IL-1α Processing, Signaling and Its Role in Cancer Progression



Abstract
1. Introduction
2. The Biology of IL-1α
2.1. IL-1α Expression
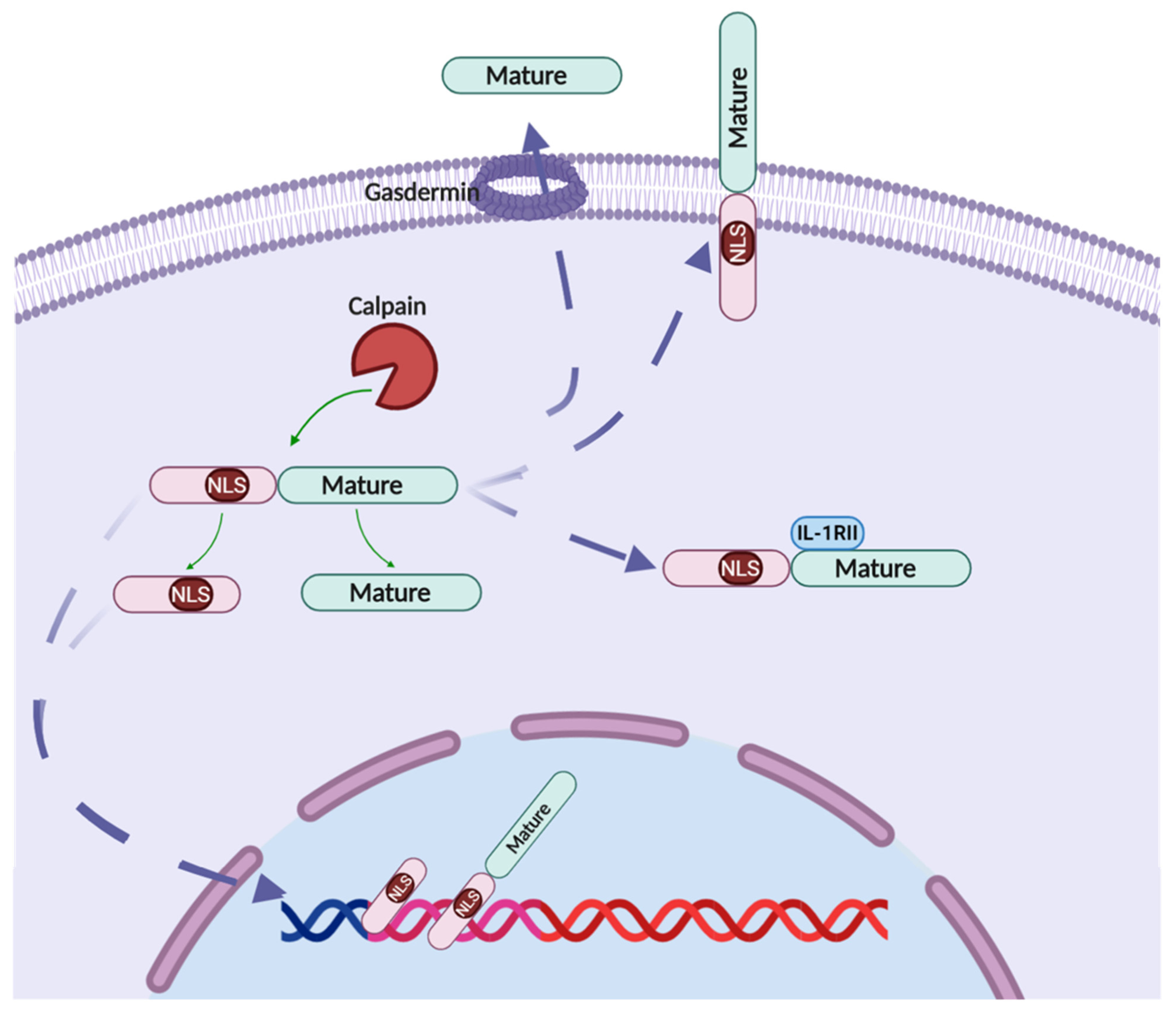
2.2. IL-1α Processing
2.2.1. Elastase, Cathepsin G and Proteinase-3
2.2.2. Calpain
2.2.3. Granzyme B
2.2.4. Chymase
2.2.5. Caspase-5 and Caspase-11
2.2.6. Thrombin
2.3. IL-1α Signaling
2.4. IL-1α Cellular Localization
2.4.1. Nucleus Localization
2.4.2. Membrane IL-1α
2.4.3. Cytosolic IL-1α
2.4.4. Secreted IL-1α
3. IL-1α in Cancer Development
3.1. Breast Cancer
3.2. Pancreatic Cancer
3.3. Leukemia
3.4. Ovarian Cancer
3.5. Head and Neck Squamous Carcinoma
3.6. Liver Cancer
3.7. Lung Cancer
3.8. Fibrosarcoma
3.9. Gastric Cancer
3.10. Prostate Cancer
3.11. Oral Squamous Cell Carcinoma
3.12. Cervical Cancer
3.13. Skin Cancer
3.14. Kidney Cancer
4. Further Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Menkin, V. Biology of inflammation; chemical mediators and cellular injury. Science 1956, 123, 527–534. [Google Scholar] [CrossRef]
- Atkins, E. Pathogenesis of fever. Physiol. Rev. 1960, 40, 580–646. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The interleukin-1 family: 10 years of discovery. FASEB J. Off. Public Fed. Am. Soc. Exp. Biol. 1994, 8, 1314–1325. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Goldin, N.P.; Wolff, S.M. Demonstration and characterization of two distinct human leukocytic pyrogens. J. Exp. Med. 1974, 139, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [PubMed]
- Lomedico, P.T.; Gubler, U.; Hellmann, C.P.; Dukovich, M.; Giri, J.G.; Pan, Y.C.; Collier, K.; Semionow, R.; Chua, A.O.; Mizel, S.B. Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature 1984, 312, 458–462. [Google Scholar] [CrossRef]
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K.; et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647. [Google Scholar] [CrossRef]
- Voronov, E.; Dotan, S.; Krelin, Y.; Song, X.; Elkabets, M.; Carmi, Y.; Rider, P.; Idan, C.; Romzova, M.; Kaplanov, I.; et al. Unique Versus Redundant Functions of IL-1alpha and IL-1beta in the Tumor Microenvironment. Front. Immunol. 2013, 4, 177. [Google Scholar] [CrossRef]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef]
- Beuscher, H.U.; Nickells, M.W.; Colten, H.R. The precursor of interleukin-1 alpha is phosphorylated at residue serine 90. J. Biol. Chem. 1988, 263, 4023–4028. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Appella, E.; Yamada, M.; Copeland, T.D.; Oppenheim, J.J.; Matsushima, K. Phosphorylation of intracellular precursors of human IL-1. J. Immunol. 1988, 140, 2279–2287. [Google Scholar] [PubMed]
- Stevenson, F.T.; Bursten, S.L.; Fanton, C.; Locksley, R.M.; Lovett, D.H. The 31-kDa precursor of interleukin 1 alpha is myristoylated on specific lysines within the 16-kDa N-terminal propiece. Proc. Natl. Acad. Sci. USA 1993, 90, 7245–7249. [Google Scholar] [CrossRef]
- Cohen, I.; Rider, P.; Vornov, E.; Tomas, M.; Tudor, C.; Wegner, M.; Brondani, L.; Freudenberg, M.; Mittler, G.; Ferrando-May, E.; et al. IL-1alpha is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci. Rep. 2015, 5, 14756. [Google Scholar] [CrossRef]
- Gross, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef]
- Mosley, B.; Urdal, D.L.; Prickett, K.S.; Larsen, A.; Cosman, D.; Conlon, P.J.; Gillis, S.; Dower, S.K. The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J. Biol. Chem. 1987, 262, 2941–2944. [Google Scholar] [CrossRef]
- Afonina, I.S.; Muller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, T.; Sperandio, M.; Mocsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Kronheim, S.R.; Cantrell, M.; Deeley, M.C.; March, C.J.; Prickett, K.S.; Wignall, J.; Conlon, P.J.; Cosman, D.; Hopp, T.P.; et al. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J. Biol. Chem. 1988, 263, 9437–9442. [Google Scholar] [CrossRef]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Luthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Sorimachi, H. Calpains: An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain research for drug discovery: Challenges and potential. Nat. Rev. Drug Discov. 2016, 15, 854–876. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamamoto, K.; Saido, T.; Kawasaki, H.; Oppenheim, J.J.; Matsushima, K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc. Natl. Acad. Sci. USA 1990, 87, 5548–5552. [Google Scholar] [CrossRef] [PubMed]
- Carruth, L.M.; Demczuk, S.; Mizel, S.B. Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J. Biol. Chem. 1991, 266, 12162–12167. [Google Scholar] [CrossRef]
- Zheng, Y.; Humphry, M.; Maguire, J.J.; Bennett, M.R.; Clarke, M.C. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 2013, 38, 285–295. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [PubMed]
- Pasternack, M.S.; Eisen, H.N. A novel serine esterase expressed by cytotoxic T lymphocytes. Nature 1985, 314, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.F.; Dosseto, M.; Denizot, F.; Mattei, M.G.; Clark, W.R.; Haqqi, T.M.; Ferrier, P.; Nabholz, M.; Schmitt-Verhulst, A.M.; Luciani, M.F.; et al. The inducible cytotoxic T-lymphocyte-associated gene transcript CTLA-1 sequence and gene localization to mouse chromosome 14. Nature 1986, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Bleackley, R.C.; Duggan, B.; Ehrman, N.; Lobe, C.G. Isolation of two cDNA sequences which encode cytotoxic cell proteases. FEBS Lett. 1988, 234, 153–159. [Google Scholar] [CrossRef]
- Rousalova, I.; Krepela, E. Granzyme B-induced apoptosis in cancer cells and its regulation (review). Int. J. Oncol. 2010, 37, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Cullen, S.P.; Martin, S.J. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 2010, 235, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Rider, P.; Carmi, Y.; Braiman, A.; Dotan, S.; White, M.R.; Voronov, E.; Martin, M.U.; Dinarello, C.A.; Apte, R.N. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.H. Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 2007, 217, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, H.; Schechter, N.; Lazarus, G.; Black, R.A.; Kupper, T.S. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J. Exp. Med. 1991, 174, 821–825. [Google Scholar] [CrossRef]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef]
- Kono, H.; Karmarkar, D.; Iwakura, Y.; Rock, K.L. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J. Immunol. 2010, 184, 4470–4478. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Wiggins, K.A.; Parry, A.J.; Cassidy, L.D.; Humphry, M.; Webster, S.J.; Goodall, J.C.; Narita, M.; Clarke, M.C.H. IL-1alpha cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell 2019, 18, e12946. [Google Scholar] [CrossRef]
- Crawley, J.T.; Zanardelli, S.; Chion, C.K.; Lane, D.A. The central role of thrombin in hemostasis. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 95–101. [Google Scholar] [CrossRef]
- Burzynski, L.C.; Humphry, M.; Pyrillou, K.; Wiggins, K.A.; Chan, J.N.E.; Figg, N.; Kitt, L.L.; Summers, C.; Tatham, K.C.; Martin, P.B.; et al. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1alpha by Thrombin. Immunity 2019, 50, 1033–1042 e1036. [Google Scholar] [CrossRef] [PubMed]
- Lamacchia, C.; Rodriguez, E.; Palmer, G.; Gabay, C. Endogenous IL-1alpha is a chromatin-associated protein in mouse macrophages. Cytokine 2013, 63, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, X.; Lin, D.; Lei, L.; Hu, B.; Cao, F.; Mei, Y.; Wu, D.; Liu, H. Propiece IL-1alpha facilitates the growth of acute T-lymphocytic leukemia cells through the activation of NF-kappaB and SP1. Oncotarget 2017, 8, 15677–15688. [Google Scholar] [CrossRef] [PubMed]
- Pollock, A.S.; Turck, J.; Lovett, D.H. The prodomain of interleukin 1alpha interacts with elements of the RNA processing apparatus and induces apoptosis in malignant cells. FASEB J. Off. Public Fed. Am. Soc. Exp. Biol. 2003, 17, 203–213. [Google Scholar] [CrossRef]
- Buryskova, M.; Pospisek, M.; Grothey, A.; Simmet, T.; Burysek, L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J. Biol. Chem. 2004, 279, 4017–4026. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Beller, D.I.; Mizel, S.B.; Unanue, E.R. Identification of a membrane-associated interleukin 1 in macrophages. Proc. Natl. Acad. Sci. USA 1985, 82, 1204–1208. [Google Scholar] [CrossRef]
- Beuscher, H.U.; Colten, H.R. Structure and function of membrane IL-1. Mol. Immunol. 1988, 25, 1189–1199. [Google Scholar] [CrossRef]
- Sasu, S.; Cooper, A.L.; Beasley, D. Juxtacrine effects of IL-1 alpha precursor promote iNOS expression in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1615–H1623. [Google Scholar] [CrossRef]
- Kumar, S.; Kishimoto, H.; Chua, H.L.; Badve, S.; Miller, K.D.; Bigsby, R.M.; Nakshatri, H. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am. J. Pathol. 2003, 163, 2531–2541. [Google Scholar] [CrossRef]
- Sgagias, M.K.; Kasid, A.; Danforth, D.N., Jr. Interleukin-1 alpha and tumor necrosis factor-alpha (TNF alpha) inhibit growth and induce TNF messenger RNA in MCF-7 human breast cancer cells. Mol. Endocrinol. 1991, 5, 1740–1747. [Google Scholar] [CrossRef][Green Version]
- Slamon, D.J.; Clark, G.M. In Reply: Amplification c-erbB-2 and Aggressive Human Breast Tumors? Science 1988, 240, 1796–1798. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lee, J.S.; Jie, C.; Park, M.H.; Iwakura, Y.; Patel, Y.; Soni, M.; Reisman, D.; Chen, H. HER2 Overexpression Triggers an IL1alpha Proinflammatory Circuit to Drive Tumorigenesis and Promote Chemotherapy Resistance. Cancer Res. 2018, 78, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.F.; Hudelist, G.; Gschwantler-Kaulich, D.; Fink-Retter, A.; Mueller, R.; Walter, I.; Czerwenka, K.; Kubista, E. Interleukin-1alpha protein secretion in breast cancer is associated with poor differentiation and estrogen receptor alpha negativity. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2006, 16 (Suppl. 2), 556–559. [Google Scholar] [CrossRef]
- Wessendorf, J.H.; Garfinkel, S.; Zhan, X.; Brown, S.; Maciag, T. Identification of a nuclear localization sequence within the structure of the human interleukin-1 alpha precursor. J. Biol. Chem. 1993, 268, 22100–22104. [Google Scholar] [PubMed]
- Rider, P.; Carmi, Y.; Voronov, E.; Apte, R.N. Interleukin-1alpha. Semin. Immunol. 2013, 25, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Grzybowska, E. HAX-1: A multifunctional protein with emerging roles in human disease. Biochim. Biophys. Acta 2009, 1790, 1139–1148. [Google Scholar] [CrossRef]
- Yin, H.; Morioka, H.; Towle, C.A.; Vidal, M.; Watanabe, T.; Weissbach, L. Evidence that HAX-1 is an interleukin-1 alpha N-terminal binding protein. Cytokine 2001, 15, 122–137. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Nishimagi, E.; Tochimoto, A.; Kawamoto, M.; Katsumata, Y.; Soejima, M.; Kanno, T.; Kamatani, N.; Hara, M. Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc. Natl. Acad. Sci. USA 2006, 103, 14501–14506. [Google Scholar] [CrossRef]
- Dagenais, M.; Dupaul-Chicoine, J.; Douglas, T.; Champagne, C.; Morizot, A.; Saleh, M. The Interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology 2017, 6, e1287247. [Google Scholar] [CrossRef]
- Xu, D.; Matsuo, Y.; Ma, J.; Koide, S.; Ochi, N.; Yasuda, A.; Funahashi, H.; Okada, Y.; Takeyama, H. Cancer cell-derived IL-1alpha promotes HGF secretion by stromal cells and enhances metastatic potential in pancreatic cancer cells. J. Surg. Oncol. 2010, 102, 469–477. [Google Scholar] [CrossRef]
- Melisi, D.; Niu, J.; Chang, Z.; Xia, Q.; Peng, B.; Ishiyama, S.; Evans, D.B.; Chiao, P.J. Secreted interleukin-1alpha induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-kappaB. Mol. Cancer Res. 2009, 7, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Locati, M.; Vecchi, A.; Sozzani, S.; Allavena, P. Decoy receptors: A strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 2001, 22, 328–336. [Google Scholar] [CrossRef]
- Colotta, F.; Dower, S.K.; Sims, J.E.; Mantovani, A. The type II ‘decoy’ receptor: A novel regulatory pathway for interleukin 1. Immunol. Today 1994, 15, 562–566. [Google Scholar] [CrossRef]
- Morrison, R.N.; Young, N.D.; Nowak, B.F. Description of an Atlantic salmon (Salmo salar L.) type II interleukin-1 receptor cDNA and analysis of interleukin-1 receptor expression in amoebic gill disease-affected fish. Fish Shellfish Immunol. 2012, 32, 1185–1190. [Google Scholar] [CrossRef]
- Symons, J.A.; Young, P.R.; Duff, G.W. Soluble type II interleukin 1 (IL-1) receptor binds and blocks processing of IL-1 beta precursor and loses affinity for IL-1 receptor antagonist. Proc. Natl. Acad. Sci. USA 1995, 92, 1714–1718. [Google Scholar] [CrossRef]
- Orlando, S.; Sironi, M.; Bianchi, G.; Drummond, A.H.; Boraschi, D.; Yabes, D.; Mantovani, A. Role of Metalloproteases in the Release of the IL-1 type II Decoy Receptor. J. Biol. Chem. 1997, 272, 31764–31769. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Lokau, J.; Düsterhöft, S.; Trad, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Grötzinger, J. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 2012, 586, 1093–1100. [Google Scholar] [CrossRef]
- Dawson, J.; Engelhardt, P.; Kastelic, T.; Cheneval, D.; Mackenzie, A.; Ramage, P. Effects of soluble interleukin-1 type II receptor on rabbit antigen-induced arthritis: Clinical, biochemical and histological assessment. Rheumatology 1999, 38, 401–406. [Google Scholar] [CrossRef][Green Version]
- Simeoni, E.; Dudler, J.; Fleury, S.; Li, J.; Pagnotta, M.; Pascual, M.; von Segesser, L.K.; Vassalli, G. Gene transfer of a soluble IL-1 type 2 receptor-Ig fusion protein improves cardiac allograft survival in rats. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2007, 31, 222–228. [Google Scholar] [CrossRef]
- Petzold, T.; Massberg, S. Thrombin: A Gas Pedal Driving Innate Immunity. Immunity 2019, 50, 1024–1026. [Google Scholar] [CrossRef]
- Mori, N.; Shirakawa, F.; Murakami, S.; Oda, S.; Eto, S. Interleukin-1 alpha as an autocrine growth factor for acute lymphoblastic leukaemia cells. Br. J. Haematol. 1994, 86, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Mandinova, A. S100A13 mediates the copper-dependent stress-induced release of IL-1alpha from both human U937 and murine NIH 3T3 cells. J. Cell Sci. 2003, 116, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.D.; Cousens, L.S.; Barr, P.J.; Sprang, S.R. Three-dimensional structure of human basic fibroblast growth factor, a structural homolog of interleukin 1 beta. Proc. Natl. Acad. Sci. USA 1991, 88, 3446–3450. [Google Scholar] [CrossRef]
- Prudovsky, I. The non-classical export routes: FGF1 and IL-1 point the way. J. Cell Sci. 2003, 116, 4871–4881. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.T.; Turck, J.; Locksley, R.M.; Lovett, D.H. The N-terminal propiece of interleukin 1 alpha is a transforming nuclear oncoprotein. Proc Natl Acad Sci USA 1997, 94, 508–513. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, L.; Lin, X.; Wang, Y.; Li, Y.; Guo, Q.; Li, S.; Sun, Y.; Tao, X.; Zhang, D.; et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell 2020, 181, 637–652 e615. [Google Scholar] [CrossRef]
- Tjomsland, V.; Spangeus, A.; Valila, J.; Sandstrom, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Watari, K.; Shibata, T.; Kawahara, A.; Sata, K.; Nabeshima, H.; Shinoda, A.; Abe, H.; Azuma, K.; Murakami, Y.; Izumi, H.; et al. Tumor-derived interleukin-1 promotes lymphangiogenesis and lymph node metastasis through M2-type macrophages. PLoS ONE 2014, 9, e99568. [Google Scholar] [CrossRef]
- Kuan, E.L.; Ziegler, S.F. A tumor-myeloid cell axis, mediated via the cytokines IL-1alpha and TSLP, promotes the progression of breast cancer. Nat. Immunol. 2018, 19, 366–374. [Google Scholar] [CrossRef]
- Leon, X.; Bothe, C.; Garcia, J.; Parreno, M.; Alcolea, S.; Quer, M.; Vila, L.; Camacho, M. Expression of IL-1alpha correlates with distant metastasis in patients with head and neck squamous cell carcinoma. Oncotarget 2015, 6, 37398–37409. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Cotton, M.; Rodman Iii, S.N.; Ross, K.A.; Jensen, I.J.; Sangodeyi-Miller, K.; McLaren, A.J.; Dahl, R.A.; Gibson-Corley, K.N.; Koch, A.T.; Fu, Y.X.; et al. Interleukin-1 alpha increases anti-tumor efficacy of cetuximab in head and neck squamous cell carcinoma. J. Immunother. Cancer 2019, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, S.; Ichikura, T.; Mochizuki, H. Significant correlation between expression of interleukin-1alpha and liver metastasis in gastric carcinoma. Cancer 2001, 91, 1272–1276. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, B.; Block, M.S.; Bamlet, W.R.; Vierkant, R.A.; Kalli, K.R.; Fogarty, Z.; Rider, D.N.; Sellers, T.A.; Tworoger, S.S.; Poole, E.; et al. Risk of ovarian cancer and the NF-kappaB pathway: Genetic association with IL1A and TNFSF10. Cancer Res. 2014, 74, 852–861. [Google Scholar] [CrossRef]
- Shirakawa, F.; Tanaka, Y.; Oda, S.; Eto, S.; Yamashita, U. Autocrine stimulation of interleukin 1 alpha in the growth of adult human T-cell leukemia cells. Cancer Res. 1989, 49, 1143–1147. [Google Scholar]
- Wano, Y.; Hattori, T.; Matsuoka, M.; Takatsuki, K.; Chua, A.O.; Gubler, U.; Greene, W.C. Interleukin 1 gene expression in adult T cell leukemia. J. Clin. Investig. 1987, 80, 911–916. [Google Scholar] [CrossRef]
- Voronov, E.; Weinstein, Y.; Benharroch, D.; Cagnano, E.; Ofir, R.; Dobkin, M.; White, R.M.; Zoller, M.; Barak, V.; Segal, S.; et al. Antitumor and immunotherapeutic effects of activated invasive T lymphoma cells that display short-term interleukin 1alpha expression. Cancer Res. 1999, 59, 1029–1035. [Google Scholar]
- Elkabets, M.; Krelin, Y.; Dotan, S.; Cerwenka, A.; Porgador, A.; Lichtenstein, R.G.; White, M.R.; Zoller, M.; Iwakura, Y.; Dinarello, C.A.; et al. Host-derived interleukin-1alpha is important in determining the immunogenicity of 3-methylcholantrene tumor cells. J. Immunol. 2009, 182, 4874–4881. [Google Scholar] [CrossRef]
- Douvdevani, A.; Huleihel, M.; Zoller, M.; Segal, S.; Apte, R.N. Reduced tumorigenicity of fibrosarcomas which constitutively generate IL-1 alpha either spontaneously or following IL-1 alpha gene transfer. Int. J. Cancer 1992, 51, 822–830. [Google Scholar] [CrossRef]
- Apte, R.N.; Douvdevani, A.; Zoller, M.; White, R.M.; Dvorkin, T.; Shimoni, N.; Huleihel, M.; Fima, E.; Hacham, M.; Voronov, E.; et al. Involvement of immune responses in the eradication of IL-1 alpha gene-transduced tumour cells: Mechanisms of tumour rejection and immunotherapeutical implications. Folia Biol. 1994, 40, 1–18. [Google Scholar]
- Dvorkin, T.; Song, X.; Argov, S.; White, R.M.; Zoller, M.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Immune phenomena involved in the in vivo regression of fibrosarcoma cells expressing cell-associated IL-1alpha. J. Leukoc. Biol. 2006, 80, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sawai, H.; Matsuo, Y.; Ochi, N.; Yasuda, A.; Takahashi, H.; Wakasugi, T.; Funahashi, H.; Sato, M.; Okada, Y.; et al. Interleukin-1alpha enhances angiogenesis and is associated with liver metastatic potential in human gastric cancer cell lines. J. Surg. Res. 2008, 148, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010, 138, 1022–1034 e1021-1010. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hikiba, Y.; Nakagawa, H.; Hayakawa, Y.; Yanai, A.; Akanuma, M.; Ogura, K.; Hirata, Y.; Kaestner, K.H.; Omata, M.; et al. Inhibitor of kappaB kinase beta regulates gastric carcinogenesis via interleukin-1alpha expression. Gastroenterology 2010, 139, 226–238 e226. [Google Scholar] [CrossRef]
- Jemaa, A.B.; Bouraoui, Y.; Rais, N.B.; Nouira, Y.; Oueslati, R. Cytokine profiling identifies an interaction of IL-6 and IL-1alpha to drive PSMA-PSA prostate clones. Immunobiology 2016, 221, 1424–1431. [Google Scholar] [CrossRef]
- Bae, J.Y.; Kim, E.K.; Yang, D.H.; Zhang, X.; Park, Y.J.; Lee, D.Y.; Che, C.M.; Kim, J. Reciprocal interaction between carcinoma-associated fibroblasts and squamous carcinoma cells through interleukin-1alpha induces cancer progression. Neoplasia 2014, 16, 928–938. [Google Scholar] [CrossRef]
- Murphy, J.E.; Morales, R.E.; Scott, J.; Kupper, T.S. IL-1 alpha, innate immunity, and skin carcinogenesis: The effect of constitutive expression of IL-1 alpha in epidermis on chemical carcinogenesis. J. Immunol. 2003, 170, 5697–5703. [Google Scholar] [CrossRef]
- Chirivi, R.G.; Chiodoni, C.; Musiani, P.; Garofalo, A.; Bernasconi, S.; Colombo, M.P.; Giavazzi, R. IL-1alpha gene-transfected human melanoma cells increase tumor-cell adhesion to endothelial cells and their retention in the lung of nude mice. Int. J. Cancer 1996, 67, 856–863. [Google Scholar] [CrossRef]
- Tian, T.; Lofftus, S.; Pan, Y.; Stingley, C.A.; King, S.L.; Zhao, J.; Pan, T.Y.; Lock, R.; Marglous, J.W.; Liu, K.; et al. IL1alpha Antagonizes IL1beta and Promotes Adaptive Immune Rejection of Malignant Tumors. Cancer Immunol. Res. 2020, 8, 660–671. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tochimoto, A.; Hara, M.; Kawamoto, M.; Sugiura, T.; Saito, S.; Kamatani, N. Contribution of single nucleotide polymorphisms of the IL1A gene to the cleavage of precursor IL-1alpha and its transcription activity. Immunogenetics 2007, 59, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Seibert, S.; Walch-Ruckheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. Correction: RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1alpha release, and efficient paracrine dendritic cell activation. Oncotarget 2019, 10, 4503–4504. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Tumorigenesis | Mechanism of Action | IL-1α Form | Source of IL-1α | Study Type | Reference |
|---|---|---|---|---|---|---|
| Lung Cancer | Promote | Activates angiogenesis and lymphangiogenesis | Mature | Endogenous | In vitro and in vivo | [79] |
| Breast Cancer | Promote | Increases cell proliferation | Mature | Transduced | In vitro and in vivo | [49] |
| Breast Cancer | Promote | Induces TSLP expression from tumor-infiltrating myeloid cells to increase cancer survival and metastasis spread | Mature | Endogenous | In vitro, in vivo, and patient data | [80] |
| Breast Cancer | Promote | Increases activation of NF-kB and STAT3 to generate and maintain cancer stem cells. | Mature | Endogenous and Exogenous | In vitro, in vivo, and patient data | [52] |
| Breast Cancer | Promote | Associated with dedifferentiation and malignancy | Mature | Endogenous | In vitro and patient data | [53] |
| Breast Cancer | Suppress | Inhibits cell growth at G0/G1 phase | Mature | Exogenous | In vitro | [50] |
| Breast Cancer | Suppress | Suppresses cell proliferation through IL-1R signaling | Mature | Endogenous | In vitro and in vivo | [59] |
| Head and Neck Squamous Carcinoma | Promote | Associated with distant metastasis in patients | Mature | Endogenous | Patient data | [81] |
| Head and Neck Squamous Carcinoma | Suppress | Activates T-cell dependent anti-tumor response | Mature | Exogenous | In vitro, in vivo, and patient data | [82] |
| Liver Cancer | Promote | Activates inflammation and compensatory proliferation in liver | Mature | Endogenous | In vitro and in vivo | [83] |
| Liver Cancer | Suppress | Promotes T- and NK-cell activation | Membrane | Transduced | In vitro and in vivo | [84] |
| Pancreatic Cancer | Promote | Promotes HGF secretion by fibroblasts to promote cancer invasion, proliferation, and angiogenesis | Mature | Endogenous and Exogenous | In vitro | [60] |
| Pancreatic Cancer | Promote | Constitutively activates NF-kB to induce metastatic behavior | Mature | Transduced | In vitro and in vivo | [61] |
| Pancreatic Cancer | Promote | Sustains expression of inflammatory factors in tumor microenvironment beneficial for tumor survival | Mature | Endogenous | In vitro and patient data | [77] |
| Ovarian Cancer | Promote | IL-1α SNP (rs17561) associated with increase risk, possibly due to it being more readily cleaved to form mature IL-1α | Mature | Endogenous | Patient data | [85] |
| Acute Lymphocytic Leukemia | Promote | Facilitates growth of T-ALL cells through activation of NF-kB and SP1 | N-Terminal | Transduced | In vitro and in vivo | [43] |
| Acute Lymphocytic Leukemia | Promote | Induces ALL cell growth in an IL-1α dose dependent manner | Mature | Endogenous and Exogenous | In vitro and patient data | [71] |
| Adult T-cell Leukemia | Promote | Induces ATL cell growth in an IL-1α dose dependent manner, possibly via autocrine mechanism | Mature | Exogenous | In vitro and patient data | [86] |
| Adult T-cell Leukemia | Promote | Associated with ATL patient samples and cell lines | Mature | Endogenous | In vitro and patient data | [87] |
| Adult T-cell Leukemia | Suppress | Stimulates anti-tumor immune responses | Mature | Endogenous | In vitro and in vivo | [88] |
| Fibrosarcoma | Promote | Involved in controlling the immune-surveillance of developing tumor | Mature | Endogenous | In vitro and in vivo | [89] |
| Fibrosarcoma | Suppress | Reduces tumorigenicity, increases immunogenicity, and regression of tumor | Cytosolic Full Length and Membrane | Endogenous and Transduced | In vitro and in vivo | [90] |
| Fibrosarcoma | Suppress | Increases immunogenicity, induces regression of tumor and development of systemic immunity | Cytosolic Full Length and Membrane | Endogenous and Transduced | In vitro and in vivo | [91] |
| Fibrosarcoma | Suppress | Stimulates anti-tumor immune responses and regression of tumor | Cytosolic Full Length and Membrane | Transduced | In vitro and in vivo | [92] |
| Gastric Cancer | Promote | Increases metastasis and tumor differentiation | Mature | Endogenous | Patient data | [83] |
| Gastric Cancer | Promote | Associated with enhance angiogenesis and metastasis | Mature | Endogenous | In vitro | [93] |
| Gastric Cancer | Promote | Associated with rapid progression to gastric pre-neoplasia | Full Length and Mature | Endogenous | In vitro and in vivo | [94] |
| Gastric Cancer | Promote | Positively correlated with carcinogenesis | Mature | Endogenous | In vitro and in vivo | [95] |
| Prostate Cancer | Promote | Correlated to increased serum PSA levels and progression of disease | Mature | Endogenous | Patient data | [96] |
| Oral Squamous Cell Carcinoma | Promote | Stimulates CAF proliferation and cytokine (CCL7, CXCL1, IL-8) secretion to promote OSCC cancer progression | Mature | Endogenous | In vitro, in vivo, and patient data | [97] |
| Cervical Cancer | Suppress | Activates dendritic cells to produce IL-12 for anti-tumor response | Mature | Endogenous | In vitro and patient data | [43] |
| Skin Cancer | Promote and Suppress | Suppresses carcinoma formation from prior papilloma. | Mature | Transduced | In vitro and in vivo | [98] |
| Promotes carcinoma formation not from prior papilloma | ||||||
| Skin Cancer | Promote | Induces adhesion molecules on endothelial cells, increasing tumor retention. As well as inducing potent inflammation, enhancing metastasis. | Mature | Transduced | In vitro and in vivo | [99] |
| Kidney Cancer | Promote | Induces malignant transformation | N-Terminal | Transduced | In vitro and in vivo | [75] |
| Multiple Cancers (T cell lymphoma, melanoma, lung carcinoma) | Suppress | Inhibits tumor growth by enhancing T-cell mediated antitumor immunity | Mature | Endogenous | In vitro and in vivo | [100] |
| Multiple Cancers (Various malignant human tumor cell lines) | Suppress | Induces cell apoptosis, possibly involving RNA processing apparatus | N-Terminal | Transduced | In vitro | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, J.W.; Binte Hanafi, Z.; Chew, L.C.Y.; Mei, Y.; Liu, H. IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells 2021, 10, 92. https://doi.org/10.3390/cells10010092
Chiu JW, Binte Hanafi Z, Chew LCY, Mei Y, Liu H. IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells. 2021; 10(1):92. https://doi.org/10.3390/cells10010092
Chicago/Turabian StyleChiu, Jing Wen, Zuhairah Binte Hanafi, Lionel Chin Yong Chew, Yu Mei, and Haiyan Liu. 2021. "IL-1α Processing, Signaling and Its Role in Cancer Progression" Cells 10, no. 1: 92. https://doi.org/10.3390/cells10010092
APA StyleChiu, J. W., Binte Hanafi, Z., Chew, L. C. Y., Mei, Y., & Liu, H. (2021). IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells, 10(1), 92. https://doi.org/10.3390/cells10010092