
QTL Mapping of Mesocotyl Elongation and Confirmation of a QTL in Dongxiang Common Wild Rice in China

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Evaluation of ME
2.3. Genotypic Data of the F2 Segregating Population and QTL Analyses
3. Results
3.1. Phenotypic Variation
3.2. QTL Analysis of the ME
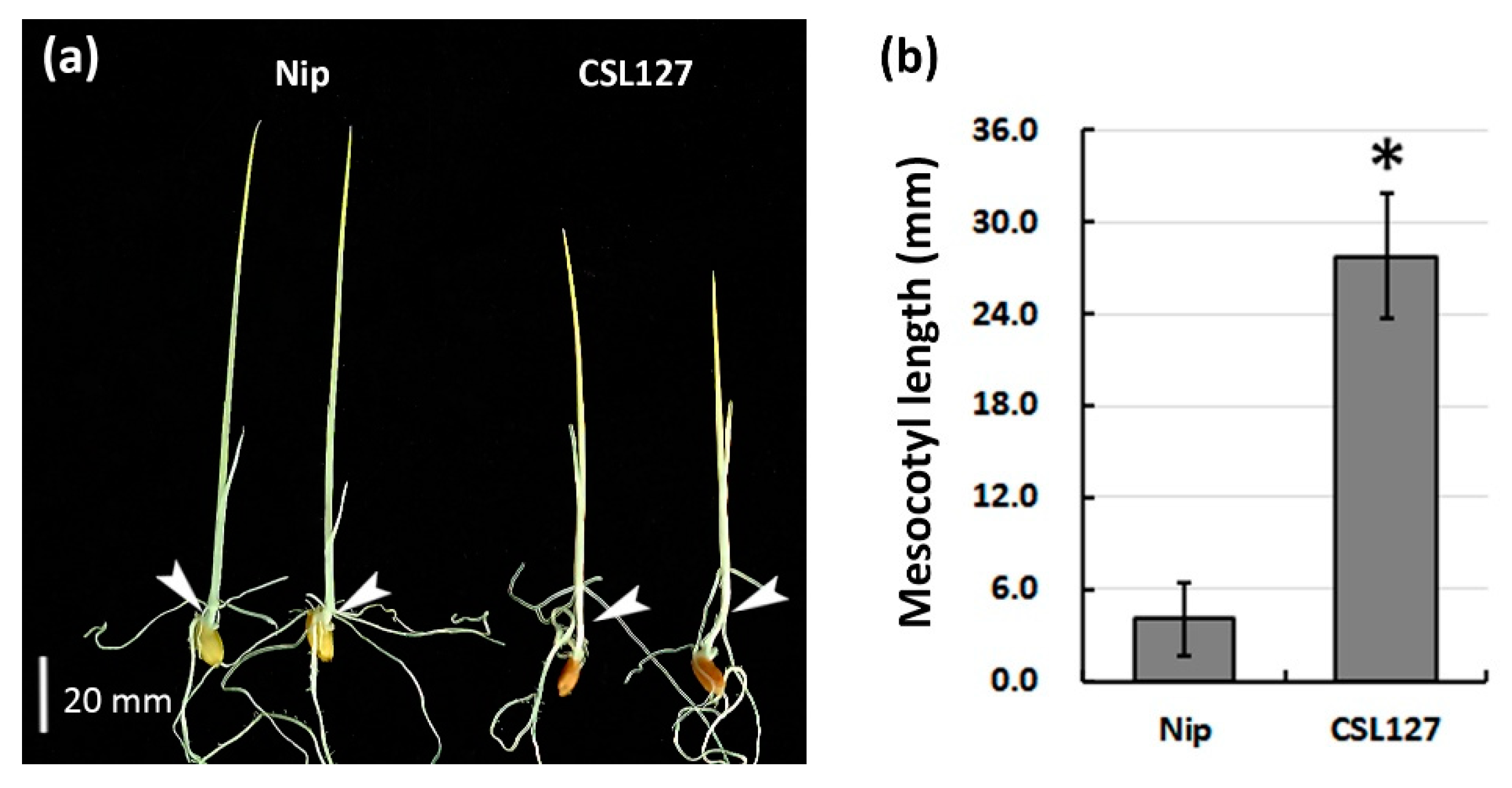
3.3. Confirmation of the Largest QTL, qML6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peng, S.; Tang, Q.; Zou, Y. Current status and challenges of rice production in China. Plant Prod. Sci. 2009, 12, 3–8. [Google Scholar] [CrossRef]
- Kato, Y.; Katsura, K. Rice adaptation to aerobic soils: Physiological considerations and implications for agronomy. Plant Prod. Sci. 2014, 17, 1–12. [Google Scholar] [CrossRef]
- Kumar, V.; Ladha, J.K. Direct seeding of rice: Recent developments and future research needs. Adv. Agron. 2011, 111, 297–413. [Google Scholar]
- Liu, H.; Hussain, S.; Zheng, M.; Peng, S.; Huang, J.; Cui, K.; Nie, L. Dry direct-seeded rice as an alternative to transplanted-flooded rice in central China. Agron. Sustain. Dev. 2015, 35, 285–294. [Google Scholar] [CrossRef]
- Farooq, M.; Siddique, K.H.M.; Rehman, H.; Aziz, T.; Lee, D.J.; Wahid, A. Rice direct seeding: Experiences, challenges and opportunities. Soil Tillage Res. 2011, 111, 87–98. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, D. Production Status and Prospects of Direct-seeded Rice in China. China Rice 1996, 2, 1–4, (In Chinese with English Abstract). [Google Scholar]
- Feng, F.; Mei, H.; Fan, P.; Li, Y.; Xu, X.; Wei, H.; Ying, M.; Luo, L. Dynamic transcriptome and phytohormone profiling along the time of light exposure in the mesocotyl of rice seedling. Sci. Rep. 2017, 7, 11961. [Google Scholar] [CrossRef]
- Kutschera, U.; Wang, Z. Growth-limiting proteins in maize coleoptiles and the auxin-brassinosteroid hypothesis of mesocotyl elongation. Protoplasma 2016, 253, 3–14. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, P.; Zhao, Z.; Zhao, G.; Tian, B.; Wang, J.; Wang, G. Mapping QTL controlling maize deep-seeding tolerance-related traits and confirmation of a major QTL for mesocotyl length. Theor. Appl. Genet. 2012, 124, 223–232. [Google Scholar] [CrossRef]
- Huang, C.; Jiang, S.; Feng, L.; Xu, Z.; Chen, W. Analysis of QTLs for mesocotyl length in rice (Oryza sativa L.). Acta Agron. Sin. 2010, 36, 1108–1113, (In Chinese with English Abstract). [Google Scholar] [CrossRef]
- Dang, X.; Thi, T.G.T.; Dong, G.; Wang, H.; Edzesi, W.M.; Hong, D. Genetic diversity and association mapping of seed vigor in rice (Oryza sativa L.). Planta 2014, 239, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lin, J.; Wu, M.; Cao, L.; Cheng, S. Analysis on germinating dynamic source of rice. Chin. J. Rice Sci. 2005, 19, 59–62, (In Chinese with English Abstract). [Google Scholar]
- Takahashi, N. Adaptive importance of mesocotyl and coleoptile growth in rice under different moisture regimes. Aust. J. Plant Physiol. 1978, 5, 511–517. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, D.; Chen, W. Preliminary study on mesocotyl elongation characteristics of northern weedy rice. China Rice 2008, 3, 47–49, (In Chinese with English Abstract). [Google Scholar]
- Turner, F.T.; Chen, C.; Bollich, C.N. Coleoptile and mesocotyl lengths in semi-dwarf rice seedlings. Crop Sci. 1982, 22, 43–46. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, G.; Lin, J.; Cheng, S. Screening for rice germplasms with specially-elongated mesocotyl. Rice Sci. 2005, 12, 226–228. [Google Scholar]
- Redoña, E.D.; Mackill, D.J. Mapping quantitative trait loci for seedling vigor in rice using RFLPs. Theor. Appl. Genet. 1996, 92, 395–402. [Google Scholar] [CrossRef]
- Katsuta-Seki, M.; Ebana, K.; Okuno, K. QTL analysis for mesocotyl elongation in rice. Rice Genet. Newsl. 1996, 13, 126. [Google Scholar]
- Cao, L.; Zhu, J.; Yan, Q.; He, L.; Wei, X.; Cheng, S. Mapping QTLs with epistasis for mesocotyl length in a DH population from indica-japonica cross of rice (Oryza sativa). Chin. J. Rice Sci. 2002, 16, 221–224, (In Chinese with English Abstract). [Google Scholar]
- Ou-yang, Y.; Zhang, Q.; Zhang, K.; Yu, S.; Zhuang, J.; Jin, Q.; Cheng, S. QTL mapping and interaction analysis of genotype × environment (Fe2+-concentrations) for mesocotyl length in Rice (Oryza sativa L.). Acta Genet. Sin. 2005, 32, 712–718, (In Chinese with English Abstract). [Google Scholar]
- Lee, H.S.; Sasaki, K.; Higashitani, A.; Ahn, S.N.; Sato, T. Mapping and characterization of quantitative trait loci for mesocotyl elongation in rice (Oryza sativa L.). Rice 2012, 5, 13. [Google Scholar] [CrossRef]
- Lee, H.S.; Sasaki, K.; Kang, J.W.; Sato, T.; Song, W.Y.; Ahn, S.N. Mesocotyl elongation is essential for seedling emergence under deep-seeding condition in rice. Rice 2017, 10, 32. [Google Scholar] [CrossRef]
- Niu, S.; Lv, Y.; Wu, Y.; Wei, X.; Sheng, Z.; Jiao, G.; Hu, S.; Tang, S. QTLs Mapping for Mesocotyl Length in Rice. China Rice 2019, 25, 55–59, (In Chinese with English Abstract). [Google Scholar]
- Wu, J.; Feng, F.; Lian, X.; Teng, X.; Wei, H.; Yu, H.; Xie, W.; Yan, M.; Fan, P.; Li, Y.; et al. Genome-wide Association Study (GWAS) of mesocotyl elongation based on re-sequencing approach in rice. BMC Plant Biol. 2015, 15, 218. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, M.; Niu, X.; Wang, C.; Xu, Q.; Feng, Y.; Wang, S.; Yuan, X.; Yu, H.; Wang, Y.; et al. Uncovering novel loci for mesocotyl elongation and shoot length in indica rice through genome-wide association mapping. Planta 2016, 243, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhan, J.; Li, J.; Lu, X.; Liu, J.; Wang, Y.; Zhao, Q.; Ye, G. Genome-wide Association Study (GWAS) for Mesocotyl Elongation in Rice (Oryza sativa L.) under Multiple Culture Conditions. Genes 2019, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.-G.; Park, S.-Y.; Lar, S.M.; Zhang, H.; Lee, A.-R.; Cao, F.-Y.; Seo, J.; Ham, T.-H.; Lee, J.; Kwon, S.-W. Genome-Wide Association Study (GWAS) of Mesocotyl Length for Direct Seeding in Rice. Agronomy 2021, 11, 2527. [Google Scholar] [CrossRef]
- Ma, X.; Han, B.; Tang, J.; Zhang, J.; Cui, D.; Geng, L.; Zhou, H.; Li, M.; Han, L. Construction of chromosome segment substitution lines of Dongxiang common wild rice (Oryza rufifipogon Griff.) in the background of the japonica rice cultivar Nipponbare (Oryza sativa L.). Plant Physiol. Biochem. 2019, 144, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tang, J.; Zhang, J.; Cui, D.; Li, H.; Li, M.; Han, L. Development of molecular markers polymorphic between Dongxiang wild rice and Geng rice cultivar ‘Nipponbare’. Acta Agron. Sin. 2019, 45, 316–321, (In Chinese with English Abstract). [Google Scholar] [CrossRef]
- Wang, J.; Wan, X.; Crossa, J.; Crouch, J.; Weng, J.; Zhai, H.; Wan, J. QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet. Res. 2006, 88, 93–104. [Google Scholar] [CrossRef]
- Qi, L.; Sun, Y.; Li, J.; Su, L.; Zheng, X.; Wang, X.; Li, K.; Yang, Q.; Qiao, W. Identify QTLs for grain size and weight in common wild rice using chromosome segment substitution lines across six environments. Breed. Sci. 2017, 67, 472–482. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in bi-parental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- McCouch, S.R.; Kochert, G.; Yu, Z.; Wang, Z.; Khush, G.S.; Coffman, W.R.; Tanksley, S.D. Molecular mapping of rice chromosomes. Theor. Appl. Genet. 1988, 76, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ye, G.; Wang, J. A modified algorithm for the improvement of composite interval mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Alibu, S.; Saito, Y.; Shiwachi, H.; Irie, K. Genotypic variation in coleoptile or mesocotyl lengths of upland rice (Oryza sativa L.) and seedling emergence in deep sowing. Afr. J. Agric. Res. 2012, 7, 6239–6248. [Google Scholar]
- Nghi, K.N.; Tondelli, A.; Valè, G.; Tagliani, A.; Marè, C.; Perata, P.; Pucciariello, C. Dissection of coleoptile elongation in japonica rice under submergence through integrated genome-wide association mapping and transcriptional analyses. Plant Cell Environ. 2019, 42, 1832–1846. [Google Scholar] [CrossRef]
- Helms, R.S.; Dilday, R.H.; Mgonja, M.A.; Amonsilpa, S. Genetic and plant growth regulator manipulation of rice (Oryza sativa L.) mesocotyl and coleoptile lengths. Proc. Ark. Acad. Sci. 1989, 43, 42–45. [Google Scholar]
- Zhan, J.; Lu, X.; Liu, H.; Zhao, Q.; Ye, G. Mesocotyl elongation, an essential trait for dry-seeded rice (Oryza sativa L.): A review of physiological and genetic basis. Planta 2019, 251, 27. [Google Scholar] [CrossRef]
- Liu, C.; Meng, Y.; Liu, J.; Wang, Y.; Ye, G. Combining QTL-seq and linkage analysis to identify the QTL of mesocotyl elongation in rice (Oryza sativa L.). Acta Agron. Sin. 2021, 47, 2036–2044, (In Chinese with English Abstract). [Google Scholar]
- Cai, H.W.; Morishima, H. QTL clusters reflect character associations in wild and cultivated rice. Theor. Appl. Genet. 2002, 104, 1217–1228. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Chromosome | Marker Name | Position | LOD | PVE (%) a | Additive Effect b | Positive Allele c |
|---|---|---|---|---|---|---|---|
| qML3 | 3 | DX-C3-27 | 28.36 Mb | 6.04 | 15.95 | 9.5 | DX |
| qML6 | 6 | DX-C6-4 | 8.15 Mb | 5.47 | 16.79 | 4.1 | DX |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Ju, C.; Cheng, Y.; Cui, D.; Han, B.; Zhao, Z.; Ma, X.; Han, L. QTL Mapping of Mesocotyl Elongation and Confirmation of a QTL in Dongxiang Common Wild Rice in China. Agronomy 2022, 12, 1800. https://doi.org/10.3390/agronomy12081800
Huang Q, Ju C, Cheng Y, Cui D, Han B, Zhao Z, Ma X, Han L. QTL Mapping of Mesocotyl Elongation and Confirmation of a QTL in Dongxiang Common Wild Rice in China. Agronomy. 2022; 12(8):1800. https://doi.org/10.3390/agronomy12081800
Chicago/Turabian StyleHuang, Qian, Chunyan Ju, Yibing Cheng, Di Cui, Bing Han, Zhengwu Zhao, Xiaoding Ma, and Longzhi Han. 2022. "QTL Mapping of Mesocotyl Elongation and Confirmation of a QTL in Dongxiang Common Wild Rice in China" Agronomy 12, no. 8: 1800. https://doi.org/10.3390/agronomy12081800
APA StyleHuang, Q., Ju, C., Cheng, Y., Cui, D., Han, B., Zhao, Z., Ma, X., & Han, L. (2022). QTL Mapping of Mesocotyl Elongation and Confirmation of a QTL in Dongxiang Common Wild Rice in China. Agronomy, 12(8), 1800. https://doi.org/10.3390/agronomy12081800