Graphene Supported Tungsten Carbide as Catalyst for Electrochemical Reduction of CO2



Abstract

1. Introduction
- Adjusting bandgap and band position to capture solar energy effectively, thereby improving the efficiency of CO2 reduction. For example, the potential of the valence band of various metal oxide catalysts is around 3 eV vs. NHE (normal hydrogen electrode), utilizing only ultraviolet light. Replacing the O atom with N or C will narrow the bandgap thereby efficiently absorbing the solar radiations [4,5].
- The use of nanostructured semiconductors in the form of nanorods, nanowires, nanotubes, nanobelts, etc. decreases the electron-hole recombination rate as these are usually in a single crystalline phase that eliminates the possibility of grain boundaries and defects in the materials as they act as recombination sites for the electron-hole pair in polycrystalline materials. At the same time, one-dimensional nanostructures improve electron transport by improving the separation of electron-hole pairs. For instance, Zn2GeO4 nanoribbons are proven to show improved photocatalytic activity towards CH4 formation when compared to bulk Zn2GeO4 [8,9]
- Use of co-catalysts in the process to promote the separation and movement of charge carriers. This will minimize the recombination of electron-hole pairs due to the barrier between semiconductor and co-catalyst. Metal nanoparticles, such as Pt, Rh, Pd, Cu, Ag, Au, supported on semiconductor are proven to work efficiently when compared to pure semiconducting photocatalysts [10,11].
- Alloys to combine the electronic properties of WC with other metal(s), for example, Tantalum doped WC displayed better activity towards hydrogen evolution (HER) when compared to unmodified WC [23].
- Co-catalyst to the catalytic system where strong electronic interactions between them might modify (maximize) the electrocatalytic activity. As an example, Ni with WC nanocluster for urea electro-oxidation showed high tolerance towards CO poisoning, and high stability thereby enhancing catalyst activity [30].
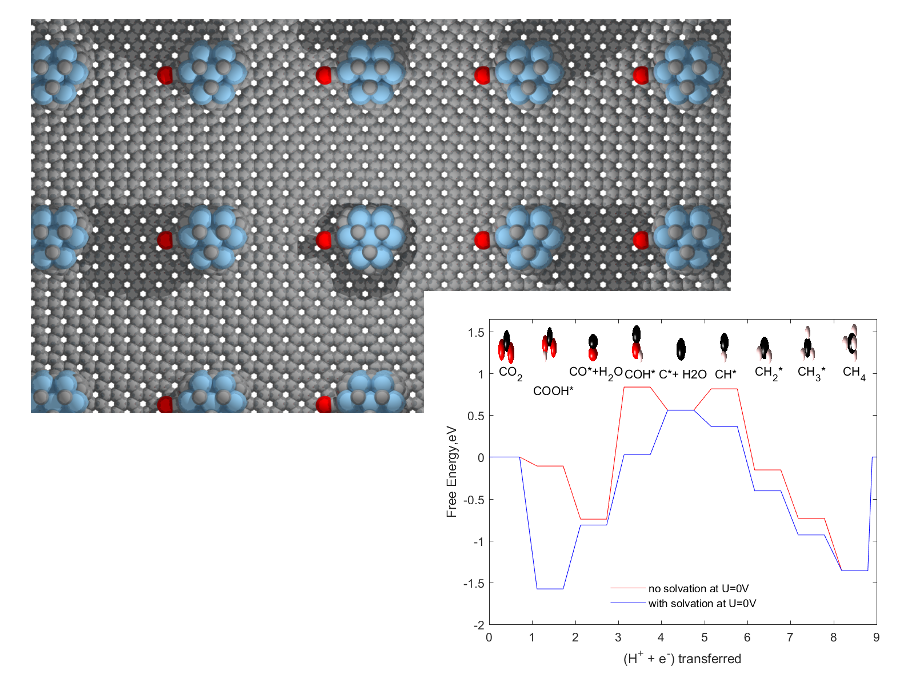
2. Results and Discussion
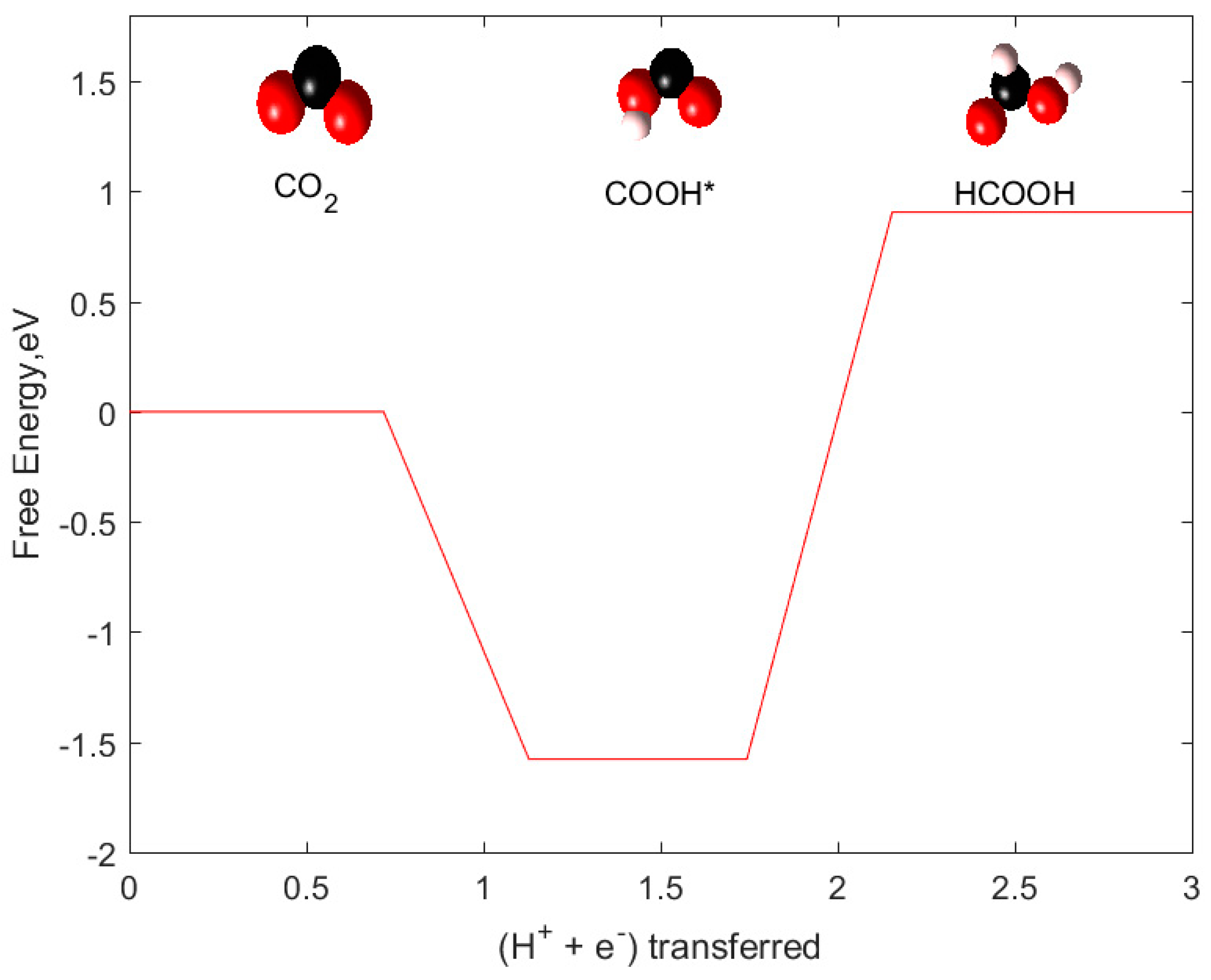
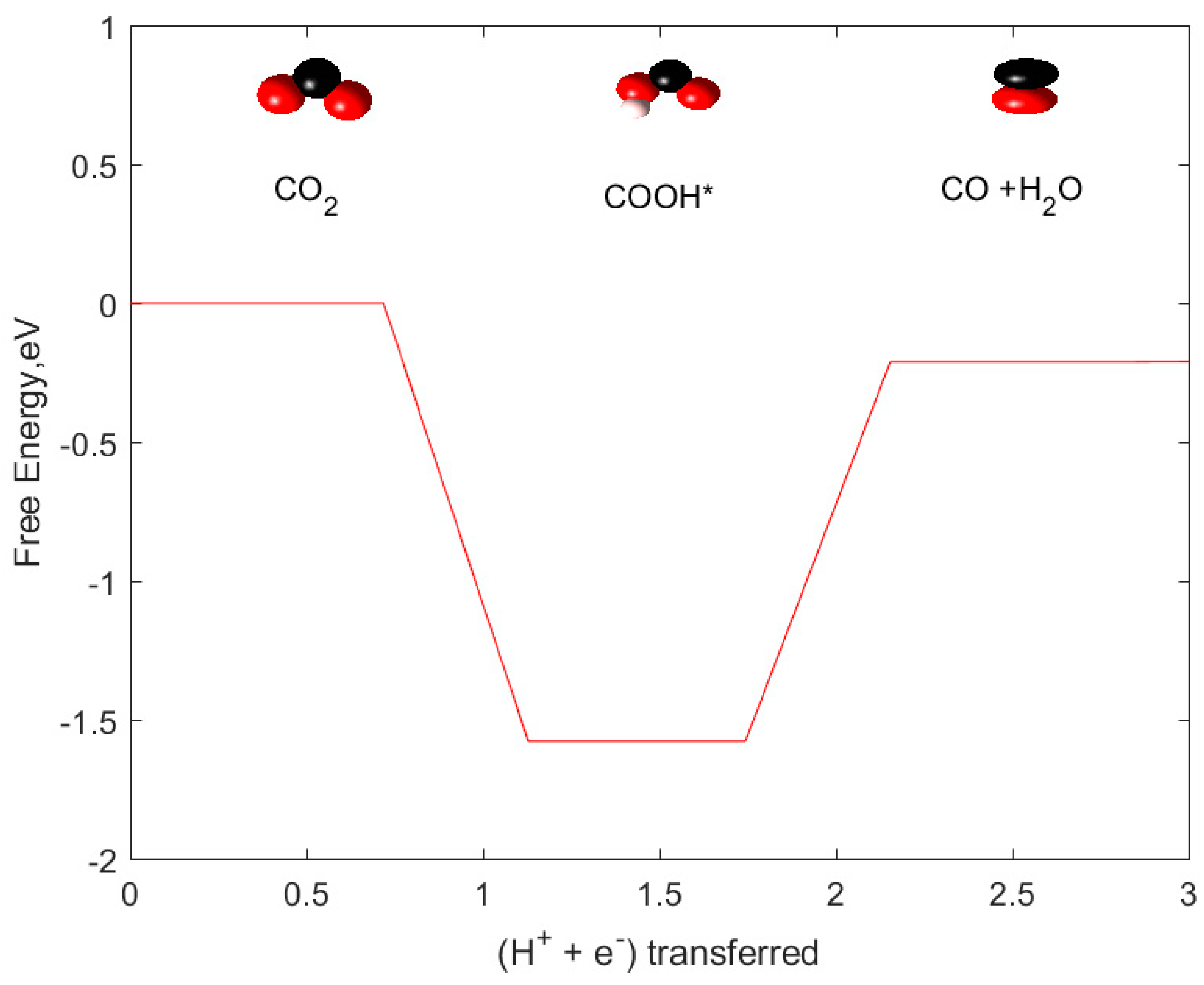
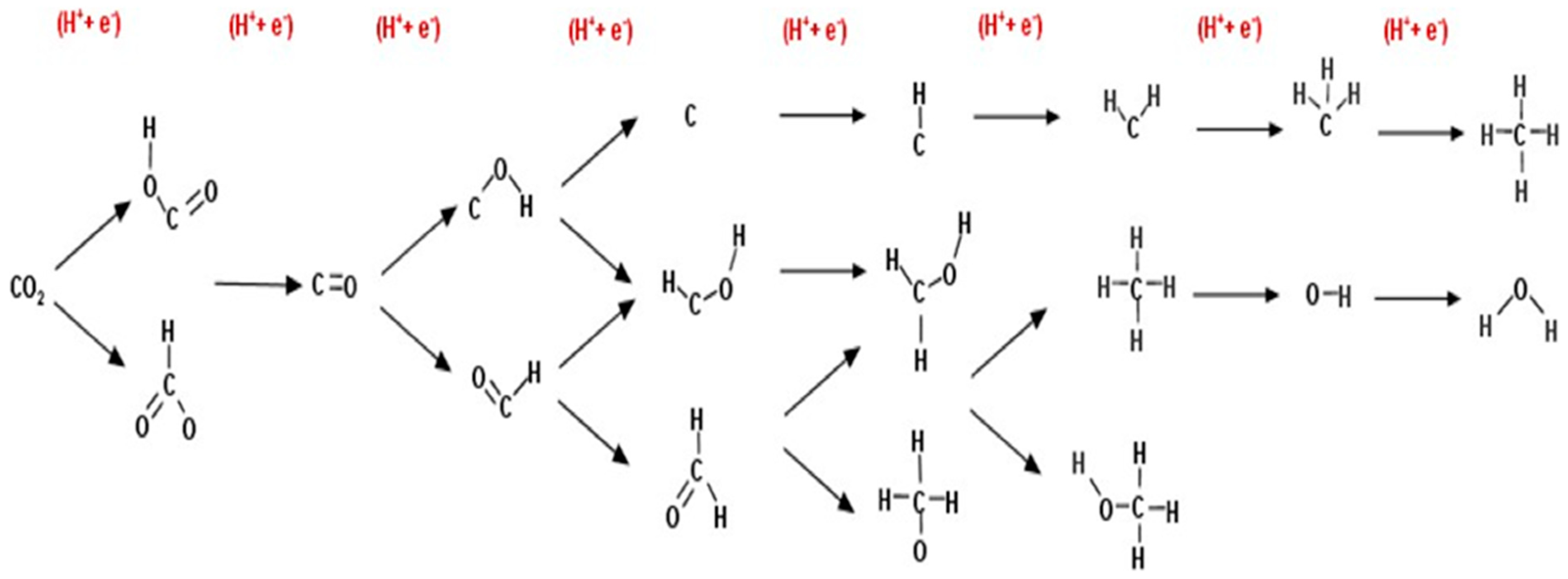
2.1. CO and HCOOH as Products
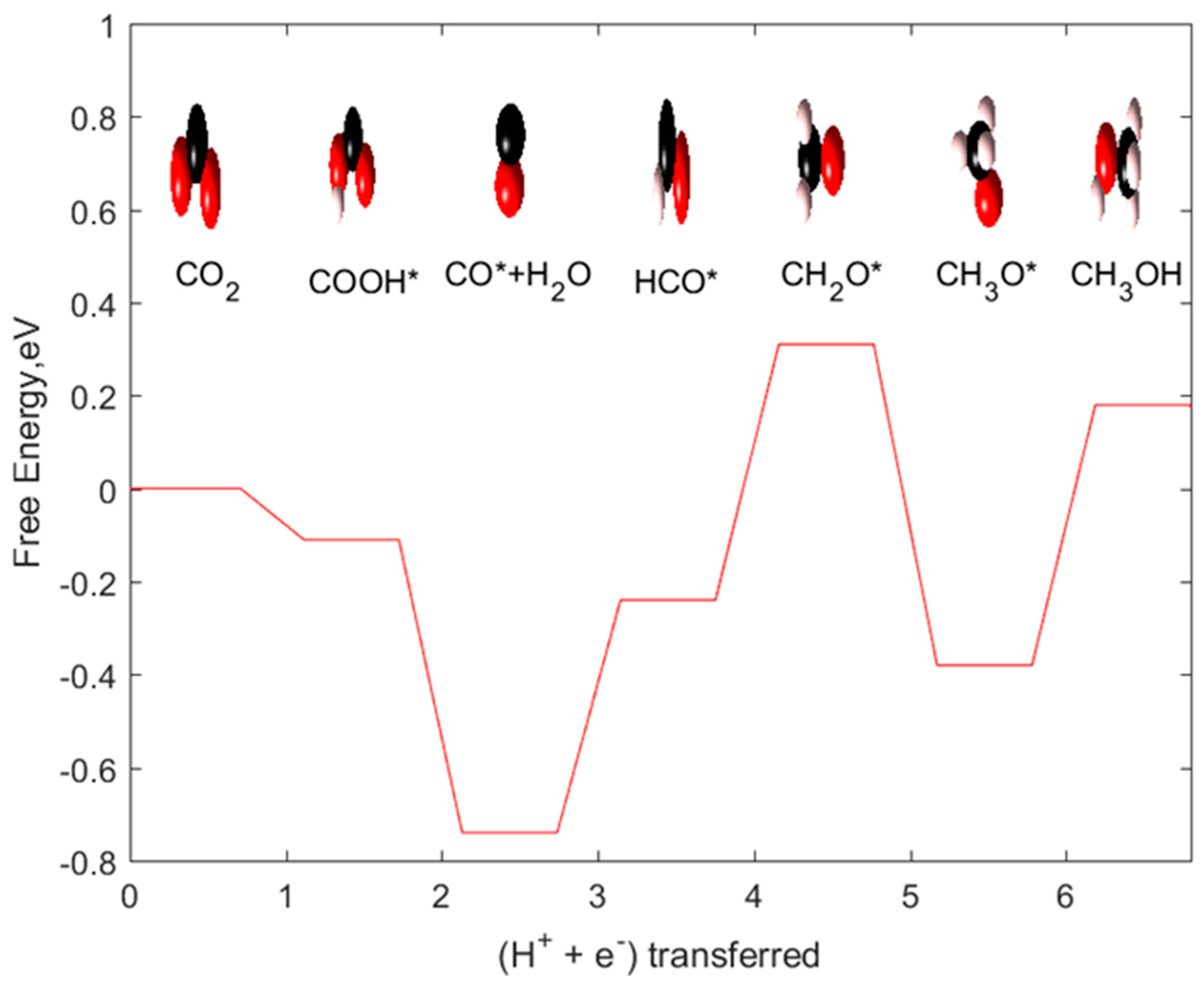
2.2. CH3OH and CH4 as Products
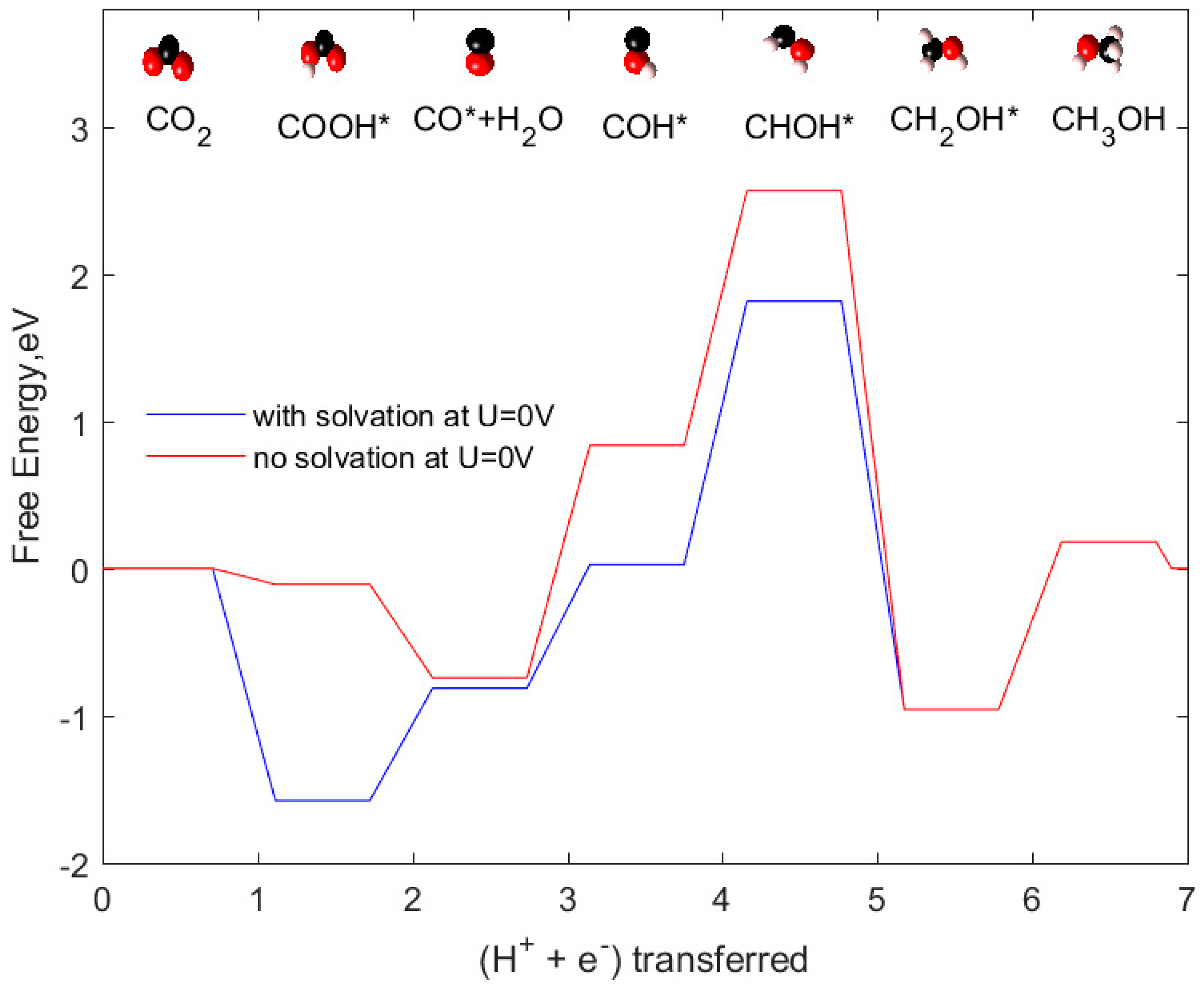
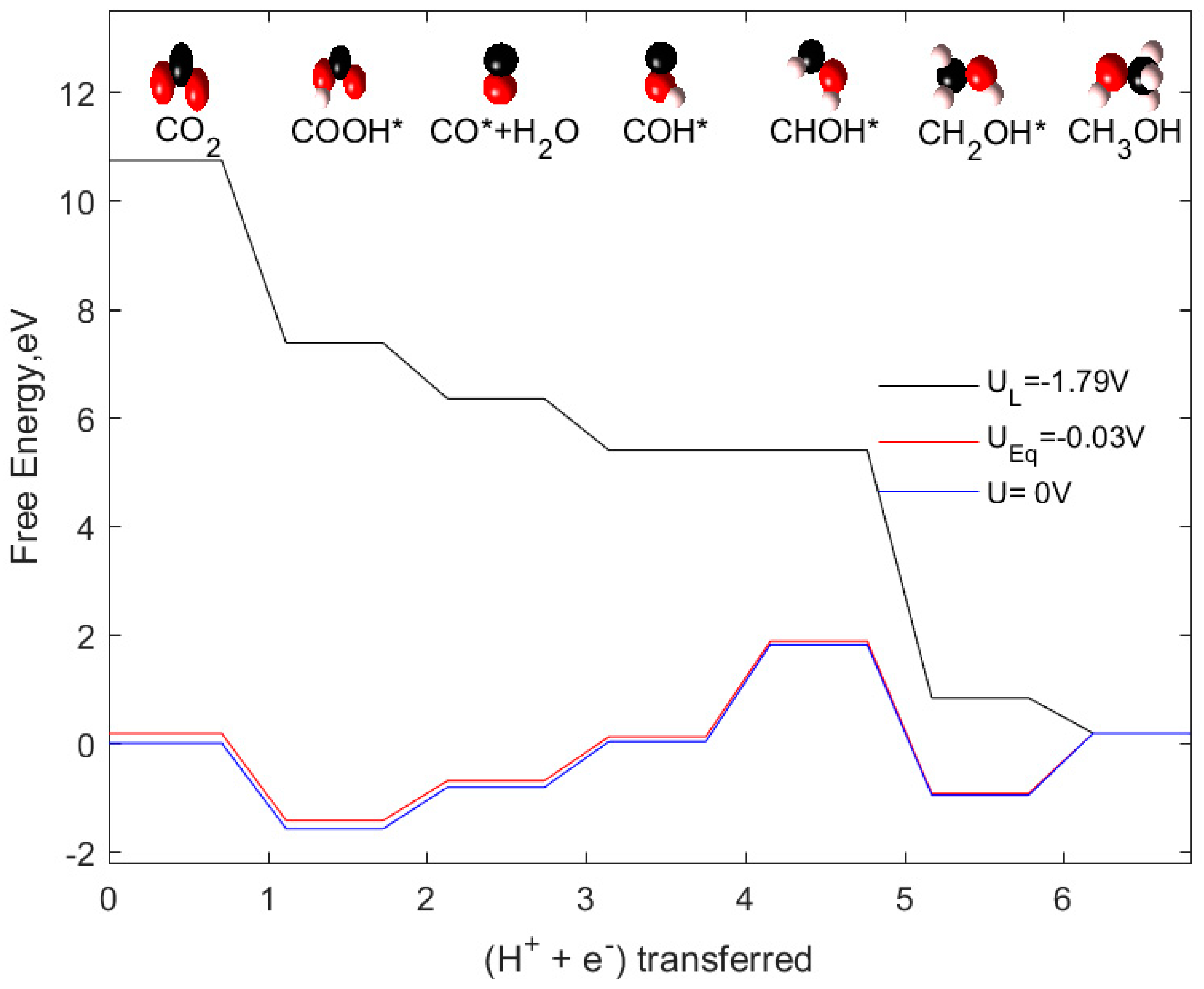
2.3. CH3OH as Product
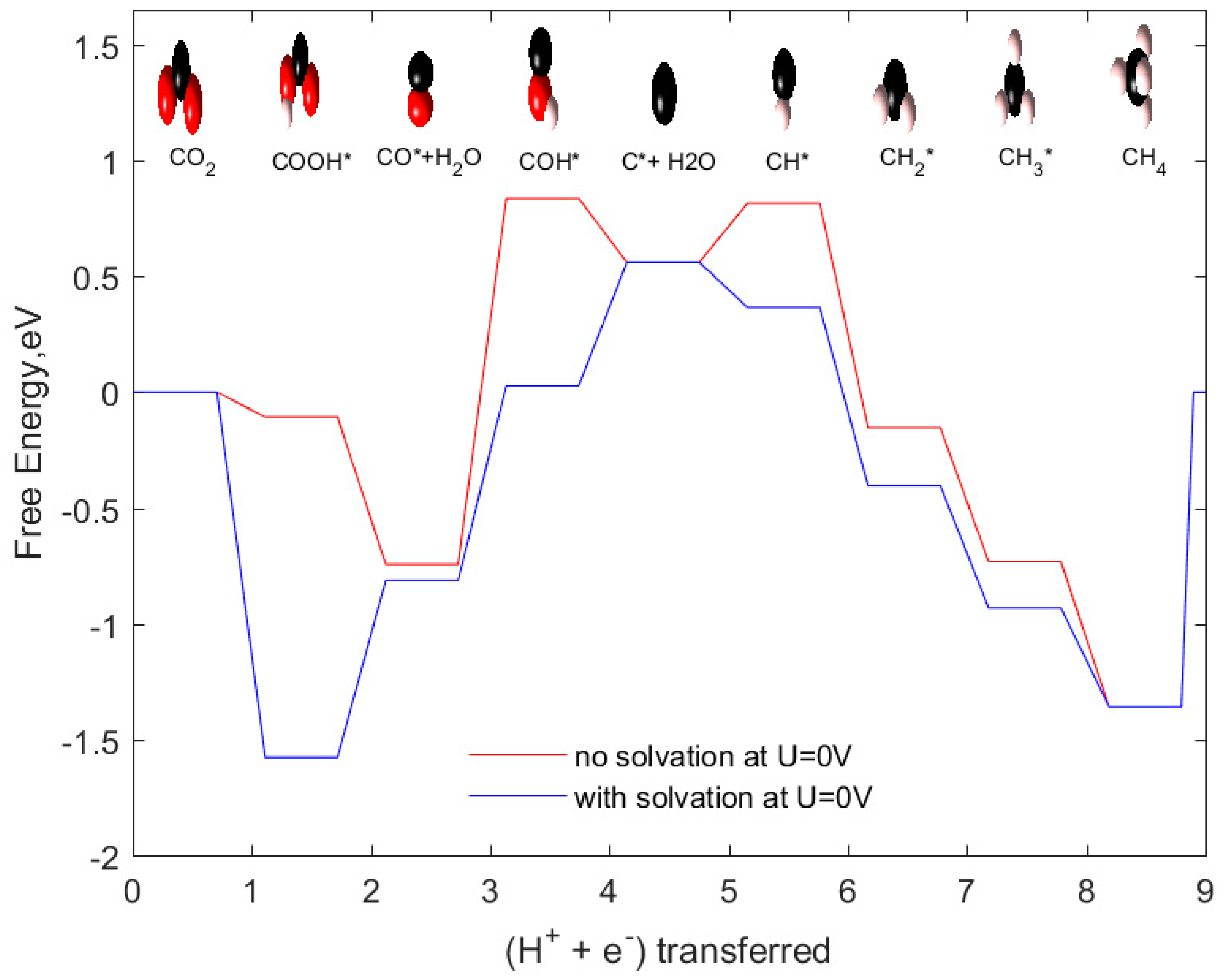
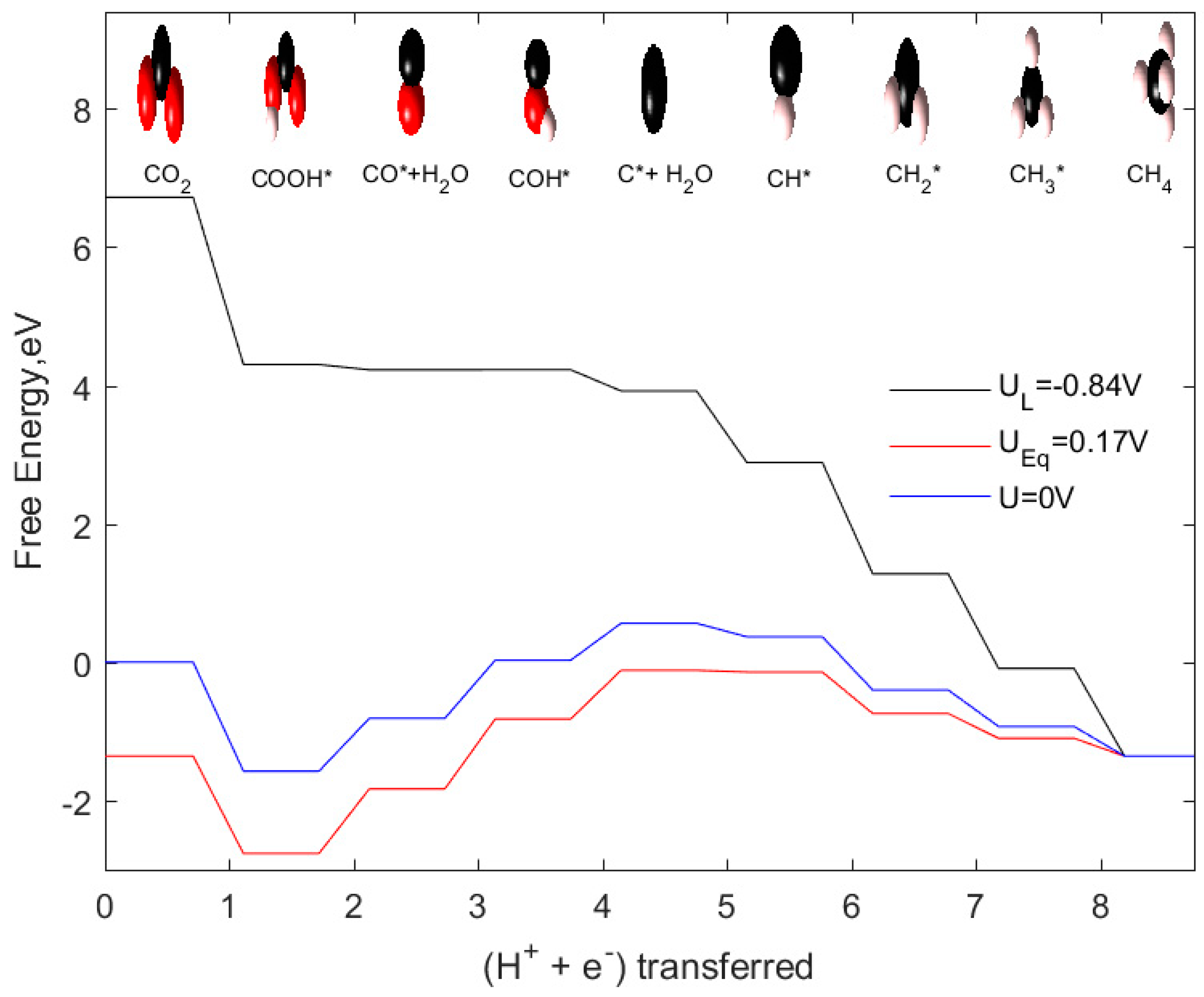
2.4. CH4 as Product
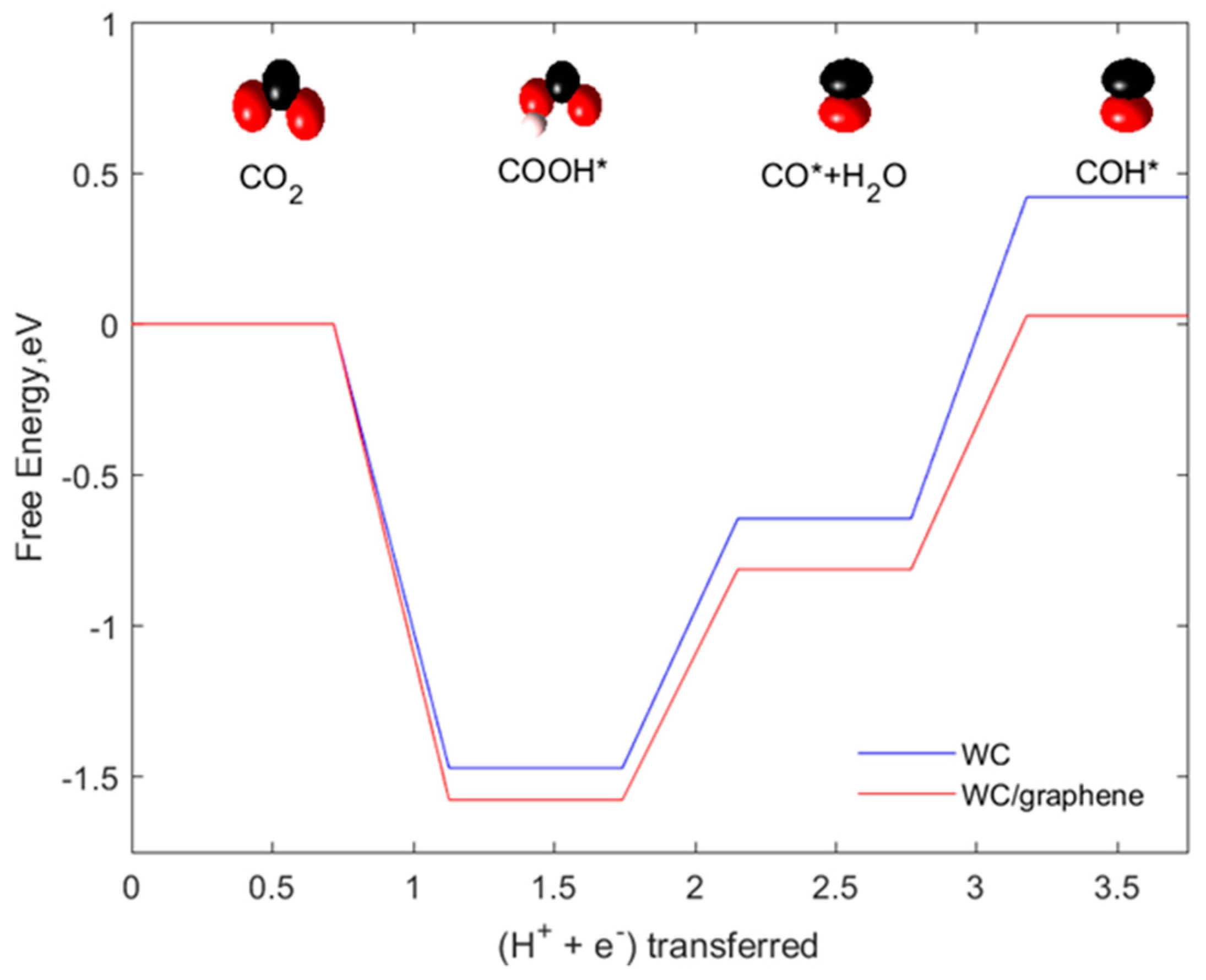
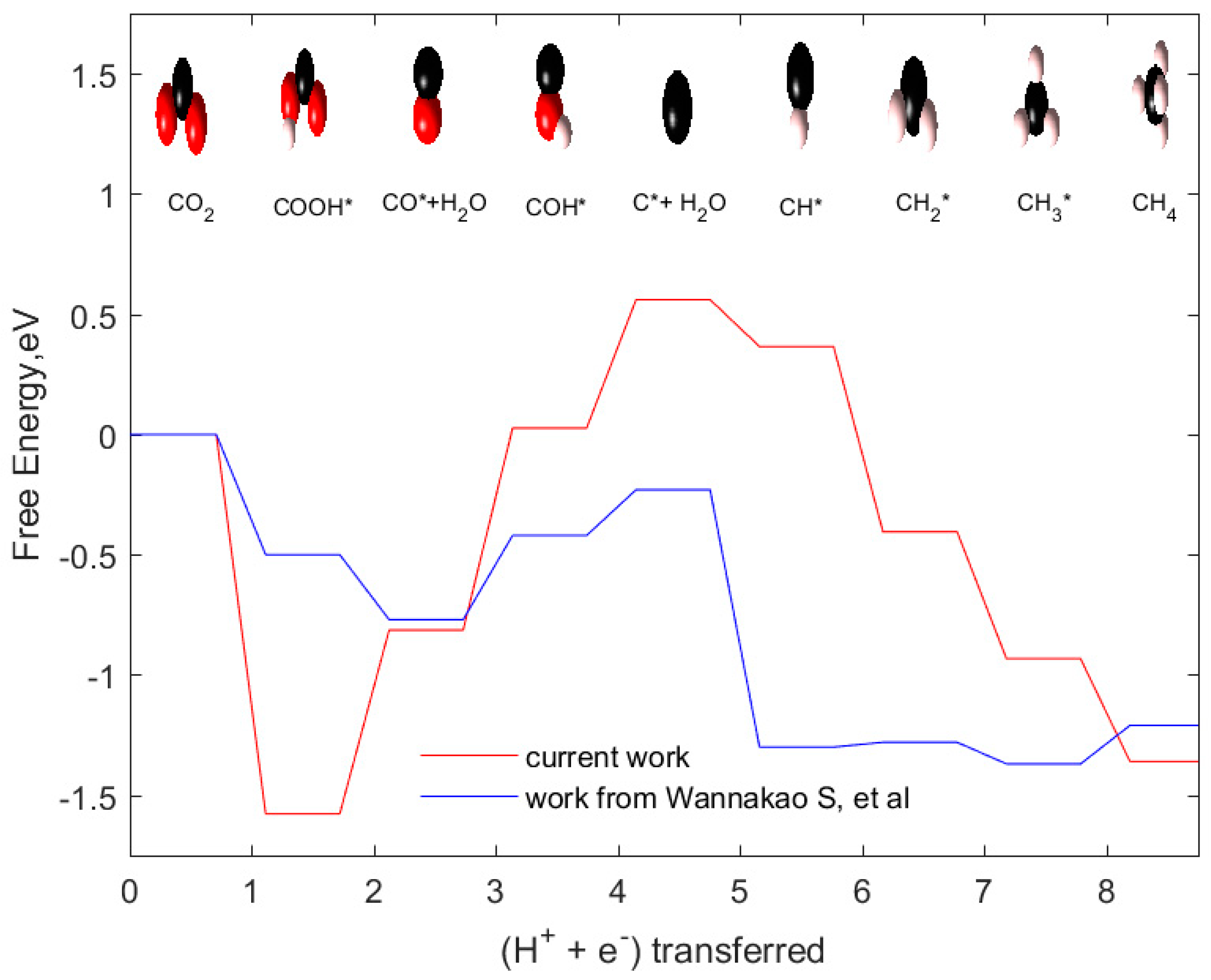
2.5. Comparison of CO2 Reduction to CH4 and CH3OH on Graphene Supported WC Nanocluster and WC (0001)
- Employing different functionals in the DFT calculations would result in differences in binding free energies of intermediates. In the work of Wannakao S. et al., it is proven that
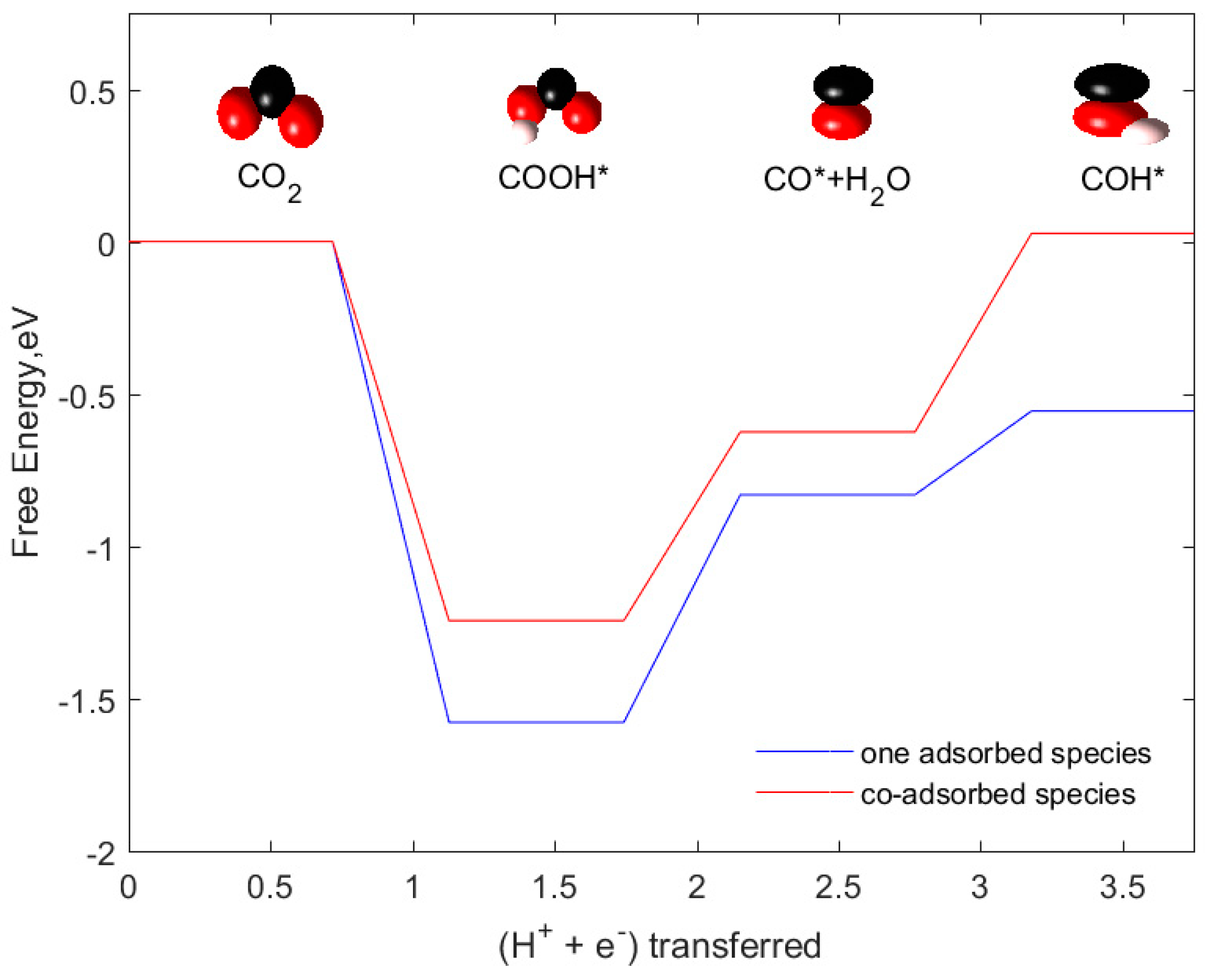
- Another reason is predicted to be due to the coverage of adsorbed intermediates on the surface of the catalyst. Our catalyst system is designed by placing only one adsorbate species on a single nanocluster which is approximately equal to 1/6 or 1/9 monolayer (ML) coverage of adsorbate species on the surface (assuming each side of the cluster mimics 3 × 2 or 3 × 3 slab surface). However, in the work of Wannakao S. et al., WC (0001) was modeled by 1/6 to 1/9 ML coverage of adsorbate species which means these are placed comparatively closer than our adsorbate species arrangement. We predict that the influence of lateral interactions between adsorbed intermediates could also lead to a difference in reaction free energies. To investigate this, we vary (increase) the surface coverage of intermediate species by placing two species instead of one in our catalyst system as neighboring atoms/moieties near the active site. In other words, this system is modified to try to approximate the effects of 1/6 to 1/9 ML coverage of adsorbate species in their work. Figure 8 shows how the reaction free energies vary when the proportion of adsorbate coverage on the catalyst system varies. Here, we have computed the free energies of initial steps in the reaction network (COOH*, CO*, COH*) as these are the pathway determining intermediates. Co-adsorption of these species generated an upward shift of binding free energies of all the initial three steps. Consequently, the rate-limiting step in the case of co-adsorbed species shifted to the COOH* protonation step from the CO* protonation step. This analysis also explains that the surface coverage of the adsorbed species plays an equally important role in determining the energetics of CO2 reduction reaction.
2.6. Role of Graphene
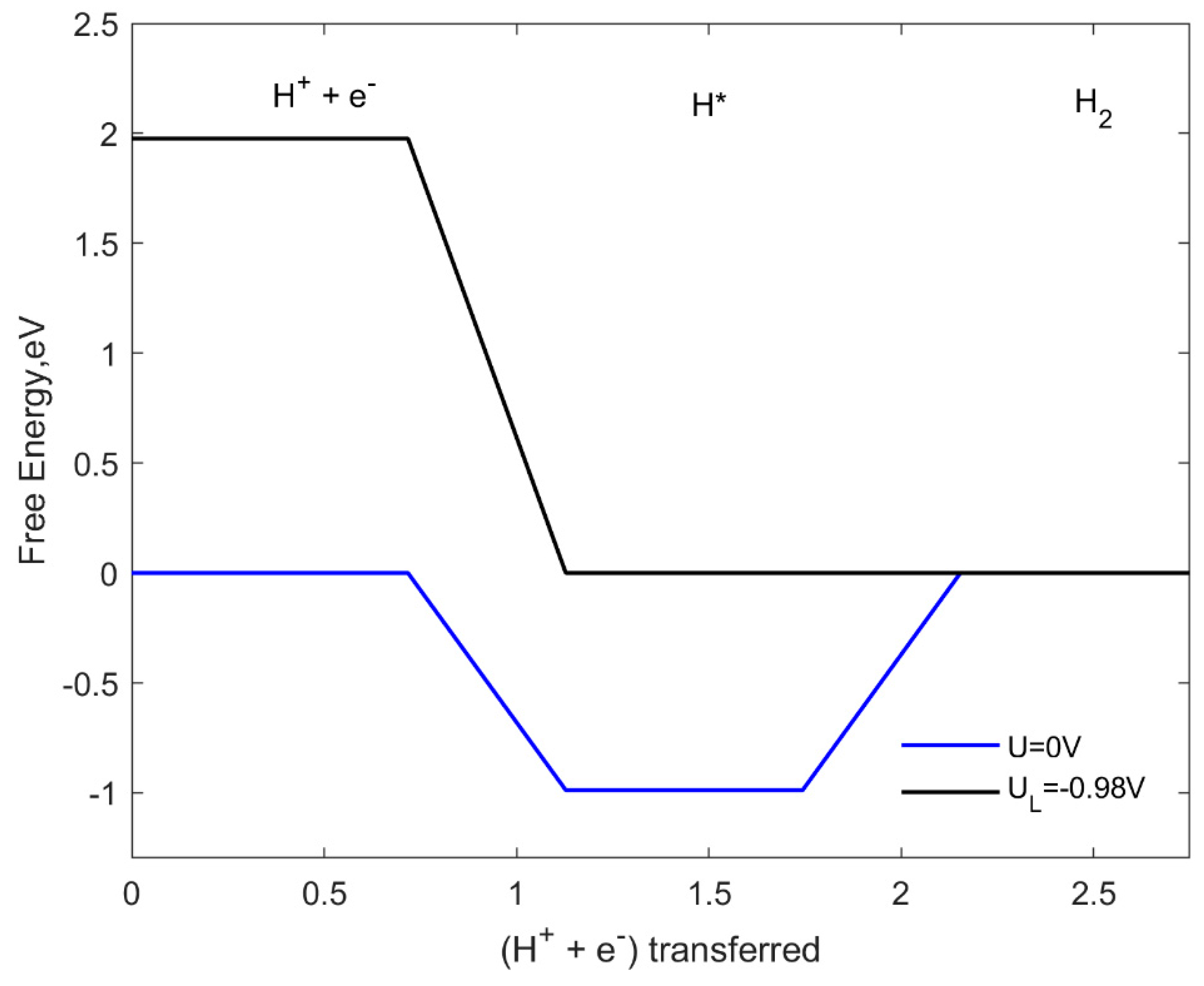
2.7. HER
3. (Computational) Materials and Methods
4. Conclusions and Future Work
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Ajmal, S.; Zheng, X.; Zhang, L. Efficient nanomaterials for harvesting clean fuels from electrochemical and photoelectrochemical CO2 reduction. Sustain. Energy Fuels 2018, 2, 510–537. [Google Scholar] [CrossRef]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects. Adv. Mater 2014, 26, 4607–4626. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, Y.; Ye, W.; Li, Y. CO2 reduction: From the electrochemical to photochemical approach. Adv. Sci. 2017, 4, 1700194. [Google Scholar] [CrossRef] [PubMed]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.; Tian, Z.; Chen, X.; Gao, J.; Zou, Z. Zn2 GeO4 crystal splitting toward sheaf-like, hyperbranched nanostructures and photocatalytic reduction of CO2 into CH 4 under visible light after nitridation. J. Mater. Chem. 2012, 22, 2033–2038. [Google Scholar] [CrossRef]
- Indrakanti, V.P.; Kubicki, J.D.; Schobert, H.H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2009, 2, 745–758. [Google Scholar] [CrossRef]
- Indrakanti, V.P.; Schobert, H.H.; Kubicki, J.D. Quantum mechanical modeling of CO2 interactions with irradiated stoichiometric and oxygen-deficient anatase TiO2 surfaces: Implications for the photocatalytic reduction of CO2. Energy Fuels 2009, 23, 5247–5256. [Google Scholar] [CrossRef]
- Yan, S.; Wan, L.; Li, Z.; Zou, Z. Facile temperature-controlled synthesis of hexagonal Zn2GeO4 nanorods with different aspect ratios toward improved photocatalytic activity for overall water splitting and photoreduction of CO2. Chem. Commun. 2011, 47, 5632–5634. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Grätzel, M.; Kostecki, R.; Mao, S.S. Nanomaterials for renewable energy production and storage. Chem. Soc. Rev. 2012, 41, 7909–7937. [Google Scholar] [CrossRef]
- Anpo, M.; Yamashita, H.; Ichihashi, Y.; Fujii, Y.; Honda, M. Photocatalytic reduction of CO2 with H2O on titanium oxides anchored within micropores of zeolites: Effects of the structure of the active sites and the addition of Pt. J. Phys. Chem. B 1997, 101, 2632–2636. [Google Scholar] [CrossRef]
- Tong, H.; Ouyang, S.; Bi, Y.; Umezawa, N.; Oshikiri, M.; Ye, J. Nano-photocatalytic materials: Possibilities and challenges. Adv. Mater. 2012, 24, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Kikuchi, K.; Suzuki, S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Gonçalves, M.R.; Gomes, A.; Condeço, J.; Fernandes, R.; Pardal, T.; Sequeira, C.A.C.; Branco, J.B. Selective electrochemical conversion of CO2 to C2 hydrocarbons. Energy Convers. Manag. 2010, 51, 30–32. [Google Scholar] [CrossRef]
- Tackett, B.M.; Sheng, W.; Chen, J.G. Opportunities and Challenges in Utilizing Metal-Modified Transition Metal Carbides as Low-Cost Electrocatalysts. Joule 2017, 1, 253–263. [Google Scholar] [CrossRef]
- Levy, R.B.; Boudart, M. Platinum-like behavior of tungsten carbide in surface catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xia, X.; Shi, F.; Zhan, J.; Tu, J.; Fan, H.J. Transition metal carbides and nitrides in energy storage and conversion. Adv. Sci. 2016, 3, 1500286. [Google Scholar] [CrossRef]
- Liang, C.; Ying, P.; Li, C. Nanostructured β-Mo2C Prepared by Carbothermal Hydrogen Reduction on Ultrahigh Surface Area Carbon Material. Chem. Mater. 2002, 14, 3148–3151. [Google Scholar] [CrossRef]
- Michalsky, R.; Zhang, Y.J.; Medford, A.J.; Peterson, A.A. Departures from the Adsorption Energy Scaling Relations for Metal Carbide Catalysts. J. Phys. Chem. C 2014, 118, 13026–13034. [Google Scholar] [CrossRef]
- Wannakao, S.; Artrith, N.; Limtrakul, J.; Kolpak, A.M. Engineering Transition-Metal-Coated Tungsten Carbides for Efficient and Selective Electrochemical Reduction of CO2 to Methane. ChemSusChem 2015, 8, 2745–2751. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. Density functional theory study of iron and cobalt carbides for Fischer− Tropsch synthesis. J. Phys. Chem. C 2009, 114, 1085–1093. [Google Scholar] [CrossRef]
- Stottlemyer, A.L.; Weigert, E.C.; Chen, J.G. Tungsten Carbides as Alternative Electrocatalysts: From Surface Science Studies to Fuel Cell Evaluation. Ind. Eng. Chem. Res. 2011, 50, 16–22. [Google Scholar] [CrossRef]
- Hunt, S.T.; Kokumai, T.M.; Zanchet, D.; Román-Leshkov, Y. Alloying tungsten carbide nanoparticles with tantalum: Impact on electrochemical oxidation resistance and hydrogen evolution activity. J. Phys. Chem. C 2015, 24, 13691–13699. [Google Scholar] [CrossRef]
- Hunt, S.T.; Milina, M.; Alba-Rubio, A.C.; Hendon, C.H.; Dumesic, J.A.; Román-Leshkov, Y. Self-assembly of noble metal monolayers on transition metal carbide nanoparticle catalysts. Science 2016, 352, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Obradović, M.D.; Gojković, S.L.; Elezović, N.R.; Ercius, P.; Radmilović, V.R.; Vračar, L.D.; Krstajić, N.V. The kinetics of the hydrogen oxidation reaction on WC/Pt catalyst with low content of Pt nano-particles. J. Electroanal. Chem. 2012, 671, 24–32. [Google Scholar] [CrossRef]
- Esposito, D.V.; Hunt, S.T.; Kimmel, Y.C.; Chen, J.G. A New Class of Electrocatalysts for Hydrogen Production from Water Electrolysis: Metal Monolayers Supported on Low-Cost Transition Metal Carbides. J. Am. Chem. Soc. 2012, 134, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Wannakao, S.; Artrith, N.; Limtrakul, J.; Kolpak, A.M. Catalytic Activity and Product Selectivity Trends for Carbon Dioxide Electroreduction on Transition Metal-Coated Tungsten Carbides. J. Phys. Chem. C 2017, 121, 20306–20314. [Google Scholar] [CrossRef]
- Chhina, H.; Campbell, S.; Kesler, O. Thermal and electrochemical stability of tungsten carbide catalyst supports. J. Power Sources 2007, 164, 431–440. [Google Scholar] [CrossRef]
- Liu, Y.; Kelly, T.G.; Chen, J.G.; Mustain, W.E. Metal carbides as alternative electrocatalyst supports. ACS Catal. 2013, 3, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, M.; Huang, Z.; Li, Y.; Qi, S.; Yi, C.; Yang, B. Ni–WC/C nanocluster catalysts for urea electrooxidation. J. Power Sources 2014, 264, 282–289. [Google Scholar] [CrossRef]
- Antolini, E. Graphene as a new carbon support for low-temperature fuel cell catalysts. Appl. Catal. B Environ. 2012, 123, 52–68. [Google Scholar] [CrossRef]
- Ross, P.N., Jr.; Stonehart, P. The relation of surface structure to the electrocatalytic activity of tungsten carbide. J. Catal. 1977, 48, 42–59. [Google Scholar] [CrossRef]
- Ooka, H.; Figueiredo, M.C.; Koper, M.T. Competition between hydrogen evolution and carbon dioxide reduction on copper electrodes in mildly acidic media. Langmuir 2017, 33, 9307–9313. [Google Scholar] [CrossRef] [PubMed]
- MedeA, M. Exploration and Design Analysis; Materials Design Inc.: New York, NY, USA, 2016. [Google Scholar]
- Kresse, G.; Furthmüller, J. Software VASP, vienna. Rev. B 1996, 54, 169. [Google Scholar]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, M.J.; Ungureanu, C.G.; Tereshchuk, P.; Batista, K.E.; Chaves, A.S.; Guedes-Sobrinho, D.; Da Silva, J.L. Theoretical Study of the Structural, Energetic, and Electronic Properties of 55-Atom Metal Nanoclusters: A DFT Investigation within van der Waals Corrections, Spin–Orbit Coupling, and PBE+U of 42 Metal Systems. J. Phys. Chem. C 2016, 120, 28844–28856. [Google Scholar] [CrossRef]
- Bucko, T.; Hafner, J.; Lebegue, S.; Angyán, J.G. Improved Description of the Structure of Molecular and Layered Crystals: Ab Initio DFT Calculations with van der Waals Corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef]
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef]
- Watson, G.W.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. π adsorption of ethene on to the {111} surface of copper: A periodic ab initio study of the effect of k-point sampling on the energy, atomic and electronic structure. Surf. Sci. 2000, 459, 93–103. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.R.K.J.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Yoo, J.S.; Christensen, R.; Vegge, T.; Nørskov, J.K.; Studt, F. Theoretical Insight into the Trends that Guide the Electrochemical Reduction of Carbon Dioxide to Formic Acid. ChemSusChem 2016, 9, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef] [PubMed]
- Hirunsit, P. Electroreduction of Carbon Dioxide to Methane on Copper, Copper–Silver, and Copper–Gold Catalysts: A DFT Study. J. Phys. Chem. C 2013, 117, 8262–8268. [Google Scholar] [CrossRef]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chem. Rev. 1999, 99, 2161–2600. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | U (V vs. RHE) |
|---|---|
| 2(H+ + e−) → H2 | 0 |
| CO2 + 2(H+ + e−) → CO + H2O | −0.10 |
| CO2 + 2(H+ + e−) → HCOOH | −0.20 |
| CO2 + 6(H+ + e−) → CH3OH + H2O | −0.03 |
| CO2 + 8(H+ + e−) → CH4 + H2O | 0.17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ananthaneni, S.; Smith, Z.; Rankin, R.B. Graphene Supported Tungsten Carbide as Catalyst for Electrochemical Reduction of CO2. Catalysts 2019, 9, 604. https://doi.org/10.3390/catal9070604
Ananthaneni S, Smith Z, Rankin RB. Graphene Supported Tungsten Carbide as Catalyst for Electrochemical Reduction of CO2. Catalysts. 2019; 9(7):604. https://doi.org/10.3390/catal9070604
Chicago/Turabian StyleAnanthaneni, Sahithi, Zachery Smith, and Rees B. Rankin. 2019. "Graphene Supported Tungsten Carbide as Catalyst for Electrochemical Reduction of CO2" Catalysts 9, no. 7: 604. https://doi.org/10.3390/catal9070604
APA StyleAnanthaneni, S., Smith, Z., & Rankin, R. B. (2019). Graphene Supported Tungsten Carbide as Catalyst for Electrochemical Reduction of CO2. Catalysts, 9(7), 604. https://doi.org/10.3390/catal9070604