Elucidation of the Structure of the 2-amino-3,5-Dibromochalcone Epoxides in Solution and Solid State


Abstract
1. Introduction
2. Materials and Methods
2.1. General Notes
2.2. Typical Procedure for Epoxidation of the 2-Aminochalcones 2a–c
2.2.1. (2-Amino-3,5-dibromophenyl)(3-phenyloxiran-2-yl)methanone (2a)
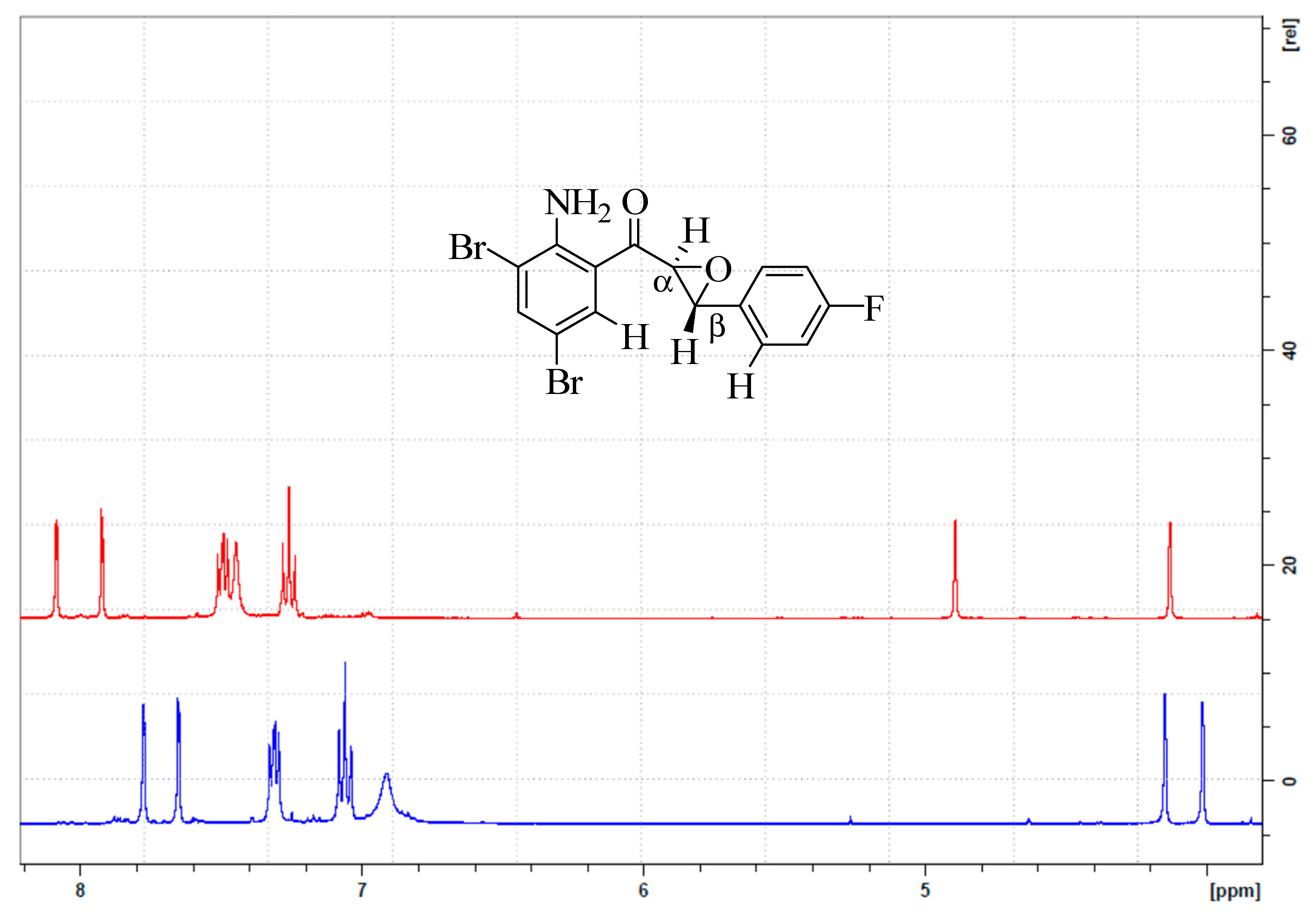
2.2.2. (2-Amino-3,5-dibromophenyl)(3-(4-fluorophenyl)oxiran-2-yl)methanone (2b)
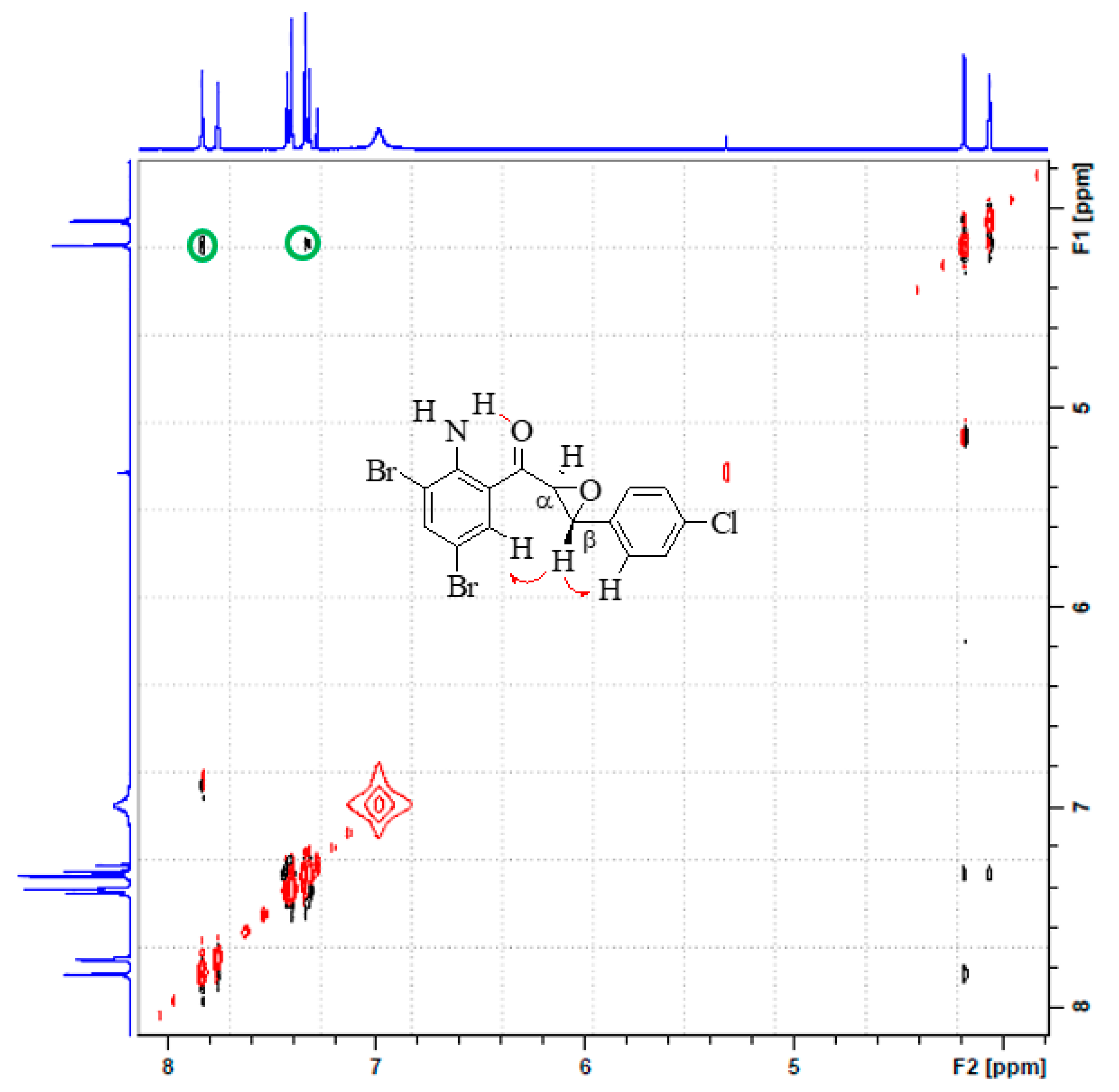
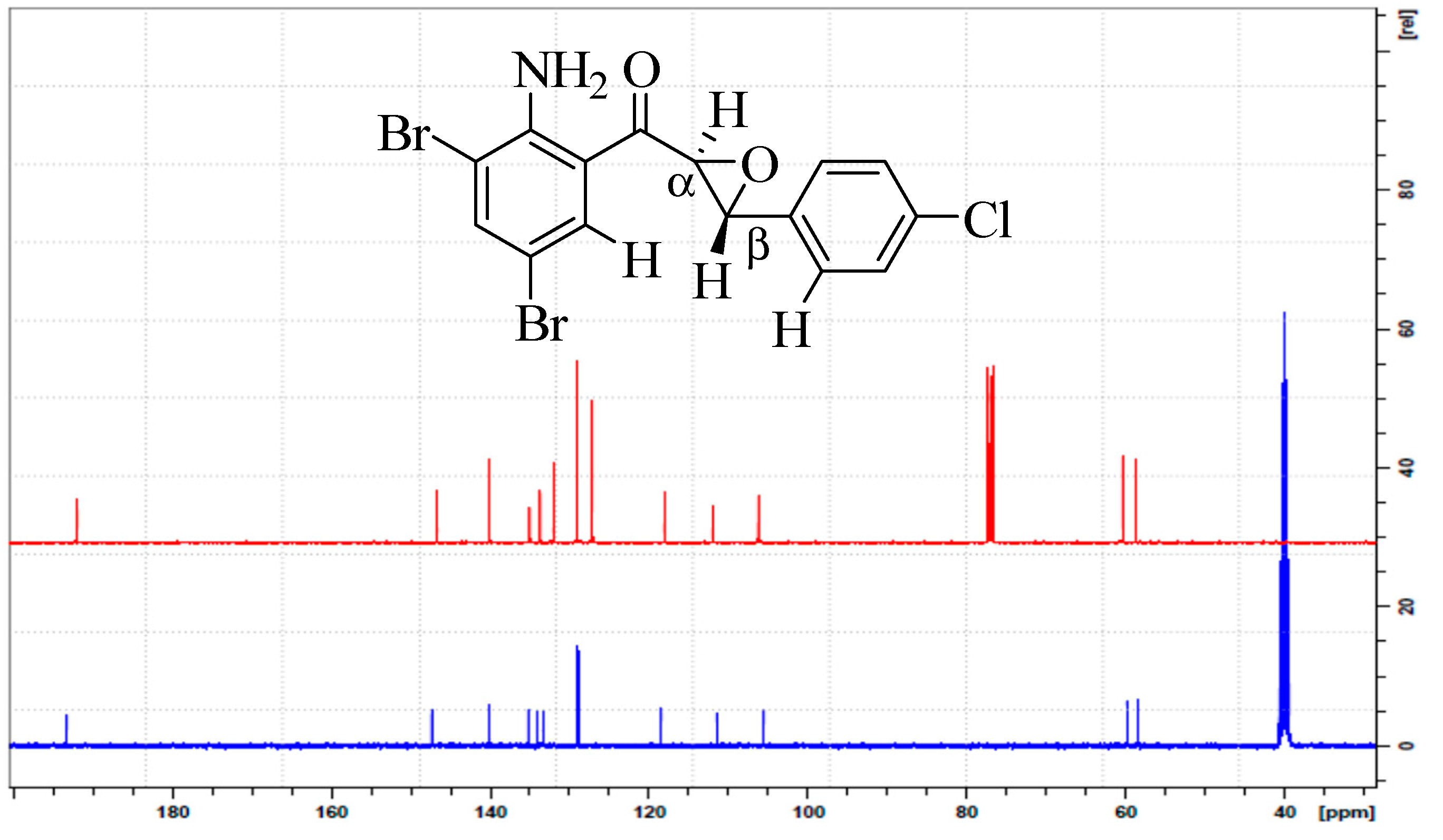
2.2.3. (2-Amino-3,5-dibromophenyl)(3-(4-chlorophenyl)oxiran-2-yl)methanone (2c)
2.3. Typical Procedure for the Reaction of 2a–c with Sulfuric Acid to Afford 3a–c
2.3.1. 6,8-Dibromo-3-phenylquinolin-4(1H)-one (3a)
2.3.2. 6,8-Dibromo-3-(4-fluorophenyl)quinolin-4(1H)-one (3b)
2.3.3. 6,8-Dibromo-3-(4-chlorophenyl)quinolin-4(1H)-one (3c)
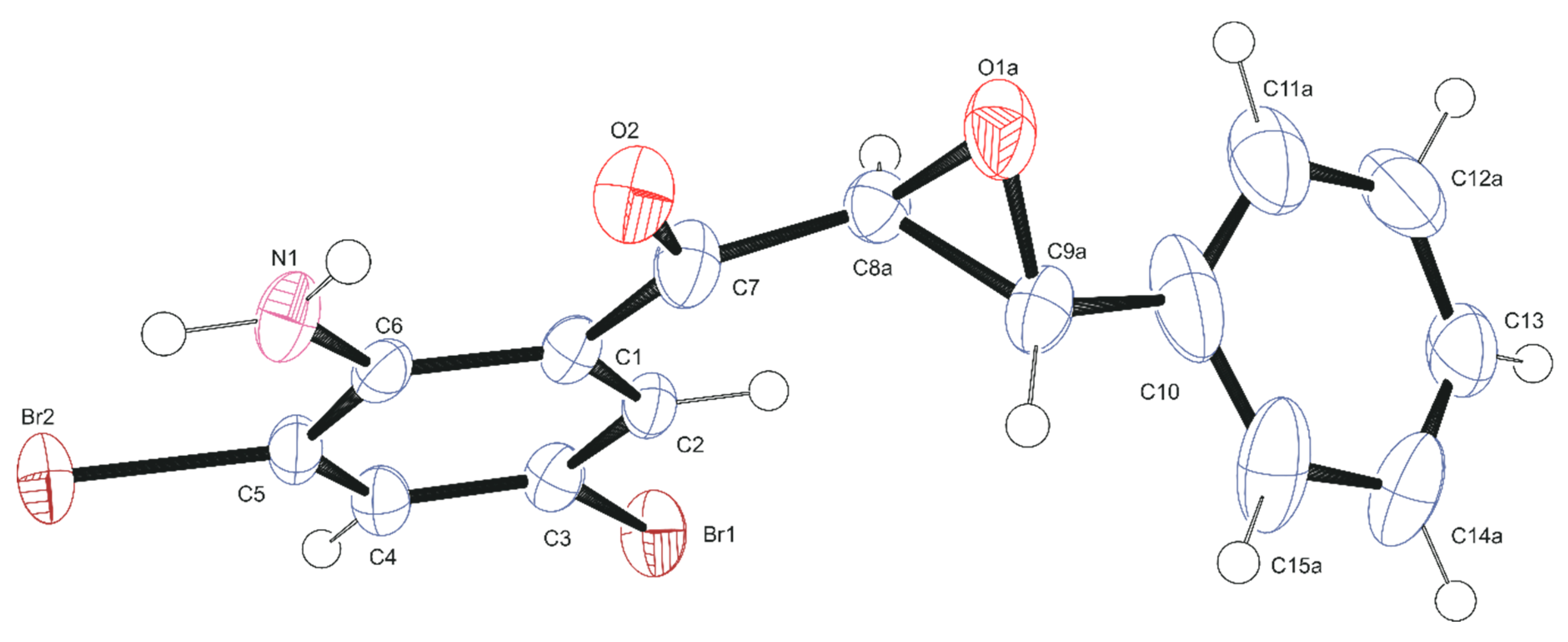
2.4. Data Collection and Refinement of 2a
2.5. Computational Methods
2.6. Hirshfeld Surface Analyses
3. Results and Discussion
3.1. Synthesis and Solution Phase Structural Analysis
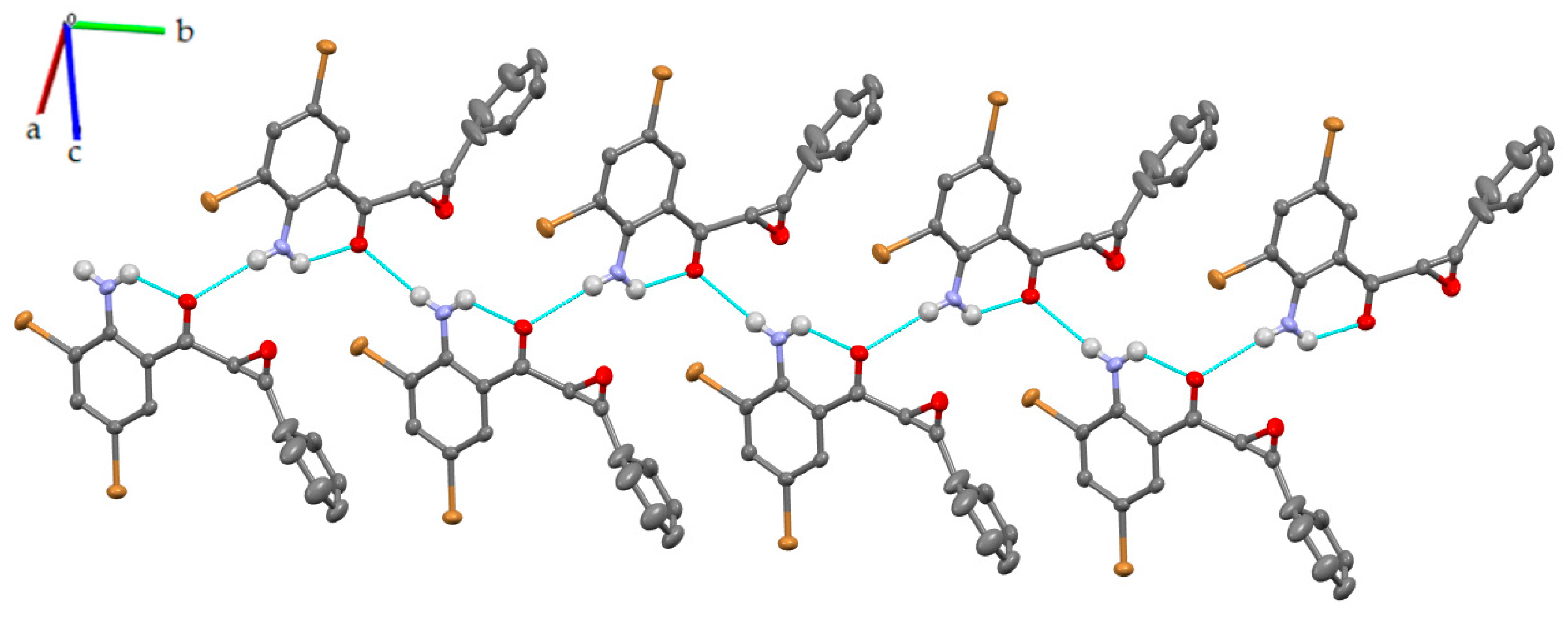
3.2. Solid-State Analysis
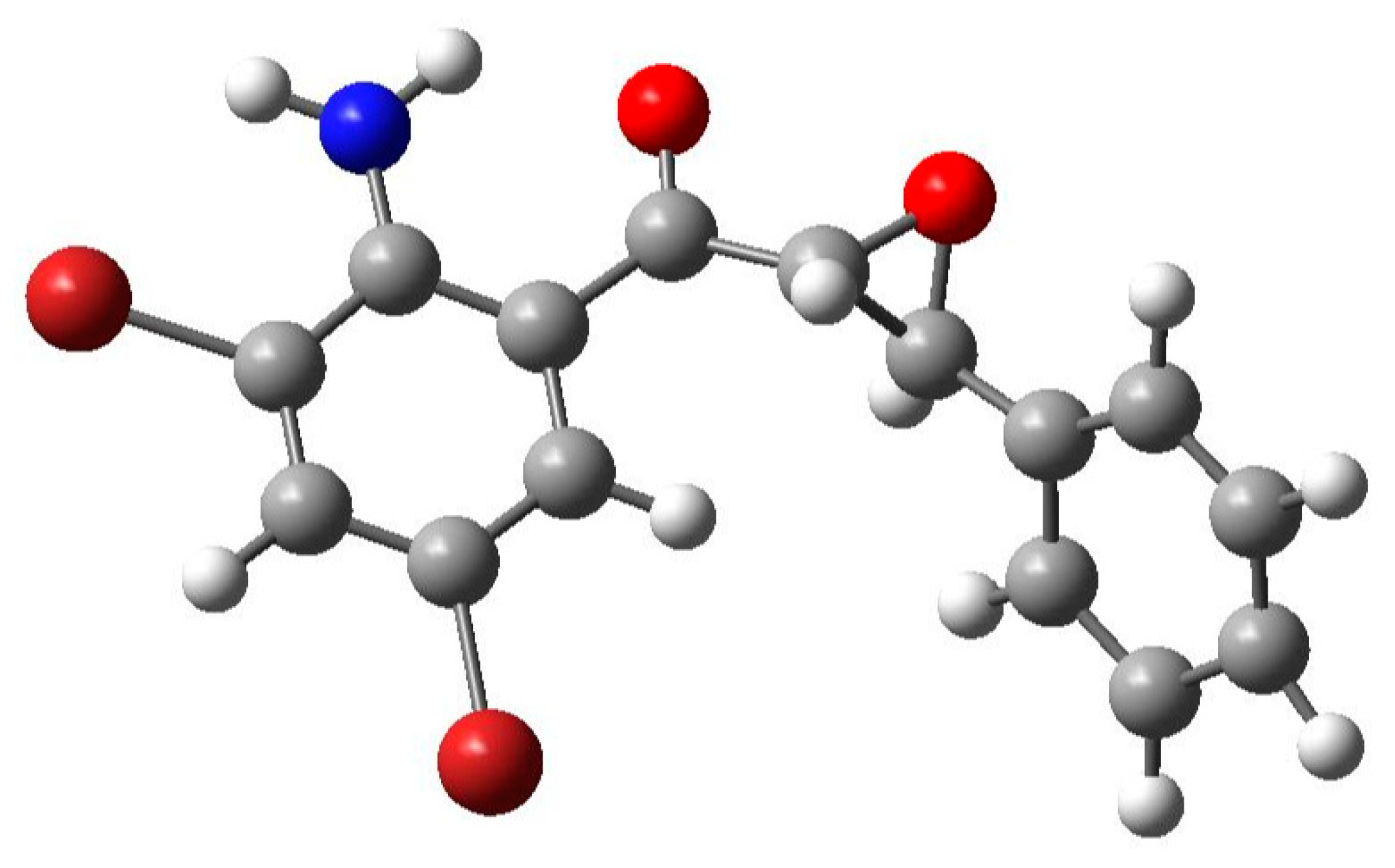
3.3. Optimized Structure from Density Functional Theory (DFT) Analysis
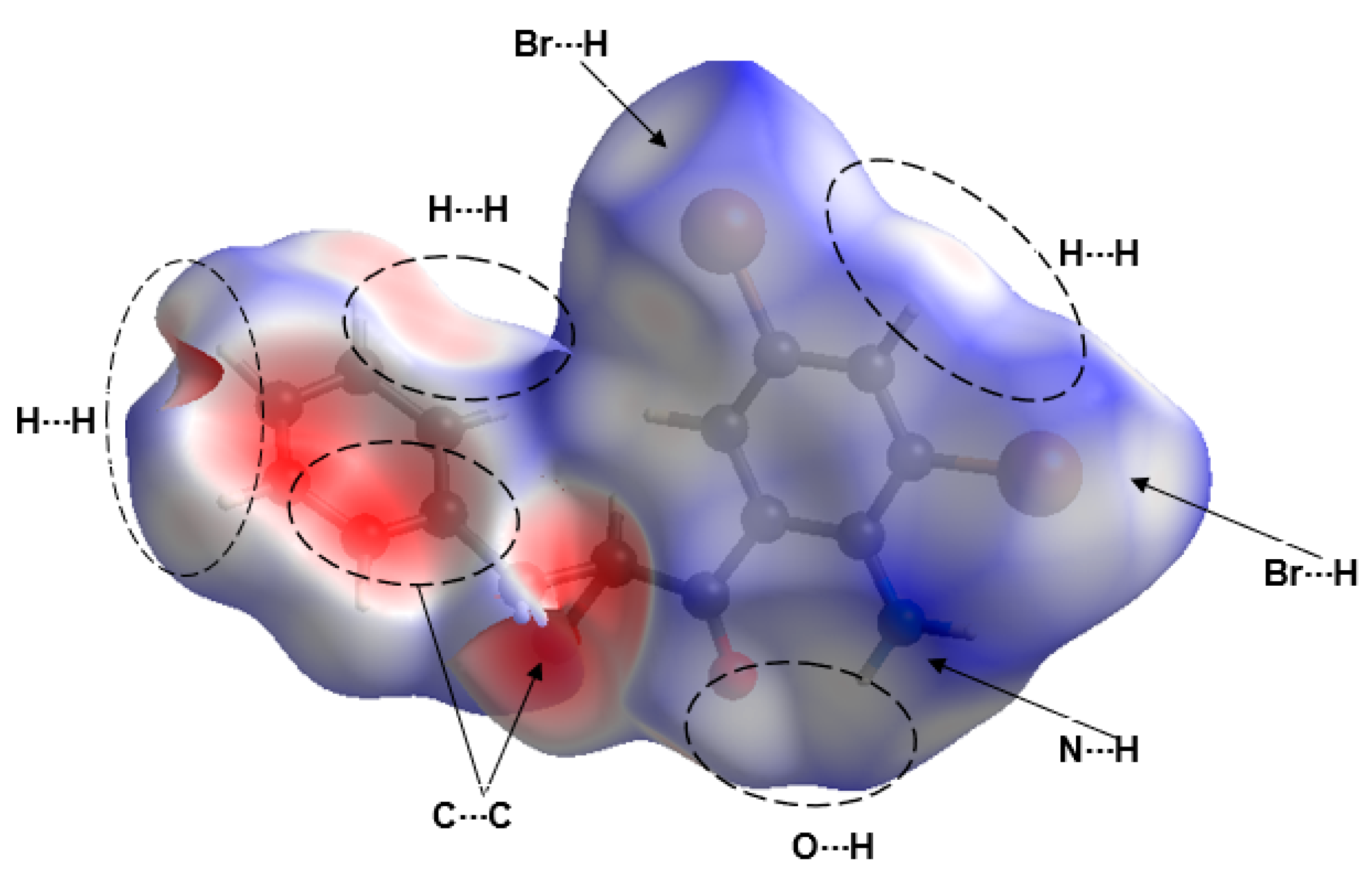
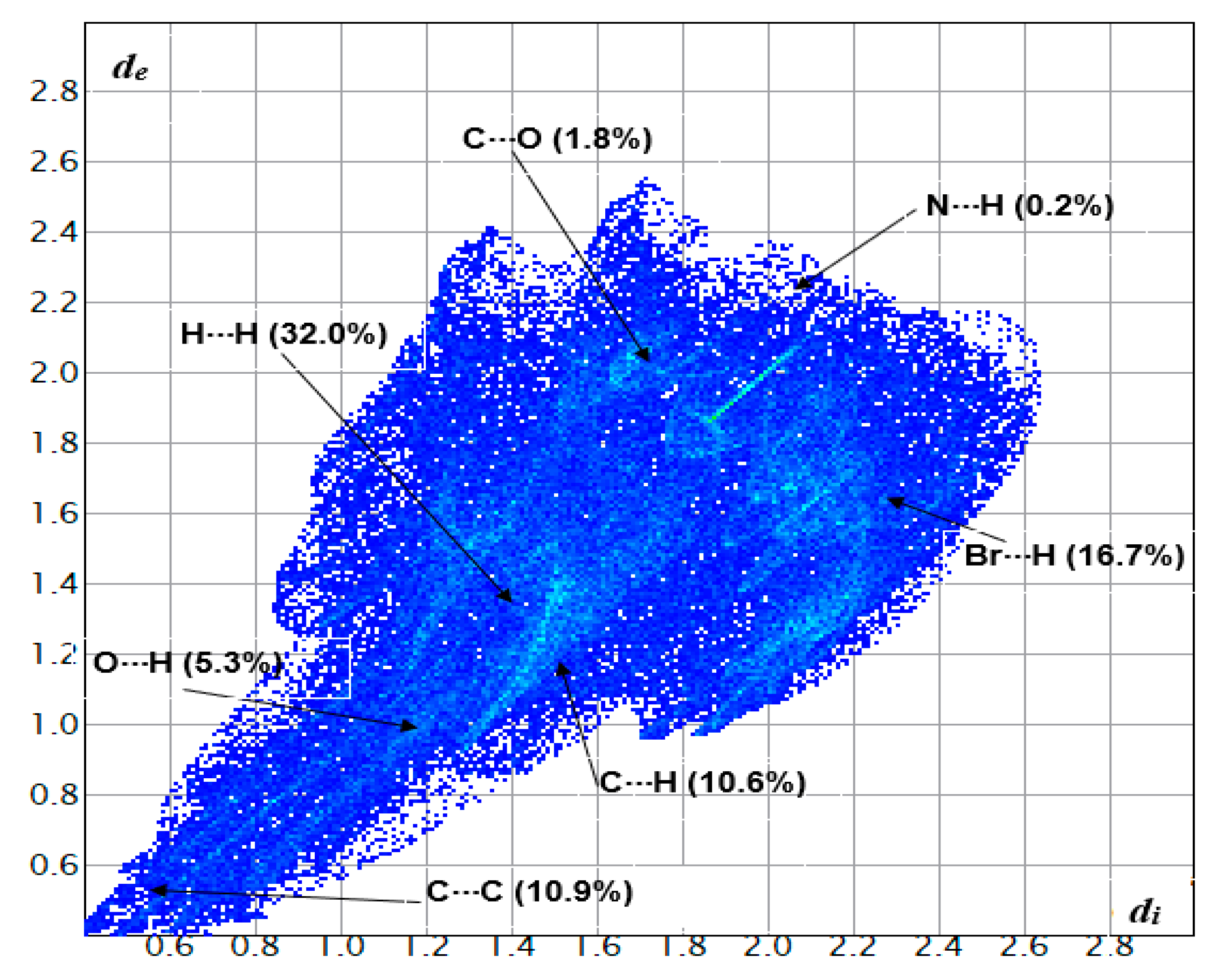
3.4. Hirshfeld Surface Analysis of Compound 2a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lattanzi, A.; Russo, A. Diaryl-2-pyrrolidinemethanols catalyzed enantioselective epoxidation of α,β-enones: New insight into the effect of structural modification of the catalyst on reaction efficiency. Tetrahedron 2006, 62, 12264–12269. [Google Scholar] [CrossRef]
- Adger, B.M.; Barkley, J.V.; Bergeron, S.; Cappi, M.W.; Flowerdew, B.E.; Jackson, M.P.; McCague, R.; Nugent, T.C.; Roberts, S.M. Improved procedure for Juliá–Colonna asymmetric epoxidation of α,β-unsaturated ketones: Total synthesis of diltiazem and TaxolTM side-chain. J. Chem. Soc. Perkin Trans. 1 1997, 3501–3507. [Google Scholar] [CrossRef]
- Border, Z.-M.; Marais, C.; Bezuidenhoudt, B.C.B.; Steenkamp, J.A. Studies towards the stereoselective α-hydroxylation of flavanones. Biosynthetic significance. Aus. J. Chem. 2008, 61, 122–130. [Google Scholar] [CrossRef]
- Bonollo, S.; Lanari, D.; Caccaro, L. Ring-Opening of Epoxides in Water. Eur. J. Org. Chem. 2011, 2587–2598. [Google Scholar] [CrossRef]
- Grabowsky, S.; Schirmeister, T.; Paulmann, C.; Pfeuffer, T.; Luger, P. Effect of electron-withdrawing substituents on the epoxide ring: An experimental and theoretical electron density analysis of a series of epoxide derivatives. J. Org. Chem. 2011, 76, 1305–1318. [Google Scholar] [CrossRef]
- Pillai, U.R.; Sahle-Demessie, E.; Varma, R.S. Microwave-expedited olefin epoxidation over hydrotalcites using hydrogen peroxide and acetonitrile. Tetrahedron Lett. 2002, 43, 2909–2911. [Google Scholar] [CrossRef]
- Adams, C.A.; Main, L. Synthesis of 2′-hydroxychalcone epoxides. Tetrahedron 1991, 47, 4959–4978. [Google Scholar] [CrossRef]
- Gormley, T.R.; O′Sullivan, W.I. Flavanoid epoxides-XIII: Acid and base catalysed reactions of 2′-tosyloxychalcone epoxides. Mechanism of the Algar-Flynn-Oyamada reaction. Tetrahedron 1972, 29, 369–373. [Google Scholar] [CrossRef]
- Adams, C.J.; Main, L. Cyclisation and subsequent reactions of 2′-hydroxy-6′-methoxychalcone epoxide and related compounds. Tetrahedron 1991, 47, 4979–4990. [Google Scholar] [CrossRef]
- Tðkés, A.L.; Janzsó, G. Reactions of 2′-aminochalcones. Synthetic Commun. 1989, 19, 3159–3168. [Google Scholar]
- Donnelly, J.A.; Farrell, D.F. Chalcone derivatives as precursors of 1,2,3,4-tetrahydro-4-quinolones. Tetrahedron 1990, 46, 885–894. [Google Scholar] [CrossRef]
- Litkei, G.; Tðkes, A.L. Oxidation of the 2′-NHR analogues of 2′-OR-chalcones; conversion of 2′-NHR-chalcone epoxides. Synthetic Commun. 1991, 21, 1597–1609. [Google Scholar] [CrossRef]
- Praveen, C.; Parthasarathy, K.; Perumal, P.T. Triflic acid promoted tandem ring-closure-aryl-migration of 2′-amino chalcone epoxide: A new synthetic route to azaisoflavones. SynLett 2010, 1635–1640. [Google Scholar] [CrossRef]
- Ahmed, N.; Kumar, H.; Babu, B.V. Intramolecular aminolysis of 2′-aminochalcone epoxides using InBr3 or BiCl3 as efficient catalysts. Synthetic Commun. 2013, 43, 567–581. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, C.; Liu, T.; Yu, L.; Yang, F.; Tang, J. Efficient construction of 3-arylquinolin-4(1H)-ones via in situ Meinwald rearrangement/intramolecular reductive cyclization of 2′-nitrochalcone epoxides. Tetrahedron 2016, 72, 7025–7031. [Google Scholar] [CrossRef]
- Ruan, L.; Shi, M.; Mao, S.; Yu, L.; Yang, F.; Tang, J. An efficient approach to construct 2-arylbenzo[b]furans from 2-methoxychalcone epoxides. Tetrahedron 2014, 70, 1065–1070. [Google Scholar] [CrossRef]
- Khoza, T.A.; Maluleka, M.M.; Mama, N.; Mphahlele, M.J. Synthesis and photophysical properties of the 2-aryl-6,8-bis(arylethenyl)-4-methoxyquinolines. Molecules 2012, 17, 14186–14204. [Google Scholar] [CrossRef]
- APEX-3, SAINT+, version 6.02 (Includes XPREP and SADABS); Bruker AXS Inc.: Madison, WI, USA, 2016.
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Short history of SHELX. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 3.0, University of Western Australia: Perth, Australia, 2001.
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Ohtsuru, M.; Tori, K.; Fukuyama, M. Proton magnetic resonance spectra of ethylene episulfoxides. Anomaly in magnitudes of spin-coupling constants between ring protons. Tetrahedron Lett. 1970, 11, 2877–2879. [Google Scholar] [CrossRef]
- Lien, J.-C.; Chen, S.-C.; Huang, L.-J.; Kuo, S.-C. Solvent effect of dimethyl sulfoxide on the chemical shifts of phenyl vinyl ketones. J. Chin. Chem. Soc. 2004, 51, 847–852. [Google Scholar] [CrossRef]
- Orlov, V.D.; Korotkov, S.A.; Sukach, Y.A.; Lavrushin, V.F. Investigation of the conformation of epoxychalcones by IR spectroscopy. Chem. Heterocyclic Comp. 1975, 11, 264–266. [Google Scholar] [CrossRef]
- Obregón-Mendoza, M.A.; Escobedo-Martínez, C.; Lozada, M.C.; Gnecco, D.; Soriano-García, M.; Enríquez, R.G. Investigation of three diasteromeric chalcone epoxides derivatives by NMR spectroscopy and X-ray crystallography. J. Chem. Crystallogr. 2014, 44, 512–519. [Google Scholar] [CrossRef]
- Kumar, A.; Bhakuni, V. Enantioselective epoxidation using liposomised m-chloroperbenzoic acid (LIP MCPBA). Tetrahedron Lett. 1996, 37, 4751–4754. [Google Scholar] [CrossRef]
- Praveena, N.; Hosamani, A.A.; Praveen, M.; Nagendrappa, G.; Row, T.N.G. cis-[3-(2-Chloro-6-methylquinolin-3-yl)oxiran-2-yl]-(p-tolyl)methanone. IUCrData 2017, 2, x170434. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley: Hoboken, NJ, USA, 1977. [Google Scholar]
- Honarparvar, B.; Govender, T.; Maguire, G.E.; Soliman, M.E.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2013, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Mphahlele, M.J.; El-Nahas, A.M. Tautomeric 2-arylquinolin-4(1H)-one derivatives- spectroscopic, X-ray and quantum chemical structural property studies. J. Mol. Struct. 2004, 688, 129–136. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
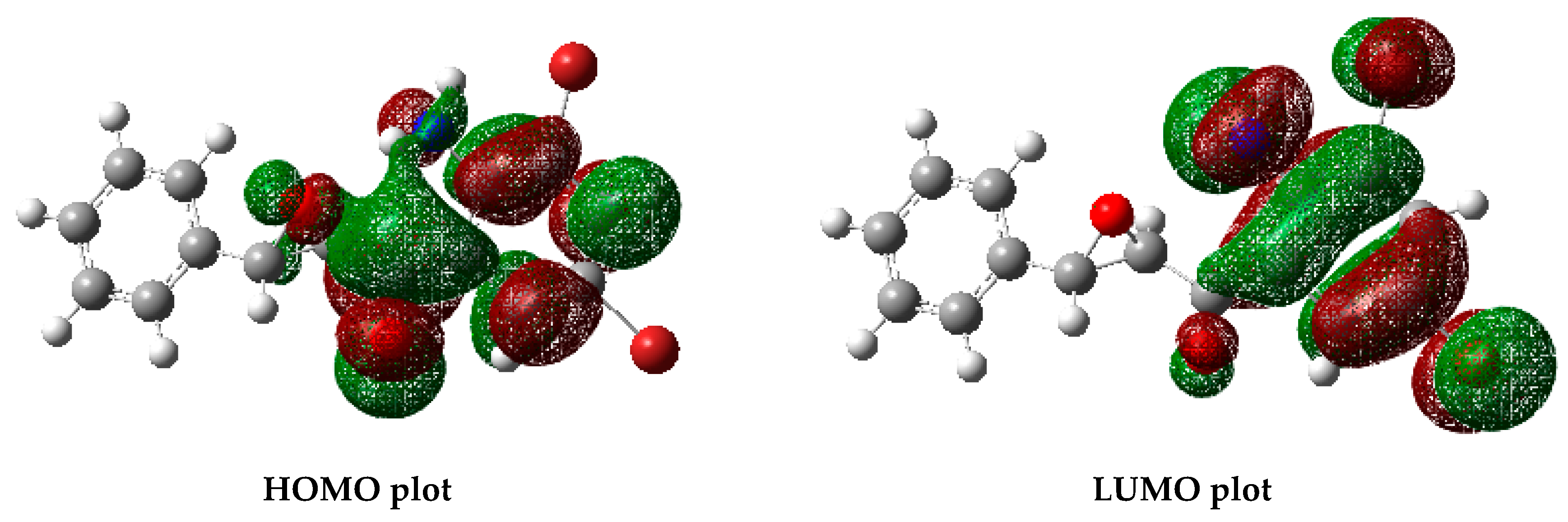{kind=link}
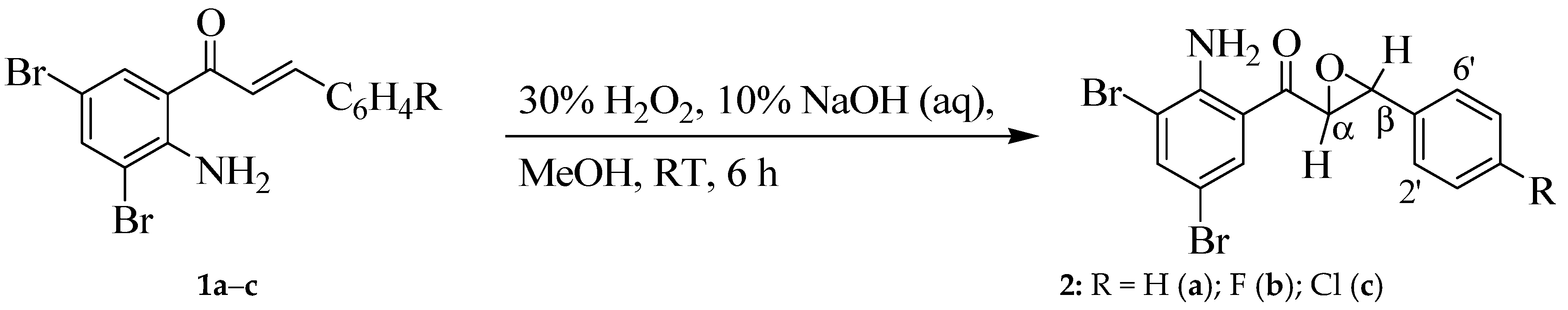{kind=link}
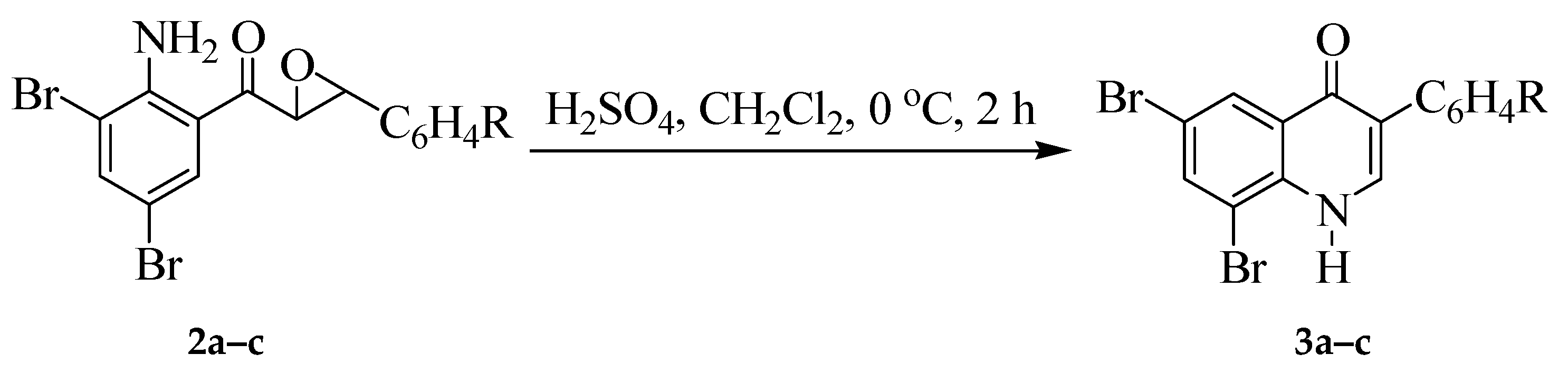{kind=link}
| CCDC 1906917 | |||
|---|---|---|---|
| Formula | C15H11Br2NO2 | Crystal size | 0.344 × 0.198 × 0.036 mm−3 |
| Formula weight | 397.07 | F(000) | 776 |
| Temperature | 173(2) K | Refinement method | Full-matrix-least-squares on F2 |
| Wavelength | 0.71073 Å | Data/restraints/parameters | 3444/293/246 |
| Crystal system | Monoclinic | Measured reflections | 18264 |
| Space group | P21/n | θmin/θmax | 2.905/27.997° |
| Unit cell dimensions | a = 4.8274(9) Å b = 9.728(2) Å c = 30.377(6) Å α = 90° β = 92.175(5)° γ = 90° | Rint | 0.0457 |
| Volume | 1425.5(5) Å3 | Goodness-of-fit on F2 | 1.039 |
| Z | 4 | Final R indices [I > 2sigma(I)] | R1 = 0.0379, wR2 = 0.0892 |
| Density (calculated) | 1.850 Mg/m3 | R indices (all data) | R1 = 0.0479, wR2 = 0.0945 |
| Absorption coefficient | 5.686 mm−1 | Largest diff. peak and hole | 0.922/−0.922 e.Å3 |
| Geometric Parameters | XRD | DFT |
|---|---|---|
| Bond Lengths (Å) | ||
| C(7)–O(2) | 1.204(4) | 1.216 |
| C(8A)–O(1A) | 1.418(6) | 1.446 |
| C(9A)–O(1A) | 1.428(6) | 1.446 |
| C(8A)–C(9A) | 1.468(7) | 1.471 |
| C(7)–C(8A) | 1.544(6) | 1.521 |
| C(9A)–C(10) | 1.554(7) | 1.490 |
| Bond Angles (°) | ||
| C(1)–C(7)–C(8A) | 115.5(3) | 118.9 |
| C(9A)–C(8A)–C(7) | 117.5(5) | 119.9 |
| C(8A)–C(9A)–C(10) | 111.0(5) | 122.3 |
| Torsion Angles (°) | ||
| C(7)–C(8a)–C(9a)–C(10) | 154.8° | 158.9 |
| C(2)–C(1)–C(7)–C(8A) | 14.8(5) | 14.3 |
| C(8A)–C(9A)–C(10)–C(15A) | −144.6(6) | −133.9 |
| C(8A)–C(9A)–C(10)–C(11A) | 61.7(9) | 62.2 |
| C(7)–C(1)–C(6)–N(1) | −0.8(5) | −0.64 |
| O(2)–C(7)–C(8A)–O(1A) | 20.1(6) | 28.8 |
| O(1A)–C(9A)–C(10)–C(11A) | −1.6(10) | −0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mphahlele, M.J.; Maluleka, M.M.; Mampa, R.M. Elucidation of the Structure of the 2-amino-3,5-Dibromochalcone Epoxides in Solution and Solid State. Crystals 2019, 9, 277. https://doi.org/10.3390/cryst9060277
Mphahlele MJ, Maluleka MM, Mampa RM. Elucidation of the Structure of the 2-amino-3,5-Dibromochalcone Epoxides in Solution and Solid State. Crystals. 2019; 9(6):277. https://doi.org/10.3390/cryst9060277
Chicago/Turabian StyleMphahlele, Malose J., Marole M. Maluleka, and Richard M. Mampa. 2019. "Elucidation of the Structure of the 2-amino-3,5-Dibromochalcone Epoxides in Solution and Solid State" Crystals 9, no. 6: 277. https://doi.org/10.3390/cryst9060277
APA StyleMphahlele, M. J., Maluleka, M. M., & Mampa, R. M. (2019). Elucidation of the Structure of the 2-amino-3,5-Dibromochalcone Epoxides in Solution and Solid State. Crystals, 9(6), 277. https://doi.org/10.3390/cryst9060277