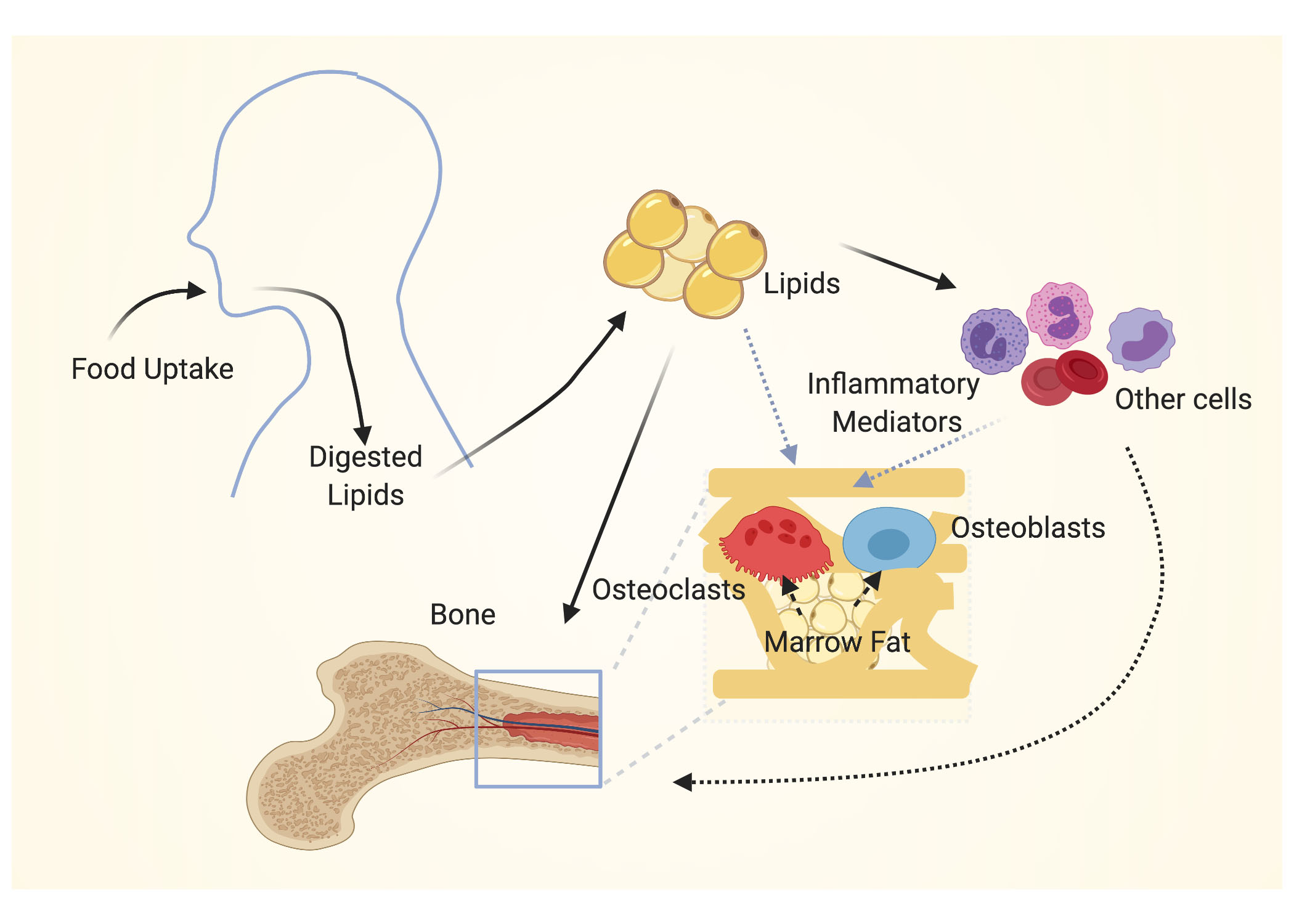
1. Introduction
2. Lipid Metabolism
3. Regulation by SREBPs
4. Cholesterol Synthesis Pathway
5. The Role of Cholesterol in Osteoclasts
6. LXRs and RXRs
7. Fatty Acid Synthesis and Fatty Acid Oxidation
8. Function of Fatty Acid in Osteoclasts
9. Dyslipidemia and Bone Metabolism
10. Statins
10.1. Preclinical Studies
10.2. Clinical Studies
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, E.F. Cellular mechanisms of bone remodeling. Rev. Endocr. Metab. Disord. 2010, 11, 219–227. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Park-Min, K.H. Mechanisms involved in normal and pathological osteoclastogenesis. Cell. Mol. Life Sci. 2018, 75, 2519–2528. [Google Scholar] [CrossRef]
- Novack, D.V.; Teitelbaum, S.L. The osteoclast: Friend or foe? Annu. Rev. Pathol. 2008, 3, 457–484. [Google Scholar] [CrossRef] [PubMed]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell ... and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- During, A.; Penel, G.; Hardouin, P. Understanding the local actions of lipids in bone physiology. Prog. Lipid Res. 2015, 59, 126–146. [Google Scholar] [CrossRef]
- Shapses, S.A.; Sukumar, D. Bone metabolism in obesity and weight loss. Annu. Rev. Nutr. 2012, 32, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Douchi, T.; Yamamoto, S.; Oki, T.; Maruta, K.; Kuwahata, R.; Yamasaki, H.; Nagata, Y. Difference in the effect of adiposity on bone density between pre- and postmenopausal women. Maturitas 2000, 34, 261–266. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Rosen, C.J. Lipids in the Bone Marrow: An Evolving Perspective. Cell Metab. 2020, 31, 219–231. [Google Scholar] [CrossRef]
- Lund, P.K.; Abadi, D.M.; Mathies, J.C. Lipid composition of normal human bone marrow as determined by column chromatography. J. Lipid Res. 1962, 3, 95–98. [Google Scholar]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Bhatt, A.; Rohatgi, A. HDL Cholesterol Efflux Capacity: Cardiovascular Risk Factor and Potential Therapeutic Target. Curr. Atheroscler. Rep. 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Zeng, S.; Park-Min, K.H. Nuclear receptors in osteoclasts. Curr. Opin. Pharm. 2020, 53, 8–17. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Hua, X.; Wu, J.; Goldstein, J.L.; Brown, M.S.; Hobbs, H.H. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics 1995, 25, 667–673. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef]
- Hua, X.; Sakai, J.; Brown, M.S.; Goldstein, J.L. Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 1996, 271, 10379–10384. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Inoue, K.; Imai, Y. Fatostatin, an SREBP inhibitor, prevented RANKL-induced bone loss by suppression of osteoclast differentiation. Biochim. Biophys. Acta 2015, 1852, 2432–2441. [Google Scholar] [CrossRef]
- Jie, Z.; Xie, Z.; Xu, W.; Zhao, X.; Jin, G.; Sun, X.; Huang, B.; Tang, P.; Wang, G.; Shen, S.; et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 115–125. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Brown, A.J.; Ikonen, E.; Olkkonen, V.M. Cholesterol precursors: More than mere markers of biosynthesis. Curr. Opin. Lipidol. 2014, 25, 133–139. [Google Scholar] [CrossRef]
- Schoenheimer, R.; Breusch, F. Synthesis and destuction of cholesterol in the organism. J. Biol. Chem. 1933, 103, 439–448. [Google Scholar] [CrossRef]
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, L.J.; Brown, A.J. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 2013, 288, 18707–18715. [Google Scholar] [CrossRef] [PubMed]
- Roitelman, J.; Simoni, R.D. Distinct sterol and nonsterol signals for the regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 1992, 267, 25264–25273. [Google Scholar] [CrossRef]
- Russell, D.W. Cholesterol biosynthesis and metabolism. Cardiovasc. Drugs 1992, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef]
- Ryu, J.; Kim, H.; Chang, E.J.; Kim, H.J.; Lee, Y.; Kim, H.H. Proteomic analysis of osteoclast lipid rafts: The role of the integrity of lipid rafts on V-ATPase activity in osteoclasts. J. Bone Miner. Metab. 2010, 28, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.D.; Yoon, S.H.; Park, C.K.; Lee, J.; Lee, Z.H.; Kim, H.H. Caveolin-1 regulates osteoclastogenesis and bone metabolism in a sex-dependent manner. J. Biol. Chem. 2015, 290, 6522–6530. [Google Scholar] [CrossRef]
- Oikawa, T.; Kuroda, Y.; Matsuo, K. Regulation of osteoclasts by membrane-derived lipid mediators. Cell. Mol. Life Sci. 2013, 70, 3341–3353. [Google Scholar] [CrossRef]
- Pelton, K.; Krieder, J.; Joiner, D.; Freeman, M.R.; Goldstein, S.A.; Solomon, K.R. Hypercholesterolemia promotes an osteoporotic phenotype. Am. J. Pathol. 2012, 181, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Potin, I.; Roman-Blas, J.A.; Martinez-Calatrava, M.J.; Gomez, R.; Largo, R.; Herrero-Beaumont, G. Hypercholesterolemia boosts joint destruction in chronic arthritis. An experimental model aggravated by foam macrophage infiltration. Arthritis Res. Ther. 2013, 15, R81. [Google Scholar] [CrossRef]
- Sanbe, T.; Tomofuji, T.; Ekuni, D.; Azuma, T.; Tamaki, N.; Yamamoto, T. Oral administration of vitamin C prevents alveolar bone resorption induced by high dietary cholesterol in rats. J. Periodontol. 2007, 78, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Morita, I.; Murota, S. Involvement of cholesterol in osteoclast-like cell formation via cellular fusion. Bone 1998, 23, 135–140. [Google Scholar] [CrossRef]
- Okayasu, M.; Nakayachi, M.; Hayashida, C.; Ito, J.; Kaneda, T.; Masuhara, M.; Suda, N.; Sato, T.; Hakeda, Y. Low-density lipoprotein receptor deficiency causes impaired osteoclastogenesis and increased bone mass in mice because of defect in osteoclastic cell-cell fusion. J. Biol. Chem. 2012, 287, 19229–19241. [Google Scholar] [CrossRef] [PubMed]
- Luegmayr, E.; Glantschnig, H.; Wesolowski, G.A.; Gentile, M.A.; Fisher, J.E.; Rodan, G.A.; Reszka, A.A. Osteoclast formation, survival and morphology are highly dependent on exogenous cholesterol/lipoproteins. Cell Death Differ. 2004, 11 (Suppl. 1), S108–S118. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Lv, Y.; He, P.; Wang, Z.; Xiong, F.; He, L.; Zheng, X.; Zhang, D.; Cao, Q.; Tang, C. HDL impairs osteoclastogenesis and induces osteoclast apoptosis via upregulation of ABCG1 expression. Acta Biochim. Biophys. Sin. 2018, 50, 853–861. [Google Scholar] [CrossRef]
- Huszar, D.; Varban, M.L.; Rinninger, F.; Feeley, R.; Arai, T.; Fairchild-Huntress, V.; Donovan, M.J.; Tall, A.R. Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Atheroscler. Thromb. Vasc. Biol. 2000, 20, 1068–1073. [Google Scholar] [CrossRef]
- Tintut, Y.; Morony, S.; Demer, L.L. Hyperlipidemia promotes osteoclastic potential of bone marrow cells ex vivo. Atheroscler. Thromb. Vasc. Biol. 2004, 24, e6–e10. [Google Scholar] [CrossRef]
- Sjogren, U.; Mukohyama, H.; Roth, C.; Sundqvist, G.; Lerner, U.H. Bone-resorbing activity from cholesterol-exposed macrophages due to enhanced expression of interleukin-1alpha. J. Dent. Res. 2002, 81, 11–16. [Google Scholar] [CrossRef]
- Wei, W.; Schwaid, A.G.; Wang, X.; Wang, X.; Chen, S.; Chu, Q.; Saghatelian, A.; Wan, Y. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. [Google Scholar] [CrossRef]
- Wei, W.; Wang, X.; Yang, M.; Smith, L.C.; Dechow, P.C.; Sonoda, J.; Evans, R.M.; Wan, Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010, 11, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, M.J.; Mun, S.H.; Giannopoulou, E.G.; Yong-Gonzalez, V.; Cross, J.R.; Murata, K.; Giguere, V.; van der Meulen, M.; Park-Min, K.H. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha. J. Clin. Investig. 2017, 127, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, S.; Zhang, J.; Yi, T.; Shen, W.; Nguyen, D.; Pereira, J.P.; Guerini, D.; Baumgarten, B.U.; Roggo, S.; Wen, B.; et al. Oxysterols direct immune cell migration via EBI2. Nature 2011, 475, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Nevius, E.; Pinho, F.; Dhodapkar, M.; Jin, H.; Nadrah, K.; Horowitz, M.C.; Kikuta, J.; Ishii, M.; Pereira, J.P. Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J. Exp. Med. 2015, 212, 1931–1946. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef]
- Robertson, K.M.; Norgard, M.; Windahl, S.H.; Hultenby, K.; Ohlsson, C.; Andersson, G.; Gustafsson, J.A. Cholesterol-sensing receptors, liver X receptor alpha and beta, have novel and distinct roles in osteoclast differentiation and activation. J. Bone Miner. Res. 2006, 21, 1276–1287. [Google Scholar] [CrossRef]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, K.A.; Yoon, H.J.; Hong, J.M.; Lee, M.J.; Lee, I.K.; Kim, S.Y. Liver X receptor activation inhibits osteoclastogenesis by suppressing NF-kappaB activity and c-Fos induction and prevents inflammatory bone loss in mice. J. Leukoc. Biol. 2013, 94, 99–107. [Google Scholar] [CrossRef]
- Remen, K.M.; Henning, P.; Lerner, U.H.; Gustafsson, J.A.; Andersson, G. Activation of liver X receptor (LXR) inhibits receptor activator of nuclear factor kappaB ligand (RANKL)-induced osteoclast differentiation in an LXRbeta-dependent mechanism. J. Biol. Chem. 2011, 286, 33084–33094. [Google Scholar] [CrossRef] [PubMed]
- Glaría, E.; Letelier, N.A.; Valledor, A.F. Integrating the roles of liver X receptors in inflammation and infection: Mechanisms and outcomes. Curr. Opin. Pharmacol. 2020, 53, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gutierrez, M.P.; Roszer, T.; Fuentes, L.; Nunez, V.; Escolano, A.; Redondo, J.M.; De Clerck, N.; Metzger, D.; Valledor, A.F.; Ricote, M. Retinoid X receptors orchestrate osteoclast differentiation and postnatal bone remodeling. J. Clin. Investig. 2015, 125, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, J.H.; Lee, J.; Jin, H.M.; Kook, H.; Kim, K.K.; Lee, S.Y.; Kim, N. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 2007, 109, 3253–3259. [Google Scholar] [CrossRef]
- Kim, S.P.; Li, Z.; Zoch, M.L.; Frey, J.L.; Bowman, C.E.; Kushwaha, P.; Ryan, K.A.; Goh, B.C.; Scafidi, S.; Pickett, J.E.; et al. Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight 2017, 2, e92704. [Google Scholar] [CrossRef]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- Maier, T.; Leibundgut, M.; Ban, N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321, 1315–1322. [Google Scholar] [CrossRef]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Bao, M.; Zhang, K.; Wei, Y.; Hua, W.; Gao, Y.; Li, X.; Ye, L. Therapeutic potentials and modulatory mechanisms of fatty acids in bone. Cell Prolif. 2020, 53, e12735. [Google Scholar] [CrossRef] [PubMed]
- Brenna, J.T.; Salem, N., Jr.; Sinclair, A.J.; Cunnane, S.C. α-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fat. Acids 2009, 80, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Jones, A.E.; Wootton, S.A. Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men. Br. J. Nutr. 2002, 88, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Wootton, S.A. Conversion of alpha-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br. J. Nutr. 2002, 88, 411–420. [Google Scholar] [CrossRef]
- Brossard, N.; Croset, M.; Pachiaudi, C.; Riou, J.P.; Tayot, J.L.; Lagarde, M. Retroconversion and metabolism of [13C]22:6n-3 in humans and rats after intake of a single dose of [13C]22:6n-3-triacylglycerols. Am. J. Clin. Nutr. 1996, 64, 577–586. [Google Scholar] [CrossRef]
- Al, M.D.; van Houwelingen, A.C.; Hornstra, G. Long-chain polyunsaturated fatty acids, pregnancy, and pregnancy outcome. Am. J. Clin. Nutr. 2000, 71, 285S–291S. [Google Scholar] [CrossRef]
- Mangano, K.M.; Sahni, S.; Kerstetter, J.E.; Kenny, A.M.; Hannan, M.T. Polyunsaturated fatty acids and their relation with bone and muscle health in adults. Curr. Osteoporos. Rep. 2013, 11, 203–212. [Google Scholar] [CrossRef]
- Kasonga, A.E.; Deepak, V.; Kruger, M.C.; Coetzee, M. Arachidonic acid and docosahexaenoic acid suppress osteoclast formation and activity in human CD14+ monocytes, in vitro. PLoS ONE 2015, 10, e0125145. [Google Scholar] [CrossRef]
- Sun, D.; Krishnan, A.; Zaman, K.; Lawrence, R.; Bhattacharya, A.; Fernandes, G. Dietary n-3 fatty acids decrease osteoclastogenesis and loss of bone mass in ovariectomized mice. J. Bone Miner. Res. 2003, 18, 1206–1216. [Google Scholar] [CrossRef]
- Kim, H.J.; Ohk, B.; Yoon, H.J.; Kang, W.Y.; Seong, S.J.; Kim, S.Y.; Yoon, Y.R. Docosahexaenoic acid signaling attenuates the proliferation and differentiation of bone marrow-derived osteoclast precursors and promotes apoptosis in mature osteoclasts. Cell. Signal. 2017, 29, 226–232. [Google Scholar] [CrossRef]
- Boeyens, J.C.; Deepak, V.; Chua, W.H.; Kruger, M.C.; Joubert, A.M.; Coetzee, M. Effects of omega3- and omega6-polyunsaturated fatty acids on RANKL-induced osteoclast differentiation of RAW264.7 cells: A comparative in vitro study. Nutrients 2014, 6, 2584–2601. [Google Scholar] [CrossRef]
- Appleton, K.M.; Fraser, W.D.; Rogers, P.J.; Ness, A.R.; Tobias, J.H. Supplementation with a low-moderate dose of n-3 long-chain PUFA has no short-term effect on bone resorption in human adults. Br. J. Nutr. 2011, 105, 1145–1149. [Google Scholar] [CrossRef]
- Lavado-Garcia, J.; Roncero-Martin, R.; Moran, J.M.; Pedrera-Canal, M.; Aliaga, I.; Leal-Hernandez, O.; Rico-Martin, S.; Canal-Macias, M.L. Long-chain omega-3 polyunsaturated fatty acid dietary intake is positively associated with bone mineral density in normal and osteopenic Spanish women. PLoS ONE 2018, 13, e0190539. [Google Scholar] [CrossRef]
- Kishikawa, A.; Kitaura, H.; Kimura, K.; Ogawa, S.; Qi, J.; Shen, W.R.; Ohori, F.; Noguchi, T.; Marahleh, A.; Nara, Y.; et al. Docosahexaenoic Acid Inhibits Inflammation-Induced Osteoclast Formation and Bone Resorption in vivo Through GPR120 by Inhibiting TNF-alpha Production in Macrophages and Directly Inhibiting Osteoclast Formation. Front. Endocrinol. 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.M.; Gomes-Filho, J.E.; Ervolino, E.; Pipa, C.B.; Cardoso, C.B.M.; Andrada, A.C.; Kawai, T.; Cintra, L.T.A. Omega 3 Fatty Acids Reduce Bone Resorption While Promoting Bone Generation in Rat Apical Periodontitis. J. Endod. 2017, 43, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Muhlhausler, B.S.; Gibson, R.A.; Xian, C.J. Perinatal maternal dietary supplementation of omega3-fatty acids transiently affects bone marrow microenvironment, osteoblast and osteoclast formation, and bone mass in male offspring. Endocrinology 2012, 153, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; MacGibbon, A.; Lin, J.M.; Watson, M.; Callon, K.E.; Tong, P.C.; Dunford, J.E.; van der Does, Y.; Williams, G.A.; Grey, A.B.; et al. Modulation of osteoclastogenesis by fatty acids. Endocrinology 2008, 149, 5688–5695. [Google Scholar] [CrossRef]
- Wauquier, F.; Philippe, C.; Leotoing, L.; Mercier, S.; Davicco, M.J.; Lebecque, P.; Guicheux, J.; Pilet, P.; Miot-Noirault, E.; Poitout, V.; et al. The free fatty acid receptor G protein-coupled receptor 40 (GPR40) protects from bone loss through inhibition of osteoclast differentiation. J. Biol. Chem. 2013, 288, 6542–6551. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, H.J.; Kim, B.K.; Kang, W.Y.; Seong, S.J.; Lim, M.S.; Kim, S.Y.; Yoon, Y.R. G Protein-Coupled Receptor 120 Signaling Negatively Regulates Osteoclast Differentiation, Survival, and Function. J. Cell. Physiol. 2016, 231, 844–851. [Google Scholar] [CrossRef]
- Philippe, C.; Wauquier, F.; Leotoing, L.; Coxam, V.; Wittrant, Y. GW9508, a free fatty acid receptor agonist, specifically induces cell death in bone resorbing precursor cells through increased oxidative stress from mitochondrial origin. Exp. Cell Res. 2013, 319, 3035–3041. [Google Scholar] [CrossRef]
- Garcia, C.; Boyce, B.F.; Gilles, J.; Dallas, M.; Qiao, M.; Mundy, G.R.; Bonewald, L.F. Leukotriene B4 stimulates osteoclastic bone resorption both in vitro and in vivo. J. Bone Miner. Res. 1996, 11, 1619–1627. [Google Scholar] [CrossRef]
- Kasonga, A.; Kruger, M.C.; Coetzee, M. Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes. Int. J. Mol. Sci. 2019, 20, 1798. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.C.; Coetzee, M.; Haag, M.; Weiler, H. Long-chain polyunsaturated fatty acids: Selected mechanisms of action on bone. Prog. Lipid Res. 2010, 49, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.R.; Sul, O.J.; Kim, Y.Y.; Kim, H.J.; Yu, R.; Suh, J.H.; Choi, H.S. Saturated fatty acids enhance osteoclast survival. J. Lipid Res. 2010, 51, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Drosatos-Tampakaki, Z.; Drosatos, K.; Siegelin, Y.; Gong, S.; Khan, S.; Van Dyke, T.; Goldberg, I.J.; Schulze, P.C.; Schulze-Spate, U. Palmitic acid and DGAT1 deficiency enhance osteoclastogenesis, while oleic acid-induced triglyceride formation prevents it. J. Bone Miner. Res. 2014, 29, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- van Heerden, B.; Kasonga, A.; Kruger, M.C.; Coetzee, M. Palmitoleic Acid Inhibits RANKL-Induced Osteoclastogenesis and Bone Resorption by Suppressing NF-kappaB and MAPK Signalling Pathways. Nutrients 2017, 9, 441. [Google Scholar] [CrossRef]
- Li, J.Y.; Chassaing, B.; Tyagi, A.M.; Vaccaro, C.; Luo, T.; Adams, J.; Darby, T.M.; Weitzmann, M.N.; Mulle, J.G.; Gewirtz, A.T.; et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Investig. 2016, 126, 2049–2063. [Google Scholar] [CrossRef]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Lucas, S.; Omata, Y.; Hofmann, J.; Bottcher, M.; Iljazovic, A.; Sarter, K.; Albrecht, O.; Schulz, O.; Krishnacoumar, B.; Kronke, G.; et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 2018, 9, 55. [Google Scholar] [CrossRef]
- Kwon, J.O.; Jin, W.J.; Kim, B.; Kim, H.H.; Lee, Z.H. Myristoleic acid inhibits osteoclast formation and bone resorption by suppressing the RANKL activation of Src and Pyk2. Eur. J. Pharm. 2015, 768, 189–198. [Google Scholar] [CrossRef]
- Pinals, R.S.; Jabbs, J.M. Type-IV hyperlipoproteinaemia and transient osteoporosis. Lancet 1972, 2, 929. [Google Scholar] [CrossRef]
- Tanko, L.B.; Bagger, Y.Z.; Christiansen, C. Low bone mineral density in the hip as a marker of advanced atherosclerosis in elderly women. Calcif. Tissue Int. 2003, 73, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sugimoto, T.; Yano, S.; Yamauchi, M.; Sowa, H.; Chen, Q.; Chihara, K. Plasma lipids and osteoporosis in postmenopausal women. Endocr. J. 2002, 49, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pagnotti, G.M.; Styner, M.; Uzer, G.; Patel, V.S.; Wright, L.E.; Ness, K.K.; Guise, T.A.; Rubin, J.; Rubin, C.T. Combating osteoporosis and obesity with exercise: Leveraging cell mechanosensitivity. Nat. Rev. Endocrinol. 2019, 15, 339–355. [Google Scholar] [CrossRef]
- Zhao, L.J.; Liu, Y.J.; Liu, P.Y.; Hamilton, J.; Recker, R.R.; Deng, H.W. Relationship of obesity with osteoporosis. J. Clin. Endocrinol. Metab. 2007, 92, 1640–1646. [Google Scholar] [CrossRef]
- Zhou, Y.; Deng, T.; Zhang, H.; Guan, Q.; Zhao, H.; Yu, C.; Shao, S.; Zhao, M.; Xu, J. Hypercholesterolaemia increases the risk of highturnover osteoporosis in men. Mol. Med. Rep. 2019, 19, 4603–4612. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Venners, S.A.; Terwedow, H.A.; Feng, Y.; Niu, T.; Li, Z.; Laird, N.; Brain, J.D.; Cummings, S.R.; Bouxsein, M.L.; et al. Relation of body composition, fat mass, and serum lipids to osteoporotic fractures and bone mineral density in Chinese men and women. Am. J. Clin. Nutr. 2006, 83, 146–154. [Google Scholar] [CrossRef]
- Syed, F.A.; Oursler, M.J.; Hefferanm, T.E.; Peterson, J.M.; Riggs, B.L.; Khosla, S. Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos. Int. 2008, 19, 1323–1330. [Google Scholar] [CrossRef]
- Wend, K.; Wend, P.; Drew, B.G.; Hevener, A.L.; Miranda-Carboni, G.A.; Krum, S.A. ERalpha regulates lipid metabolism in bone through ATGL and perilipin. J. Cell. Biochem. 2013, 114, 1306–1314. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef]
- Wardlaw, G.M. Putting body weight and osteoporosis into perspective. Am. J. Clin. Nutr. 1996, 63, 433S–436S. [Google Scholar] [CrossRef] [PubMed]
- Radak, T.L. Caloric restriction and calcium’s effect on bone metabolism and body composition in overweight and obese premenopausal women. Nutr. Rev. 2004, 62, 468–481. [Google Scholar] [CrossRef]
- Faienza, M.F.; D’Amato, G.; Chiarito, M.; Colaianni, G.; Colucci, S.; Grano, M.; Corbo, F.; Brunetti, G. Mechanisms Involved in Childhood Obesity-Related Bone Fragility. Front. Endocrinol. 2019, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Baxter-Jones, A.D.; Kontulainen, S.A.; Faulkner, R.A.; Bailey, D.A. A longitudinal study of the relationship of physical activity to bone mineral accrual from adolescence to young adulthood. Bone 2008, 43, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, A.; Celi, M.; Volpe, S.L.; Sorge, R.; Tarantino, U. Long-term effect of exercise on bone mineral density and body composition in post-menopausal ex-elite athletes: A retrospective study. Eur. J. Clin. Nutr. 2012, 66, 69–74. [Google Scholar] [CrossRef][Green Version]
- Follin, S.L.; Hansen, L.B. Current approaches to the prevention and treatment of postmenopausal osteoporosis. Am. J. Health Syst. Pharm. 2003, 60, 883–901, quiz 903-884. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Atkinson, E.J.; Riggs, B.L.; Melton, L.J., 3rd. Relationship between body composition and bone mass in women. J. Bone Miner. Res. 1996, 11, 857–863. [Google Scholar] [CrossRef]
- Ilesanmi-Oyelere, B.L.; Coad, J.; Roy, N.; Kruger, M.C. Lean Body Mass in the Prediction of Bone Mineral Density in Postmenopausal Women. BioRes. Open Access 2018, 7, 150–158. [Google Scholar] [CrossRef]
- Bierhals, I.O.; Dos Santos Vaz, J.; Bielemann, R.M.; de Mola, C.L.; Barros, F.C.; Goncalves, H.; Wehrmeister, F.C.; Assuncao, M.C.F. Associations between body mass index, body composition and bone density in young adults: Findings from a southern Brazilian cohort. BMC Musculoskelet. Disord. 2019, 20, 322. [Google Scholar] [CrossRef]
- Valderrabano, R.J.; Linares, M.I. Diabetes mellitus and bone health: Epidemiology, etiology and implications for fracture risk stratification. Clin. Diabetes Endocrinol. 2018, 4, 9. [Google Scholar] [CrossRef]
- Lapmanee, S.; Charoenphandhu, N.; Aeimlapa, R.; Suntornsaratoon, P.; Wongdee, K.; Tiyasatkulkovit, W.; Kengkoom, K.; Chaimongkolnukul, K.; Seriwatanachai, D.; Krishnamra, N. High dietary cholesterol masks type 2 diabetes-induced osteopenia and changes in bone microstructure in rats. Lipids 2014, 49, 975–986. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Pereira Coutinho, M.; Bandeira, E.; de Almeida, J.M.; Godoi, E.T.; Vasconcelos, G.; Bandeira, F. Low Bone Mass is Associated with Increased Carotid Intima Media Thickness in Men with Type 2 Diabetes Mellitus. Clin. Med. Insights Endocrinol. Diabetes 2013, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Janghorbani, M.; Van Dam, R.M.; Willett, W.C.; Hu, F.B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 2007, 166, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.N.; Shah, C.S.; Snell-Bergeon, J.K. Type 1 diabetes and risk of fracture: Meta-analysis and review of the literature. Diabet. Med. 2015, 32, 1134–1142. [Google Scholar] [CrossRef]
- Fan, Y.; Wei, F.; Lang, Y.; Liu, Y. Diabetes mellitus and risk of hip fractures: A meta-analysis. Osteoporos. Int. 2016, 27, 219–228. [Google Scholar] [CrossRef]
- Wang, H.; Ba, Y.; Xing, Q.; Du, J.L. Diabetes mellitus and the risk of fractures at specific sites: A meta-analysis. BMJ Open 2019, 9, e024067. [Google Scholar] [CrossRef]
- Petit, C.; Batool, F.; Bugueno, I.M.; Schwinte, P.; Benkirane-Jessel, N.; Huck, O. Contribution of Statins towards Periodontal Treatment: A Review. Mediat. Inflamm. 2019, 2019, 6367402. [Google Scholar] [CrossRef]
- Fentoglu, O.; Kirzioglu, F.Y.; Ozdem, M.; Kocak, H.; Sutcu, R.; Sert, T. Proinflammatory cytokine levels in hyperlipidemic patients with periodontitis after periodontal treatment. Oral Dis. 2012, 18, 299–306. [Google Scholar] [CrossRef]
- Catalfamo, D.L.; Britten, T.M.; Storch, D.L.; Calderon, N.L.; Sorenson, H.L.; Wallet, S.M. Hyperglycemia induced and intrinsic alterations in type 2 diabetes-derived osteoclast function. Oral Dis. 2013, 19, 303–312. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, T.R.; Tobert, J.A. Simvastatin: A review. Expert Opin. Pharm. 2004, 5, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Larsson, B.A.M.; Sundh, D.; Mellstrom, D.; Axelsson, K.F.; Nilsson, A.G.; Lorentzon, M. Association Between Cortical Bone Microstructure and Statin Use in Older Women. J. Clin. Endocrinol. Metab. 2019, 104, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Scranton, R.E.; Young, M.; Lawler, E.; Solomon, D.; Gagnon, D.; Gaziano, J.M. Statin use and fracture risk: Study of a US veterans population. Arch. Intern. Med. 2005, 165, 2007–2012. [Google Scholar] [CrossRef]
- Chan, K.A.; Andrade, S.E.; Boles, M.; Buist, D.S.; Chase, G.A.; Donahue, J.G.; Goodman, M.J.; Gurwitz, J.H.; LaCroix, A.Z.; Platt, R. Inhibitors of hydroxymethylglutaryl-coenzyme A reductase and risk of fracture among older women. Lancet 2000, 355, 2185–2188. [Google Scholar] [CrossRef]
- Morse, L.R.; Coker, J.; Battaglino, R.A. Statins and Bone Health: A Mini Review. Actual. Osteol. 2018, 14, 31–35. [Google Scholar]
- Bauer, D.C. HMG CoA reductase inhibitors and the skeleton: A comprehensive review. Osteoporos. Int. 2003, 14, 273–282. [Google Scholar] [CrossRef]
- Zheng, J.; Brion, M.J.; Kemp, J.P.; Warrington, N.M.; Borges, M.C.; Hemani, G.; Richardson, T.G.; Rasheed, H.; Qiao, Z.; Haycock, P.; et al. The Effect of Plasma Lipids and Lipid-Lowering Interventions on Bone Mineral Density: A Mendelian Randomization Study. J. Bone Miner. Res. 2020, 35, 1224–1235. [Google Scholar] [CrossRef]
- Mundy, G.; Garrett, R.; Harris, S.; Chan, J.; Chen, D.; Rossini, G.; Boyce, B.; Zhao, M.; Gutierrez, G. Stimulation of bone formation in vitro and in rodents by statins. Science 1999, 286, 1946–1949. [Google Scholar] [CrossRef]
- Ohnaka, K.; Shimoda, S.; Nawata, H.; Shimokawa, H.; Kaibuchi, K.; Iwamoto, Y.; Takayanagi, R. Pitavastatin enhanced BMP-2 and osteocalcin expression by inhibition of Rho-associated kinase in human osteoblasts. Biochem. Biophys. Res. Commun. 2001, 287, 337–342. [Google Scholar] [CrossRef]
- Viereck, V.; Grundker, C.; Blaschke, S.; Frosch, K.H.; Schoppet, M.; Emons, G.; Hofbauer, L.C. Atorvastatin stimulates the production of osteoprotegerin by human osteoblasts. J. Cell. Biochem. 2005, 96, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Staal, A.; Frith, J.C.; French, M.H.; Swartz, J.; Gungor, T.; Harrity, T.W.; Tamasi, J.; Rogers, M.J.; Feyen, J.H. The ability of statins to inhibit bone resorption is directly related to their inhibitory effect on HMG-CoA reductase activity. J. Bone Miner. Res. 2003, 18, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Haneji, T. Stimulation of osteoclast formation by RANKL requires interferon regulatory factor-4 and is inhibited by simvastatin in a mouse model of bone loss. PLoS ONE 2013, 8, e72033. [Google Scholar] [CrossRef] [PubMed]
- Oxlund, H.; Andreassen, T.T. Simvastatin treatment partially prevents ovariectomy-induced bone loss while increasing cortical bone formation. Bone 2004, 34, 609–618. [Google Scholar] [CrossRef]
- Tobert, J.A. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef]
- Grasser, W.A.; Baumann, A.P.; Petras, S.F.; Harwood, H.J., Jr.; Devalaraja, R.; Renkiewicz, R.; Baragi, V.; Thompson, D.D.; Paraklar, V.M. Regulation of osteoclast differentiation by statins. J. Musculoskelet. Neuronal Interact. 2003, 3, 53–62. [Google Scholar]
- Edwards, C.J.; Hart, D.J.; Spector, T.D. Oral statins and increased bone-mineral density in postmenopausal women. Lancet 2000, 355, 2218–2219. [Google Scholar] [CrossRef]
- Lupattelli, G.; Scarponi, A.M.; Vaudo, G.; Siepi, D.; Roscini, A.R.; Gemelli, F.; Pirro, M.; Latini, R.A.; Sinzinger, H.; Marchesi, S.; et al. Simvastatin increases bone mineral density in hypercholesterolemic postmenopausal women. Metabolism 2004, 53, 744–748. [Google Scholar] [CrossRef]
- Chuengsamarn, S.; Rattanamongkoulgul, S.; Suwanwalaikorn, S.; Wattanasirichaigoon, S.; Kaufman, L. Effects of statins vs. non-statin lipid-lowering therapy on bone formation and bone mineral density biomarkers in patients with hyperlipidemia. Bone 2010, 46, 1011–1015. [Google Scholar] [CrossRef]
- Safaei, H.; Janghorbani, M.; Aminorroaya, A.; Amini, M. Lovastatin effects on bone mineral density in postmenopausal women with type 2 diabetes mellitus. Acta Diabetol. 2007, 44, 76–82. [Google Scholar] [CrossRef]
- Uysal, A.R.; Delibasi, T.; Erdogan, M.F.; Kamel, N.; Baskal, N.; Tonyukuk, V.; Corapcioglu, D.; Gullu, S.; Erdogan, G. Effect of simvastatin use on bone mineral density in women with type 2 diabetes. Endocr. Pract. 2007, 13, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.; Ertugrul, D.T.; Cil, H.; Ata, N.; Akin, K.O.; Yalcin, A.A.; Kucukazman, M.; Dal, K.; Hokkaomeroglu, M.S.; Yavuz, B.B.; et al. Increased levels of 25 hydroxyvitamin D and 1,25-dihydroxyvitamin D after rosuvastatin treatment: A novel pleiotropic effect of statins? Cardiovasc. Drugs Ther. 2009, 23, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Nakashima, R.; Ito, T.; Masaki, T.; Yorioka, N. HMG-CoA reductase inhibitors prevent bone loss in patients with Type 2 diabetes mellitus. Diabet. Med. J. Br. Diabet. Assoc. 2004, 21, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.S.; Lee, M.D.; Lee, S.K.; Kim, H.M.; Fitzpatrick, L.A. HMG-CoA reductase inhibitors increase BMD in type 2 diabetes mellitus patients. J. Clin. Endocrinol. Metab. 2000, 85, 1137–1142. [Google Scholar] [CrossRef]
- Kanazawa, I.; Yamaguchi, T.; Yamauchi, M.; Sugimoto, T. Rosuvastatin increased serum osteocalcin levels independent of its serum cholesterol-lowering effect in patients with type 2 diabetes and hypercholesterolemia. Intern. Med. 2009, 48, 1869–1873. [Google Scholar] [CrossRef]
- Bahammam, M.A.; Attia, M.S. Effects of Systemic Simvastatin on the Concentrations of Visfatin, Tumor Necrosis Factor-alpha, and Interleukin-6 in Gingival Crevicular Fluid in Patients with Type 2 Diabetes and Chronic Periodontitis. J. Immunol. Res. 2018, 2018, 8481735. [Google Scholar] [CrossRef]
- Kumari, M.; Martande, S.S.; Pradeep, A.R. Subgingivally delivered 1.2% atorvastatin in the treatment of chronic periodontitis among smokers: A randomized, controlled clinical trial. J. Investig. Clin. Dent. 2017, 8, e12213. [Google Scholar] [CrossRef]
- Pradeep, A.R.; Garg, V.; Kanoriya, D.; Singhal, S. 1.2% Rosuvastatin Versus 1.2% Atorvastatin Gel Local Drug Delivery and Redelivery in Treatment of Intrabony Defects in Chronic Periodontitis: A Randomized Placebo-Controlled Clinical Trial. J. Periodontol. 2016, 87, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Braatvedt, G.D.; Bagg, W.; Gamble, G.; Davidson, J.; Reid, I.R. The effect of atorvastatin on markers of bone turnover in patients with type 2 diabetes. Bone 2004, 35, 766–770. [Google Scholar] [CrossRef]
- Rejnmark, L.; Buus, N.H.; Vestergaard, P.; Heickendorff, L.; Andreasen, F.; Larsen, M.L.; Mosekilde, L. Effects of simvastatin on bone turnover and BMD: A 1-year randomized controlled trial in postmenopausal osteopenic women. J. Bone Miner. Res. 2004, 19, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Berthold, H.K.; Unverdorben, S.; Zittermann, A.; Degenhardt, R.; Baumeister, B.; Unverdorben, M.; Krone, W.; Vetter, H.; Gouni-Berthold, I. Age-dependent effects of atorvastatin on biochemical bone turnover markers: A randomized controlled trial in postmenopausal women. Osteoporos. Int. 2004, 15, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Park-Min, K.H. Metabolic reprogramming in osteoclasts. Semin. Immunopathol. 2019, 41, 565–572. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Oros, I.; Sola, R.; Valls, R.M.; Brea, A.; Mozas, P.; Puzo, J.; Pocovi, M. Genetic Variants of LDLR and PCSK9 Associated with Variations in Response to Antihypercholesterolemic Effects of Armolipid Plus with Berberine. PLoS ONE 2016, 11, e0150785. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, W.; Gao, L.; Liu, S. Therapeutic targets of hypercholesterolemia: HMGCR and LDLR. Diabetes Metab. Syndr. Obes. 2019, 12, 1543–1553. [Google Scholar] [CrossRef]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Subjects | Compound | Group Size (# of Treatment) | Effects | Duration of Supplementation | Ref. |
|---|---|---|---|---|---|
| Elderly women | Statins | 3028 (803) | lower cortical porosity and higher cortical bone density | Not reported | [133] |
| Healthy periodontium, chronic periodontitis + T2DM, chronic periodontitis, chronic periodontitis + T2DM+simvastatin | Simvastatin | 80 (20) | Improved clinical parameters of periodontitis (also lowers IL-6 & TNFa). | 5 to 10 years | [156] |
| T2DM with chronic periodontitis | Atorvastatin (Subgingivally local delivery) | 75 | Improved in reducing chronic periodontitis. | 3, 6 and 9 months | [157] |
| T2DM with chronic periodontitis | Rosuvastatin (RSV) or atorvastatin (ATV) (Subgingivally local delivery) | 90 (38) | Improved clinical parameters of periodontitis in RSV group relative to ATV and placebo groups | 6 and 9 months | [158] |
| Hyperlipidemia with osteopenia | Simvastatin gemfibrozil fibrate | 212 (106) | Improved bone mass and significantly higher bone turnover markers | 18 months | [149] |
| Patients with T2DM and hypercholesterolemia | Rosuvastatin or ezetimibe | 36 (18) | Increased serum osteocalcin | 3 months | [155] |
| Postmenopausal women with T2DM | Lovastatin | 55 (28) | Increased BMD in Lumbar spine and Ward’s triangle | 18 months | [150] |
| Women with T2DM | Simvastatin | 111 (37) | * Improved BMD-no significant changes | Mean duration 46 months | [151] |
| US veterans populations | Statins | 91,052 (28,063) | Significant reduction of fracture risk | Not specified, but excluded from the statin group if individuals stopped statin for more than 12 months | [134] |
| Hypercholesterolemic postmenopausal women | Simvastatin | 60 (20) | Significant increase of BMD at the spine and femoral hip | 2 years | [148] |
| T2DM patients | Atorvastatin | 25 | No significant effect on bone turnover markers | 12 weeks | [159] |
| Elderly women over 60 years old | Statins | 3675 (928) | Decreased risk of non-pathological fracture | 1 year or more | [135] |
| Postmenopausal women | Atorvastatin, Pravastatin, Fluvastatin, or Simvastatin | 141 (41) | Improved BMD at the spine and hip | 9 to 78 months | [147] |
| T2DM patients | Lovastatin, Pravastatin, or Simvastatin | 69 (36) | Improved BMD at femoral neck, ward triangles, trochanter and total hip | 15 months | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Oh, B.; Park-Min, K.-H. Regulation of Osteoclast Differentiation and Activity by Lipid Metabolism. Cells 2021, 10, 89. https://doi.org/10.3390/cells10010089
Kim H, Oh B, Park-Min K-H. Regulation of Osteoclast Differentiation and Activity by Lipid Metabolism. Cells. 2021; 10(1):89. https://doi.org/10.3390/cells10010089
Chicago/Turabian StyleKim, Haemin, Brian Oh, and Kyung-Hyun Park-Min. 2021. "Regulation of Osteoclast Differentiation and Activity by Lipid Metabolism" Cells 10, no. 1: 89. https://doi.org/10.3390/cells10010089
APA StyleKim, H., Oh, B., & Park-Min, K.-H. (2021). Regulation of Osteoclast Differentiation and Activity by Lipid Metabolism. Cells, 10(1), 89. https://doi.org/10.3390/cells10010089