
Structural and Electrochemical Properties of F-Doped RbTiOPO4 (RTP:F) Predicted from First Principles


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
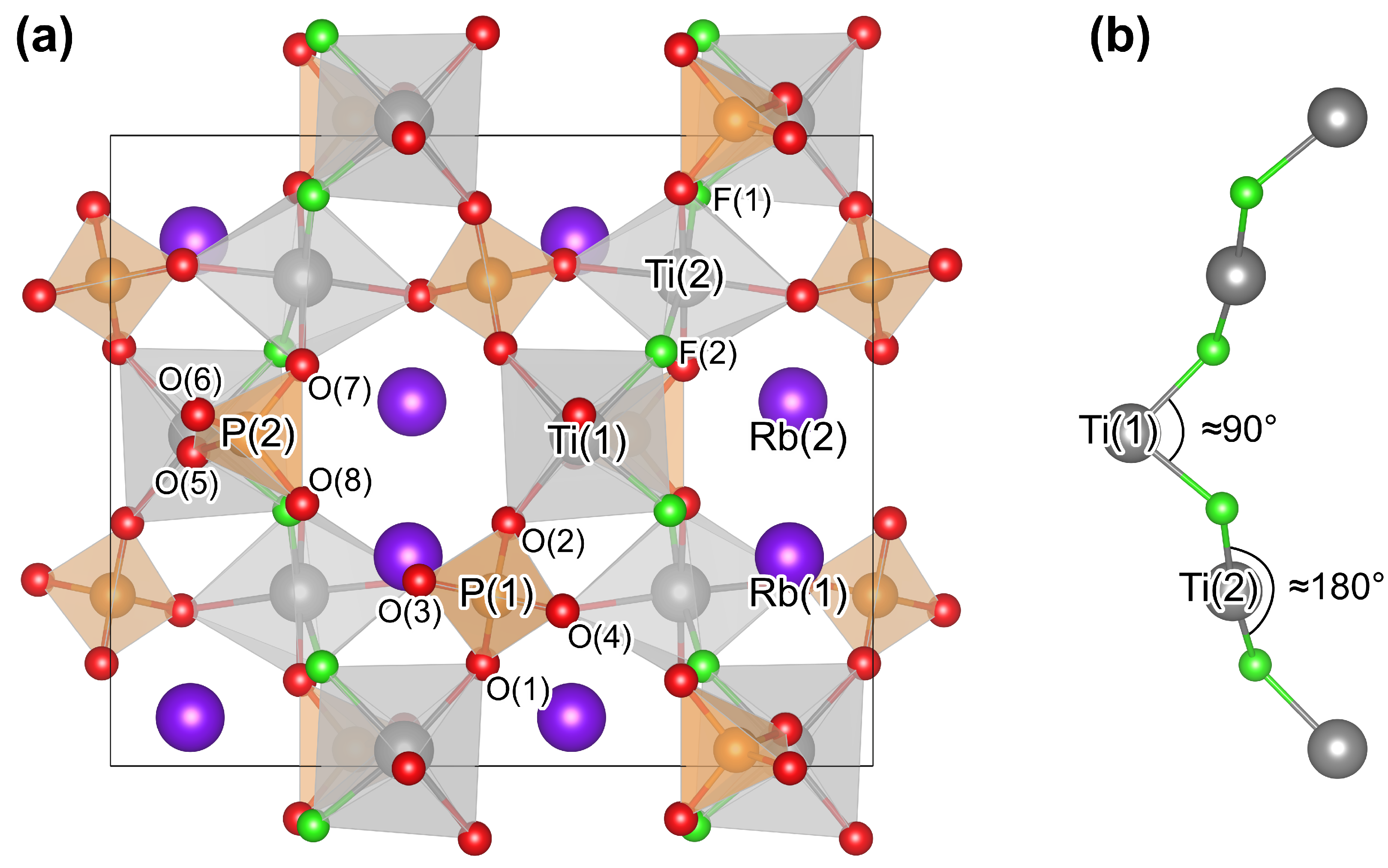
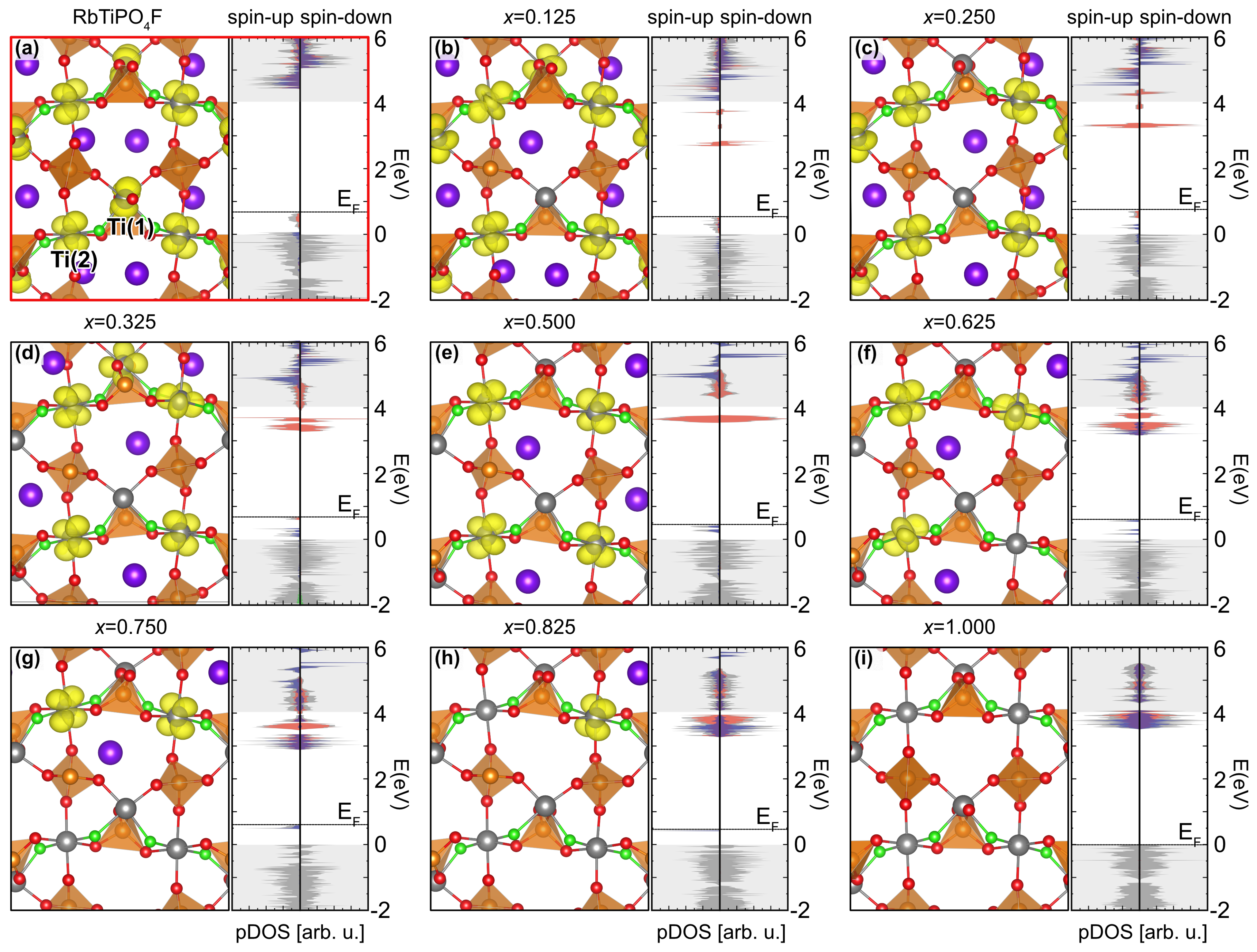
3.1. Crystallographic and Electric Properties of Stoichiometric RTP:F
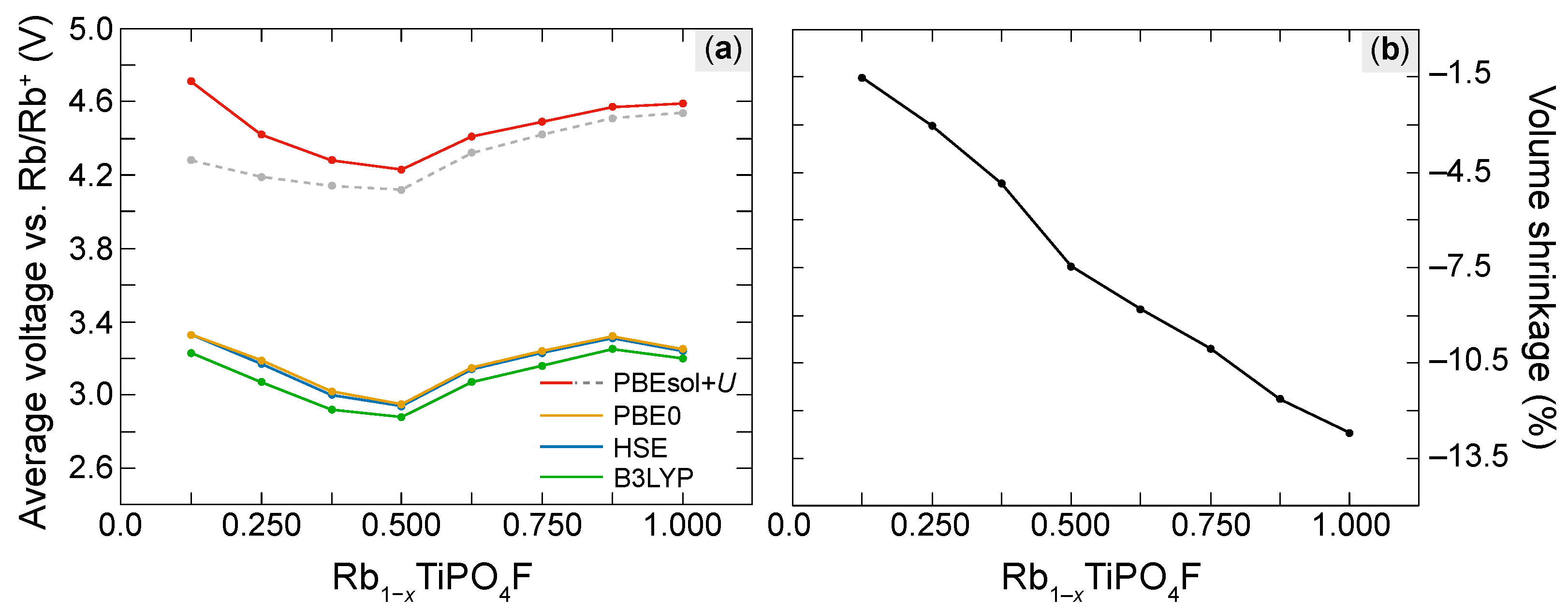
3.2. Electrochemical Performance of RTP:F
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Placke, T.; Kloepsch, R.; Dühnen, S.; Winter, M. Lithium ion, lithium metal, and alternative rechargeable battery technologies: The odyssey for high energy density. J. Solid State Electrochem. 2017, 21, 1939–1964. [Google Scholar] [CrossRef]
- Blomgren, G.E. The Development and Future of Lithium Ion Batteries. J. Electrochem. Soc. 2016, 164, A5019. [Google Scholar] [CrossRef]
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [PubMed]
- Andre, D.; Kim, S.J.; Lamp, P.; Lux, S.F.; Maglia, F.; Paschos, O.; Stiaszny, B. Future generations of cathode materials: An automotive industry perspective. J. Mater. Chem. A 2015, 3, 6709–6732. [Google Scholar] [CrossRef]
- Patry, G.; Romagny, A.; Martinet, S.; Froelich, D. Cost modeling of lithium-ion battery cells for automotive applications. Energy Sci. Eng. 2015, 3, 71–82. [Google Scholar] [CrossRef]
- Slater, M.D.; Kim, D.; Lee, E.; Johnson, C.S. Sodium-Ion Batteries. Adv. Funct. Mater. 2013, 23, 947–958. [Google Scholar] [CrossRef]
- Rudola, A.; Rennie, A.J.R.; Heap, R.; Seyyed Meysami, S.; Lowbridge, A.; Mazzali, F.; Sayers, R.; Wright, C.J.; Barker, J. Commercialisation of high energy density sodium-ion batteries: Faradion’s journey and outlook. J. Mater. Chem. A 2021, 9, 8279–8302. [Google Scholar] [CrossRef]
- Komaba, S.; Murata, W.; Ishikawa, T.; Yabuuchi, N.; Ozeki, T.; Nakayama, T.; Ogata, A.; Gotoh, K.; Fujiwara, K. Electrochemical Na Insertion and Solid Electrolyte Interphase for Hard-Carbon Electrodes and Application to Na-Ion Batteries. Adv. Funct. Mater. 2011, 21, 3859–3867. [Google Scholar] [CrossRef]
- Kim, S.W.; Seo, D.H.; Ma, X.; Ceder, G.; Kang, K. Electrode Materials for Rechargeable Sodium-Ion Batteries: Potential Alternatives to Current Lithium-Ion Batteries. Adv. Energy Mater. 2012, 2, 710–721. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Myung, S.T.; Sun, Y.K. Sodium-ion batteries: Present and future. Chem. Soc. Rev. 2017, 46, 3529–3614. [Google Scholar] [CrossRef]
- Pan, H.; Hu, Y.S.; Chen, L. Room-temperature stationary sodium-ion batteries for large-scale electric energy storage. Energy Environ. Sci. 2013, 6, 2338–2360. [Google Scholar] [CrossRef]
- Xu, Y.; Titirici, M.; Chen, J.; Cora, F.; Cullen, P.L.; Edge, J.S.; Fan, K.; Fan, L.; Feng, J.; Hosaka, T.; et al. 2023 roadmap for potassium-ion batteries. J. Phys. Energy 2023, 5, 021502. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Tang, Y.; Ji, X.; Jia, C.; Wang, H. Advancements and Challenges in Potassium Ion Batteries: A Comprehensive Review. Adv. Funct. Mater. 2020, 30, 1909486. [Google Scholar] [CrossRef]
- Min, X.; Xiao, J.; Fang, M.; Wang, W.; Zhao, Y.; Liu, Y.; Abdelkader, A.M.; Xi, K.; Kumar, R.V.; Huang, Z. Potassium-ion batteries: Outlook on present and future technologies. Energy Environ. Sci. 2021, 14, 2186–2243. [Google Scholar] [CrossRef]
- Wu, X.; Leonard, D.P.; Ji, X. Emerging Non-Aqueous Potassium-Ion Batteries: Challenges and Opportunities. Chem. Mater. 2017, 29, 5031–5042. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Guo, Z. Approaching high-performance potassium-ion batteries via advanced design strategies and engineering. Sci. Adv. 2019, 5, eaav7412. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Luchinin, N.D.; Aksyonov, D.A.; Morozov, A.V.; Ryazantsev, S.V.; Gaboardi, M.; Plaisier, J.R.; Stevenson, K.J.; Abakumov, A.M.; Antipov, E.V. Titanium-based potassium-ion battery positive electrode with extraordinarily high redox potential. Nat. Commun. 2020, 11, 1484. [Google Scholar] [CrossRef] [PubMed]
- Fedotov, S.S.; Samarin, A.S.; Nikitina, V.A.; Aksyonov, D.A.; Sokolov, S.A.; Zhugayevych, A.; Stevenson, K.J.; Khasanova, N.R.; Abakumov, A.M.; Antipov, E.V. Reversible facile Rb+ and K+ ions de/insertion in a KTiOPO4-type RbVPO4F cathode material. J. Mater. Chem. A 2018, 6, 14420–14430. [Google Scholar] [CrossRef]
- Lu, B.; Ru, N.; Duan, J.; Li, Z.; Qu, J. In-Plane Porous Graphene: A Promising Anode Material with High Ion Mobility and Energy Storage for Rubidium-Ion Batteries. ACS Omega 2023, 8, 21842–21852. [Google Scholar] [CrossRef]
- Lu, B.; Liu, X.; Qu, J.; Li, Z. Monolayer H-MoS2 with high ion mobility as a promising anode for rubidium (cesium)-ion batteries. Nanoscale Adv. 2022, 4, 3756–3763. [Google Scholar] [CrossRef]
- Sultana, I.; Rahman, M.M.; Ramireddy, T.; Chen, Y.; Glushenkov, A.M. High capacity potassium-ion battery anodes based on black phosphorus. J. Mater. Chem. A 2017, 5, 23506–23512. [Google Scholar] [CrossRef]
- McGilligan, J.P.; Moore, K.R.; Kang, S.; Mott, R.; Mis, A.; Roper, C.; Donley, E.A.; Kitching, J. Dynamic Characterization of an Alkali-Ion Battery as a Source for Laser-Cooled Atoms. Phys. Rev. Appl. 2020, 13, 044038. [Google Scholar] [CrossRef]
- Kang, S.; Mott, R.P.; Gilmore, K.A.; Sorenson, L.D.; Rakher, M.T.; Donley, E.A.; Kitching, J.; Roper, C.S. A low-power reversible alkali atom source. Appl. Phys. Lett. 2017, 110, 244101. [Google Scholar] [CrossRef]
- Kang, S.; Mott, R.P.; Mis, A.V.; Roper, C.S.; Donley, E.A.; Kitching, J. Active stabilization of alkali-atom vapor density with a solid-state electrochemical alkali-atom source. Opt. Express 2018, 26, 3696–3701. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Moore, K.R.; McGilligan, J.P.; Mott, R.; Mis, A.; Roper, C.; Donley, E.A.; Kitching, J. Magneto-optic trap using a reversible, solid-state alkali-metal source. Opt. Lett. 2019, 44, 3002–3005. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cai, X.; Yin, H.; Li, Y.; Lin, W.; Huang, S.; Zhang, Y. A new candidate in polyanionic compounds for a potassium-ion battery cathode: KTiOPO4. J. Phys. Chem. Lett. 2021, 12, 2721–2726. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cai, X.; Li, Y.; Fang, Z.; Li, Y.; Lin, W.; Huang, S.; Zhang, Y. DFT investigations of KTiOPO4Mx (M = K, Na, and Li) anodes for alkali-ion battery. J. Chem. Phys. 2022, 156, 204702. [Google Scholar] [CrossRef] [PubMed]
- Fedotov, S.S.; Samarin, A.S.; Antipov, E.V. KTiOPO4-structured electrode materials for metal-ion batteries: A review. J. Power Sources 2020, 480, 228840. [Google Scholar] [CrossRef]
- Katorova, N.S.; Fedotov, S.S.; Rupasov, D.P.; Luchinin, N.D.; Delattre, B.; Chiang, Y.M.; Abakumov, A.M.; Stevenson, K.J. Effect of Concentrated Diglyme-Based Electrolytes on the Electrochemical Performance of Potassium-Ion Batteries. ACS Appl. Energy Mater. 2019, 2, 6051–6059. [Google Scholar] [CrossRef]
- Tan, H.; Du, X.; Huang, J.Q.; Zhang, B. KVPO4F as a novel insertion-type anode for potassium ion batteries. Chem. Commun. 2019, 55, 11311–11314. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Khasanova, N.R.; Samarin, A.S.; Drozhzhin, O.A.; Batuk, D.; Karakulina, O.M.; Hadermann, J.; Abakumov, A.M.; Antipov, E.V. AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries. Chem. Mater. 2016, 28, 411–415. [Google Scholar] [CrossRef]
- Chihara, K.; Katogi, A.; Kubota, K.; Komaba, S. KVPO4F and KVOPO4 toward 4 volt-class potassium-ion batteries. Chem. Commun. 2017, 53, 5208–5211. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lin, Y.C.; Liu, J.; Rana, J.; Zhang, H.; Zhou, H.; Chu, I.H.; Wiaderek, K.M.; Omenya, F.; Chernova, N.A.; et al. KVOPO4: A New High Capacity Multielectron Na-Ion Battery Cathode. Adv. Energy Mater. 2018, 8, 1800221. [Google Scholar] [CrossRef]
- Mu, L.; Ben, L.; Hu, Y.S.; Li, H.; Chen, L.; Huang, X. Novel 1.5 V anode materials, ATiOPO4 (A = NH4, K, Na), for room-temperature sodium-ion batteries. J. Mater. Chem. A 2016, 4, 7141–7147. [Google Scholar] [CrossRef]
- Galceran, M.; Rikarte, J.; Zarrabeitia, M.; Pujol, M.C.; Aguiló, M.; Casas-Cabanas, M. Investigation of NaTiOPO4 as Anode for Sodium-Ion Batteries: A Solid Electrolyte Interphase Free Material? ACS Appl. Energy Mater. 2019, 2, 1923–1931. [Google Scholar] [CrossRef]
- Liu, S.; Shao, L.; Zhang, X.; Zhou, M.; Tao, Z.; Chen, J. KTiOPO4 as a novel anode material for sodium-ion batteries. J. Alloys Compd. 2018, 754, 147–152. [Google Scholar] [CrossRef]
- Bocchini, A.; Gerstmann, U.; Bartley, T.; Steinrück, H.G.; Henkel, G.; Schmidt, W.G. Electrochemical performance of KTiOAsO4 (KTA) in potassium-ion batteries from density-functional theory. Phys. Rev. Mater. 2022, 6, 105401. [Google Scholar] [CrossRef]
- Roth, M. Stoichiometry and Domain Structure of KTP-Type Nonlinear Optical Crystals. In Springer Handbook of Crystal Growth; Dhanaraj, G., Byrappa, K., Prasad, V., Dudley, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar] [CrossRef]
- Masse, R.; Grenier, J.C. Étude des monophosphates du type MITiOPO4 avec MI = K, Rb et Tl. Bull. Minéral. 1971, 94, 437–439. [Google Scholar]
- Bierlein, J.D.; Vanherzeele, H. Potassium titanyl phosphate: Properties and new applications. J. Opt. Soc. Am. B Opt. Phys. 1989, 6, 622–633. [Google Scholar] [CrossRef]
- Tordjman, P.I.; Masse, E.; Guitel, J.C. Structure cristalline du monophosphate KTiPO5. Z. Krist. 1974, 139, 103–115. [Google Scholar] [CrossRef]
- Rangan, K.K.; Prasad, B.R.; Subramanian, C.K.; Gopalakrishnan, J. Coupled substitution of niobium and silicon in potassium titanyl phosphate and arsenate KTiOPO4 and KTiOAsO4. Synthesis and nonlinear optical properties of KTi1-xNbxOX1-xSixO4 (X = P, As). Inorg. Chem. 1993, 32, 4291–4293. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Allan, D.R.; Loveday, J.S.; Nelmes, R.J.; Thomas, P.A. A high-pressure structural study of potassium titanyl phosphate (KTP) up to 5 GPa. J. Phys. Condens. Matter 1992, 4, 2747. [Google Scholar] [CrossRef]
- Thomas, P.A.; Duhlev, R.; Teat, S.J. A comparative structural study of a flux-grown crystal of K0.86Rb0.14TiOPO4 and an ion-exchanged crystal of K0.84Rb0.16TiOPO4. Acta Crystallogr. Sect. B 1994, 50, 538–543. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Baldereschi, A. Mean-Value Point in the Brillouin Zone. Phys. Rev. B 1973, 7, 5212–5215. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Deák, P.; Lorke, M.; Aradi, B.; Frauenheim, T. Optimized hybrid functionals for defect calculations in semiconductors. J. Appl. Phys. 2019, 126, 130901. [Google Scholar] [CrossRef]
- Janotti, A.; Varley, J.B.; Rinke, P.; Umezawa, N.; Kresse, G.; Van de Walle, C.G. Hybrid functional studies of the oxygen vacancy in TiO2. Phys. Rev. B 2010, 81, 085212. [Google Scholar] [CrossRef]
- Komsa, H.P.; Broqvist, P.; Pasquarello, A. Alignment of defect levels and band edges through hybrid functionals: Effect of screening in the exchange term. Phys. Rev. B 2010, 81, 205118. [Google Scholar] [CrossRef]
- Chevrier, V.L.; Ong, S.P.; Armiento, R.; Chan, M.K.Y.; Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B 2010, 82, 075122. [Google Scholar] [CrossRef]
- Ceder, G.; Aydinol, M.; Kohan, A. Application of first-principles calculations to the design of rechargeable Li-batteries. Comput. Mater. Sci. 1997, 8, 161–169. [Google Scholar] [CrossRef]
- Krishnamurthy, V.; Viswanathan, V. Beyond Transition Metal Oxide Cathodes for Electric Aviation: The Case of Rechargeable CFx. ACS Energy Lett. 2020, 5, 3330–3335. [Google Scholar] [CrossRef]
- Ghosh, A.; Pal, S.; Sarkar, P. Rational Design of Two-Dimensional Porous Boron Phosphide as Efficient Cathode Material for Li and Na Ion Batteries: A First-Principles Study. J. Phys. Chem. C 2022, 126, 5092–5100. [Google Scholar] [CrossRef]
- Lv, X.; Li, F.; Gong, J.; Gu, J.; Lin, S.; Chen, Z. Metallic FeSe monolayer as an anode material for Li and non-Li ion batteries: A DFT study. Phys. Chem. Chem. Phys. 2020, 22, 8902–8912. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, A.; Deng, Z.; Banerjee, S.; Ong, S.P. Rechargeable Alkali-Ion Battery Materials: Theory and Computation. Chem. Rev. 2020, 120, 6977–7019. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, S.; Schindlmayr, A.; Schmidt, W.G. Quasiparticle energies and optical response of RbTiOPO4 and KTiOAsO4. J. Phys. Mater. 2021, 5, 015002. [Google Scholar] [CrossRef]
- Zumsteg, F.C.; Bierlein, J.D.; Gier, T.E. KxRb1-xTiOPO4: A new nonlinear optical material. J. Appl. Phys. 1976, 47, 4980–4985. [Google Scholar] [CrossRef]
- Neufeld, S.; Bocchini, A.; Gerstmann, U.; Schindlmayr, A.; Schmidt, W.G. Potassium titanyl phosphate (KTP) quasiparticle energies and optical response. J. Phys. Mater. 2019, 2, 045003. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Accounts Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef]
- Ohzuku, T.; Ueda, A. Solid-State Redox Reactions of LiCoO2 (R3m) for 4 Volt Secondary Lithium Cells. J. Electrochem. Soc. 1994, 141, 2972. [Google Scholar] [CrossRef]
- Lung-Hao Hu, B.; Wu, F.Y.; Lin, C.T.; Khlobystov, A.N.; Li, L.J. Graphene-modified LiFePO4 cathode for lithium ion battery beyond theoretical capacity. Nat. Commun. 2013, 4, 1687. [Google Scholar] [CrossRef] [PubMed]
- Franger, S.; Cras, F.L.; Bourbon, C.; Rouault, H. LiFePO4 Synthesis Routes for Enhanced Electrochemical Performance. Electrochem. Solid-State Lett. 2002, 5, A231. [Google Scholar] [CrossRef]
- Shen, K.; Xu, X.; Tang, Y. Recent progress of magnetic field application in lithium-based batteries. Nano Energy 2022, 92, 106703. [Google Scholar] [CrossRef]
- Costa, C.M.; Merazzo, K.J.; Gonçalves, R.; Amos, C.; Lanceros-Méndez, S. Magnetically active lithium-ion batteries towards battery performance improvement. iScience 2021, 24, 102691. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Compound | a [Å] | b [Å] | c [Å] | V [Å] | |
|---|---|---|---|---|---|
| KTiOPO | 12.819 | 6.399 | 10.584 | 868.2 | Exp. [45] |
| 12.860 | 6.432 | 10.616 | 876.7 | PBEsol [67] | |
| KTiPOF | 13.002 | 6.434 | 10.764 | 900.5 | Exp. [17] |
| 13.30 | 6.56 | 11.04 | 894.9 | PBE [17] | |
| RbTiOPO | 12.952 | 6.500 | 10.558 | 888.9 | Exp. [66] |
| 12.986 | 6.521 | 10.568 | 894.9 | PBEsol [65] | |
| RbTiPOF | 13.236 | 6.616 | 10.945 | 958.5 | PBEsol, This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocchini, A.; Xie, Y.; Schmidt, W.G.; Gerstmann, U. Structural and Electrochemical Properties of F-Doped RbTiOPO4 (RTP:F) Predicted from First Principles. Crystals 2024, 14, 5. https://doi.org/10.3390/cryst14010005
Bocchini A, Xie Y, Schmidt WG, Gerstmann U. Structural and Electrochemical Properties of F-Doped RbTiOPO4 (RTP:F) Predicted from First Principles. Crystals. 2024; 14(1):5. https://doi.org/10.3390/cryst14010005
Chicago/Turabian StyleBocchini, Adriana, Yingjie Xie, Wolf Gero Schmidt, and Uwe Gerstmann. 2024. "Structural and Electrochemical Properties of F-Doped RbTiOPO4 (RTP:F) Predicted from First Principles" Crystals 14, no. 1: 5. https://doi.org/10.3390/cryst14010005
APA StyleBocchini, A., Xie, Y., Schmidt, W. G., & Gerstmann, U. (2024). Structural and Electrochemical Properties of F-Doped RbTiOPO4 (RTP:F) Predicted from First Principles. Crystals, 14(1), 5. https://doi.org/10.3390/cryst14010005