
Organocatalysis: Fundamentals and Comparisons to Metal and Enzyme Catalysis


Abstract
:1. Introduction
2. Acyl Group Transfers
2.1. Esterification and Ester Hydrolysis
2.2. Acid or Base-Catalyzed Acyl Transfers
2.3. Amphoteric Compounds Are Good Catalysts for Acyl Transfers
2.4. Catalysis by Nucleofugal Group Substitution
2.5. N-Heterocyclic Carbene-Catalyzed Transesterifications
2.6. Enzyme-Catalyzed Acyl Transfers
2.7. Mimics of Carbopeptidase A
2.8. Direct Amide Bond Formation from Amines and Carboxylic Acids
3. Catalysis of Nucleophilic Additions
3.1. Catalysis of Nucleophilic Additions to Aldehydes, Ketones and Imines
3.2. Bifunctional Catalysts for Nucleophilic Addition/Elimination
3.3. σ- and π-Nucleophiles as Catalysts for Nucleophilic Additions to Aldehydes and Ketones
3.4. Catalysis by Self-Assembled Encapsulation
3.5. Catalysis of 1,4- (Conjugate) Additions
4. Anionic Nucleophilic Displacement Reactions
4.1. Displacement Reactions in the Gas Phase
4.2. Pulling on the Leaving Group
4.3. Phase Transfer Catalysis
4.4. Asymmetric Ion-Pairing Catalysis
5. Catalytic Umpolung C–C Bond Forming Reactions
5.1. Benzoin Condensation: Umpolung of Aldehydes
5.2. The Stetter Reaction: Umpolung of Aldehyde
5.3. Umpolung of Enals
5.4. Umpolung of Michael Acceptors
5.5. The Rauhut–Currier Reaction
5.6. The Morita–Baylis–Hillman Reaction
5.7. Nucleophilic Catalysis of Cycloadditions
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cp | cyclopentadienyl |
| DCC | N,N'-dicyclohexycarbodiimide |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DMF | dimethylformamide |
| DHP | 3,4-dihydro-2H-pyrimido[2,1-b]benzothiazole |
| DMAO | 4-dimethylaminopyridine |
| MBH | Morita–Baylis–Hillman reaction |
| MIBA | 4-methoxy-2-iodobenzeneboronic acid |
| NHC | N-heterocyclic carbene |
| THF | tetrahydrofuran |
| OTf | triflates |
| OTs | p-toluenesulfonate |
| PTC | phase transfer catalyst |
| PYP | 4-pyrrolidinopyridine |
| pyr | pyridine |
| SET | single electron transfer |
| TBD | triazabicyclo[4.4.0]dec-5-ene |
| THTP | 2,3,6,7-tetrahydro-5H-thiazolo[3,2-a]pyrimidine |
| TON | turnover number |
| TOF | turnover frequency |
References
- Haber, J. Catalysis—Where Science and Industry Meet. Pure Appl. Chem. 1994, 66, 1597–1620. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis: A Practical Approach, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–507. [Google Scholar]
- Llyod, L. Handbook of Industrial Catalysts. In Fundamental & Applied Catalysis; Springer: New York, NY, USA, 2011; pp. 1–490. [Google Scholar]
- Dalko, P.I.; Moisan, L. In the golden age of organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, S.; Jørgensen, K.A. Organocatalysis-after the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- List, B. Emil Knoevenagel and the Roots of Aminocatalysis. Angew. Chem. Int. Ed. 2010, 49, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, L.; Jiang, H.; Jørgensen, K.A. A Simple Recipe for Sophisticated Cocktails: Organocatalytic One-Pot Reactions-Concept, Nomenclature, and Future Perspectives. Angew. Chem. Int. Ed. 2011, 50, 8492–8509. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 56, 2133–2140. [Google Scholar] [CrossRef]
- Berzelius, J. Jahres-Bericht über Fortschritte der Physischen Wissenschaften; Laupp: Tübingen, Germany, 1836; Volume 15. [Google Scholar]
- Robertson, A.J.B. The Early History of Catalysis. Platinum Met. Rev. 1975, 19, 64–69. [Google Scholar]
- Roberts, M. M. Birth of the catalytic concept (1800–1900). Catal. Lett. 2000, 67, 1–4. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels–Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Garrett, B.C. Ions at the air/water interface. Science 2004, 303, 1146–1147. [Google Scholar] [CrossRef] [PubMed]
- Tait, M.J.; Franks, F. Water in Biological Systems. Nature 1971, 230, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Chabinyc, M.L.; Craig, S.L.; Regan, C.K.; Brauman, J.I. Gas-phase ionic reactions: Dynamics and mechanism of nucleophilic displacements. Science 1998, 279, 1882–1886. [Google Scholar] [CrossRef] [PubMed]
- Aloisio, S.; Francisco, J.S. Radical-water complexes in Earth’s atmosphere. Acc. Chem. Res. 2000, 33, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.W.M.; Ravishankara, A.R. Role of hydrogen-bonded intermediates in the bimolecular reactions of the hydroxyl radical. J. Phys. Chem. A 2002, 106, 4798–4807. [Google Scholar] [CrossRef]
- Hansen, J.C.; Francisco, J.S. Radical-molecule complexes: Changing our perspective on the molecular mechanisms of radical-molecule reactions and their impact on atmospheric chemistry. ChemPhysChem 2002, 3, 833–840. [Google Scholar] [CrossRef]
- Voehringer-Martinez, E.; Hansmann, B.; Hernandez-Soto, H.; Francisco, J.S.; Troe, J.; Abel, B. Water catalysis of a radical-molecule gas-phase reaction. Science 2007, 315, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Otera, J. Esterification: Methods, Reactions and Applications; John Wiley & Sons: Chichester, UK, 2003; p. 450. [Google Scholar]
- Rehberg, C.E.; Fisher, C.H. Preparation and properties of the n-alkyl acrylates. J. Am. Chem. Soc. 1944, 66, 1203–1207. [Google Scholar] [CrossRef]
- Rehberg, C.E. N-Butyl-acrylate. Org. Synth. 1955, 3, 146–147. [Google Scholar]
- Dewolfe, R.H. Synthesis of Carboxylic and Carbonic Ortho Esters. Synthesis 1974, 153–172. [Google Scholar] [CrossRef]
- Rothman, E.S.; Silbert, L.S.; Pfeffer, P.E.; Hecht, S.S. Enol esters 15. Synthesis of highly hindered esters via isopropenyl ester intermediates. J. Org. Chem. 1972, 37, 3551–3552. [Google Scholar] [CrossRef]
- Taft, R.W., Jr.; Newman, M.S.; Verhoek, F.H. The Kinetics of the Base-Catalyzed Methanolysis of Ortho-Substituted, Meta-Substituted and Para-Substituted l-Menthyl Benzoates. J. Am. Chem. Soc. 1950, 72, 4511–4519. [Google Scholar] [CrossRef]
- Billman, J.H.; Smith, W.T.; Rendall, J.L. Anticonvulsants 5. Esters of ɣ-Diethylamino-α-Phenylbutyric Acid. J. Am. Chem. Soc. 1947, 69, 2058–2059. [Google Scholar] [CrossRef] [PubMed]
- Reimer, M.; Downes, H.R. Preparation of esters by direct replacement of alkoxyl groups. J. Am. Chem. Soc. 1921, 43, 945–951. [Google Scholar] [CrossRef]
- Rossi, R.A.; De Rossi, R.H. Preparation of Benzoate Esters of Tertiary Alcohols by Transesterification. J. Org. Chem. 1974, 39, 855–856. [Google Scholar] [CrossRef]
- Frank, R.L.; Davis, H.R.; Drake, S.S.; McPherson, J.B. Ester interchange by means of the Grignard complex. J. Am. Chem. Soc. 1944, 66, 1509–1510. [Google Scholar] [CrossRef]
- Taber, D.F.; Amedio, J.C.; Patel, Y.K. Preparation of Beta-Keto-Esters by 4-DMAP-Catalyzed Ester Exchange. J. Org. Chem. 1985, 50, 3618–3619. [Google Scholar] [CrossRef]
- Mottet, C.; Hamelin, O.; Garavel, G.; Depres, J.P.; Greene, A.E. A simple and efficient preparation of propargylic β-keto esters through transesterification. J. Org. Chem. 1999, 64, 1380–1382. [Google Scholar] [CrossRef]
- Seebach, D.; Thaler, A.; Blaser, D.; Ko, S.Y. Transesterifications with 1,8-Diazabicyclo[5.4.0]Undec-7-Ene/Lithium Bromide (DBU/LiBr)—Also Applicable to Cleavage of Peptides from Resins in Merrifield Syntheses. Helv. Chim. Acta 1991, 74, 1102–1118. [Google Scholar] [CrossRef]
- Yazawa, H.; Tanaka, K.; Kariyone, K. Reaction of Carboxylic Esters with Boron Tribromide—Convenient Method for Synthesis of Amides and Transesterification. Tetrahedron Lett. 1974, 3995–3996. [Google Scholar] [CrossRef]
- Blossey, E.C.; Turner, L.M.; Neckers, D.C. Polymer Protected Reagents 2. Esterifications with P-AlCl3. Tetrahedron Lett. 1973, 1823–1826. [Google Scholar] [CrossRef]
- Otera, J.; Yano, T.; Kawabata, A.; Nozaki, H. Novel Distannoxane-Catalyzed Transesterification and A New Entry to α,β-Unsaturated Carboxylic-Acids. Tetrahedron Lett. 1986, 27, 2383–2386. [Google Scholar] [CrossRef]
- Otera, J.; Dan-oh, N.; Nozaki, H. Unique Template Effects of Distannoxane Catalysts in Transesterification of Diol Esters. Tetrahedron 1993, 49, 3065–3074. [Google Scholar] [CrossRef]
- Pereyre, M.; Colin, G.; Delvigne, J.P. Alkoxytrialkyltin compounds as transesterification reaction catalysts. Bull. Soc. Chim. Fr. 1969, 262–263. [Google Scholar]
- Otera, J.; Dan-oh, N.; Nozaki, H. Novel Template Effects of Distannoxane Catalysts in Highly Efficient Transesterification and Esterification. J. Org. Chem. 1991, 56, 5307–5311. [Google Scholar] [CrossRef]
- Xiang, J.N.; Toyoshima, S.; Orita, A.; Otera, J. A practical and green chemical process: Fluoroalkyldistannoxane-catalyzed biphasic transesterification. Angew. Chem. Int. Ed. 2001, 40, 3670–3672. [Google Scholar] [CrossRef]
- Kunz, H.; Waldmann, H. 1,3-Dithian-2-yl Methyl-Ester as 2-Step Protecting Group for the Carboxy Function in Peptide-Synthesis. Angew. Chem. Int. Ed. Engl. 1983, 22, 62. [Google Scholar] [CrossRef]
- Waldmann, H.; Kunz, H. 1,3-Dithian-2-Ylmethyl Esters as 2-Step Carboxy-Protecting Groups in the Synthesis of N-Glycopeptides. J. Org. Chem. 1988, 53, 4172–4175. [Google Scholar] [CrossRef]
- Seebach, D.; Hungerbühler, E.; Näf, R.; Schnurrenberger, P.; Weidmann, B.; Zuger, M. Titanate-Mediated Transesterifications with Functionalized Substrates. Synthesis 1982, 138–141. [Google Scholar] [CrossRef]
- Imwinkelried, R.; Schiess, M.; Seebach, D. Diisopropyl (2S,3S)-2,3-O-isopropylidenetartrate [1,3-dioxolane-4,5-dicarboxylic acid, 2,2-dimethyl-, bis(1-methylethyl)ester, (4R-trans)-]. Org. Synth. 1987, 65, 230–235. [Google Scholar]
- Ishihara, K.; Ohara, S.; Yamamoto, H. Direct condensation of carboxylic acids with alcohols catalyzed by hafnium(IV) salts. Science 2000, 290, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Kuo, J.H.; Ku, C.H.; Weng, S.S.; Liu, C.Y. Nucleophilic acyl substitutions of esters with protic nucleophiles mediated by amphoteric, oxotitanium, and vanadyl species. J. Org. Chem. 2005, 70, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Yamamoto, T.; Yamamoto, A. Trans-Esterification Reaction Catalyzed by Alkoxo(triphenylphosphine)copper(I) Complexes. Bull. Chem. Soc. Jpn. 1979, 52, 146–150. [Google Scholar] [CrossRef]
- Kim, Y.J.; Osakada, K.; Takenaka, A.; Yamamoto, A. Alkylnickel and Alklpalladium Alkoxides Associated with Alcohols through Hydrogen-Bonding. J. Am. Chem. Soc. 1990, 112, 1096–1104. [Google Scholar] [CrossRef]
- Grasa, G.A.; Kissling, R.M.; Nolan, S.P. N-heterocyclic carbenes as versatile nucleophilic catalysts for transesterification/acylation reactions. Org. Lett. 2002, 4, 3583–3586. [Google Scholar] [CrossRef] [PubMed]
- Nyce, G.W.; Lamboy, J.A.; Connor, E.F.; Waymouth, R.M.; Hedrick, J.L. Expanding the catalytic activity of nucleophilic N-heterocyclic carbenes for transesterification reactions. Org. Lett. 2002, 4, 3587–3590. [Google Scholar] [CrossRef] [PubMed]
- Grasa, G.A.; Gueveli, T.; Singh, R.; Nolan, S.P. Efficient transesterification/acylation reactions mediated by N-heterocyclic carbene catalysts. J. Org. Chem. 2003, 68, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kissling, R.M.; Letellier, M.A.; Nolan, S.P. Transesterification/acylation of secondary alcohols mediated by N-heterocyclic carbene catalysts. J. Org. Chem. 2004, 69, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, C.K.; Li, H.G.; Sohn, C.K.; Kim, C.K.; Lee, H.W.; Lee, B.S. Acyl-transfer mechanisms involving various acyl functional groups: > X-Y with X = C, S, P and Y = O, S. J. Am. Chem. Soc. 2000, 122, 11162–11172. [Google Scholar] [CrossRef]
- Ingold, C.K. Structure and Mechanism in Organic Chemistry; Cornell Univ. Press: Ithaca NY, USA, 1969; p. 1266. [Google Scholar]
- Rao, G.V.; Balakrishnan, M.; Venkatasubramanian, N. E1Cb Pathway of Acyl Transfer. J. Sci. Ind. Res. 1978, 37, 547–557. [Google Scholar]
- Otton, J.; Ratton, S. Investigation of the Formation of Poly(Ethylene-Terephthalate) with Model Molecules—Kinetics and Mechanism of the Catalytic Esterification and Alcoholysis Reactions 1. Carboxylic-Acid Catalysis (Monofunctional Reactants). J. Polym. Sci. Part A Polym. Chem. 1988, 26, 2183–2197. [Google Scholar] [CrossRef]
- Pratt, R.C.; Lohmeijer, B.G.G.; Long, D.A.; Waymouth, R.M.; Hedrick, J.L. Triazabicyclodecene: A simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2006, 128, 4556–4557. [Google Scholar] [CrossRef] [PubMed]
- Chuma, A.; Horn, H.W.; Swope, W.C.; Pratt, R.C.; Zhang, L.; Lohmeijer, B.G.G.; Wade, C.G.; Waymouth, R.M.; Hedrick, J.L.; Rice, J.E. The Reaction Mechanism for the Organocatalytic Ring-Opening Polymerization of L-Lactide Using a Guanidine-Based Catalyst: Hydrogen-Bonded or Covalently Bound? J. Am. Chem. Soc. 2008, 130, 6749–6754. [Google Scholar] [CrossRef] [PubMed]
- Simón, L.; Goodman, J.M. The Mechanism of TBD-Catalyzed Ring-Opening Polymerization of Cyclic Esters. J. Org. Chem. 2007, 72, 9656–9662. [Google Scholar] [CrossRef] [PubMed]
- Yatsimirsky, A.K. Metal ion catalysis in acyl and phosphoryl transfer: Transition states as ligands. Coord. Chem. Rev. 2005, 249, 1997–2011. [Google Scholar] [CrossRef]
- Parac-Vogt, T.N.; Deleersnyder, K.; Binnemans, K. Lanthanide(III) tosylates as new acylation catalysts. Eur. J. Org. Chem. 2005, 1810–1815. [Google Scholar] [CrossRef]
- Nakayama, M.; Sato, A.; Ishihara, K.; Yamamoto, H. Water-tolerant and reusable catalysts for direct ester condensation between equimolar amounts of carboxylic acids and alcohols. Adv. Synth. Catal. 2004, 346, 1275–1279. [Google Scholar] [CrossRef]
- Sato, A.; Nakamura, Y.; Maki, T.; Ishihara, K.; Yamamoto, H. Zr(IV)-Fe(III), -Ga(III), and -Sn(IV) binary metal complexes as synergistic and reusable esterification catalysts. Adv. Synth. Catal. 2005, 347, 1337–1340. [Google Scholar] [CrossRef]
- Bayryamov, S.G.; Rangelov, M.A.; Mladjova, A.P.; Yomtova, V.; Petkov, D.D. Unambiguous evidence for efficient chemical catalysis of adenosine ester aminolysis by its 2′/3′-OH. J. Am. Chem. Soc. 2007, 129, 5790–5791. [Google Scholar] [CrossRef] [PubMed]
- Hoydonckx, H.E.; De Vos, D.E.; Chavan, S.A.; Jacobs, P.A. Esterification and transesterification of renewable chemicals. Top. Catal. 2004, 27, 83–96. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoo, N.; Jafari, A.A. Facile and selective preparation of esters from carboxylic acids catalyzed by aluminum dodecatungstophosphate (AlPW12O40) as a versatile, recyclable and a highly water tolerant green Lewis acid catalyst. Lett. Org. Chem. 2006, 3, 25–28. [Google Scholar] [CrossRef]
- Bose, D.S.; Satyender, A.; Das, A.P.R.; Mereyala, H.B. A facile, catalytic and environmentally benign method for esterification of carboxylic acids and transesterification of carboxylic esters with nearly equimolar amounts of alcohols. Synthesis 2006, 2392–2396. [Google Scholar] [CrossRef]
- Kawabata, T.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Highly efficient esterification of carboxylic acids with alcohols by montmorillonite-enwrapped titanium as a heterogeneous acid catalyst. Tetrahedron Lett. 2003, 44, 9205–9208. [Google Scholar] [CrossRef]
- Bossaert, W.D.; De Vos, D.E.; Van Rhijn, W.M.; Bullen, J.; Grobet, P.J.; Jacobs, P.A. Mesoporous sulfonic acids as selective heterogeneous catalysts for the synthesis of monoglycerides. J. Catal. 1999, 182, 156–164. [Google Scholar] [CrossRef]
- Bertin, J.; Kagan, H.B.; Luche, J.L.; Setton, R. Graphite Electrolytic Lamellar Reagents in Organic-Chemistry-Esterifications in Presence of Graphite Bisulfate. J. Am. Chem. Soc. 1974, 96, 8113–8115. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical + Electrochemical Electron-Transfer Theory. Ann. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron-Transfer Reactions in Chemistry—Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Albery, W.J. The Application of the Marcus Relation to Reactions in Solution. Ann. Rev. Phys. Chem. 1980, 31, 227–263. [Google Scholar] [CrossRef]
- Lin, M.H.; Rajanbabu, T.V. Metal-catalyzed acyl transfer reactions of enol esters: Role of Y5(OiPr13O and (thd)2Y(OiPr) as transesterification catalysts. Org. Lett. 2000, 2, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Litvinenko, L.M.; Kirichenko, A.I. Basicity and Stereospecificity in Nucleophile Catalysis by Tertiary Amines. Dokl. Akad. Nauk SSSR 1967, 176, 97–100. [Google Scholar]
- Steglich, W.; Höfle, G. N,N-Dimethyl-4-Pyridinamine, A Very Effective Acylation Catalyst. Angew. Chem. Int. Ed. Engl. 1969, 8, 981. [Google Scholar] [CrossRef]
- Haslam, E. Recent Developments in Methods for the Esterification and Protection of the Carboxyl Group. Tetrahedron 1980, 36, 2409–2433. [Google Scholar] [CrossRef]
- Hoefle, G.; Steglich, W.; Vorbrüggen, H. 4-Dialkylaminopyridines as Acylation Catalysts 4. 4-Dialkylaminopyridines as Highly Active Acylation Catalysts. Angew. Chem. Int. Ed. Engl. 1978, 17, 569–583. [Google Scholar] [CrossRef]
- Hassner, A.; Alexanian, V. Synthetic Methods 12. Direct Room-Temperature Esterification of Carboxylic-Acids. Tetrahedron Lett. 1978, 4475–4478. [Google Scholar] [CrossRef]
- Basel, Y.; Hassner, A. Di-tert-butyl dicarbonate and 4-(dimethylamino)pyridine revisited. Their reactions with amines and alcohols. J. Org. Chem. 2000, 65, 6368–6380. [Google Scholar] [CrossRef] [PubMed]
- Prashad, M.; Hu, B.; Har, D.; Repic, O.; Blacklock, T.J. A new, convenient and selective 4-dimethylaminopyridine-catalyzed trifluoroacetylation of anilines with ethyl trifluoroacetate. Tetrahedron Lett. 2000, 41, 9957–9961. [Google Scholar] [CrossRef]
- Singh, S.; Das, G.; Singh, O.V.; Han, H. Development of more potent 4-dimethylaminopyridine analogues. Org. Lett. 2007, 9, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Held, I.; Xu, S.J.; Zipse, H. Modular design of pyridine-based acyl-transfer catalysts. Synthesis 2007, 1185–1196. [Google Scholar]
- Baidya, M.; Kobayashi, S.; Brotzel, F.; Schmidhammer, U.; Riedle, E.; Mayr, H. DABCO and DMAP—Why are they different in organocatalysis? Angew. Chem. Int. Ed. 2007, 46, 6176–6179. [Google Scholar] [CrossRef] [PubMed]
- Vedejs, E.; Chen, X.H. Kinetic resolution of secondary alcohols. Enantioselective acylation mediated by a chiral (dimethylamino)pyridine derivative. J. Am. Chem. Soc. 1996, 118, 1809–1810. [Google Scholar] [CrossRef]
- Ruble, J.C.; Fu, G.C. Chiral π-complexes of heterocycles with transition metals: A versatile new family of nucleophilic catalysts. J. Org. Chem. 1996, 61, 7230–7231. [Google Scholar] [CrossRef] [PubMed]
- Wurz, R.P.; Lee, E.C.; Ruble, J.C.; Fu, G.C. Synthesis and resolution of planar-chiral derivatives of 4-(Dimethylamino)pyridine. Adv. Synth. Catal. 2007, 349, 2345–2352. [Google Scholar] [CrossRef]
- Wurz, R.P. Chiral dialkylaminopyridine catalysts in asymmetric synthesis. Chem. Rev. 2007, 107, 5570–5595. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Schreiner, P.R. Organocatalytic Enantioselective Acyl Transfer onto Racemic as well as meso Alcohols, Amines, and Thiols. Angew. Chem. Int. Ed. 2011, 50, 6012–6042. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Schedel, H. A catalytic one-step process for the chemo- and regioselective acylation of Monosaccharides. J. Am. Chem. Soc. 2007, 129, 12890–12895. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Satoh, K.; Sakurai, K.; Takano, S. Efficient Ester Exchange-Reaction of Phosphonoacetates. Tetrahedron Lett. 1987, 28, 2713–2716. [Google Scholar] [CrossRef]
- Hoegberg, T.; Strom, P.; Ebner, M.; Raemsby, S. Cyanide as an Efficient and Mild Catalyst in the Aminolysis of Esters. J. Org. Chem. 1987, 52, 2033–2036. [Google Scholar] [CrossRef]
- Vedejs, E.; Diver, S.T. Tributylphosphine—A Remarkable Acylation Catalyst. J. Am. Chem. Soc. 1993, 115, 3358–3359. [Google Scholar] [CrossRef]
- Vedejs, E.; Bennett, N.S.; Conn, L.M.; Diver, S.T.; Gingras, M.; Lin, S.; Oliver, P.A.; Peterson, M.J. Tributylphosphine-Catalyzed Acylations of Alcohols—Scope and Related Reactions. J. Org. Chem. 1993, 58, 7286–7288. [Google Scholar] [CrossRef]
- Vedejs, E.; Daugulis, O.; Mackay, J.A.; Rozners, E. Enantioselective acyl transfer using chiral phosphine catalysts. Synlett 2001, 1499–1505. [Google Scholar] [CrossRef]
- Mackay, J.A.; Vedejs, E. Synthesis and reactivity of new chiral bicyclic phospholanes as acyl-transfer catalysts. J. Org. Chem. 2006, 71, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Methot, J.L.; Roush, W.R. Nucleophilic phosphine organocatalysis. Adv. Synth. Catal 2004, 346, 1035–1050. [Google Scholar] [CrossRef]
- Holz, J.; Gensow, M.; Zayas, O.; Boerner, A. Synthesis of chiral heterocyclic phosphines for application in asymmetric catalysis. Curr. Org. Chem. 2007, 11, 61–106. [Google Scholar] [CrossRef]
- Spivey, A.C.; Arseniyadis, S. Amine, Alcohol and Phosphine Catalysts for Acyl Transfer Reactions. Top. Curr. Chem. 2009, 291, 233–280. [Google Scholar]
- De Kanta, C.; Klauber, E.G.; Seidel, D. Merging Nucleophilic and Hydrogen Bonding Catalysis: An Anion Binding Approach to the Kinetic Resolution of Amines. J. Am. Chem. Soc. 2009, 131, 17060–17061. [Google Scholar]
- Klauber, E.G.; de Kanta, C.; Shah, T.K.; Seidel, D. Merging Nucleophilic and Hydrogen Bonding Catalysis: An Anion Binding Approach to the Kinetic Resolution of Propargylic Amines. J. Am. Chem. Soc. 2010, 132, 13624–13626. [Google Scholar] [CrossRef] [PubMed]
- Klauber, E.G.; Mittal, N.; Shah, T.K.; Seidel, D. A Dual-Catalysis/Anion-Binding Approach to the Kinetic Resolution of Allylic Amines. Org. Lett. 2011, 13, 2464–2467. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Lippert, K.M.; De, C.K.; Klauber, E.G.; Emge, T.J.; Schreiner, P.R.; Seidel, D. A Dual-Catalysis Anion-Binding Approach to the Kinetic Resolution of Amines: Insights into the Mechanism via a Combined Experimental and Computational Study. J. Am. Chem. Soc. 2015, 137, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.; Reau, R.; Dahan, F.; Bertrand, G. DBU and DBN Are Strong Nucleophiles—X-ray Crystal-Structures of Onio-Substituted and Dionio-Substituted Phosphanes. Angew. Chem. Int. Ed. Engl. 1993, 32, 399–401. [Google Scholar] [CrossRef]
- Birman, V.B.; Li, X.; Han, Z. Nonaromatic amidine derivatives as acylation catalysts. Org. Lett. 2007, 9, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Birman, V.B.; Uffman, E.W.; Hui, J.; Li, X.M.; Kilbane, C.J. 2,3-dihydroimidazo[1,2-a]pyridines: A new class of enantioselective acyl transfer catalysts and their use in kinetic resolution of alcohols. J. Am. Chem. Soc. 2004, 126, 12226–12227. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Bumbu, V.D.; Liu, P.; Li, X.; Jiang, H.; Uffman, E.W.; Guo, L.; Zhang, W.; Jiang, X.; Houk, K.; Birman, V.B. Catalytic, Enantioselective N-Acylation of Lactams and Thiolactams Using Amidine-Based Catalysts. J. Am. Chem. Soc. 2012, 134, 17605–17612. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.E.; Bull, S.D.; Williams, J.M. Amidines, isothioureas, and guanidines as nucleophilic catalysts. Chem. Soc. Rev. 2012, 41, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Igau, A.; Grützmacher, H.; Baceiredo, A.; Bertrand, G. Analogous α,α-Bis-Carbenoid Triply Bonded Species—Synthesis of A Stable λ-3-Phosphinocarbene λ-5-Phosphaacetylene. J. Am. Chem. Soc. 1988, 110, 6463–6466. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Bakhtiar, C.; Smith, E.H. Transfer of Alkoxycarbonyl from Alkyl Imidazolium-2-Carboxylates to Benzyl Alcohol, A Cyclohexanone Enamine and Diethylamine. J. Chem. Soc. Perkin Trans. 1 1994, 3, 239–243. [Google Scholar] [CrossRef]
- Grasa, G.A.; Singh, R.; Nolan, S.P. Transesterification/acylation reactions catalyzed by molecular catalysts. Synthesis 2004, 971–985. [Google Scholar] [CrossRef]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Moeck, A.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Coulembier, O.; Lohmeijer, B.G.G.; Dove, A.P.; Pratt, R.C.; Mespouille, L.; Culkin, D.A.; Benight, S.J.; Dubois, P.; Waymouth, R.M.; Hedrick, J.L. Alcohol adducts of N-heterocyclic carbenes: Latent catalysts for the thermally-controlled living polymerization of cyclic esters. Macromolecules 2006, 39, 5617–5628. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic, carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, D.M.; Michailidis, F.R.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Lee, H.M.; Hu, C.H. Theoretical study on the mechanism of N-heterocyclic carbene catalyzed transesterification reactions. Tetrahedron Lett. 2005, 46, 6265–6270. [Google Scholar] [CrossRef]
- Singh, R.; Nolan, S.P. Synthesis of phosphorus esters by transesterification mediated by N-heterocyclic carbenes (NHCs). Chem. Commun. 2005, 5456–5458. [Google Scholar] [CrossRef] [PubMed]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. Int. Ed. 2009, 48, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yamauchi, K.; Muramatsu, K.; Sato, M. First example of chiral N-heterocyclic carbenes as catalysts for kinetic resolution. Chem. Commun. 2004, 2770–2771. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Muramatsu, K.; Yamauchi, K.; Morie, Y.; Sato, M. Chiral N-heterocyclic carbenes as asymmetric acylation catalysts. Tetrahedron 2006, 62, 302–310. [Google Scholar] [CrossRef]
- Kano, T.; Sasaki, K.; Maruoka, K. Enantioselective acylation of secondary alcohols catalyzed by chiral N-heterocyclic carbenes. Org. Lett. 2005, 7, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Candish, L.; Lupton, D.W. N-Heterocyclic Carbene-Catalyzed (4+2) Cycloaddition/Decarboxylation of Silyl Dienol Ethers with α,β-Unsaturated Acid Fluorides. J. Am. Chem. Soc. 2011, 133, 4694–4697. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Stasch, A.; Paddon-Row, M.N.; Lupton, D.W. Synthetic and Quantum Mechanical Studies into the N-Heterocyclic Carbene Catalyzed (4+2) Cycloaddition. J. Org. Chem. 2012, 77, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Candish, L.; Forsyth, C.M.; Lupton, D.W. N-tert-Butyl Triazolylidenes: Catalysts for the Enantioselective (3+2) Annulation of α,β-Unsaturated Acyl Azoliums. Angew. Chem. Int. Ed. 2013, 52, 9149–9152. [Google Scholar] [CrossRef] [PubMed]
- Candish, L.; Levens, A.; Lupton, D.W. N-Heterocyclic carbene catalysed redox isomerisation of esters to functionalised benzaldehydes. Chem. Sci. 2015, 6, 2366–2370. [Google Scholar] [CrossRef]
- Levens, A.; Zhang, C.; Candish, L.; Forsyth, C.M.; Lupton, D.W. Enantioselective N-Heterocyclic Carbene Catalyzed Diene Regenerative (4+2) Annulation. Org. Lett. 2015, 17, 5332–5335. [Google Scholar] [CrossRef] [PubMed]
- Sigler, P.B.; Blow, D.M.; Matthews, B.W.; Henderso, R. Structure of Crystalline α-Chymotrypsin 2. A Preliminary Report Including A Hypothesis for Activation Mechanism. J. Mol. Biol. 1968, 35, 143–164. [Google Scholar] [CrossRef]
- Kasserra, H.P.; Laidler, K.J. Mechanisms of Action of Trypsin and Chymotrypsin. Can. J. Chem. 1969, 47, 4031–4039. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4523. [Google Scholar] [CrossRef] [PubMed]
- Quiocho, F.A.; Lipsomb, W.N. Carboxypeptidase A: A protein and an enzyme. Adv. Protein Chem. 1971, 25, 1–78. [Google Scholar] [PubMed]
- Christianson, D.W.; Lipscomb, W.N. Carboxypeptidase-A. Acc. Chem. Res. 1989, 22, 62–69. [Google Scholar] [CrossRef]
- Satoh, T.; Imai, T.; Kitajyo, Y.; Kakuchi, T. Novel synthetic method for preparing artificial carbohydrate polymers. Curr. Top. Polym. Res. 2005, 195–231. [Google Scholar]
- Kimura, E.; Shiota, T.; Koike, T.; Shiro, M.; Kodama, M. A Zinc(II) Complex of 1,5,9-Triazacyclododecane ([12]aneN3) as A Model for Carbonic Anhydrase. J. Am. Chem. Soc. 1990, 112, 5805–5811. [Google Scholar] [CrossRef]
- Breslow, R.; Chipman, D. Mixed metal complexes as enzyme models I. Intracomplex nucleophilic catalysis by an oxime anion. J. Am. Chem. Soc. 1965, 87, 4195–4196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.L.; Breslow, R. Ester hydrolysis by a catalytic cyclodextrin dimer enzyme mimic with a metallobipyridyl linking group. J. Am. Chem. Soc. 1997, 119, 1676–1681. [Google Scholar] [CrossRef]
- Yan, J.M.; Breslow, R. An enzyme mimic that hydrolyzes an unactivated ester with catalytic turnover. Tetrahedron Lett. 2000, 41, 2059–2062. [Google Scholar] [CrossRef]
- Luzhkov, V.; Aqvist, J. Free-energy perturbation calculations of binding and transition-state energies: Hydrolysis of phenyl esters by β-cyclodextrin. Chem. Phys. Lett. 1999, 302, 267–272. [Google Scholar] [CrossRef]
- Breslow, R. The hydrophobic effect in reaction mechanism studies and in catalysis by artificial enzymes. J. Phys. Org. Chem. 2006, 19, 813–822. [Google Scholar] [CrossRef]
- Bhosale, S.V.; Bhosale, S.V. β-Cyclodextrin as a catalyst in organic synthesis. Mini-Rev. Org. Chem. 2007, 4, 231–242. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Holowka, E.P.; Sun, V.Z.; Kamei, D.T.; Deming, T.J. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater. 2007, 6, 52–57. [Google Scholar] [CrossRef] [PubMed]
- NIST Chemistry WebBook. NIST Standard Reference Database; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2009.
- Ulijn, R.V.; Moore, B.D.; Janssen, A.E.M.; Halling, P.J. A single aqueous reference equilibrium constant for amide synthesis-hydrolysis. J. Chem. Soc. Perkin Trans. 2 2002, 5, 1024–1028. [Google Scholar] [CrossRef]
- Guranda, D.T.; Ushakov, G.A.; Yolkin, P.G.; Svedas, V.K. Thermodynamics of phenylacetamides synthesis: Linear free energy relationship with the pK of amine. J. Mol. Cat. B: Enzym. 2012, 74, 48–53. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A.; Volatron, F. Solvent-free preparation of amides from acids and primary amines under microwave irradiation. Tetrahedron 2002, 58, 2155–2162. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Bode, J.W. Emerging methods in amide- and peptide-bond formation. Curr. Opin. Drug Discov. Dev. 2006, 9, 765–775. [Google Scholar] [CrossRef]
- Trost, B.M. The Atom Economy—A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. Atom Economy—A Challenge for Organic-Synthesis—Homogeneous Catalysis Leads the Way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.; Pelter, A. Trisdialkylaminoboranes—New Reagents for Synthesis of Enamines and Amides. J. Chem. Soc. 1965, 5142–5144. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Starkov, P.; Sheppard, T.D. Direct Synthesis of Amides from Carboxylic Acids and Amines Using B(OCH2CF3)3. J. Org. Chem. 2013, 78, 4512–4523. [Google Scholar] [CrossRef] [PubMed]
- Karaluka, V.; Lanigan, R.M.; Murray, P.M.; Badland, M.; Sheppard, T.D. B(OCH2CF3)3-mediated direct amidation of pharmaceutically relevant building blocks in cyclopentyl methyl ether. Org. Biomol. Chem. 2015, 13, 10888–10894. [Google Scholar] [CrossRef] [PubMed]
- Pelter, A.; Levitt, T.E.; Nelson, P. Some Amide Forming Reactions Involving Boron Reagents. Tetrahedron 1970, 26, 1539–1544. [Google Scholar] [CrossRef]
- Latta, R.; Springsteen, G.; Wang, B.H. Development and synthesis of an arylboronic acid-based solid-phase amidation catalyst. Synthesis 2001, 1611–1613. [Google Scholar] [CrossRef]
- Ishihara, K.; Ohara, S.; Yamamoto, H. 3,4,5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 1996, 61, 4196–4197. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Davies, B.; Giles, R.L.; Grosjean, C.; Smith, G.E.; Whiting, A. To catalyze or not to catalyze? Insight into direct amide bond formation from amines and carboxylic acids under thermal and catalyzed conditions. Adv. Synth. Catal. 2006, 348, 813–820. [Google Scholar] [CrossRef]
- Marcelli, T. Mechanistic Insights into Direct Amide Bond Formation Catalyzed by Boronic Acids: Halogens as Lewis Bases. Angew. Chem. Int. Ed. 2010, 49, 6840–6843. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, H.Z.; Fu, Y.; Guo, Q.X. Mechanism of arylboronic acid-catalyzed amidation reaction between carboxylic acids and amines. Org. Biomol. Chem. 2013, 11, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Ishihara, K.; Yamamoto, H. N-Alkyl-4-boronopyridinium salts as thermally stable and reusable amide condensation catalysts. Org. Lett. 2005, 7, 5043–5046. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Ishihara, K.; Yamamoto, H. N-Alkyl-4-boronopyridinium halides versus boric acid as catalysts for the esterification of α-hydroxycarboxylic acids. Org. Lett. 2005, 7, 5047–5050. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Ishihara, K.; Yamamoto, H. New boron(III)-catalyzed amide and ester condensation reactions. Tetrahedron 2007, 63, 8645–8657. [Google Scholar] [CrossRef]
- Gernigon, N.; Al-Zoubi, R.M.; Hall, D.G. Direct Amidation of Carboxylic Acids Catalyzed by ortho-Iodo Arylboronic Acids: Catalyst Optimization, Scope, and Preliminary Mechanistic Study Supporting a Peculiar Halogen Acceleration Effect. J. Org. Chem. 2012, 77, 8386–8400. [Google Scholar] [CrossRef] [PubMed]
- Gernigon, N.; Zheng, H.; Hall, D.G. Solid-supported ortho-iodoarylboronic acid catalyst for direct amidation of carboxylic acids. Tetrahedron Lett. 2013, 54, 4475–4478. [Google Scholar] [CrossRef]
- Fatemi, S.; Gernigon, N.; Hall, D.G. A multigram-scale lower E-factor procedure for MIBA-catalyzed direct amidation and its application to the coupling of α and β aminoacids. Green Chem. 2015, 17, 4016–4028. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Thao Nguyen, D.; Mecinovic, J. Zirconium-catalyzed direct amide bond formation between carboxylic esters and amines. Tetrahedron 2015, 71, 5547–5553. [Google Scholar] [CrossRef]
- Ali, M.A.; Siddiki, S.M.A.H.; Onodera, W.; Kon, K.; Shimizu, K.-i. Amidation of Carboxylic Acids with Amines by Nb2O5 as a Reusable Lewis Acid Catalyst. ChemCatChem 2015, 7, 3555–3561. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Rutjes, F.P.; Mecinovic, J. Triphenylphosphine-catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 2014, 50, 5763–5766. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, H.; Takemoto, Y. Carbonyl and Imine Activation. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 1, pp. 751–769. [Google Scholar]
- Corey, E.J.; Grogan, M.J. Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Swain, C.G.; Brown, J.F. Concerted Displacement Reactions 7. The Mechanism of Acid Base Catalysis in Non-Aqueous Solvents. J. Am. Chem. Soc. 1952, 74, 2534–2537. [Google Scholar] [CrossRef]
- Swain, C.G.; Brown, J.F. Concerted Displacement Reactions 8. Polyfunctional Catalysis. J. Am. Chem. Soc. 1952, 74, 2538–2543. [Google Scholar] [CrossRef]
- Beak, P. Energies and Alkylations of Tautomeric Heterocyclic-Compounds—Old Problems New Answers. Acc. Chem. Res. 1977, 10, 186–192. [Google Scholar] [CrossRef]
- Morpurgo, S.; Bossa, M. The epimerisation of 2-tetrahydropyranol catalysed by the tautomeric couples 2-pyridone/2-hydroxypyridine and formamide/formamidic acid as a model for the sugar’s mutarotation: A theoretical study. Phys. Chem. Chem. Phys. 2003, 5, 1181–1189. [Google Scholar] [CrossRef]
- Rony, P.R. Polyfunctional Catalysis I. Activation Parameters for Mutarotation of Tetramethyl-d-Glucose in Benzene. J. Am. Chem. Soc. 1968, 90, 2824–2831. [Google Scholar] [CrossRef]
- Arai, K. Preparation of Poly(4-Vinyl-2-Hydroxypyridine) and Its Catalytic Activity on Mutarotation of 2,3,4,6-Tetramethyl-α-d-Glucose. J. Polym. Sci. Part A: Polym. Chem. 1993, 31, 193–197. [Google Scholar] [CrossRef]
- Engdahl, K.A.; Bivehed, H.; Ahlberg, P.; Saunders, W.H., Jr. Rate-Controlling 2-Proton Transfer Coupled with Heavy-Atom Motion in the 2-Pyridinone-Catalyzed Mutarotation of Tetramethylglucose—Experimental and Calculated Deuterium-Isotope Effects. J. Am. Chem. Soc. 1983, 105, 4767–4774. [Google Scholar] [CrossRef]
- Kergomard, A.; Renard, M. Mutarotation of Tetramethylglucose Catalysed by Carboxylic Acids in Benzene Solution. Tetrahedron 1968, 24, 6643–6651. [Google Scholar] [CrossRef]
- Breslow, R. Bifunctional Acid-Base Catalysis by Imidazole Groups in Enzyme Mimics. J. Mol. Catal. 1994, 91, 161–174. [Google Scholar] [CrossRef]
- Melander, C.; Horne, D.A. Mutarotation of tetramethylglucose catalyzed by ribonucleosides. Tetrahedron Lett. 1997, 38, 713–716. [Google Scholar] [CrossRef]
- Nakamizo, N. Catalysis in Peptide Synthesis with Active Esters 3. Prominent Catalysis of Sodium 2-Pyridinolate Or 4-Pyridinolate in Aminolysis of Esters. Bull. Chem. Soc. Jpn. 1971, 44, 2006. [Google Scholar] [CrossRef]
- Chen, F.X.; Feng, X.M. Synthesis of racemic tertiary cyanohydrins. Synlett 2005, 892–899. [Google Scholar] [CrossRef]
- Kanai, M.; Kato, N.; Ichikawa, E.; Shibasaki, M. Power of cooperativity: Lewis acid-Lewis base bifunctional asymmetric catalysis. Synlett 2005, 1491–1508. [Google Scholar] [CrossRef]
- Song, J.J.; Gallou, F.; Reeves, J.T.; Tan, Z.L.; Yee, N.K.; Senanayake, C.H. Activation of TMSCN by N-heterocyclic carbenes for facile cyanosilylation of carbonyl compounds. J. Org. Chem. 2006, 71, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Brefort, J.L.; Corriu, R.; Guérin, C.; Henner, B. Hypervalent Silicon Hydrides—Evidence of Their Intermediacy in the Reaction of Optically-Active 1-NpPhMeSiH(D) with Hydrides. J. Organomet. Chem. 1989, 370, 9–15. [Google Scholar] [CrossRef]
- Gutsev, G.L. A Theoretical Investigation on the Structure of the Hypervalent Carbon and Silicon Pentahalogenides as Well as Their Singly Charged Anions. Chem. Phys. 1992, 166, 57–68. [Google Scholar] [CrossRef]
- Murthy, V.S.; Miller, J.M. Formation of Pentavalent Silicon Anions, (CH3O)2Si(OEt)3–and CH3OSiH(OEt)3–, in the Gas-Phase. Rapid Commun. Mass Spectrom. 1994, 8, 698–700. [Google Scholar] [CrossRef]
- Wang, X.; Tian, S.K. Neutral π-nucleophile-catalyzed cyanation of aldehydes and ketones. Synlett 2007, 1416–1420. [Google Scholar] [CrossRef]
- Dirksen, A.; Dirksen, S.; Hackeng, T.M.; Dawson, P.E. Nucleophilic catalysis of hydrazone formation and transimination: Implications for dynamic covalent chemistry. J. Am. Chem. Soc. 2006, 128, 15602–15603. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Hackeng, T.M.; Dawson, P.E. Nucleophilic catalysis of oxime ligation. Angew. Chem. Int. Ed. 2006, 45, 7581–7584. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Dawson, P.E. Rapid Oxime and Hydrazone Ligations with Aromatic Aldehydes for Biomolecular Labeling. Bioconjugate Chem. 2008, 19, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Miyamoto, Y.; Inoue, T.; Oguni, N. Enantioselective Trimethylsilylcyanation of Some Aldehydes Catalyzed by Chiral Schiff-Base Titanium Alkoxide Complexes. J. Org. Chem. 1993, 58, 1515–1522. [Google Scholar] [CrossRef]
- Groger, H. Catalytic enantioselective Strecker reactions and analogous syntheses. Chem. Rev. 2003, 103, 2795–2828. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Usanov, D.L.; Young, C. Lewis Acid Catalyzed Asymmetric Cyanohydrin Synthesis. Chem. Rev. 2008, 108, 5146–5226. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, J.; Wascinska, N.; Gajewy, J. Recent Progress in Lewis Base Activation and Control of Stereoselectivity in the Additions of Trimethylsilyl Nucleophiles. Chem. Rev. 2008, 108, 5227–5252. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, X.; Lin, L.; Feng, X. Recent Progress in the Chemically Catalyzed Enantioselective Synthesis of Cyanohydrins. Eur. J. Org. Chem. 2010, 4751–4769. [Google Scholar] [CrossRef]
- Kurono, N.; Ohkuma, T. Catalytic Asymmetric Cyanation Reactions. ACS Catal. 2016, 6, 989–1023. [Google Scholar] [CrossRef]
- Pluth, M.D.; Bergman, R.G.; Raymond, K.N. Acid catalysis in basic solution: A supramolecular host promotes orthoformate hydrolysis. Science 2007, 316, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Pluth, M.D.; Bergman, R.G.; Raymond, K.N. Proton-Mediated Chemistry and Catalysis in a Self-Assembled Supramolecular Host. Acc. Chem. Res. 2009, 42, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B. The Michael Reaction in March’s Adanced Organic Chemistry; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; Chapter 15.24 to 15.27; pp. 943–959. [Google Scholar]
- Csaky, A.G.; de la Herran, G.; Murcia, M.C. Conjugate addition reactions of carbon nucleophiles to electron-deficient dienes. Chem. Soc. Rev. 2010, 39, 4080–4102. [Google Scholar] [CrossRef] [PubMed]
- Helder, R.; Arends, R.; Bolt, W.; Hiemstra, H.; Wynberg, H. Alkaloid Catalyzed Asymmetric Synthesis 3. Addition of Mercaptans to 2-Cyclohexene-1-one—Determination of Enantiomeric Excess Using C-13 NMR. Tetrahedron Lett. 1977, 2181–2182. [Google Scholar] [CrossRef]
- Hiemstra, H.; Wynberg, H. Addition of Aromatic Thiols to Conjugated Cycloalkenones, Catalyzed by Chiral β-Hydroxy Amines—A Mechanistic Study on Homogeneous Catalytic Asymmetric-Synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Grayson, M.N.; Houk, K.N. Cinchona Alkaloid-Catalyzed Asymmetric Conjugate Additions: The Bifunctional Brønsted Acid-Hydrogen Bonding Model. J. Am. Chem. Soc. 2016, 138, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Sibi, M.P.; Manyem, S. Enantioselective conjugate additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Maudit, M.; Basle, O.; Clavier, H.; Crevisy, C.; Denicourt-Nowicki, A. Metal-Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Chapter 4; pp. 189–341. [Google Scholar]
- Vicario, J.L.; Reyes, E.; Carrillo, L.; Uria, U. Organocatalytic Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes in Comprehensive Organic Synthesis; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Chapter 4; pp. 119–188. [Google Scholar]
- Howell, G.P. Asymmetric and Diastereoselective Conjugate Addition Reactions: C–C Bond Formation at Large Scale. Org. Process Res. Dev. 2012, 16, 1258–1272. [Google Scholar] [CrossRef]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, M.; Sasai, H.; Arai, T. Asymmetric catalysis with heterobimetallic compounds. Angew. Chem. Int. Ed. Engl. 1997, 36, 1237–1256. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent Progress in Asymmetric Bifunctional Catalysis Using Multimetallic Systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Takada, H.; Lin, S.; Kumagai, N.; Shibasaki, M. Direct Catalytic Asymmetric Vinylogous Conjugate Addition of Unsaturated Butyrolactones to α,β-Unsaturated Thioamides. Angew. Chem. Int. Ed. 2014, 53, 5327–5331. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, Y.; Okino, T.; Takemoto, Y. Enantioselective Michael addition to α,β-unsaturated imides catalyzed by a bifunctional organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 4032–4035. [Google Scholar] [CrossRef] [PubMed]
- Zu, L.-S.; Xie, H.; Li, H.; Wang, H.; Jiang, W.; Wang, W. Chiral amine thiourea-promoted enantioselective domino Michael-Aldol reactions between 2-mercaptobenzaldehydes and maleimides. Adv. Synth. Catal. 2007, 349, 1882–1886. [Google Scholar] [CrossRef]
- Bohme, D.K.; Mackay, G.I. Bridging the Gap between the Gas-Phase and Solution—Transition in the Kinetics of Nucleophilic Displacement-Reactions. J. Am. Chem. Soc. 1981, 103, 978–979. [Google Scholar] [CrossRef]
- Ryding, M.J.; Debnarova, A.; Fernandez, I.; Uggerud, E. Nucleophilic Substitution in Reactions between Partially Hydrated Superoxide Anions and Alkyl Halides. J. Org. Chem. 2015, 80, 6133–6142. [Google Scholar] [CrossRef] [PubMed]
- Gronert, S. Mass spectrometric studies of organic ion/molecule reactions. Chem. Rev. 2001, 101, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Brauman, J.I.; Olmstead, W.N.; Lieder, C.A. Gas-Phase Nucleophilic Displacement-Reactions. J. Am. Chem. Soc. 1974, 96, 4030–4031. [Google Scholar] [CrossRef]
- Wladkowski, B.D.; Lim, K.F.; Allen, W.D.; Brauman, J.I. The SN2 Identity Exchange-Reaction ClCH2CN + Cl- →Cl– + ClCH2CN—Experiment and Theory. J. Am. Chem. Soc. 1992, 114, 9136–9153. [Google Scholar] [CrossRef]
- Vayner, G.; Houk, K.N.; Jorgensen, W.L.; Brauman, J.I. Steric retardation of SN2 reactions in the gas phase and solution. J. Am. Chem. Soc. 2004, 126, 9054–9058. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.G.; Polanyi, M. Further considerations on the thermodynamics of chemical equilibria and reaction rates. Trans. Faraday Soc. 1936, 32, 1333–1359. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11–24. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Pross, A.; Radom, L. Gas-phase non-identity SN2 reactions of halide anions with methyl halides: A high-level computational study. J. Am. Chem. Soc. 1996, 118, 6273–6284. [Google Scholar] [CrossRef]
- Dodd, J.A.; Brauman, J.I. On the Application of the Marcus Equation to Methyl Transfer (SN2) Reactions. J. Am. Chem. Soc. 1984, 106, 5356–5357. [Google Scholar] [CrossRef]
- Brønsted, J.N. Acid and basic catalysis. Chem. Rev. 1928, 5, 231–238. [Google Scholar] [CrossRef]
- Bell, R.P. The kinetics of proton transfer reactions. Trans. Faraday Soc. 1938, 34, 0229–0236. [Google Scholar] [CrossRef]
- Dimroth, O. Relationships between affinity and reactions speed. Angew. Chem. 1933, 46, 571–576. [Google Scholar] [CrossRef]
- Shaik, S.S.; Pross, A. SN2 Reactivity of CH3X Derivatives—A Valence Bond Approach. J. Am. Chem. Soc. 1982, 104, 2708–2719. [Google Scholar] [CrossRef]
- Pross, A. Relationship Between Rates and Equilibria and the Mechanistic Significance of the Bronsted Parameter—A Qualitative Valence-Bond Approach. J. Org. Chem. 1984, 49, 1811–1818. [Google Scholar] [CrossRef]
- Shaik, S.S.; Schlegel, H.B.; Wolfe, S. Theoretical Aspects of Physical Organic Chemistry. The SN2 Mechanism; Wiley: New York, NY, USA, 1992. [Google Scholar]
- Shaik, S.S.; Ioffe, A.; Reddy, A.C.; Pross, A. Is the Avoided Crossing State a Good Approximation for the Transition-State of A Chemical-Reaction—An Analysis of Menschutkin and Ionic SN2 Reactions. J. Am. Chem. Soc. 1994, 116, 262–273. [Google Scholar] [CrossRef]
- Anglada, J.M.; Besalú, E.; Bofill, J.M.; Crehuet, R. Prediction of approximate transition states by Bell-Evans-Polanyi principle: I. J. Comput. Chem. 1999, 20, 1112–1129. [Google Scholar] [CrossRef]
- Bofill, J.M.; Anglada, J.M.; Besalú, E.; Crehuet, R. Quantum chemical reactivity: Beyond the study of small molecules. In Fundamentals of Molecular Similarity; Carbó-Dorca, R., Gironés, X., Mezey, P.G., Eds.; Springer: New York, NY, USA, 2001; pp. 125–141. [Google Scholar]
- Wester, R.; Bragg, A.E.; Davis, A.V.; Neumark, D.M. Time-resolved study of the symmetric SN2-reaction I–+CH3I. J. Chem. Phys. 2003, 119, 10032–10039. [Google Scholar] [CrossRef]
- Arnaut, L.G.; Formosinho, S.J. The rates of SN2 reactions and their relation to molecular and solvent properties. Chem. Eur. J. 2007, 13, 8018–8028. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.L. Metal ion catalysis of nucleophilic organic reaction in solution. Adv. Chem. Ser. 1963, 37, 19–36. [Google Scholar]
- Pocker, Y.; Kevill, D.N. Electrophilic Catalysis in Nucleophilic Substitution and Elimination 3. Conductances of Soms Silver and Tetraethylammonium Salts in Acetonitrile and Kinetics of Reaction of 2-Octyl Bromide with Tetraethylammonium and Silver Nitrates in That Solvent. J. Am. Chem. Soc. 1965, 87, 4760–4770. [Google Scholar] [CrossRef]
- Pocker, Y.; Wong, W.H. Electrophilic Catalysis in Nucleophilic-Substitution and Elimination 7. Kinetics and Mechanism of Reaction of Neopentyl Iodide with Tetraethylammonium and Silver Nitrates and Perchlorates in Acetonitrile. J. Am. Chem. Soc. 1975, 97, 7097–7104. [Google Scholar] [CrossRef]
- Maia, A.; Landini, D.; Betti, C.; Leska, B.; Schröder, G. Catalytic activity and anion activation in SN2 reactions promoted by complexes of silicon polypodands. Comparison with traditional polyethers. New J. Chem. 2005, 29, 1195–1198. [Google Scholar] [CrossRef]
- Buncel, E.; Nagelkerke, R.; Thatcher, G.R.J. Metal ion catalysis and inhibition in nucleophilic displacement reactions at carbon, phosphorous, and sulfur centers part 10—Alkali metal ion catalysis in nucleophilic displacement by ethoxide ion on p-nitrophenyl phenylphosphonate: Evidence for multiple metal ion catalysis. Can. J. Chem. 2003, 81, 53–63. [Google Scholar]
- Takada, K.; Tanaka, S.; Nagasawa, K. Asymmetric Mannich-type reaction of aromatic α-amido sulfone with malonate using guanidine-thiourea bifunctional organocatalyst. Synlett 2009, 1643–1646. [Google Scholar] [CrossRef]
- Makosza, M.; Serafin, B. Reactions of organic anions I. Catalytic ethylation of phenylacetonitrile in aqueous medium. Rocz. Chem. 1965, 39, 1223–1230. [Google Scholar]
- Makosza, M.; Jonczyk, A. Phase-transfer alkylation of nitriles: α-phenylbutyronitrile. Org. Synth. 1976, 55, 91–95. [Google Scholar]
- Makosza, M.; Fedorynski, M. Thirty years of phase transfer catalysis. Pol. J. Chem. 1996, 70, 1093–1110. [Google Scholar]
- Makosza, M. Phase-transfer catalysis. A general green methodology in organic synthesis. Pure Appl. Chem. 2000, 72, 1399–1403. [Google Scholar] [CrossRef]
- Starks, C.M. Phase-Transfer Catalysis 1. Heterogeneous Reactions Involving Anion Transfer by Quaternary Ammonium and Phosphonium Salts. J. Am. Chem. Soc. 1971, 93, 195–199. [Google Scholar] [CrossRef]
- Starks, C.M.; Liotta, C. Phase Transfer Catalysis: Principles and Techniques; Academic Press: New York, NY, USA, 1978; p. 384. [Google Scholar]
- Herriott, A.W.; Picker, D. Phase Transfer Catalysis—Evaluation of Catalysts. J. Am. Chem. Soc. 1975, 97, 2345–2349. [Google Scholar] [CrossRef]
- Halpern, M. Phase-Transfer catalysis. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2002; pp. 495–500. [Google Scholar]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis: Principles and Techniques, 2nd ed.; Verlag Chemie: Weinheim, Germany, 1983; Volume 11, p. 386. [Google Scholar]
- Makosza, M.; Fedorynski, M. Catalysis in 2-Phase Systems—Phase-Transfer and Related Phenomena. Adv. Catal. 1987, 35, 375–422. [Google Scholar]
- Yadav, G.D. Insight into green phase transfer catalysis. Top. Catal. 2004, 29, 145–161. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shirakawa, S.; Maruoka, K. Phase-Transfer-Catalyzed Asymmetric Conjugate Cyanation of Alkylidenemalonates with KCN in the Presence of a Brønsted Acid Additive. Org. Lett. 2013, 15, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Tokuda, T.; Maruoka, K. Phase-transfer-catalyzed asymmetric desymmetrizations of cyclopentanones. Org. Chem. Front. 2015, 2, 336–339. [Google Scholar] [CrossRef]
- Shirakawa, S.; Koga, K.; Tokuda, T.; Yamamoto, K.; Maruoka, K. Catalytic Asymmetric Synthesis of 3,3′-Diaryloxindoles as Triarylmethanes with a Chiral All-Carbon Quaternary Center: Phase-Transfer-Catalyzed SNAr Reaction. Angew. Chem. Int. Ed. 2014, 53, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.J. Cyclic Polyethers and Their Complexes with Metal Salts. J. Am. Chem. Soc. 1967, 89, 7017–7036. [Google Scholar] [CrossRef]
- Pedersen, C.J. Cyclic Polyethers and Their Complexes with Metal Salts. J. Am. Chem. Soc. 1967, 89, 2495–2496. [Google Scholar] [CrossRef]
- Newcomb, M.; Gokel, G.W.; Cram, D.J. Pyridyl Unit in Host Compounds. J. Am. Chem. Soc. 1974, 96, 6810–6811. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.E.; Xie, S.; Jones, L.A.; Osterhout, M.H.; Henry, C.; Roper, T.D. Large Scale Application of Asymmetric Phase-Tansfer Catalysis for Amino Acid Synthesis. In Asymmetric Catalysis on Industrial Scale: Challenges, Approaches, and Solutions, 2nd ed.; Blaser, H.-U., Federsel, H.-J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 473–484. [Google Scholar]
- Nicolas, C.; Lacour, J. Triazatriangulenium cations: Highly stable carbocations for phase-transfer catalysis. Org. Lett. 2006, 8, 4343–4346. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.J.; Frensdorff, H.K. Macrocyclic Polyethers and Their Complexes. Angew. Chem. Int. Ed. 1972, 11, 16–25. [Google Scholar] [CrossRef] [PubMed]
- DiBiase, S.A.; Gokel, G.W. Crown-Cation Complex Effects. 8. Reactions of Crown Ether Activated tert-Butoxide Ion. J. Org. Chem. 1978, 43, 447–452. [Google Scholar] [CrossRef]
- Gokel, G.W. Crown Ethers and Cryptands; Monographs in Supramolecular Chemistry, The Royal Society of Chemistry: London, UK, 1991; Volume 3, p. 199. [Google Scholar]
- Brachvogel, R.-C.; Maid, H.; von Delius, M. NMR Studies on Li+, Na+ and K+ complexes of orthoesters Cryptand o-Me2–1.1.1. Int. J. Mol. Sci. 2015, 16, 20641–20656. [Google Scholar] [CrossRef] [PubMed]
- Izatt, R.M.; Bradshaw, J.S.; Nielsen, S.A.; Lamb, J.D.; Christensen, J.J.; Sen, D. Thermodynamic and Kinetic Data for Cation Macrocycle Interaction. Chem. Rev. 1985, 85, 271–339. [Google Scholar] [CrossRef]
- Landini, D.; Montanari, F.; Pirisi, F.M. Crown Ethers as Phase-Transfer Catalysts in 2-Phase Reactions. J. Chem. Soc. Chem. Commun. 1974, 879–880. [Google Scholar] [CrossRef]
- Wong, K.H. Kinetics of Esterification of Potassium Para-Nitrobenzoate by Benzyl Bromide Using Dicyclohexyl-18-Crown-6 as Phase-Transfer Agent. J. Chem. Soc. Chem. Commun. 1978, 282–283. [Google Scholar] [CrossRef]
- Cinquini, M.; Montanari, F.; Tundo, P. Alkyl Substituted Aza-Macrobicyclic Polyethers—Highly Efficient Catalysts in 2-Phase Reactions. J. Chem. Soc. Chem. Commun. 1975, 393–394. [Google Scholar] [CrossRef]
- Rapi, Z.; Bako, P.; Keglevich, G.; Szoellosy, A.; Drahos, L.; Botyanszki, A.; Holczbauer, T. Asymmetric phase transfer Darzens reactions catalyzed by d-glucose- and d-mannose-based chiral crown ethers. Tetrahedron: Asymmetry 2012, 23, 489–496. [Google Scholar] [CrossRef]
- Bako, P.; Keglevich, G.; Rapi, Z.; ToKe, L. The Enantiomeric Differentiation Ability of Chiral Crown Ethers Based on Carbohydrates. Curr. Org. Chem. 2012, 16, 297–304. [Google Scholar] [CrossRef]
- Rapi, Z.; Demuth, B.; Keglevich, G.; Grun, A.; Drahos, L.; Soti, P.L.; Bako, P. Enantioselective Michael addition of malonates to aromatic nitroalkenes catalyzed by monosaccharide-based chiral crown ethers. Tetrahedron: Asymmetry 2014, 25, 141–147. [Google Scholar] [CrossRef]
- Bako, P.; Rapi, Z.; Gruen, A.; Nemcsok, T.; Hegedus, L.; Keglevich, G. Asymmetric Michael Addition of Malonates to Enones Catalyzed by an α-d-Glucopyranoside-Based Crown Ether. Synlett 2015, 26, 1847–1851. [Google Scholar] [CrossRef]
- Mann, J. Modern Methods for the Introduction of Fluorine into Organic-Molecules—An Approach to Compounds with Altered Chemical and Biological-Activities. Chem. Soc. Rev. 1987, 16, 381–436. [Google Scholar] [CrossRef]
- Mikami, K.; Itoh, Y.; Yamanaka, M. Fluorinated carbonyl and olefinic compounds: Basic character and asymmetric catalytic reactions. Chem. Rev. 2004, 104, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, M. CF3-bearing aromatic and heterocyclic building blocks. Angew. Chem. Int. Ed. 2006, 45, 5432–5446. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Gronert, S.; Kass, S.R. Theoretical studies of eliminations 6. The regiochemistry and stereochemistry of the gas-phase reactions of 3-halocyclohexenes with fluoride. An ab initio study. J. Org. Chem. 1997, 62, 7991–8000. [Google Scholar] [CrossRef] [PubMed]
- Pliego, J.R.; Riveros, J.M. Theoretical study of the gas-phase reaction of fluoride and chloride ions with methyl formate. J. Phys. Chem. A 2002, 106, 371–378. [Google Scholar] [CrossRef]
- Mugnai, M.; Cardini, G.; Schettino, V. Substitution and elimination reaction of F– with C2H5Cl: An ab Initio molecular dynamics study. J. Phys. Chem. A 2003, 107, 2540–2547. [Google Scholar] [CrossRef]
- Wilkinson, J.A. Recent Advances in the Selective Formation of the C–F Bond. Chem. Rev. 1992, 92, 505–519. [Google Scholar] [CrossRef]
- Murray, C.B.; Sandford, G.; Korn, S.R. Ionic liquids as media for nucleophilic fluorination. J. Fluorine Chem. 2003, 123, 81–84. [Google Scholar] [CrossRef]
- Kim, D.W.; Song, C.E.; Chi, D.Y. New method of fluorination using potassium fluoride in ionic liquid: Significantly enhanced reactivity of fluoride and improved selectivity. J. Am. Chem. Soc. 2002, 124, 10278–10279. [Google Scholar] [CrossRef] [PubMed]
- Liotta, C.L.; Harris, H.P. Chemistry of Naked Anions 1. Reactions of 18-Crown-6 Complex of Potassium Fluoride with Organic Substrates in Aprotic Organic-Solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Cox, D.P.; Terpinski, J.; Lawrynowicz, W. Anhydrous Tetrabutylammonium Fluoride—A Mild But Highly Efficient Source of Nucleophilic Fluoride-Ion. J. Org. Chem. 1984, 49, 3216–3219. [Google Scholar] [CrossRef]
- Sun, H.R.; DiMagno, S.G. Anhydrous tetrabutylammonium fluoride. J. Am. Chem. Soc. 2005, 127, 2050–2051. [Google Scholar] [CrossRef] [PubMed]
- Landini, D.; Maia, A.; Rampoldi, A. Dramatic Effect of the Specific Solvation on the Reactivity of Quaternary Ammonium Fluorides and Poly(Hydrogen Fluorides), (HF)n•F–, in Media of Low Polarity. J. Org. Chem. 1989, 54, 328–332. [Google Scholar] [CrossRef]
- Albanese, D.; Landini, D.; Penso, M. Hydrated tetrabutylammonium fluoride as a powerful nucleophilic fluorinating agent. J. Org. Chem. 1998, 63, 9587–9589. [Google Scholar] [CrossRef]
- Pliego, J.R.; Pilo-Veloso, D. Chemoselective nucleophilic fluorination induced by selective solvation of the SN2 transition state. J. Phys. Chem. B 2007, 111, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Leadbeater, N.E.; Marco, M.; Tominack, B.J. First examples of transition-metal free Sonogashira-type couplings. Org. Lett. 2003, 5, 3919–3922. [Google Scholar] [CrossRef] [PubMed]
- Cinquini, M.; Colonna, S.; Molinari, H.; Montanari, F.; Tundo, P. Heterogeneous Phase-Transfer Catalysts: Onium Salts, Crown Ethers, and Cryptands Immobilized on Polymer Supports. J. Chem. Soc. Chem. Commun. 1976, 394–396. [Google Scholar] [CrossRef]
- He, L.N.; Yasuda, H.; Sakakura, T. New procedure for recycling homogeneous catalyst: propylene carbonate synthesis under supercritical CO2 conditions. Green Chem. 2003, 5, 92–94. [Google Scholar] [CrossRef]
- Shirakawa, S.; Tanaka, Y.; Maruoka, K. Development of a recyclable fluorous chiral phase-transfer catalyst: Application to the catalytic asymmetric synthesis of α-amino acids. Org. Lett. 2004, 6, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Stuart, A.M.; Vidal, J.A. Perfluoroalkylated 4,13-diaza-18-crown-6 ethers: Synthesis, phase-transfer catalysis, and recycling studies. J. Org. Chem. 2007, 72, 3735–3740. [Google Scholar] [CrossRef] [PubMed]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Fletcher, S.; Nelson, A. Towards phase-transfer catalysts with a chiral anion: Inducing asymmetry in the reactions of cations. Tetrahedron: Asymmetry 2003, 14, 1995–2004. [Google Scholar] [CrossRef]
- Hamilton, G.L.; Kanai, T.; Toste, F.D. Chiral Anion-Mediated Asymmetric Ring Opening of meso-Aziridinium and Episulfonium Ions. J. Am. Chem. Soc. 2008, 130, 14984–14986. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Hamilton, G.L.; Toste, F.D. The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. 2012, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Y.M.; Drljevic, A.; Rauniyar, V.; Phipps, R.J.; Toste, F.D. A combination of directing groups and chiral anion phase-transfer catalysis for enantioselective fluorination of alkenes. Proc. Nat. Acad. Sci. USA 2013, 110, 13729–13733. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.M.; Reisberg, S.H.; Shunatona, H.P.; Patel, J.S.; Toste, F.D. Chiral Anion Phase Transfer of Aryldiazonium Cations: An Enantioselective Synthesis of C3-Diazenated Pyrroloindolines. Angew. Chem. Int. Ed. 2014, 53, 5600–5603. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D. Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. Engl. 1979, 18, 239–258. [Google Scholar] [CrossRef]
- Smoliakova, I.P.; Caple, R.; Gregory, D.; Smit, W.A.; Shashkov, A.S.; Chizhov, O.S. Highly Selective Formation of a β-C-Glucosidic Bond in the Reactions of Arscl-Glucal Adducts with Silicon-Containing Nucleophiles. J. Org. Chem. 1995, 60, 1221–1227. [Google Scholar] [CrossRef]
- Han, M.M.; Smoliakova, I.P.; Koikov, L.N. Stereoselective synthesis of C-[2-S-(p-tolyl)-2-thio-β-d-galactopyranosyl] compounds using the reaction of TolSCl adducts of D-galactal with C-nucleophiles. Carbohydr. Res. 2000, 323, 202–207. [Google Scholar] [CrossRef]
- Wöhler, F.; Liebig, J. Untersuchungen über das Radikal der Benzoesäure. Ann. Pharm. 1832, 3, 249–282. [Google Scholar] [CrossRef]
- Lapworth, A. Reactions involving the addition of hydrogen cyanide to carbon compounds. J. Chem. Soc. 1903, 83, 995–1005. [Google Scholar] [CrossRef]
- Wiberg, K.B. The Deuterium Isotope Effect of Some Ionic Reactions of Benzaldehyde. J. Am. Chem. Soc. 1954, 76, 5371–5375. [Google Scholar] [CrossRef]
- Johnson, J.S. Catalyzed reactions of acyl anion equivalents. Angew. Chem. Int. Ed. 2004, 43, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- Ukai, T.; Tanaka, R.; Dokawa, T. A new catalyst for acyloin condensation I. J. Pharm. Soc. Jpn. 1943, 63, 296–300. [Google Scholar]
- Kluger, R. Lessons from thiamin-watching. Pure Appl. Chem. 1997, 69, 1957–1967. [Google Scholar] [CrossRef]
- Breslow, R. On the Mechanism of Thiamine Action 4. Evidence from Studies on Model Systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Breslow, R.; Schmuck, C. The mechanism of thiazolium catalysis. Tetrahedron Lett. 1996, 37, 8241–8242. [Google Scholar] [CrossRef]
- White, M.J.; Leeper, F.J. Kinetics of the thiazolium ion-catalyzed benzoin condensation. J. Org. Chem. 2001, 66, 5124–5131. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Kim, R. The Thiazolium Catalyzed Benzoin Condensation with Mild Base Does Not Involve a Dimer Intermediate. Tetrahedron Lett. 1994, 35, 699–702. [Google Scholar] [CrossRef]
- Teles, J.H.; Melder, J.P.; Ebel, K.; Schneider, R.; Gehrer, E.; Harder, W.; Brode, S.; Enders, D.; Breuer, K.; Raabe, G. The chemistry of stable carbenes 2. Benzoin-type condensations of formaldehyde catalyzed by stable carbenes. Helv. Chim. Acta 1996, 79, 61–83. [Google Scholar] [CrossRef]
- Enders, D.; Balensiefer, T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res. 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Butlerow, A. Bildung einer zuckerartigen Substanz durch Synthese. Liebigs Ann. Chem. 1861, 120, 295–298. [Google Scholar] [CrossRef]
- Shigemasa, Y.; Ueda, T.; Saimoto, H. Formose Reactions 26. 1St Synthesis of DL-2-C-Hydroxymethyl-3-Pentulose in the Formose Reaction. J. Carbohydr. Res. 1989, 8, 669–673. [Google Scholar] [CrossRef]
- Tajima, H.; Niitsu, T.; Inoue, H. Polymerization of Formaldehyde by an Immobilized Thiamine Catalyst on Cation-Exchange Resin. J. Chem. Eng. Jpn. 1999, 32, 776–782. [Google Scholar] [CrossRef]
- López-Calahorra, F.; Castro, E.; Ochoa, A.; Marti, J. Further evidences about the role of bis(thiazolin-2-ylidene)s as the actual catalytic species in the generalised benzoin condensation. Tetrahedron Lett. 1996, 37, 5019–5022. [Google Scholar] [CrossRef]
- Li, G.Q.; Dai, L.X.; You, S.L. Thiazolium-derived N-heterocyclic carbene-catalyzed cross-coupling of aldehydes with unactivated imines. Chem. Commun. 2007, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Lappert, M.F.; Maskell, R.K. A New Class of Benzoin Condensation Catalyst, the Bi-(1,3-Dialkylimidazolidin-2-Ylidenes). J. Chem. Soc. Chem. Commun. 1982, 580–581. [Google Scholar] [CrossRef]
- Iwamoto, K.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Benzoin reaction in water as an aqueous medium catalyzed by benzimidazolium salt. Tetrahedron Lett. 2006, 47, 7175–7177. [Google Scholar] [CrossRef]
- Reich, B.J.E.; Justice, A.K.; Beckstead, B.T.; Reibenspies, J.H.; Miller, S.A. Cyanide-catalyzed cyclizations via aldimine coupling. J. Org. Chem. 2004, 69, 1357–1359. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Kool, E. A ɣ-Cyclodextrin Thiazolium Salt Holoenzyme Mimic for the Benzoin Condensation. Tetrahedron Lett. 1988, 29, 1635–1638. [Google Scholar] [CrossRef]
- Ikeda, H.; Horimoto, Y.; Nakata, M.; Ueno, A. Artificial holoenzymes for benzoin condensation using thiazolio-appended β-cyclodextrin dimers. Tetrahedron Lett. 2000, 41, 6483–6487. [Google Scholar] [CrossRef]
- Xin, L.H.; Bausch, C.C.; Johnson, J.S. Mechanism and scope of the cyanide-catalyzed cross silyl benzoin reaction. J. Am. Chem. Soc. 2005, 127, 1833–1840. [Google Scholar]
- Brook, A.G. Some Molecular-Rearrangements of Organosilicon Compounds. Acc. Chem. Res. 1974, 7, 77–84. [Google Scholar] [CrossRef]
- Moser, W.H. The Brook rearrangement in tandem bond formation strategies. Tetrahedron 2001, 57, 2065–2084. [Google Scholar] [CrossRef]
- Bausch, C.C.; Johnson, J.S. Cyanide-catalyzed additions of acyl phosphonates to aldehydes: A new acyl donor for benzoin-type reactions. Adv. Synth. Catal. 2005, 347, 1207–1211. [Google Scholar] [CrossRef]
- Mattson, A.E.; Bharadwaj, A.R.; Zuhl, A.M.; Scheidt, K.A. Thiazolium-catalyzed additions of acylsilanes: A general strategy for acyl anion addition reactions. J. Org. Chem. 2006, 71, 5715–5724. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.S.; Biju, A.T.; Nair, V. Recent advances in N-heterocyclic carbene (NHC)-catalysed benzoin reactions. Beilstein J. Org. Chem. 2016, 12, 444–461. [Google Scholar] [CrossRef] [PubMed]
- Stetter, H.; Schreckenberg, M. Addition of aldehydes to activated double bonds. Angew. Chem. 1973, 85, 89. [Google Scholar] [CrossRef]
- Stetter, H. Catalyzed Addition of Aldehydes to Activated Double-Bonds—New Synthetic Approach. Angew. Chem. Int. Ed. Engl. 1976, 15, 639–647. [Google Scholar] [CrossRef]
- Stetter, H.; Kuhlmann, H. The catalyzed nucleophilic addition of aldehydes to electrophilic double bonds. Org. React. 1991, 40, 408–496. [Google Scholar]
- Trost, B.M.; Shuey, C.D.; Di Ninno, F. Stereocontrolled Total Synthesis of (±)-Hirsutic Acid-C. J. Am. Chem. Soc. 1979, 101, 1284–1285. [Google Scholar]
- Ciganek, E. Esters of 2,3-Dihydro-3-Oxobenzofuran-2-Acetic Acid and 3,4-Dihydro-4-Oxo-2H-1-Benzopyran-3-Acetic Acid by Intramolecular Stetter Reactions. Synthesis 1995, 1311–1314. [Google Scholar] [CrossRef]
- Zhou, Z.-Z.; Ji, F.-Q.; Cao, M.; Yang, G.-F. An Efficient Intramolecular Stetter Reaction in Room Temperature Ionic Liquids Promoted By Microwave Irradiation. Adv. Synth. Catal. 2006, 348, 1826–1830. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Runsink, J.; Teles, J.H. The first asymmetric intramolecular Stetter reaction. Helv. Chim. Acta 1996, 79, 1899–1902. [Google Scholar] [CrossRef]
- Kerr, M.S.; Rovis, T. Enantioselective synthesis of quaternary stereocenters via a catalytic asymmetric Stetter reaction. J. Am. Chem. Soc. 2004, 126, 8876–8877. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M. New developments in the asymmetric Stetter reaction. Angew. Chem. Int. Ed. 2005, 44, 2632–2634. [Google Scholar] [CrossRef] [PubMed]
- Mattson, A.E.; Bharadwaj, A.R.; Scheidt, K.A. The thiazolium-catalyzed sila-Stetter reaction: Conjugate addition of acylsilanes to unsaturated esters and ketones. J. Am. Chem. Soc. 2004, 126, 2314–2315. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Perreault, S.; Rovis, T. Catalytic Asymmetric Intermolecular Stetter Reaction of Glyoxamides with Alkylidenemalonates. J. Am. Chem. Soc. 2008, 130, 14066–14067. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.A.; Oberg, K.M.; Dalton, D.M.; Rovis, T. Catalytic Asymmetric Intermolecular Stetter Reaction of Heterocyclic Aldehydes with Nitroalkenes: Backbone Fluorination Improves Selectivity. J. Am. Chem. Soc. 2009, 131, 10872–10874. [Google Scholar] [CrossRef] [PubMed]
- Burstein, C.; Glorius, F. Organocatalyzed conjugate umpolung of α,β-unsaturated aldehydes for the synthesis of ɣ-butyrolactones. Angew. Chem. Int. Ed. 2004, 43, 6205–6208. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.S.; Rosen, E.L.; Bode, J.W. N-Heterocyclic carbene-catalyzed generation of homoenolates: ɣ-butyrolactones by direct annulations of enals and aldehydes. J. Am. Chem. Soc. 2004, 126, 14370–14371. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A. N-Heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Burstein, C.; Tschan, S.; Xie, X.; Glorius, F. N-Heterocyclic carbene-catalyzed conjugate umpolung for the synthesis of ɣ-butyrolactones. Synthesis 2006, 2418–2439. [Google Scholar]
- Nair, V.; Vellalath, S.; Poonoth, M.; Suresh, E. N-Heterocyclic carbene-catalyzed reaction of chalcones and enals via homoenolate: An efficient synthesis of 1,3,4-trisubstituted cyclopentenes. J. Am. Chem. Soc. 2006, 128, 8736–8737. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Bode, J.W. Enantioselective, NHC-Catalyzed bicyclo-β-lactam formation via direct annulations of enals and unsaturated N-sulfonyl ketimines. J. Am. Chem. Soc. 2008, 130, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R.; Blaschke, H.; Brunn, E. Zur 1.3-Dipolaren Cycloaddition Aromatischer Nitriloxide an Azodicarbonsaureester. Tetrahedron Lett. 1966, 405–409. [Google Scholar] [CrossRef]
- White, D.A.; Baizer, M.M. Catalysis of Michael Reaction by Tertiary Phosphines. Tetrahedron Lett. 1973, 3597–3600. [Google Scholar] [CrossRef]
- Gomez-Bengoa, E.; Cuerva, J.M.; Mateo, C.; Echavarren, A.M. Michael reaction of stabilized carbon nucleophiles catalyzed by [RuH2(PPh3)4]. J. Am. Chem. Soc. 1996, 118, 8553–8565. [Google Scholar] [CrossRef]
- Stewart, I.C.; Bergman, R.G.; Toste, F.D. Phosphine-catalyzed hydration and hydroalkoxylation of activated olefins: Use of a strong nucleophile to generate a strong base. J. Am. Chem. Soc. 2003, 125, 8696–8697. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Kazmaier, U. Internal Redox Catalyzed by Triphenylphosphine. J. Am. Chem. Soc. 1992, 114, 7933–7935. [Google Scholar] [CrossRef]
- Trost, B.M.; Li, C.J. Novel Umpolung in C–C Bond Formation Catalyzed by Triphenylphosphine. J. Am. Chem. Soc. 1994, 116, 3167–3168. [Google Scholar] [CrossRef]
- Trost, B.M.; Li, C.J. Phosphine-Catalyzed Isomerization-Addition of Oxygen Nucleophiles to 2-Alkynoates. J. Am. Chem. Soc. 1994, 116, 10819–10820. [Google Scholar] [CrossRef]
- Alvarez-Ibarra, C.; Csákÿ, A.G.; de la Oliva, C.G.; Gomez, C. Carboxylates as pronucleophiles in the phosphine-catalyzed ɣ-addition reaction. Tetrahedron Lett. 1999, 40, 8465–8467. [Google Scholar] [CrossRef]
- Trost, B.M.; Dake, G.R. Nitrogen pronucleophiles in the phosphine-catalyzed ɣ-addition reaction. J. Org. Chem. 1997, 62, 5670–5671. [Google Scholar] [CrossRef]
- Fischer, C.; Smith, S.W.; Powell, D.A.; Fu, G.C. Umpolung of Michael acceptors catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc. 2006, 128, 1472–1473. [Google Scholar] [CrossRef] [PubMed]
- Virieux, D.; Guillouzic, A.F.; Cristau, H.J. Phosphines catalyzed nucleophilic addition of azoles to allenes: synthesis of allylazoles and indolizines. Tetrahedron 2006, 62, 3710–3720. [Google Scholar] [CrossRef]
- Rauhut, M.M.; Currier, H. Dialkyl 2-Methyleneglutarates. U.S. Patent 3074999, 22 January 1963. [Google Scholar]
- McClure, J.D. Dimerization of Acrylonitrile. U.S. Patent 3225083, 21 December 1965. [Google Scholar]
- Baizer, M.M.; Anderson, J.D. Electrolytic Reductive Coupling 8. Utilization and a New Preparation of α-Methyleneglutaronitrile. J. Org. Chem. 1965, 30, 1357–1360. [Google Scholar] [CrossRef]
- Wang, L.C.; Luis, A.L.; Agapito, F.; Jang, H.Y.; Krische, M.J. Organocatalytic Michael cycloisomerization of bis(enones): The intramolecular Rauhut–Currier reaction. J. Am. Chem. Soc. 2002, 124, 2402–2403. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.A.; Mergott, D.J.; Roush, W.R. The vinylogous intramolecular Morita–Baylis–Hillman reaction: Synthesis of functionalized cyclopentenes and cyclohexenes with trialkylphosphines as nucleophilic catalysts. J. Am. Chem. Soc. 2002, 124, 2404–2405. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M.E.; Haxell, T.F.N. Organomediated Morita–Baylis–Hillman cyclization reactions. J. Am. Chem. Soc. 2005, 127, 10168–10169. [Google Scholar] [CrossRef] [PubMed]
- Tran, Y.S.; Kwon, O. Phosphine-catalyzed [4+2] annulation: Synthesis of cyclohexenes. J. Am. Chem. Soc. 2007, 129, 12632–12633. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Hosseinpour, R. An Efficient Synthesis of Tetraalkyl 2,3,4,5-Thiophenetetracarboxylate Derivatives. Synthesis 2009, 1960–1962. [Google Scholar] [CrossRef]
- Dong, X.; Liang, L.; Li, E.; Huang, Y. Highly Enantioselective Intermolecular Cross Rauhut–Currier Reaction Catalyzed by a Multifunctional Lewis Base Catalyst. Angew. Chem. Int. Ed. 2015, 54, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Suzuki, Z.; Hirose, H. A Tertiary Phosphine-Catalyzed Reaction of Acrylic Compounds with Aldehydes. Bull. Chem. Soc. Jpn. 1968, 41, 2815. [Google Scholar] [CrossRef]
- Baylis, A.B.; Hillman, M.E.D. Acrylic Compounds. Ger. Offen DE 2155113, 10 May 1972. Chem. Abstr. 1972, 77, 34174. [Google Scholar]
- Basavaiah, D.; Rao, A.J.; Satyanarayana, T. Recent advances in the Baylis–Hillman reaction and applications. Chem. Rev. 2003, 103, 811–891. [Google Scholar] [CrossRef] [PubMed]
- Langer, P. New strategies for the development of an asymmetric version of the Baylis–Hillman reaction. Angew. Chem. Int. Ed. 2000, 39, 3049–3052. [Google Scholar] [CrossRef]
- Basavaiah, D.; Kumaragurubaran, N.; Sharada, D.S. Baylis–Hillman chemistry: A novel synthesis of functionalized 1,4-pentadienes. Tetrahedron Lett. 2001, 42, 85–87. [Google Scholar] [CrossRef]
- Basavaiah, D.; Sharada, D.S.; Kumaragurubaran, N.; Reddy, R.M. The Baylis–Hillman reaction: One-pot facile synthesis of 2,4-functionalized 1,4-pentadienes. J. Org. Chem. 2002, 67, 7135–7137. [Google Scholar] [CrossRef] [PubMed]
- Koech, P.K.; Krische, M.J. Phosphine catalyzed α-arylation of enones and enals using hypervalent bismuth reagents: Regiospecific enolate arylation via nucleophilic catalysis. J. Am. Chem. Soc. 2004, 126, 5350–5351. [Google Scholar] [CrossRef] [PubMed]
- Robiette, R.; Aggarwal, V.K.; Harvey, J.N. Mechanism of the Morita–Baylis–Hillman reaction: A computational investigation. J. Am. Chem. Soc. 2007, 129, 15513–15525. [Google Scholar] [CrossRef] [PubMed]
- Plata, R.E.; Singleton, D.A. A Case Study of the Mechanism of Alcohol-Mediated Morita Baylis–Hillman Reactions. The Importance of Experimental Observations. J. Am. Chem. Soc. 2015, 137, 3811–3826. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M.E.; Haxell, T.F.N.; Seibert, K.A.; Abboud, K.A. Mechanistic implications in the Morita–Baylis–Hillman alkylation: Isolation and characterization of an intermediate. J. Am. Chem. Soc. 2006, 128, 4174–4175. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.W.; Krische, M.J. Regio- and stereoselective construction of ɣ-butenolides through phosphine-catalyzed substitution of Morita–Baylis–Hillman acetates: An organocatalytic allylic alkylation. Angew. Chem. Int. Ed. 2004, 43, 6689–6691. [Google Scholar] [CrossRef] [PubMed]
- Masson, G.; Housseman, C.; Zhu, J. The enantioselective morita-baylis-hiliman reaction and its aza counterpart. Angew. Chem. Int. Ed. 2007, 46, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, M. Recent Advances in Organocatalytic Asymmetric Morita–Baylis–Hillman/aza-Morita–Baylis–Hillman Reactions. Chem. Rev. 2013, 113, 6659–6690. [Google Scholar] [CrossRef] [PubMed]
- Gergelitsova, I.; Tauchman, J.; Cisarova, I.; Vesely, J. Bifunctional (Thio)urea-Phosphine Organocatalysts Derived from d-Glucose and α-Amino Acids and Their Application to the Enantioselective Morita–Baylis–Hillman Reaction. Synlett 2015, 26, 2690–2696. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, Q.; Shi, M. Enantioselective Synthesis of Polycyclic Indole Derivatives Based on aza-Morita–Baylis–Hillman Reaction. ACS Catal. 2015, 5, 6608–6614. [Google Scholar] [CrossRef]
- Krishna, P.R.; Kannan, V.; Reddy, P.V.N. N-Methylprolinol catalysed asymmetric Baylis–Hillman reaction. Adv. Synth. Catal. 2004, 346, 603–606. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. Organocatalytic Asymmetric Synthesis of Substituted 3-Hydroxy-2-oxindoles via Morita–Baylis–Hillman Reaction. J. Am. Chem. Soc. 2010, 132, 15176–15178. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chen, L.H.; Teng, W.D. Asymmetric aza-Morita–Baylis–Hillman reaction of N-sulfonated imines with methyl vinyl ketone catalyzed by chiral phosphine Lewis bases bearing perfluoroalkanes as pony tails. Adv. Synth. Catal. 2005, 347, 1781–1789. [Google Scholar] [CrossRef]
- Shi, M.; Chen, L.H.; Li, C.Q. Chiral phosphine Lewis bases catalyzed asymmetric aza-Baylis-Hiliman reaction of N-sulfonated imines with activated olefins. J. Am. Chem. Soc. 2005, 127, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-L.; Yuan, K.; Wu, X.-Y. Chiral phosphine-squaramides as enantioselective catalysts for the intramolecular Morita-Baylis-Hillman reaction. Chem. Commun. 2011, 47, 1012–1014. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Takizawa, S.; Sasai, H. Bifunctional organocatalysts for enantioselective aza-Morita–Baylis–Hillman reaction. J. Am. Chem. Soc. 2005, 127, 3680–3681. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Yu, X.H.; Zu, L.S.; Wang, W. Chiral binaphthyl-derived amine-thiourea organocatalyst-promoted asymmetric Morita–Baylis–Hillman reaction. Org. Lett. 2005, 7, 4293–4296. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, S.; Rémond, E.; Arteaga Arteaga, F.; Yoshida, Y.; Sridharan, V.; Bayardon, J.; Jugé, S.; Sasai, H. P-chirogenic organocatalysts: Application to the aza-Morita–Baylis–Hillman (aza-MBH) reaction of ketimines. Chem. Commun. 2013, 49, 8392–8394. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Lan, J.; Kwon, O. An expedient phosphine-catalyzed [4+2] annulation: Synthesis of highly functionalized tetrahydropyridines. J. Am. Chem. Soc. 2003, 125, 4716–4717. [Google Scholar] [CrossRef] [PubMed]
- Sunden, H.; Ibrahem, I.; Eriksson, L.; Cordova, A. Direct catalytic enantioselective aza-Diels–Alder reactions. Angew. Chem. Int. Ed. 2005, 44, 4877–4880. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Komiyama, S.; Ishitani, H. The first enantioselective aza-Diels–Alder reactions of imino dienophiles on use of a chiral zirconium catalyst. Angew. Chem. Int. Ed. 1998, 37, 979–981. [Google Scholar] [CrossRef]
- Yao, S.L.; Saaby, S.; Hazell, R.G.; Jørgensen, K.A. Catalytic enantioselective aza-Diels–Alder reactions of imines—An approach to optically active nonproteinogenic α-amino acids. Chem. Eur. J. 2000, 6, 2435–2448. [Google Scholar] [CrossRef]
- Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral Bronsted acid catalyzed enantioselective aza-Diels–Alder reaction of Brassard’s diene with imines. Angew. Chem. Int. Ed. 2006, 45, 4796–4798. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Xin, J.; Liu, Y.; Zhou, X.; Liu, X.; Feng, X. Enantioselective aza-Diels–Alder reaction of aldimines with Danishefsky-Type Diene catalyzed by chiral scandium(III)-N,N’-dioxide complexes. J. Org. Chem. 2008, 73, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Jurcik, V.; Arai, K.; Salter, M.; Yamashita, Y.; Kobayashi, S. Niobium-catalyzed highly enantioselective aza-Diels–Alder reactions. Adv. Synth. Catal. 2008, 350, 647–651. [Google Scholar] [CrossRef]
- He, M.; Struble, J.R.; Bode, J.W. Highly enantioselective azadiene Diels–Alder reactions catalyzed by chiral N-heterocyclic carbenes. J. Am. Chem. Soc. 2006, 128, 8418–8420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ruhl, K.E.; Rovis, T. N-Heterocyclic-Carbene-Catalyzed Asymmetric Oxidative Hetero-Diels–Alder Reactions with Simple Aliphatic Aldehydes. Angew. Chem. Int. Ed. 2012, 51, 12330–12333. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Ni, Q.; Zhu, C.; Raabe, G.; Enders, D. Asymmetric N-Heterocyclic Carbene Catalyzed Annulation of 2-Alkenylbenzothiazoles with α-Chloro Aldehydes. Synthesis 2015, 47, 421–428. [Google Scholar] [CrossRef]
- France, S.; Weatherwax, A.; Taggi, A.E.; Lectka, T. Advances in the catalytic, asymmetric synthesis of β-lactams. Acc. Chem. Res. 2004, 37, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.R.; He, L.; Wu, X.; Shao, P.L.; Ye, S. Chiral N-heterocyclic carbene catalyzed Staudinger reaction of ketenes with imines: Highly enantioselective synthesis of N-Boc β-lactams. Org. Lett. 2008, 10, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.M.; Lu, X.Y. Phosphine-Catalyzed Cycloaddition of 2,3-Butadienoates Or 2-Butynoates with Electron-Deficient Olefins—A Novel [3+2]-Annulation Approach to Cyclopentenes. J. Org. Chem. 1995, 60, 2906–2908. [Google Scholar] [CrossRef]
- Pham, T.Q.; Pyne, S.G.; Skelton, B.W.; Whitet, A.H. Synthesis of carbocyclic hydantocidins via regioselective and diastereoselective phosphine-catalyzed [3+2]-cycloadditions to 5-methylenehydantoins. J. Org. Chem. 2005, 70, 6369–6377. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.R.; Williams, M.C.; Pyne, S.G.; Ung, A.T.; Skelton, B.W.; White, A.H.; Turner, P. Synthesis of 2-azaspiro[4.4]nonan-1-ones via phosphine-catalysed [3+2]-cycloadditions. Tetrahedron 2005, 61, 8120–8129. [Google Scholar] [CrossRef]
- Shao, P.L.; Chen, X.Y.; Ye, S. Formal [3+2] Cycloaddition of Ketenes and Oxaziridines Catalyzed by Chiral Lewis Bases: Enantioselective Synthesis of Oxazolin-4-ones. Angew. Chem. Int. Ed. 2010, 49, 8412–8416. [Google Scholar] [CrossRef] [PubMed]
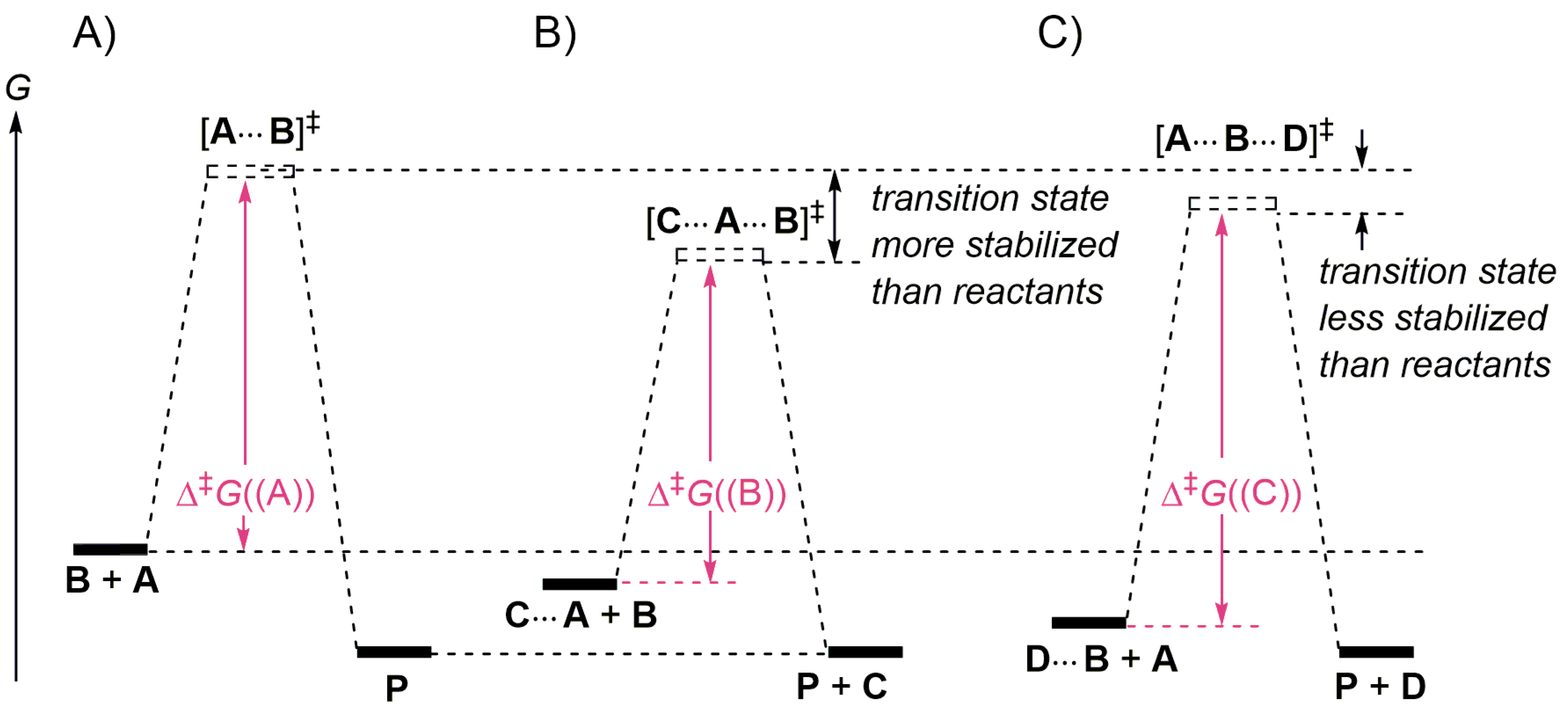
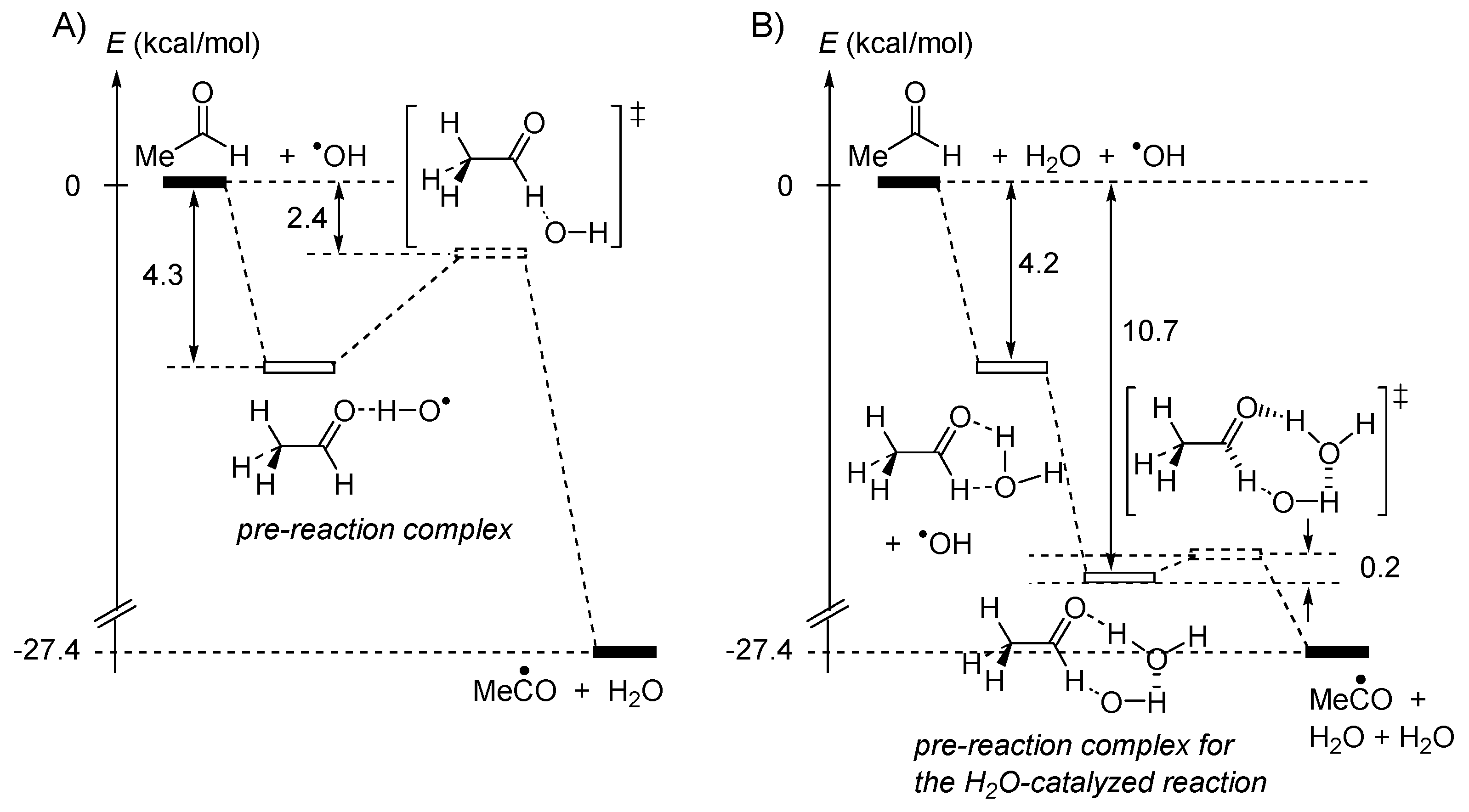


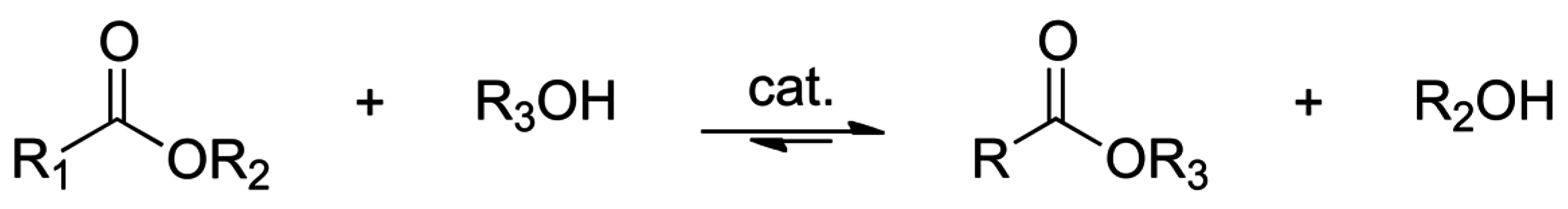

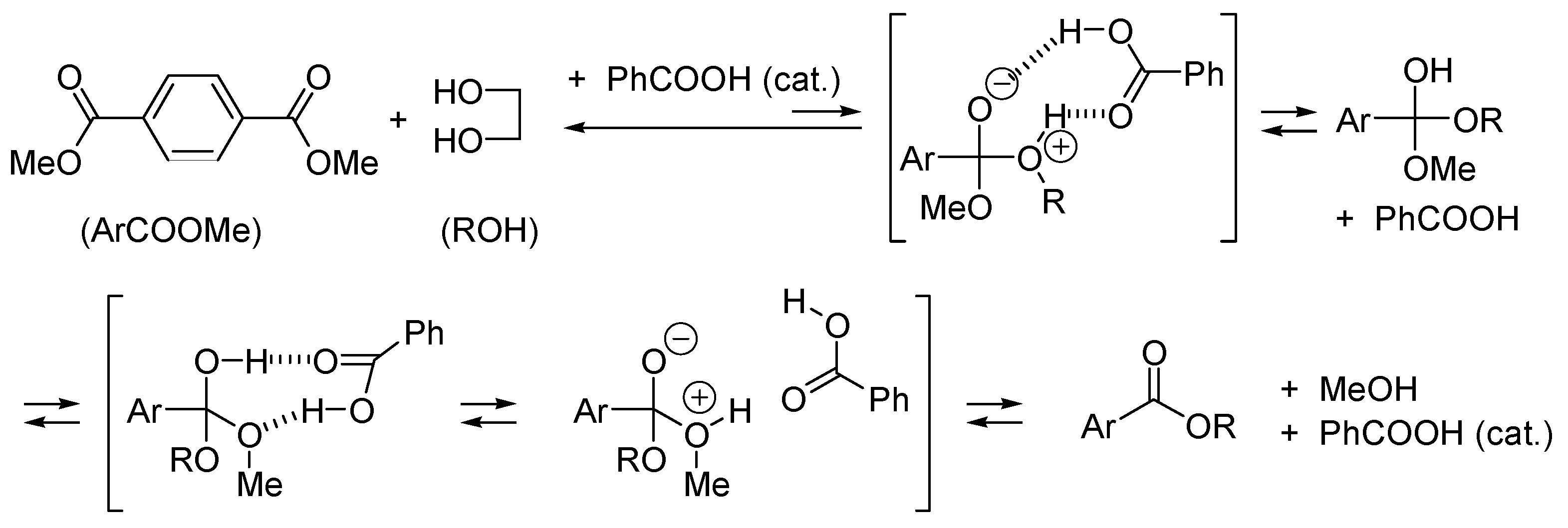
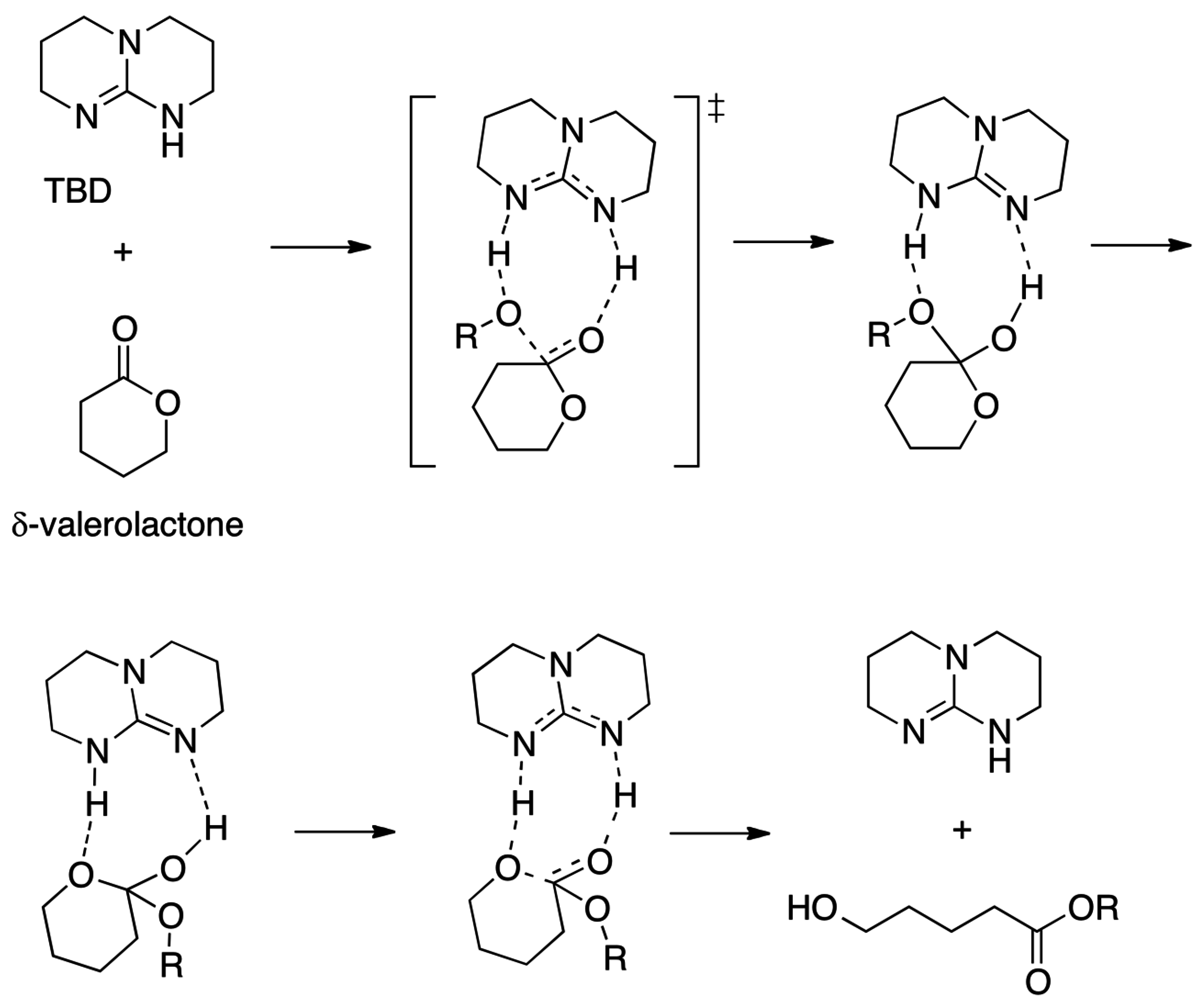
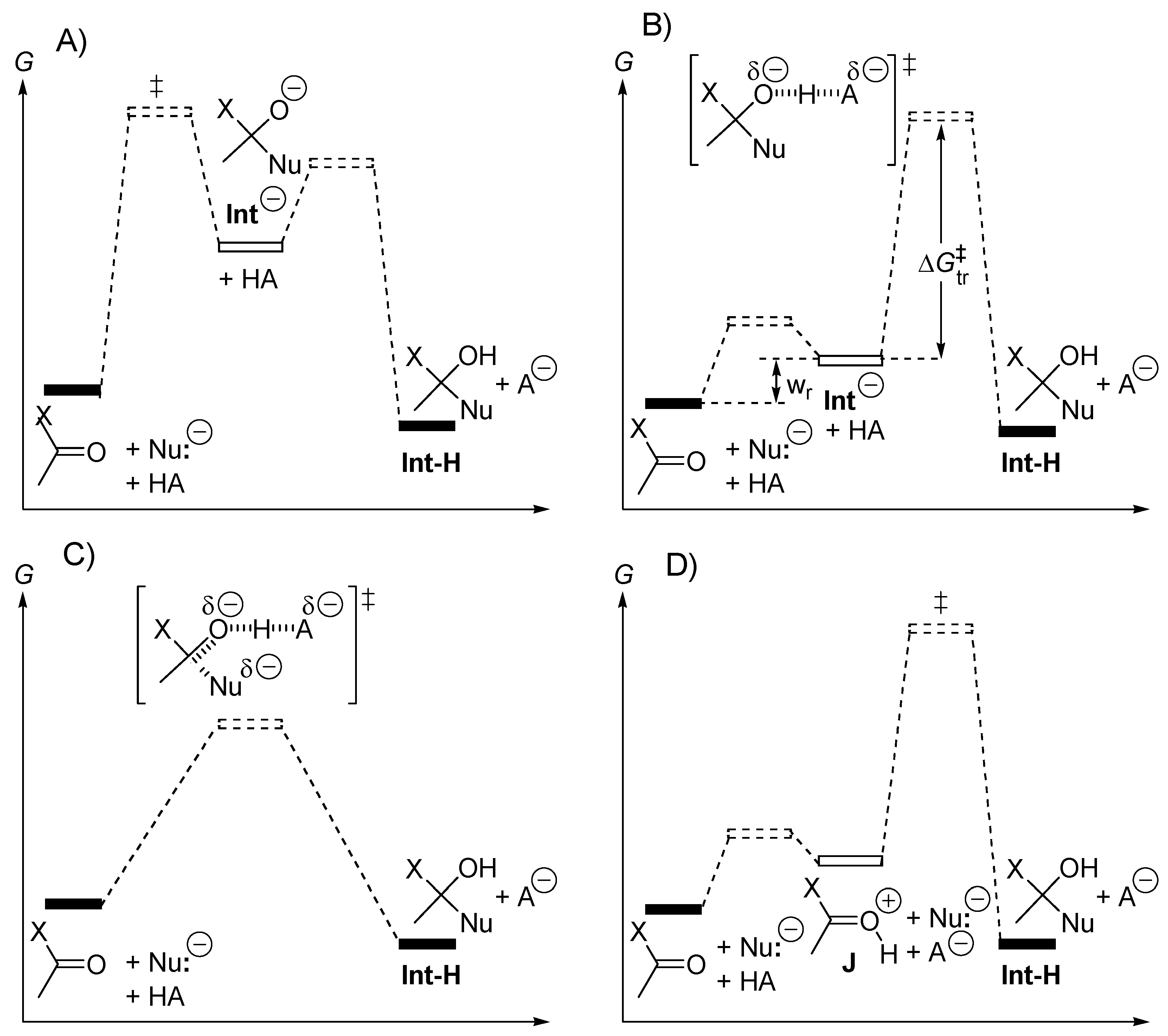
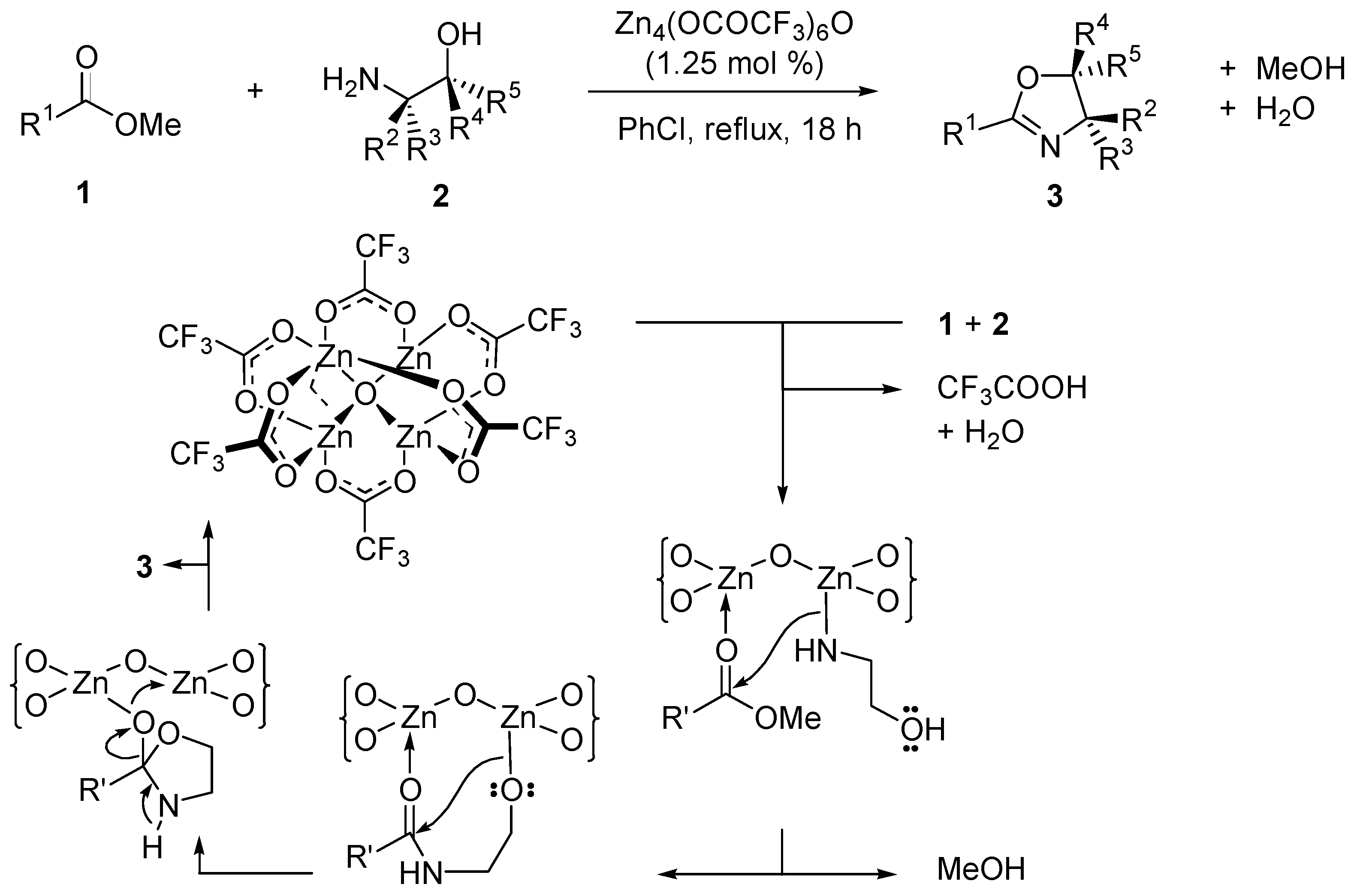
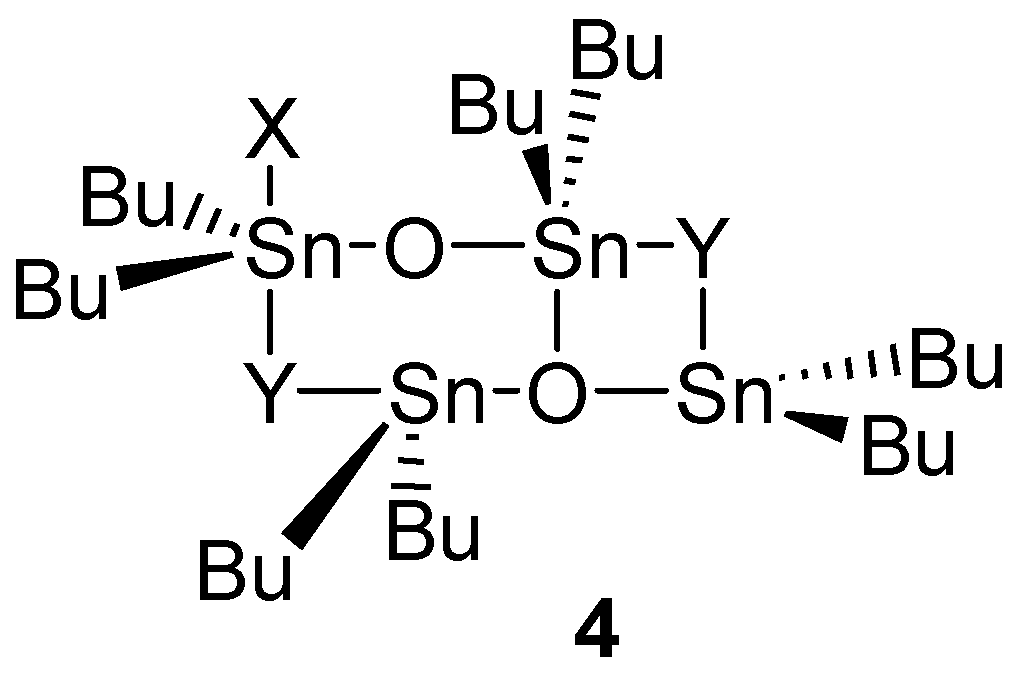
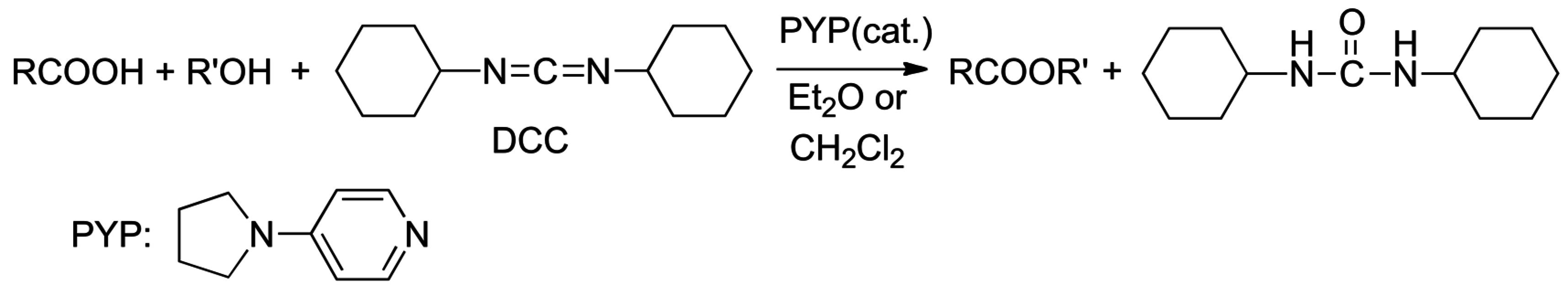

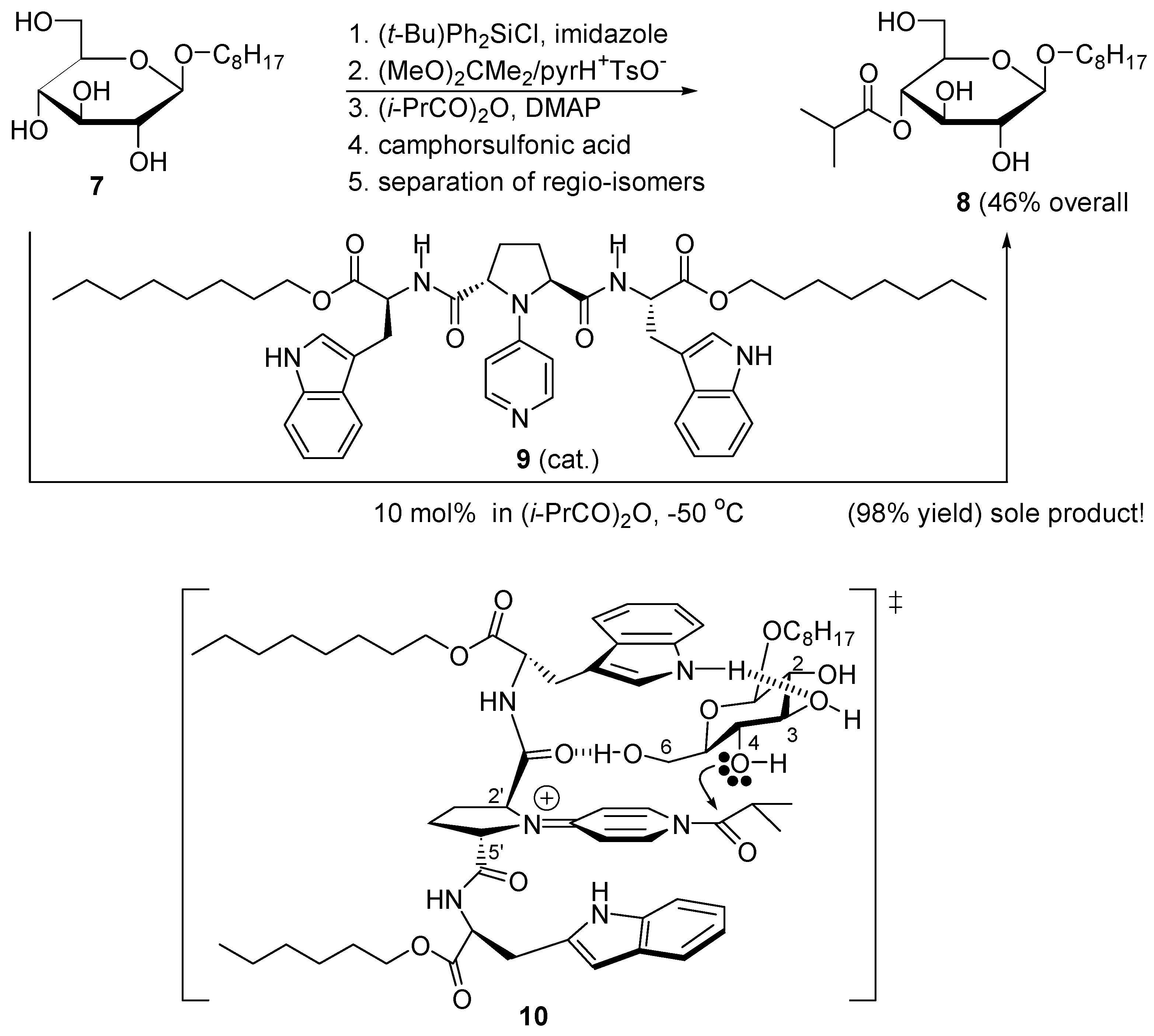
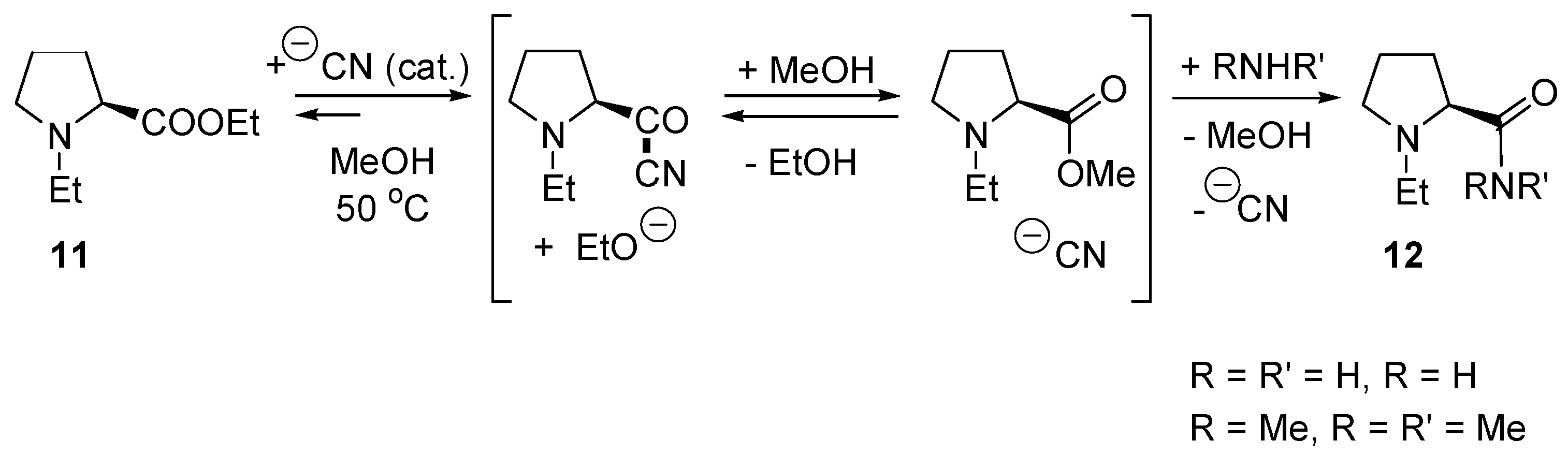
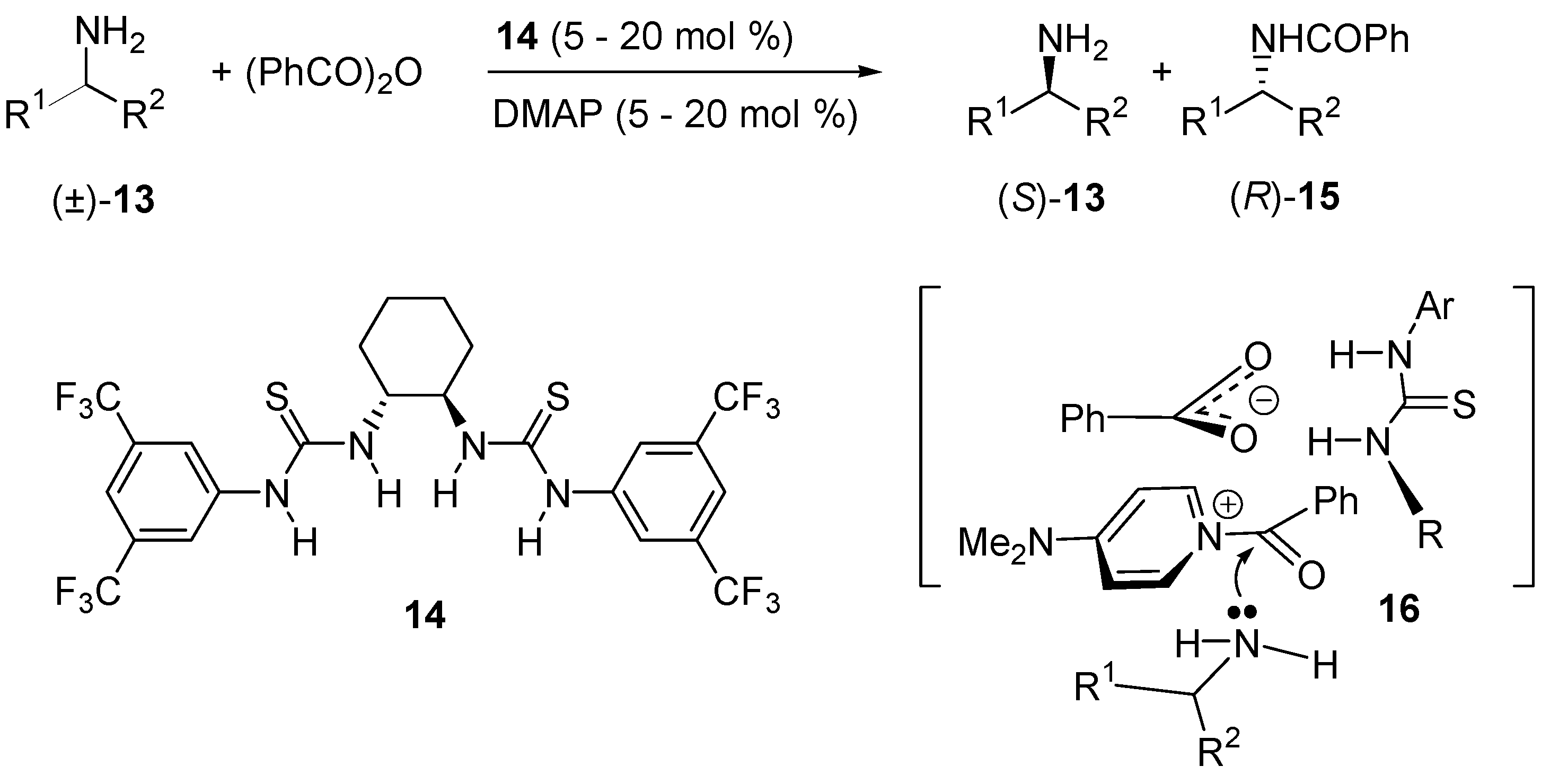
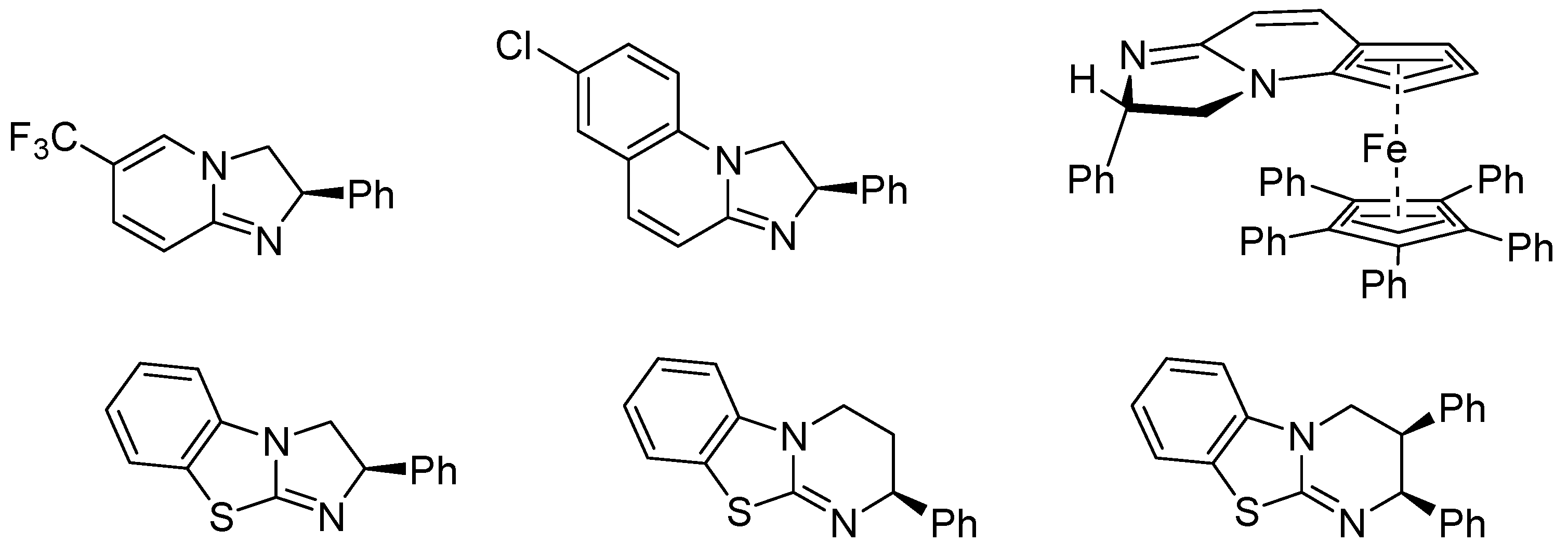
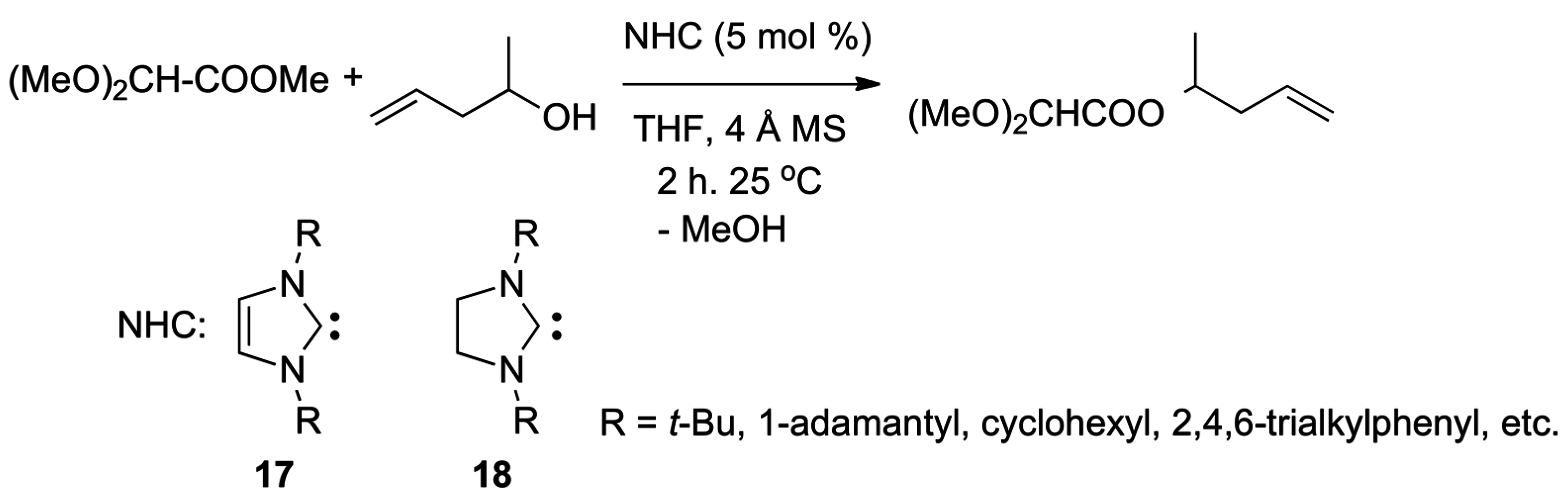
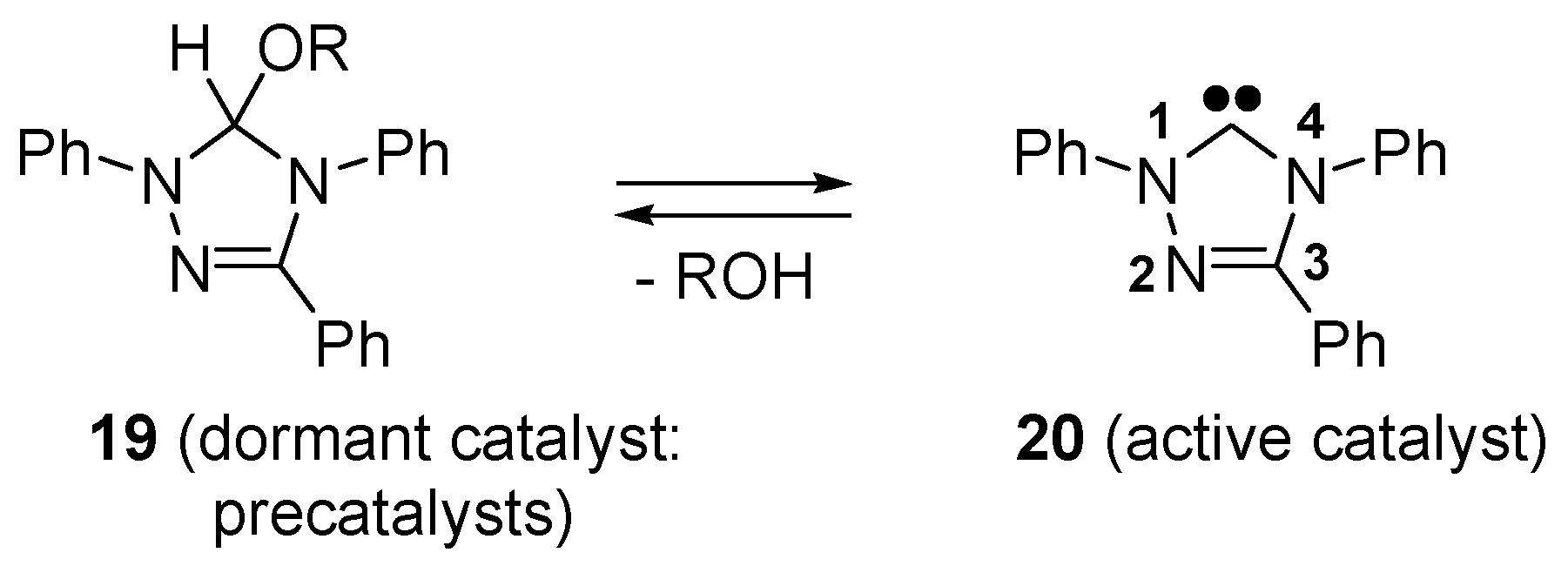
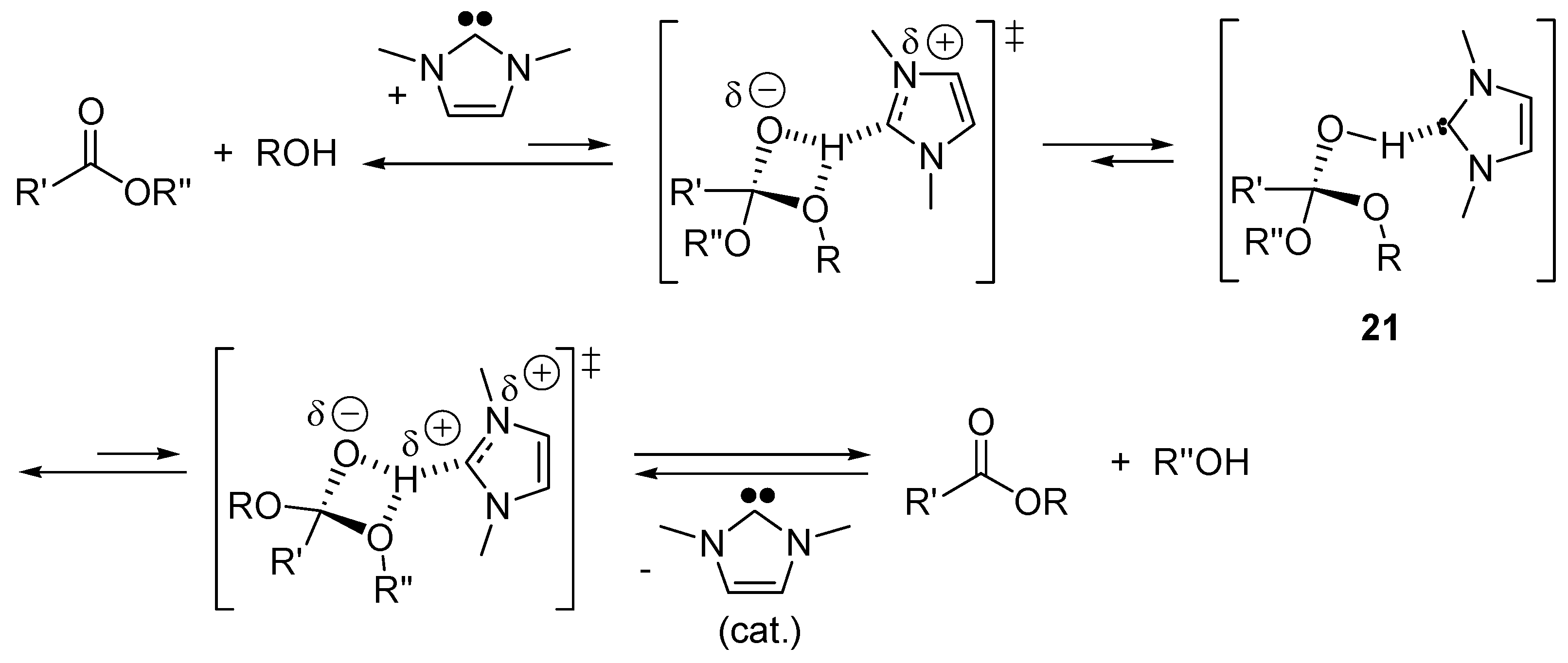
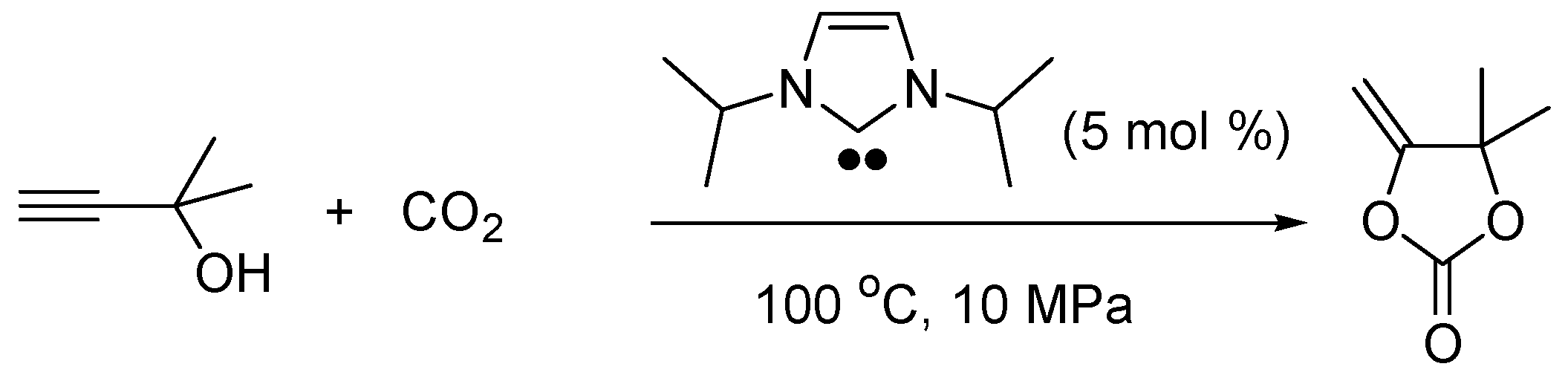
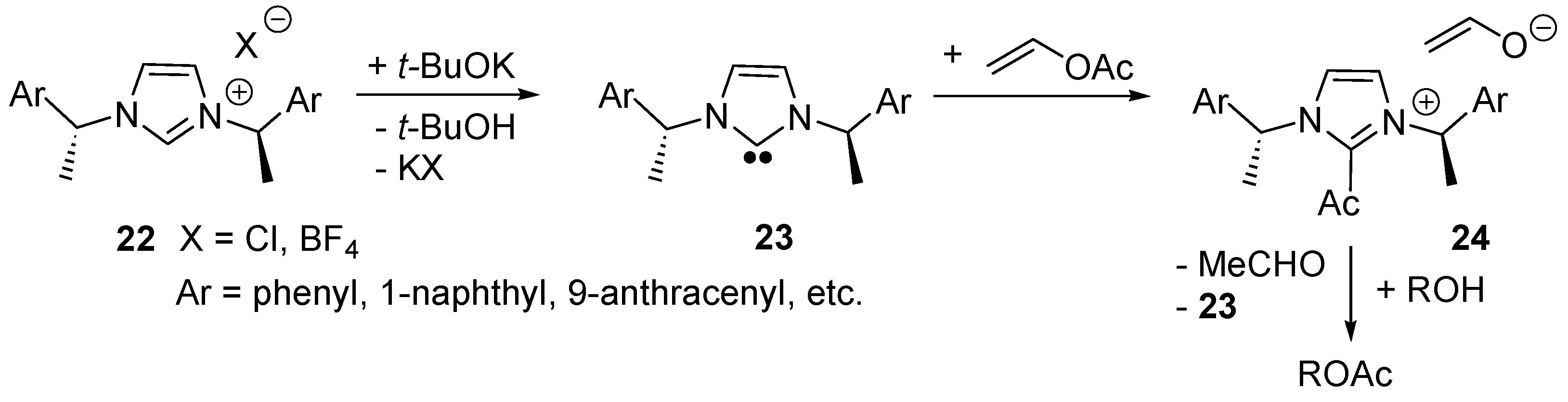

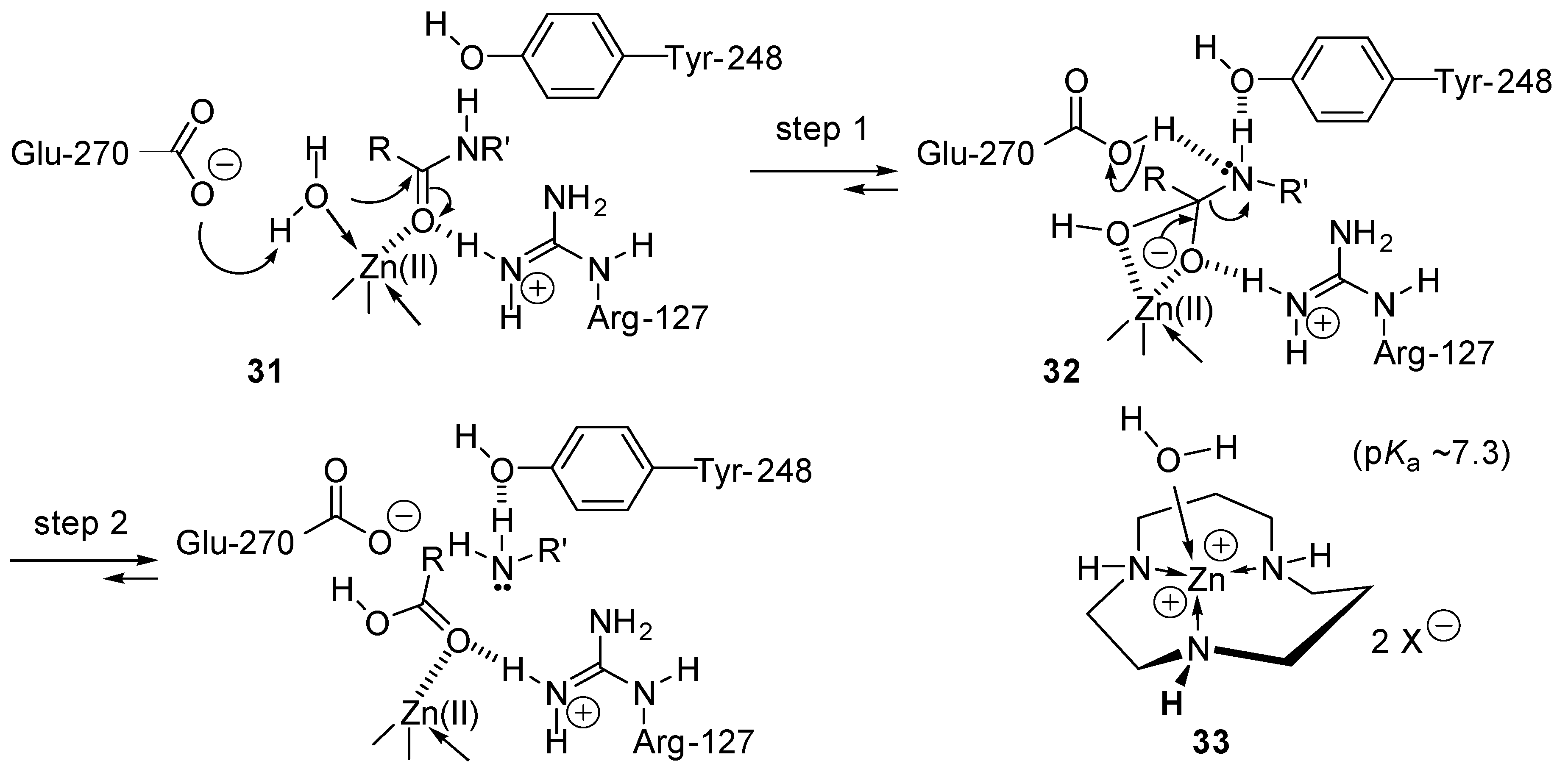
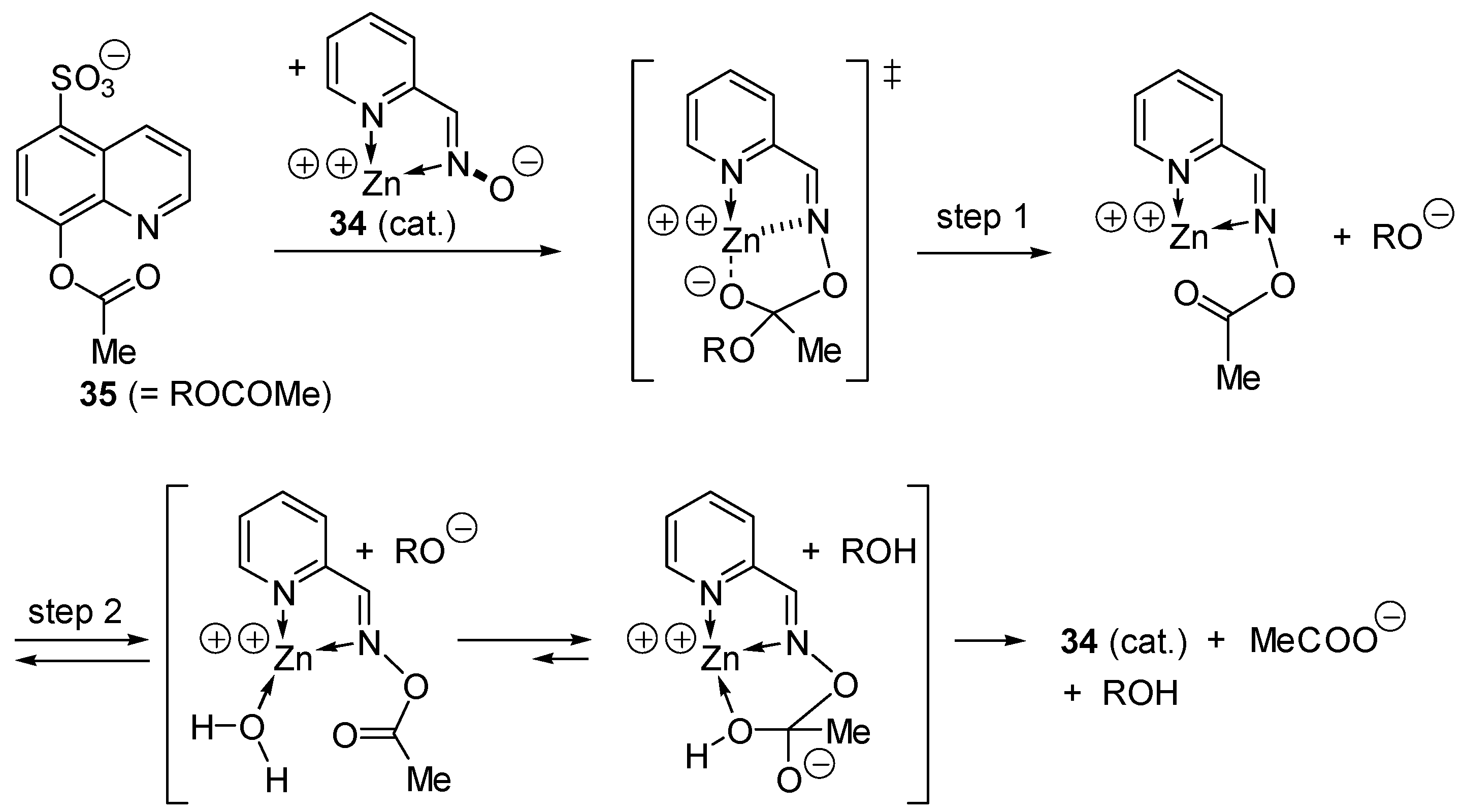

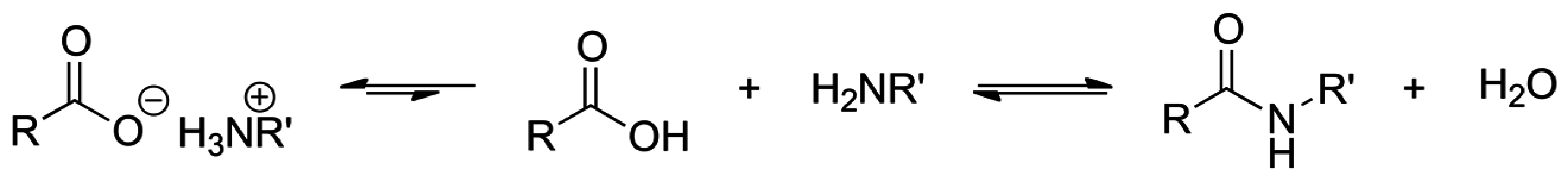
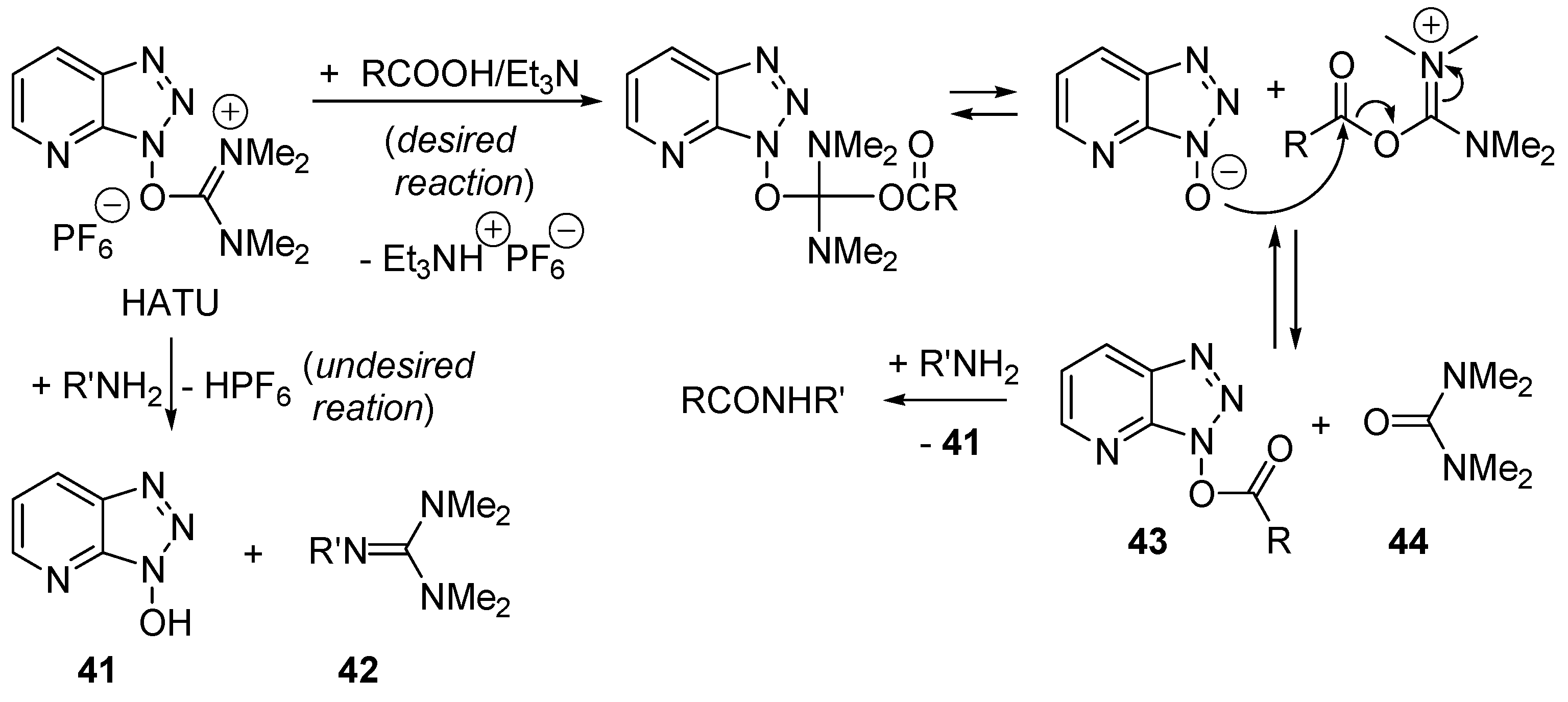
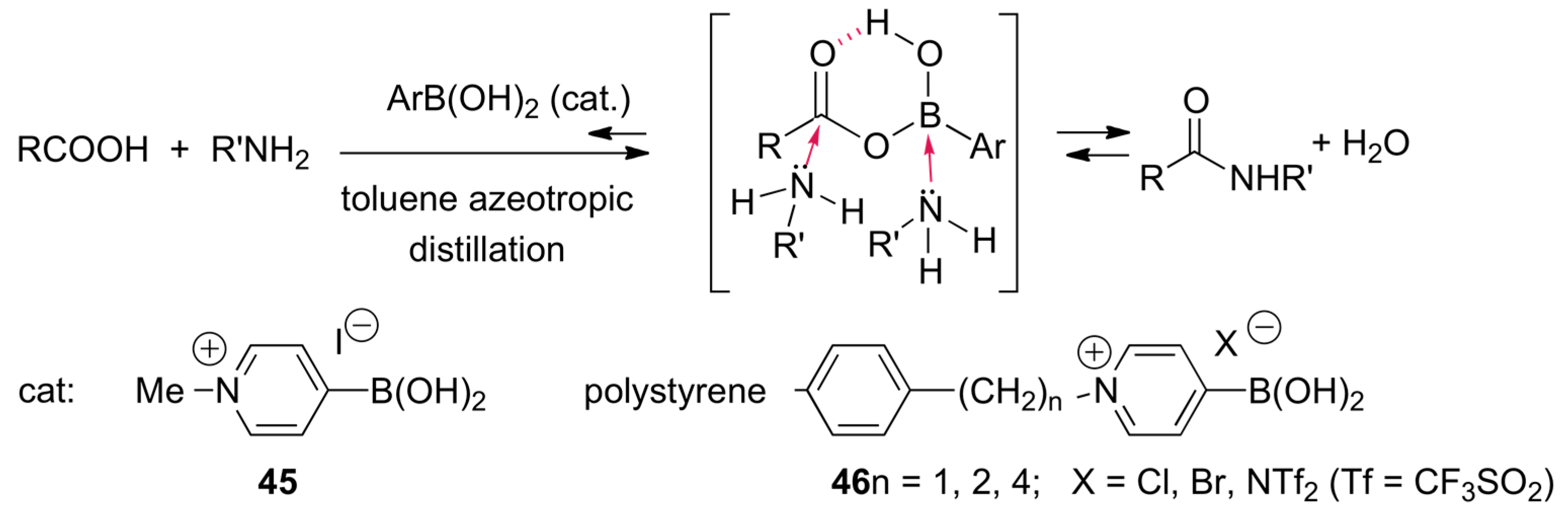

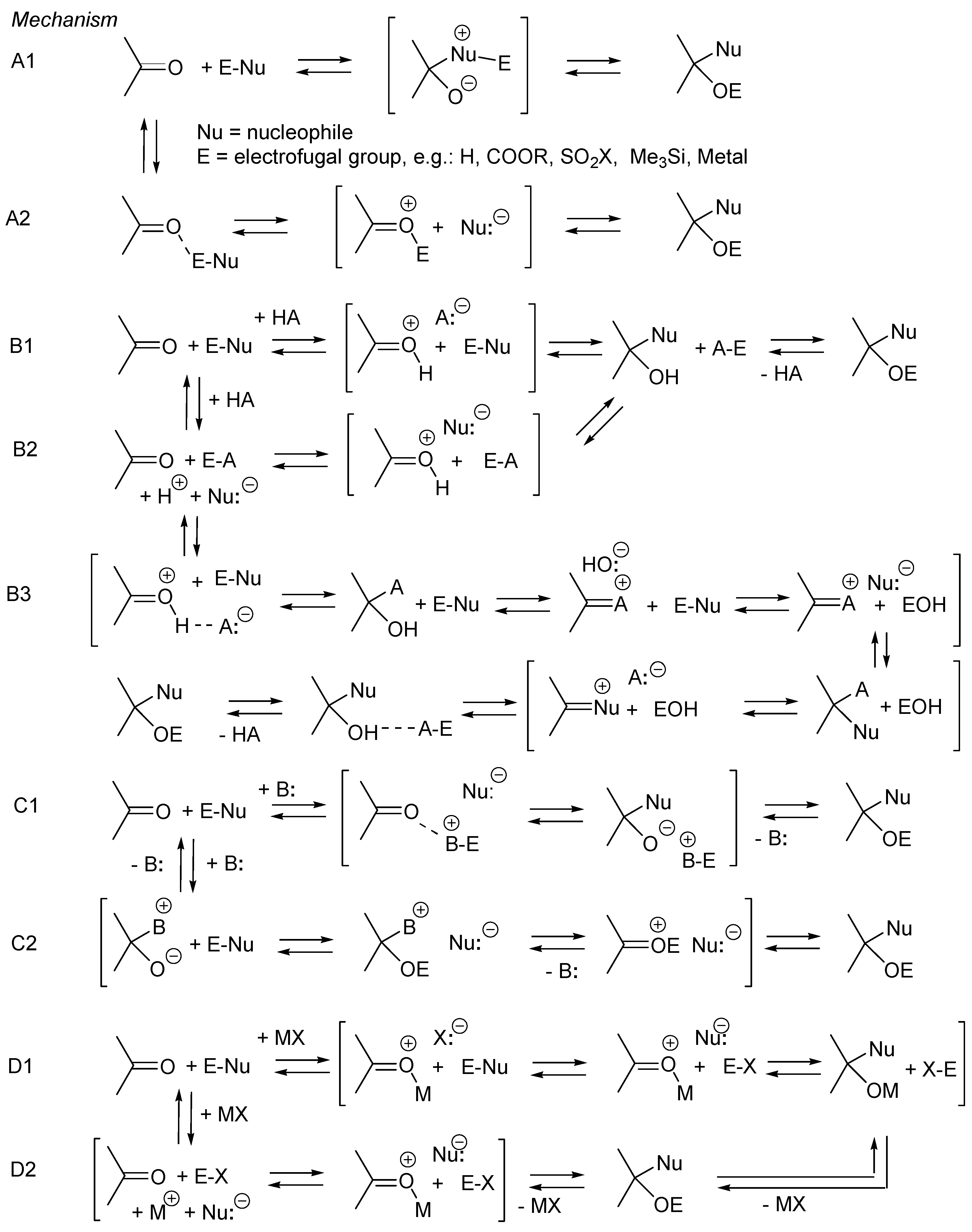
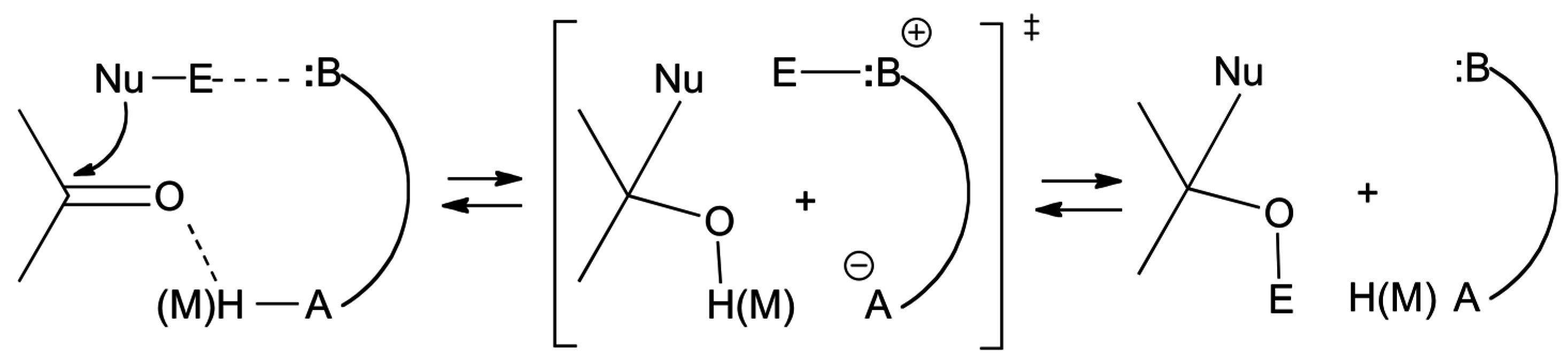
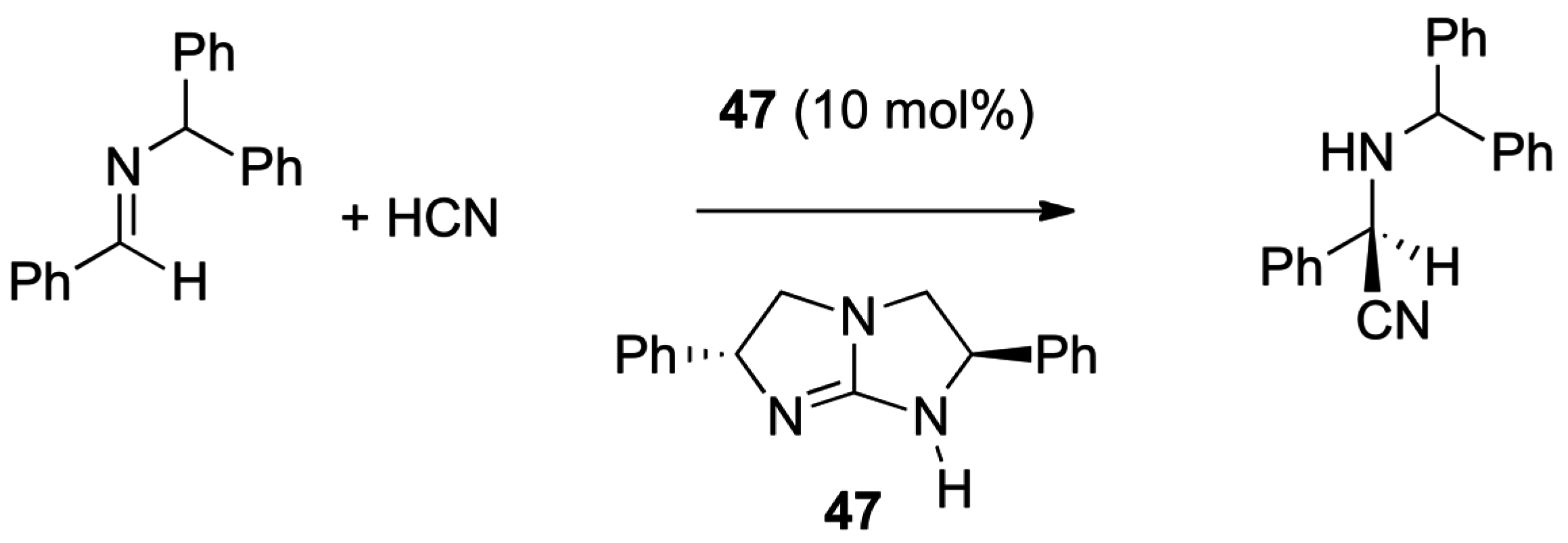
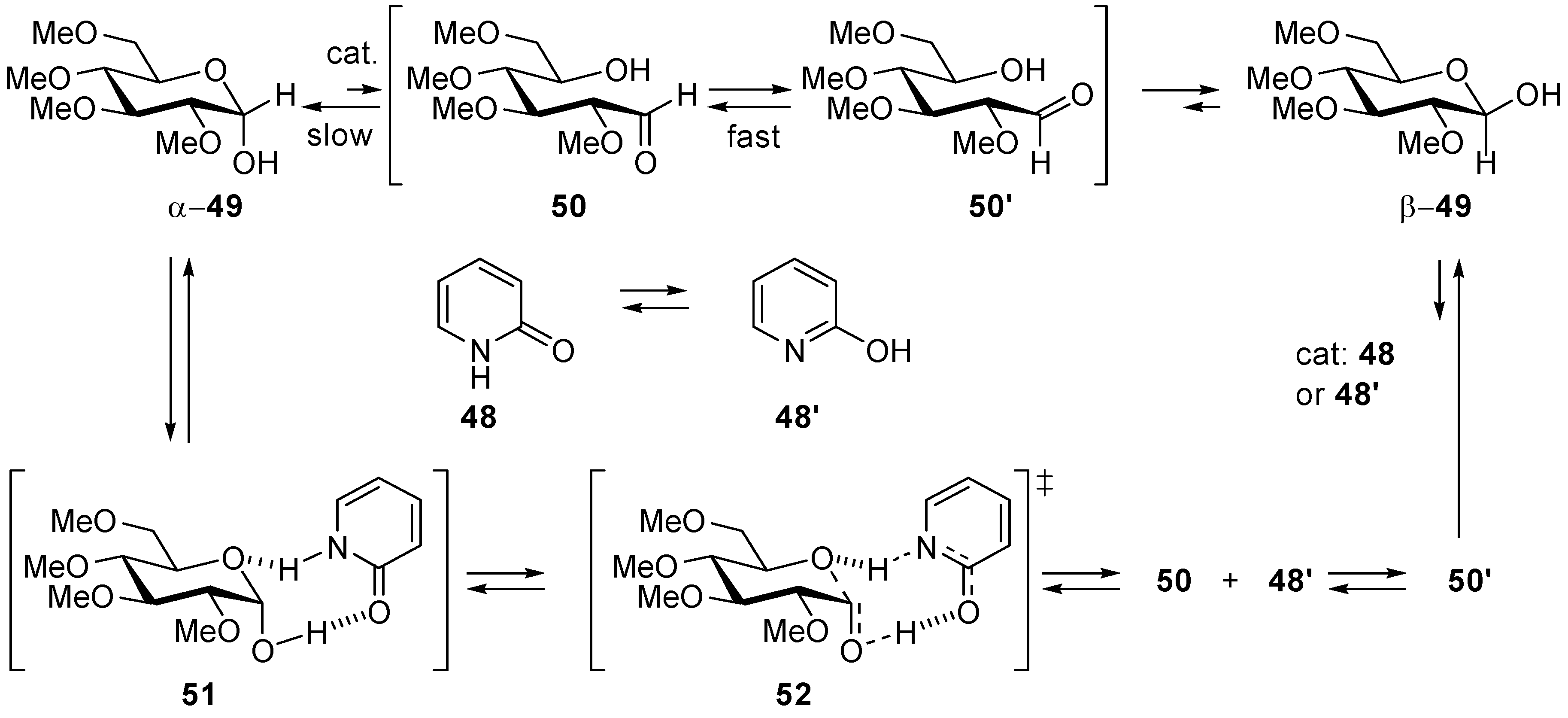
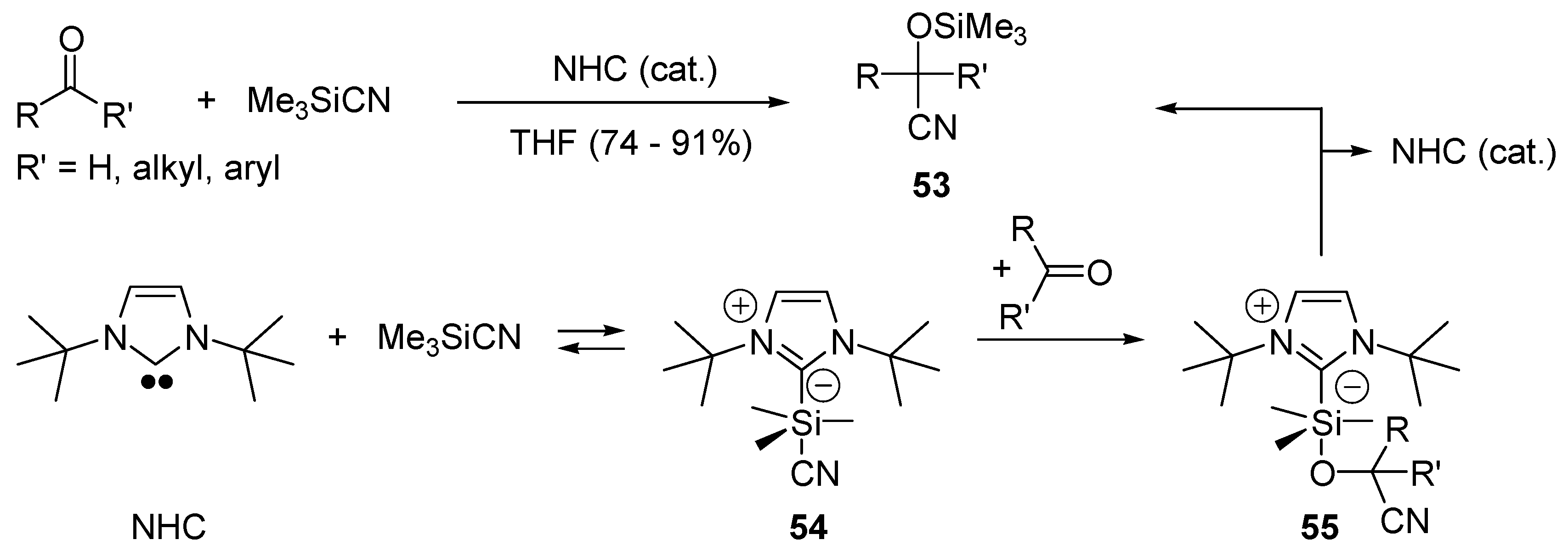
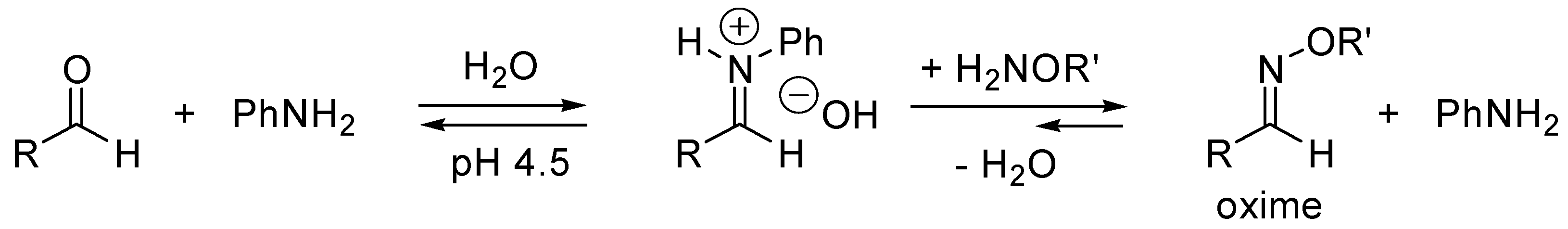
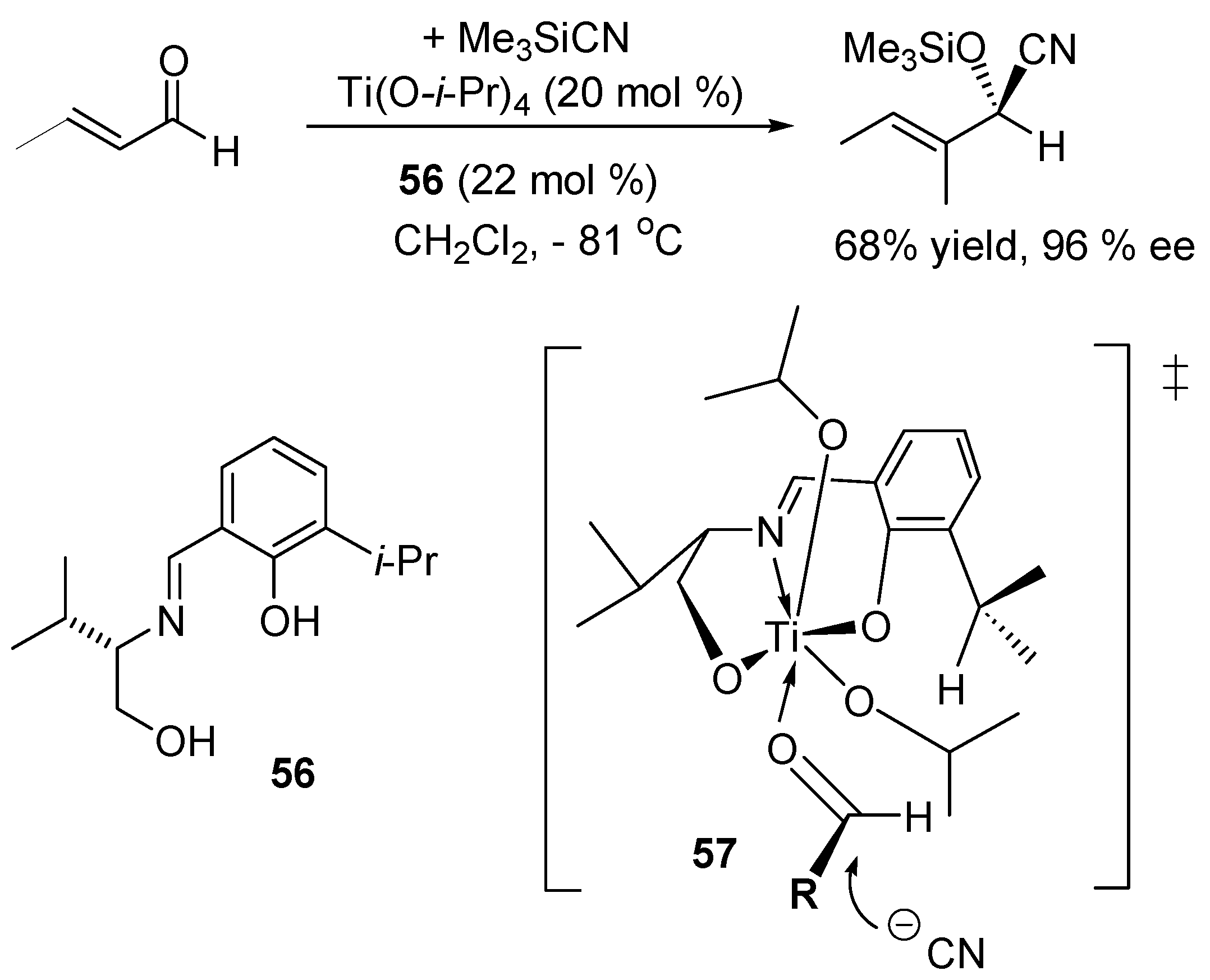
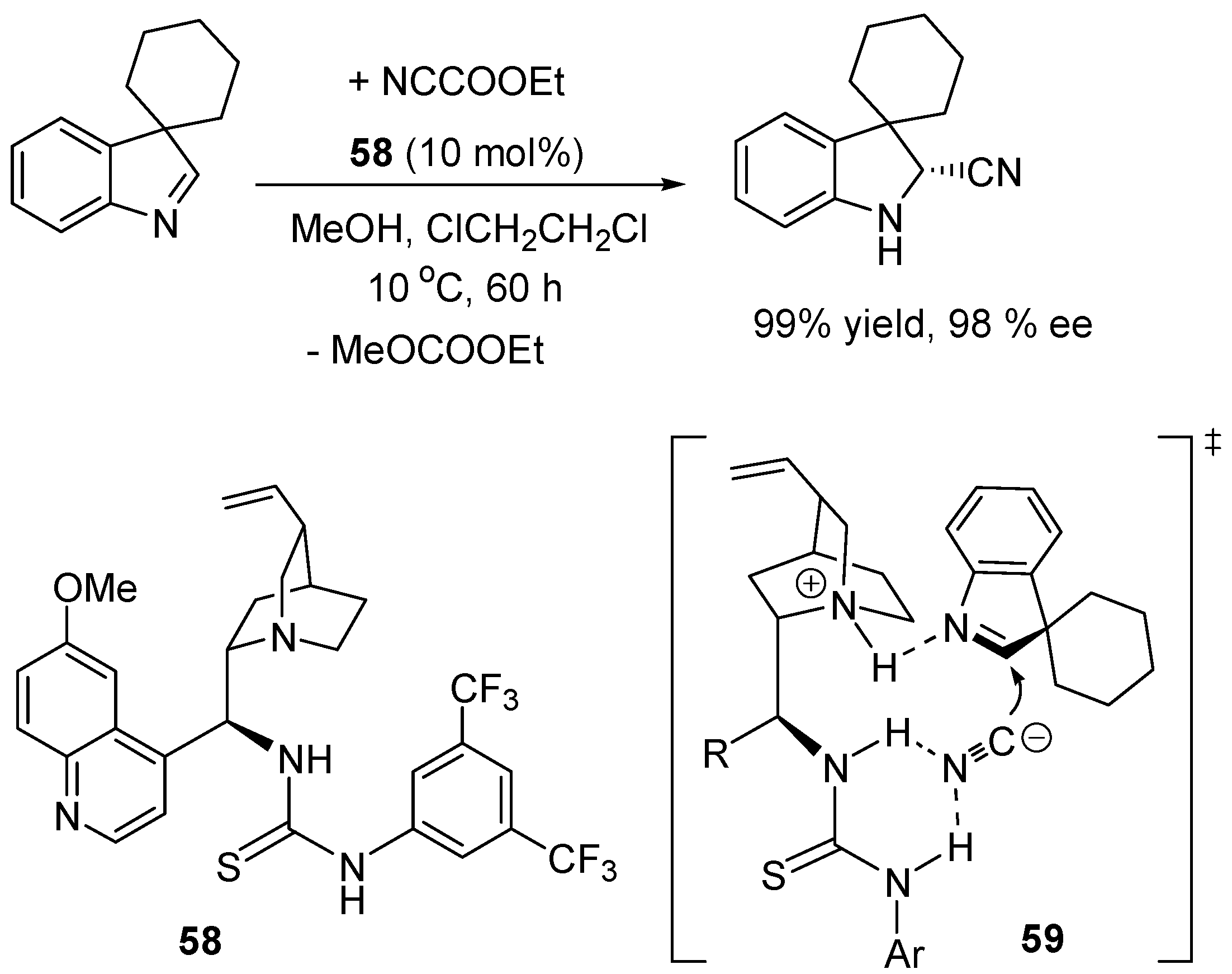
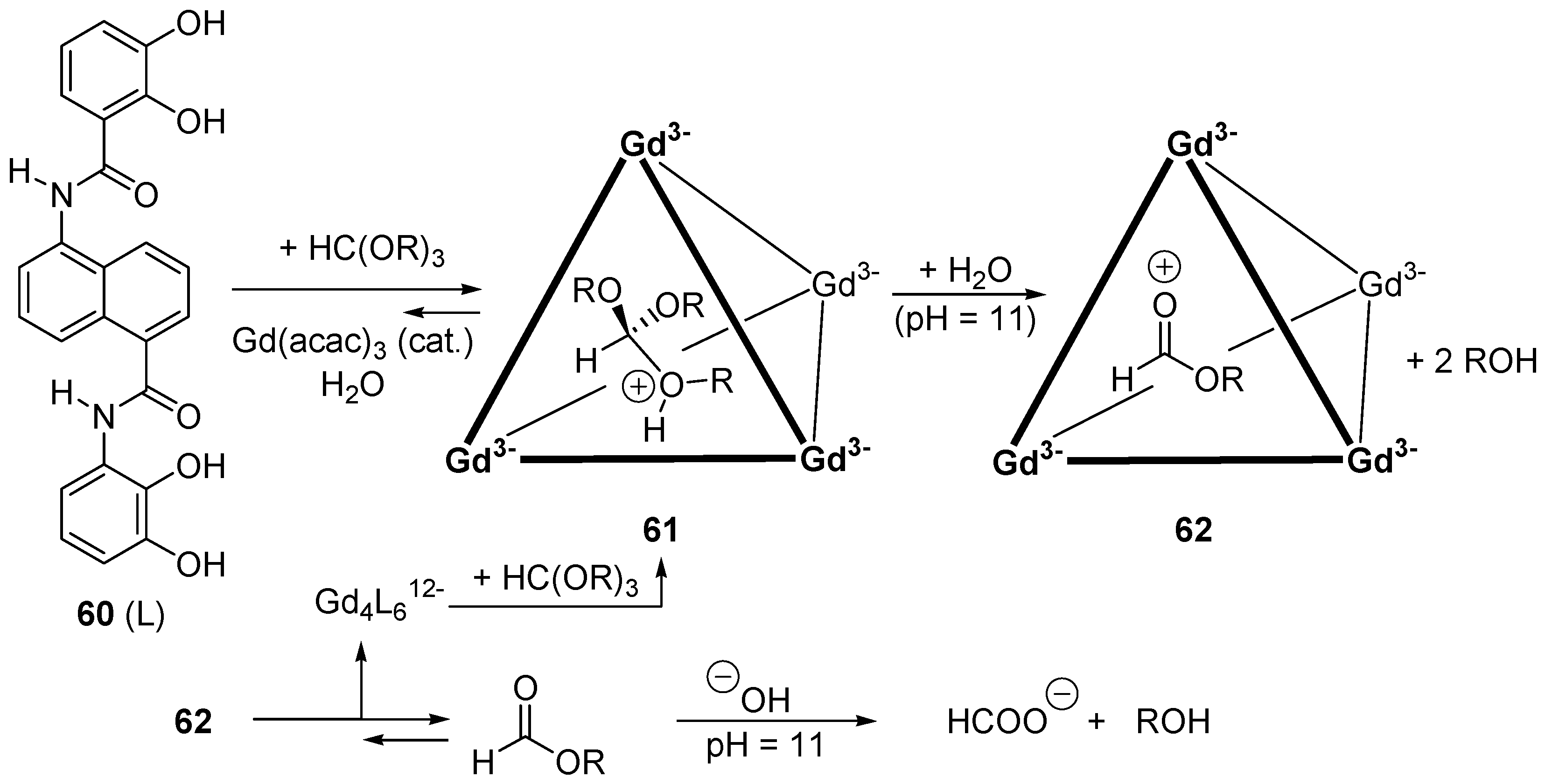
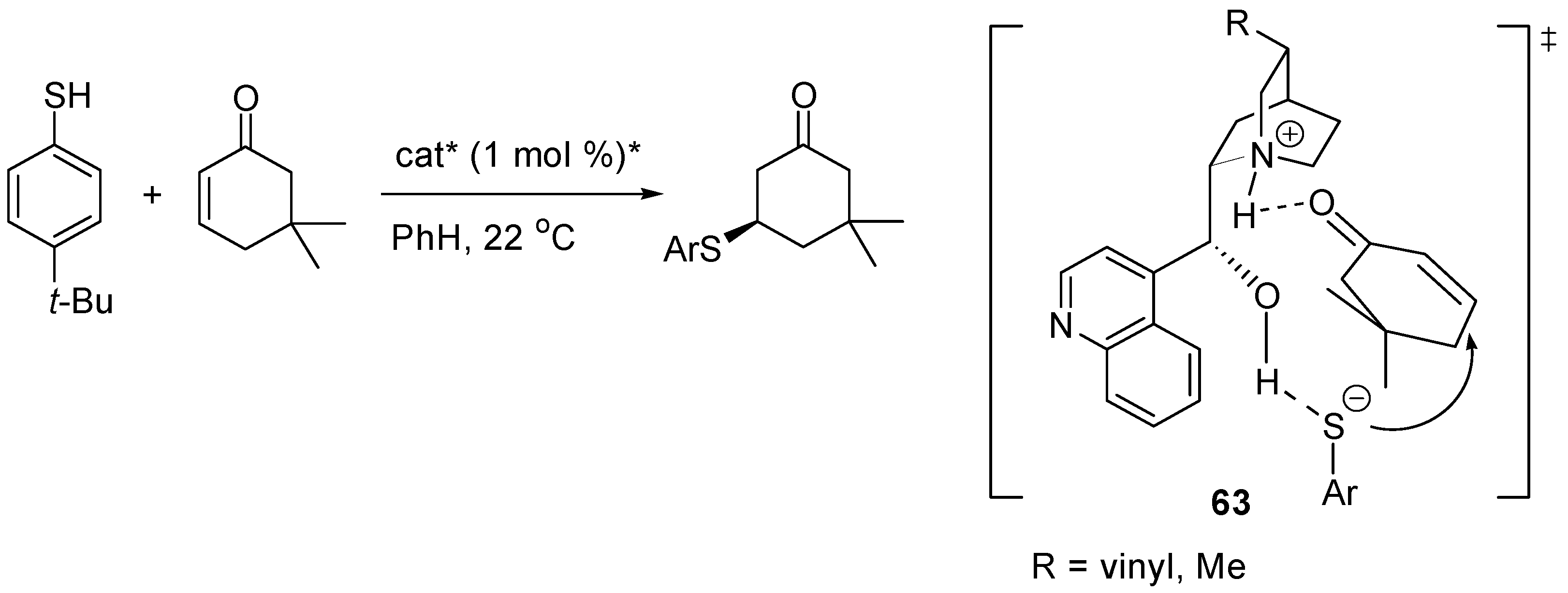
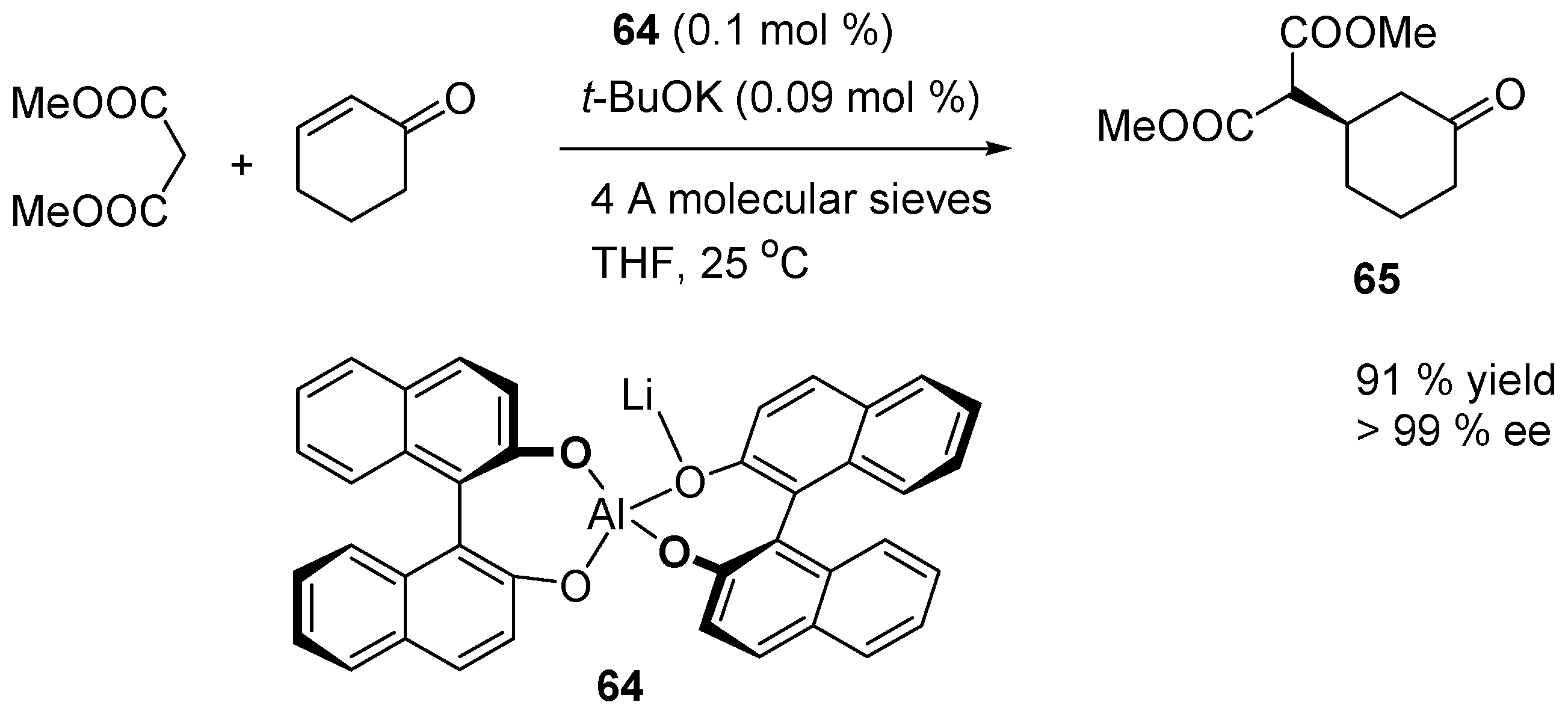
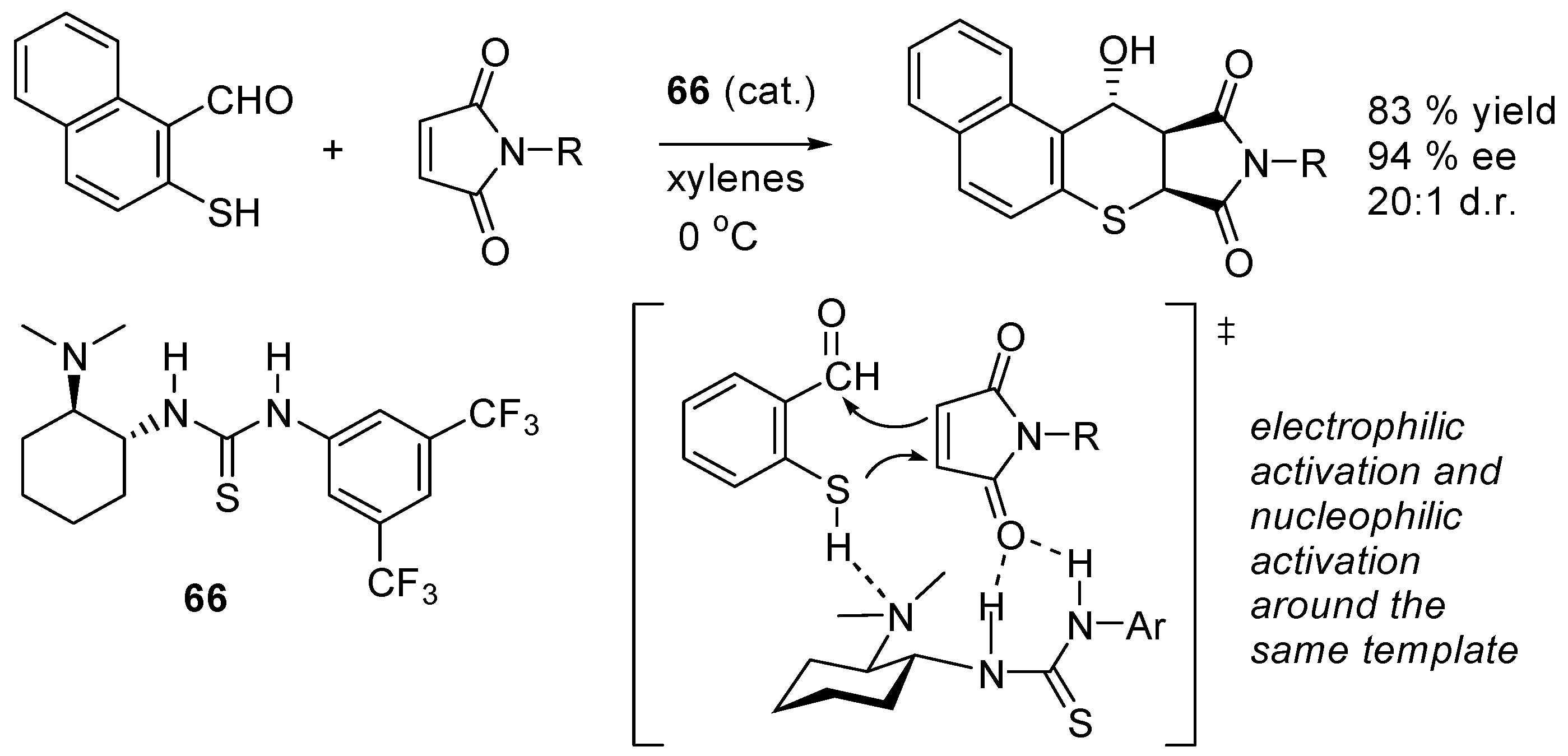
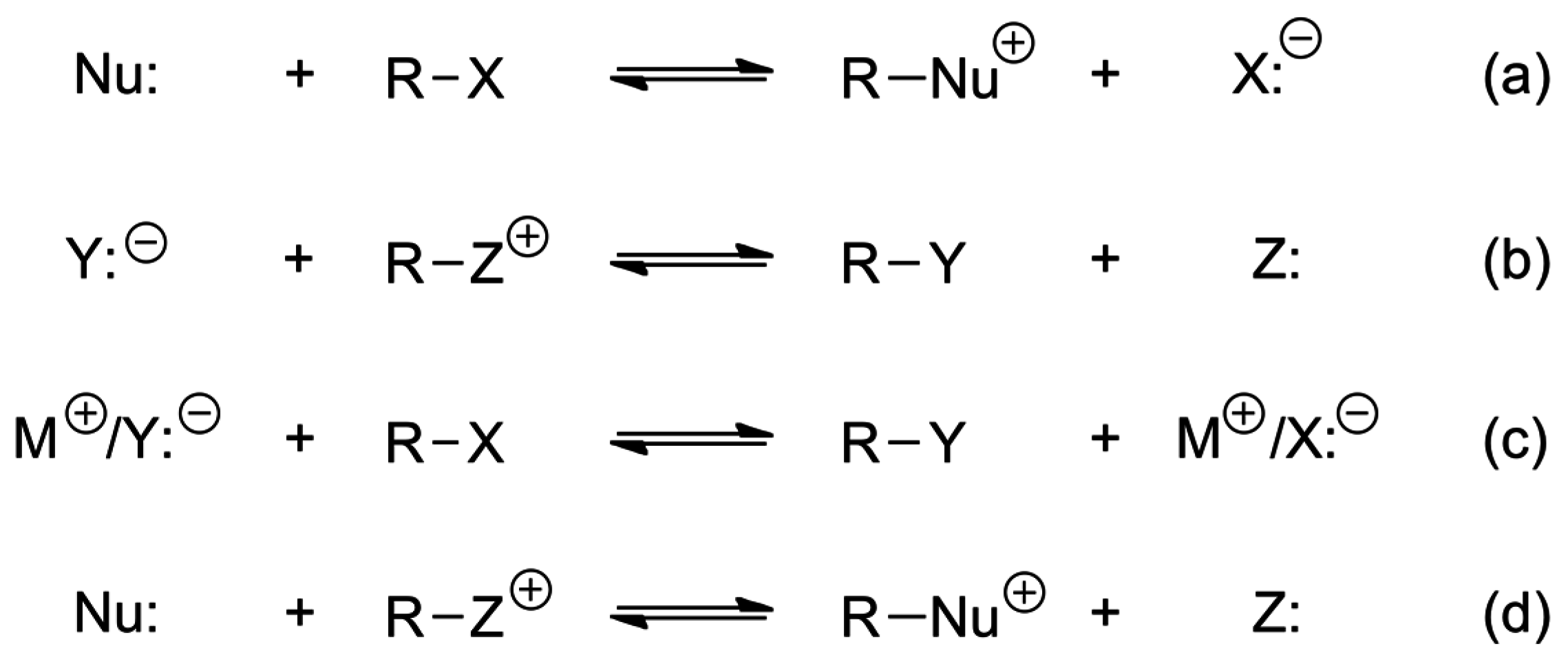
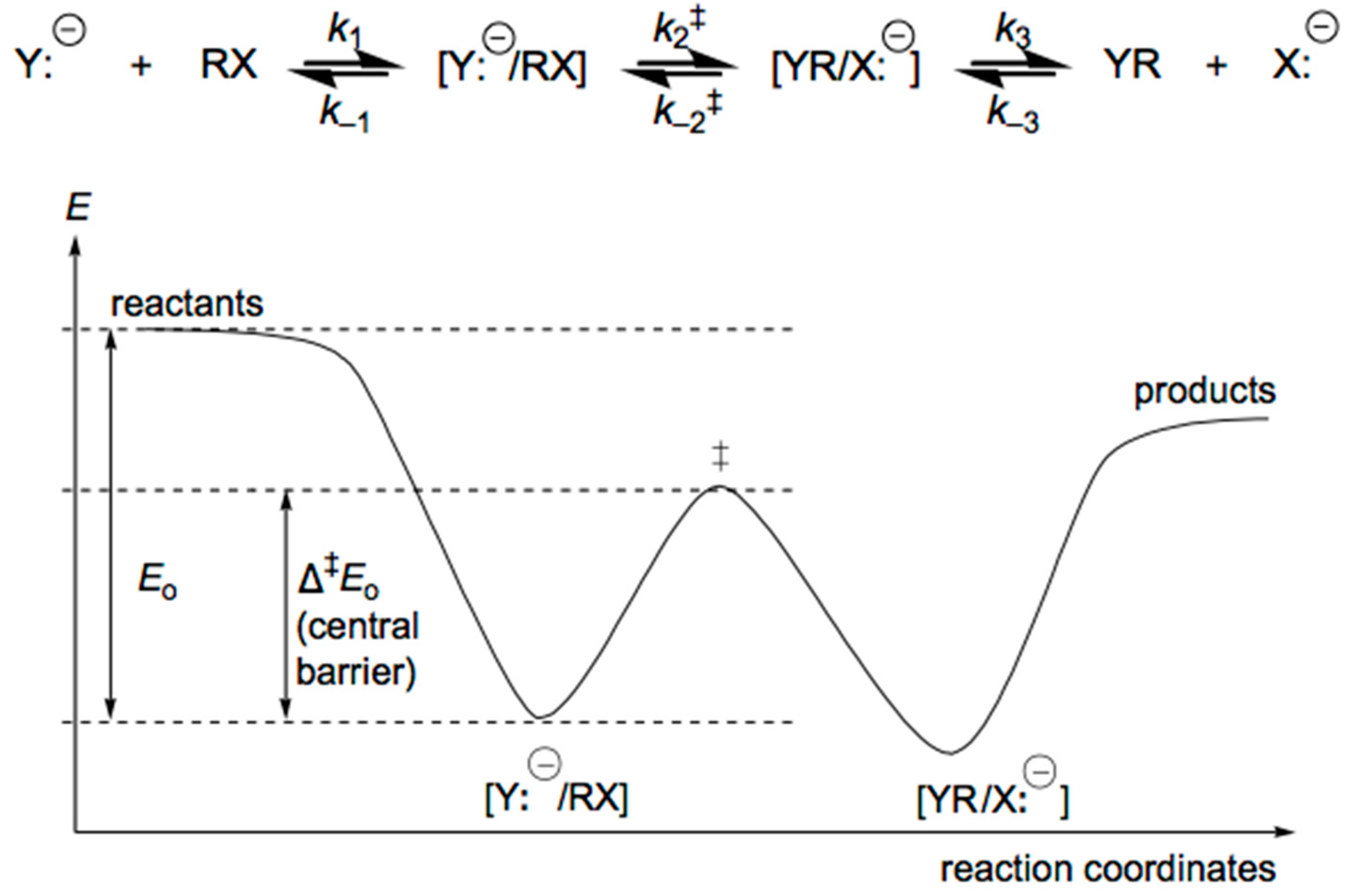
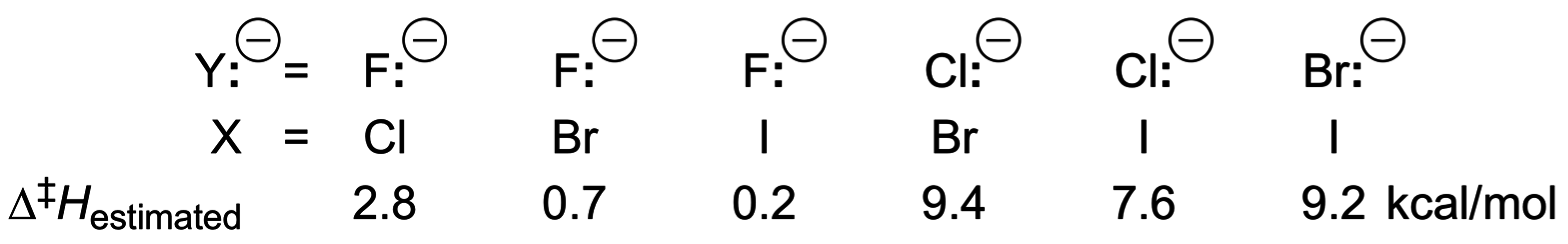
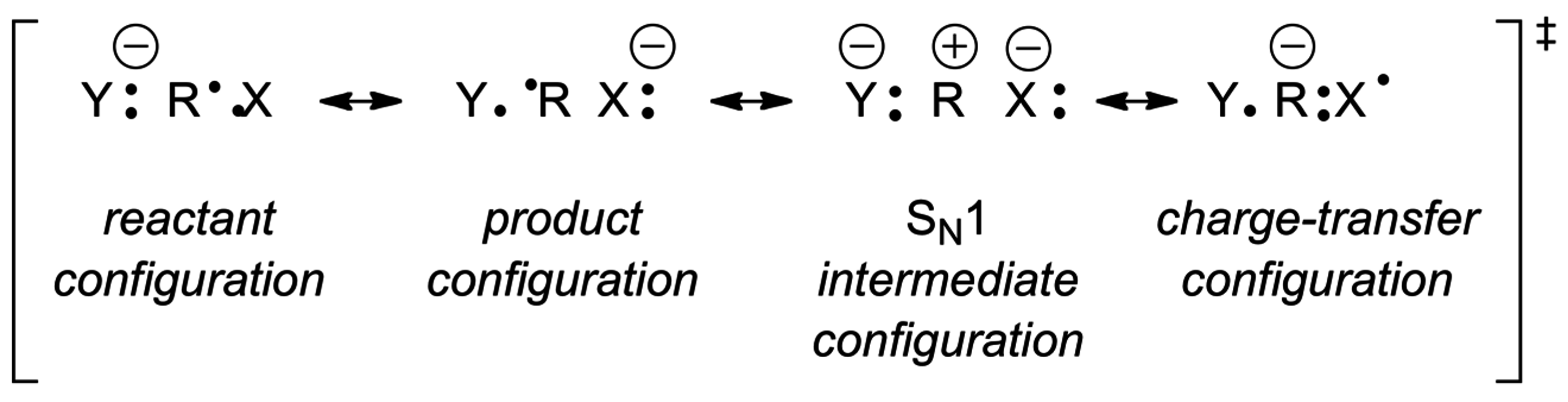
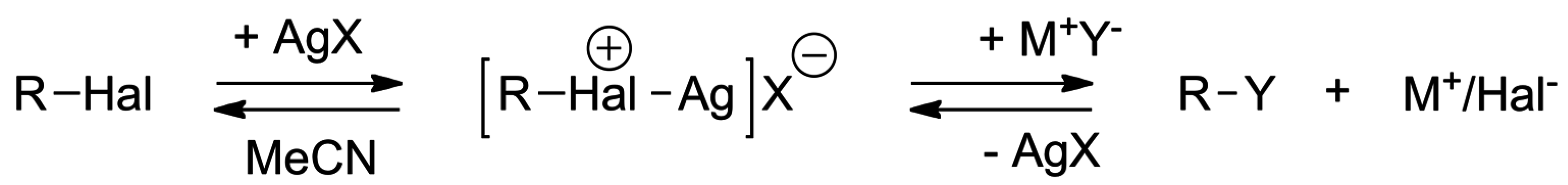
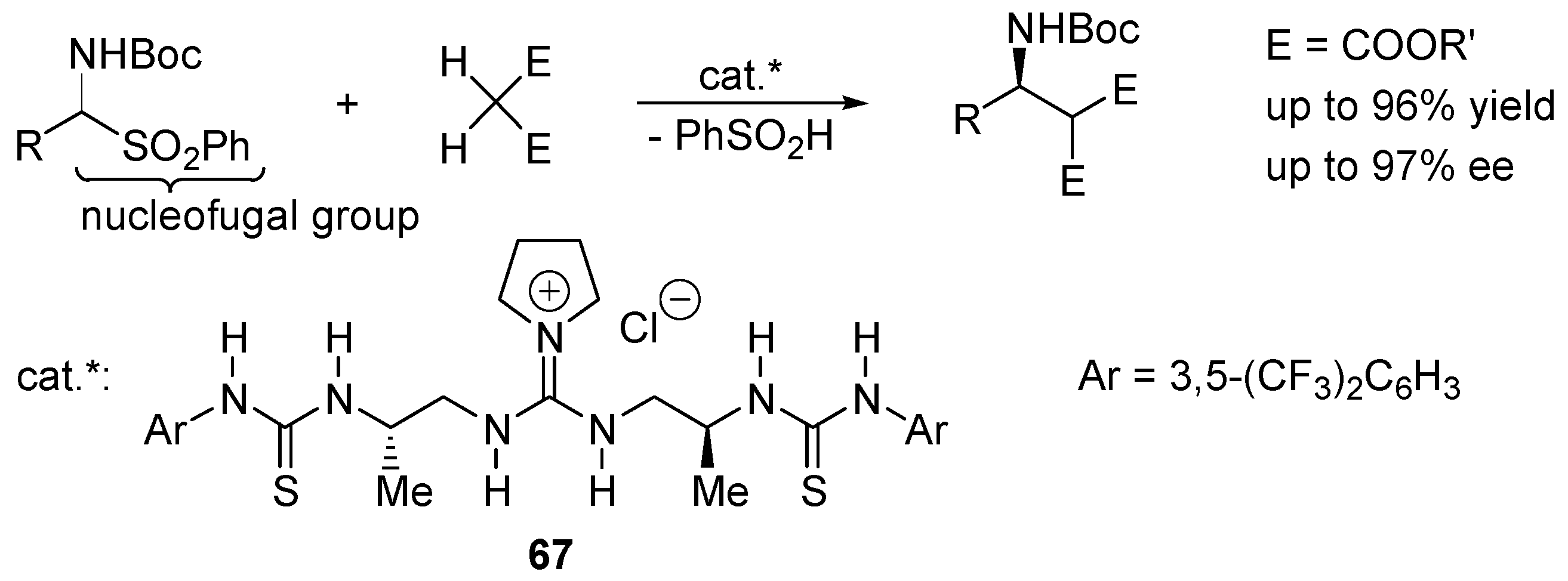
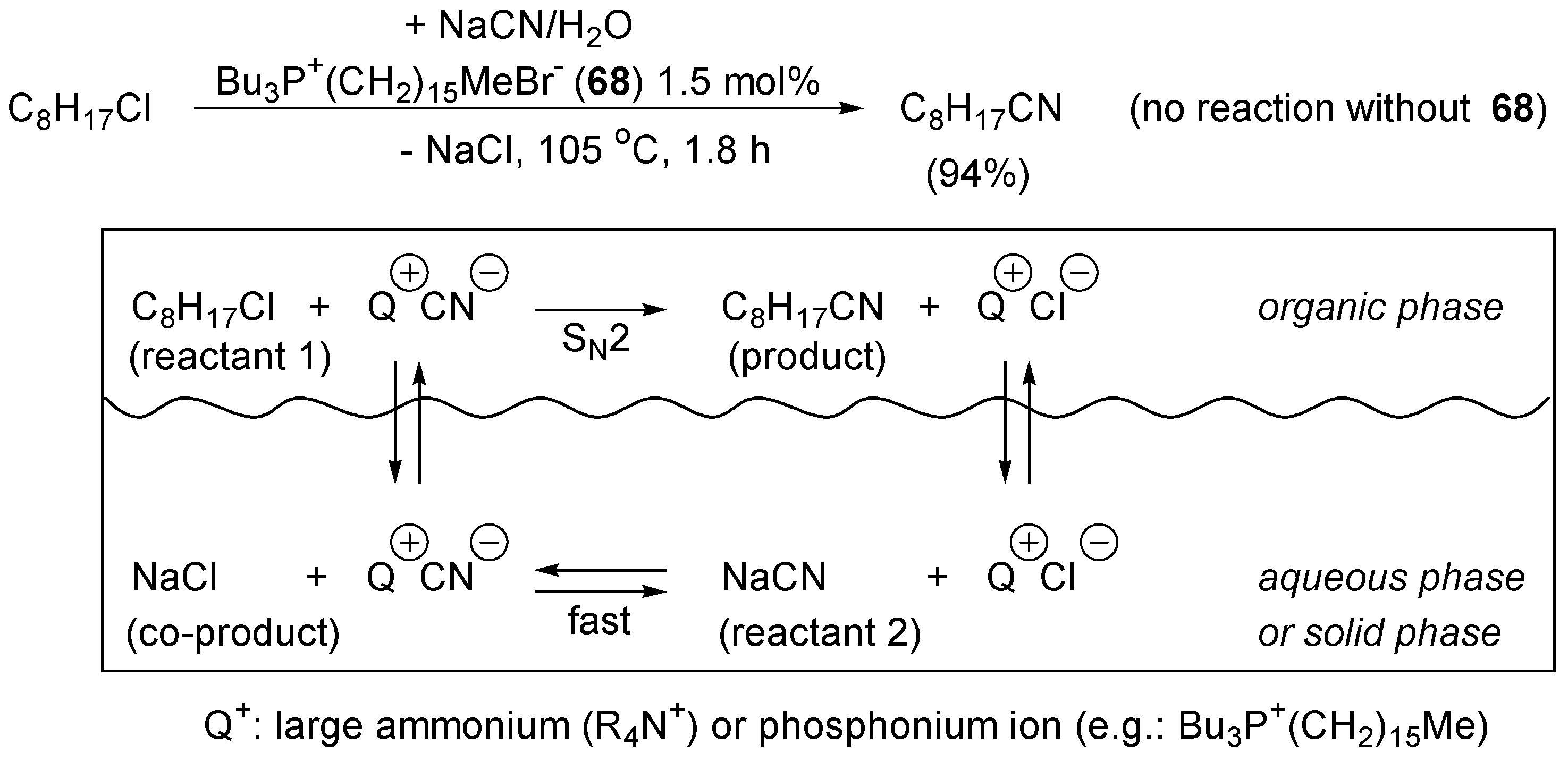
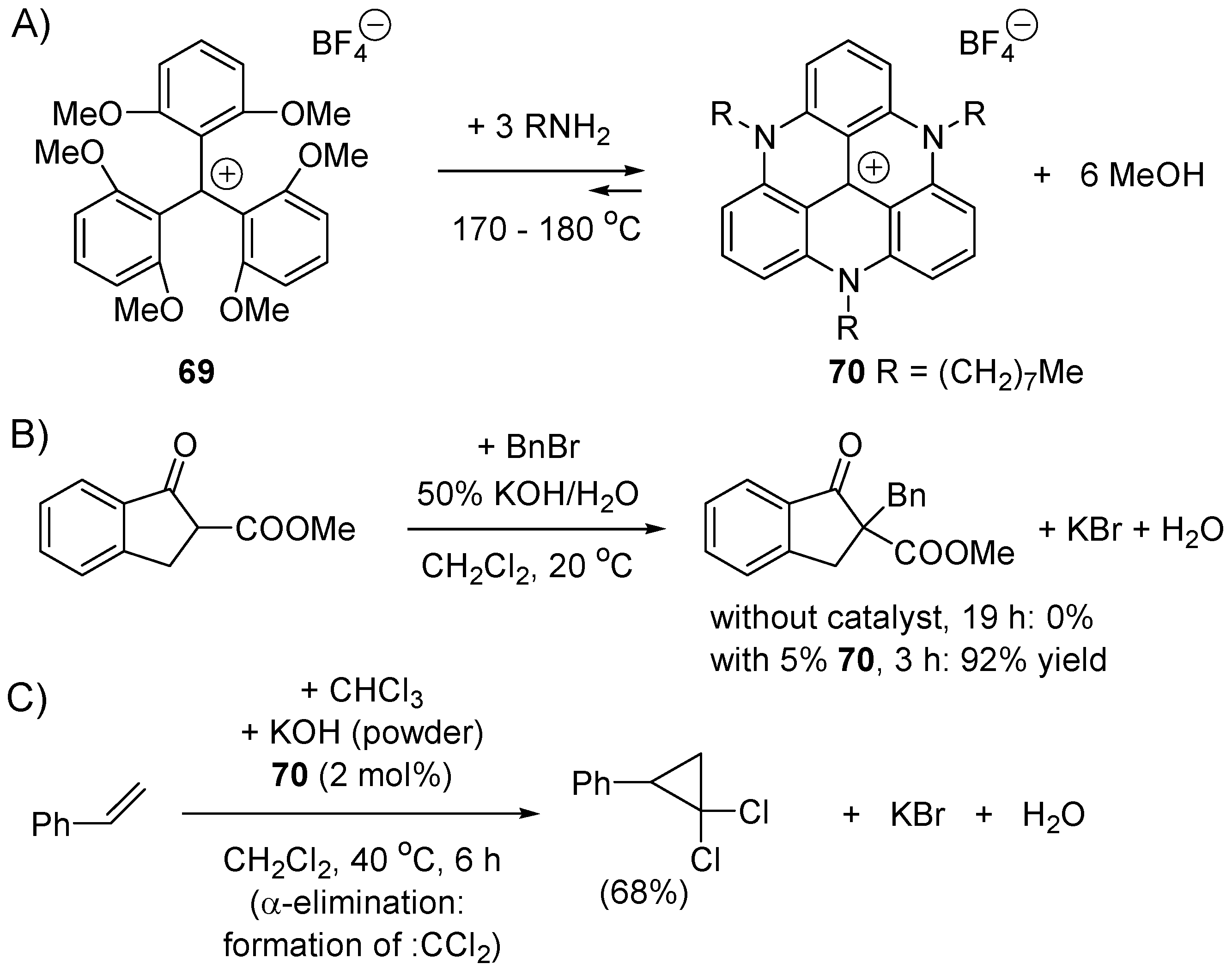
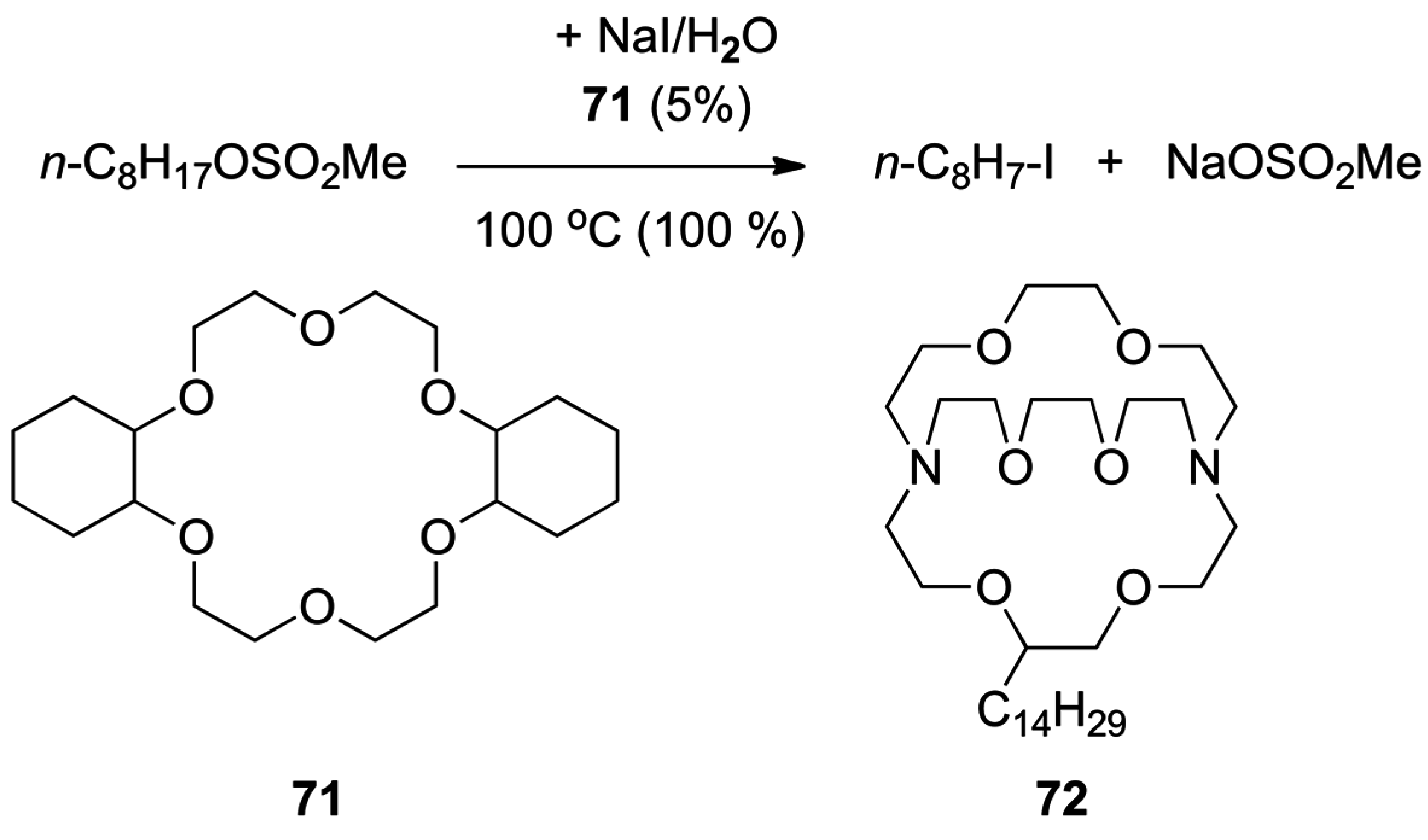
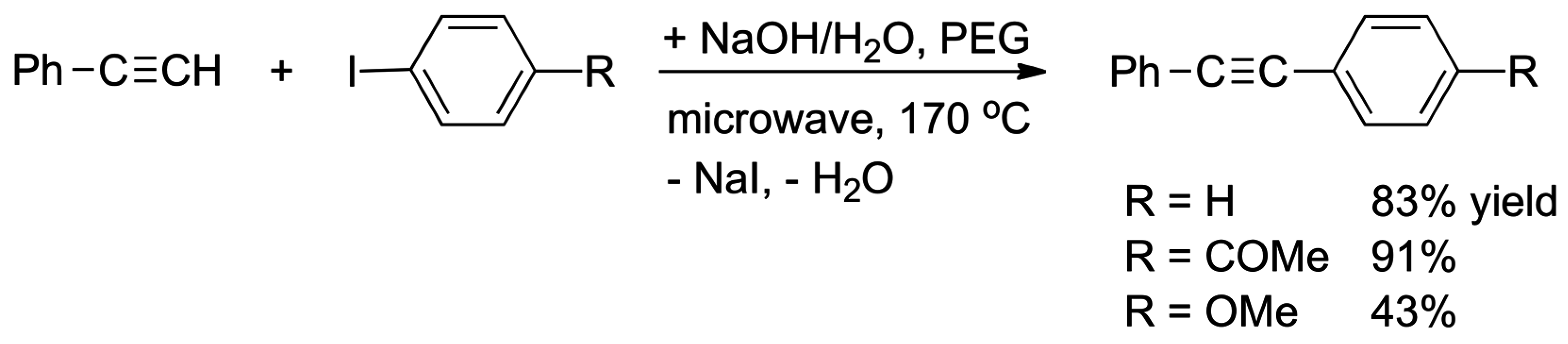
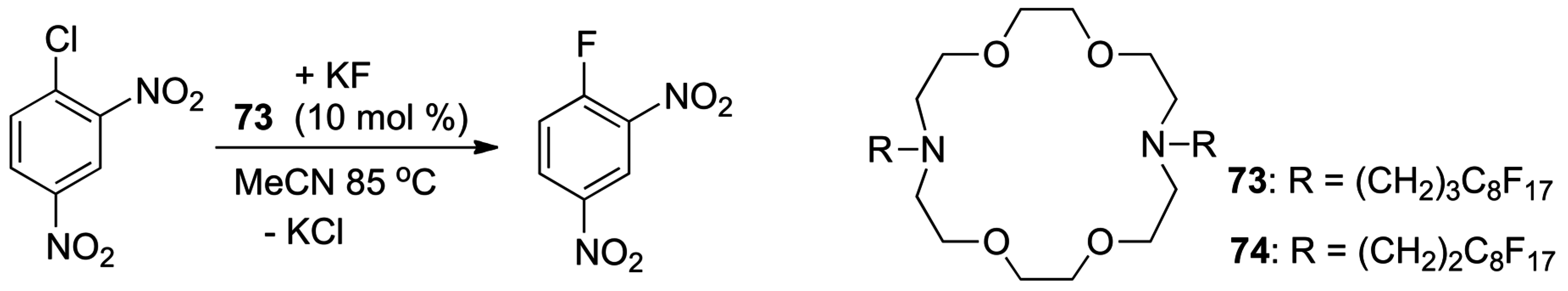
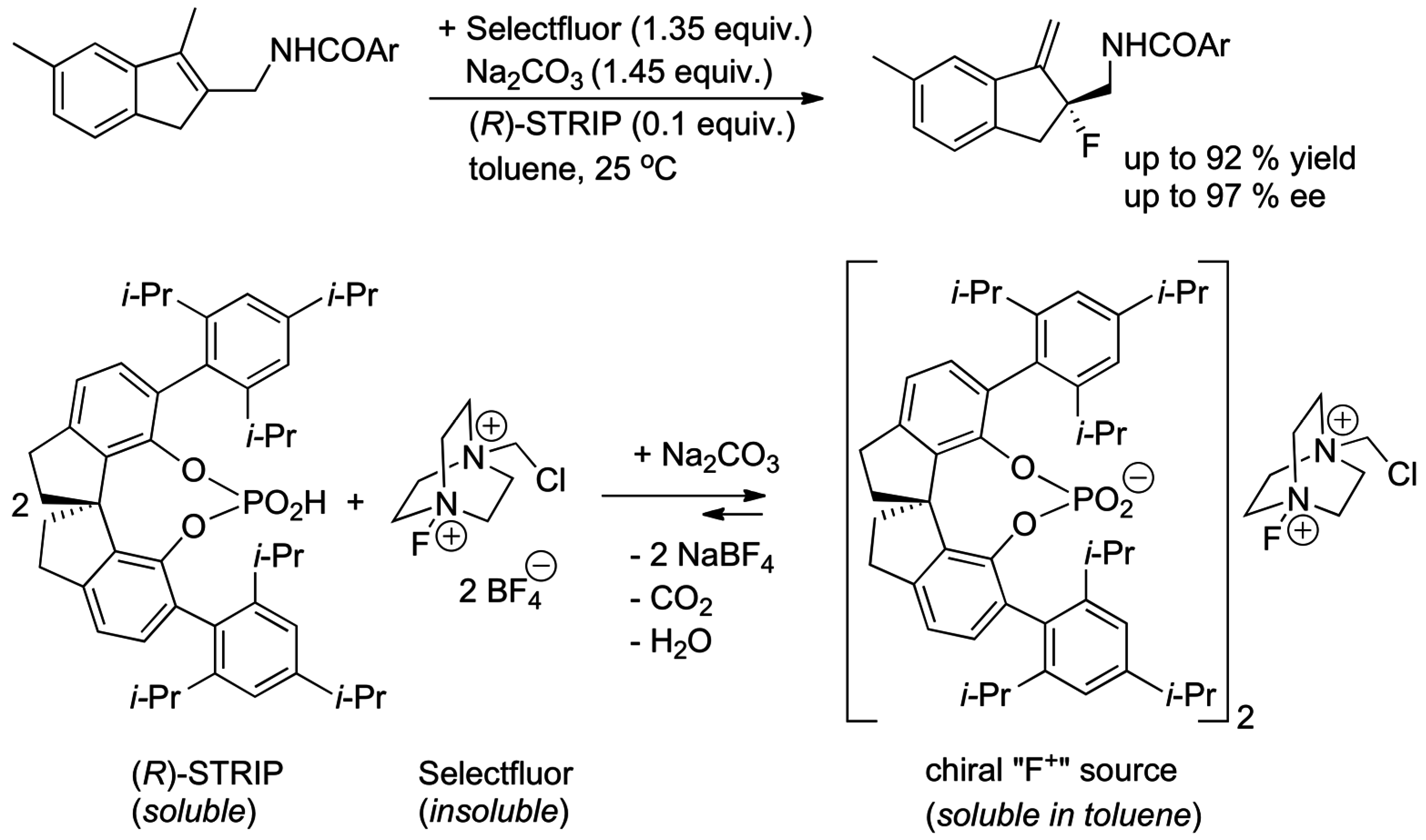
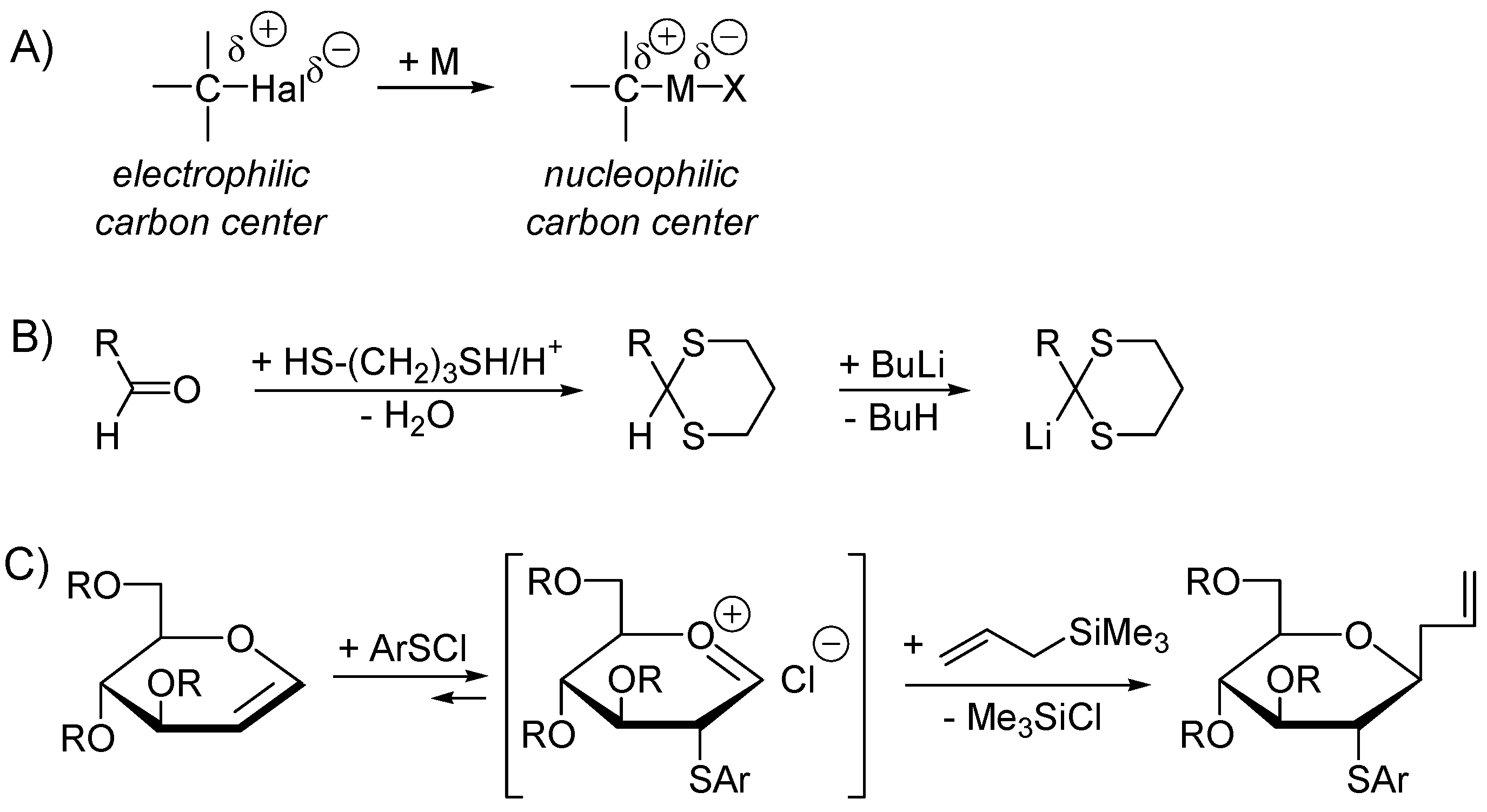
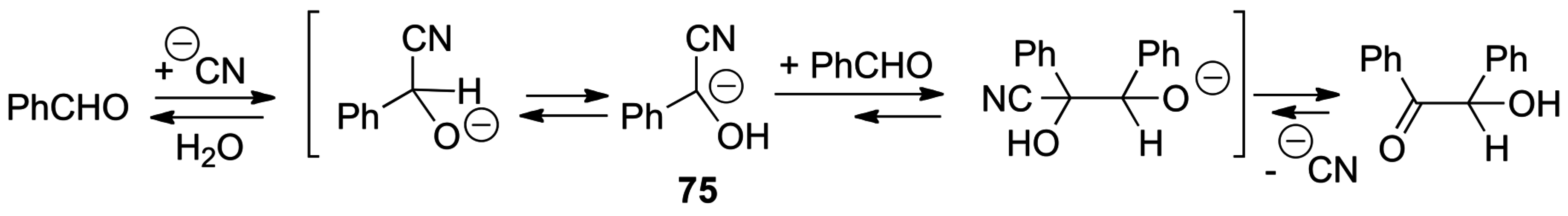
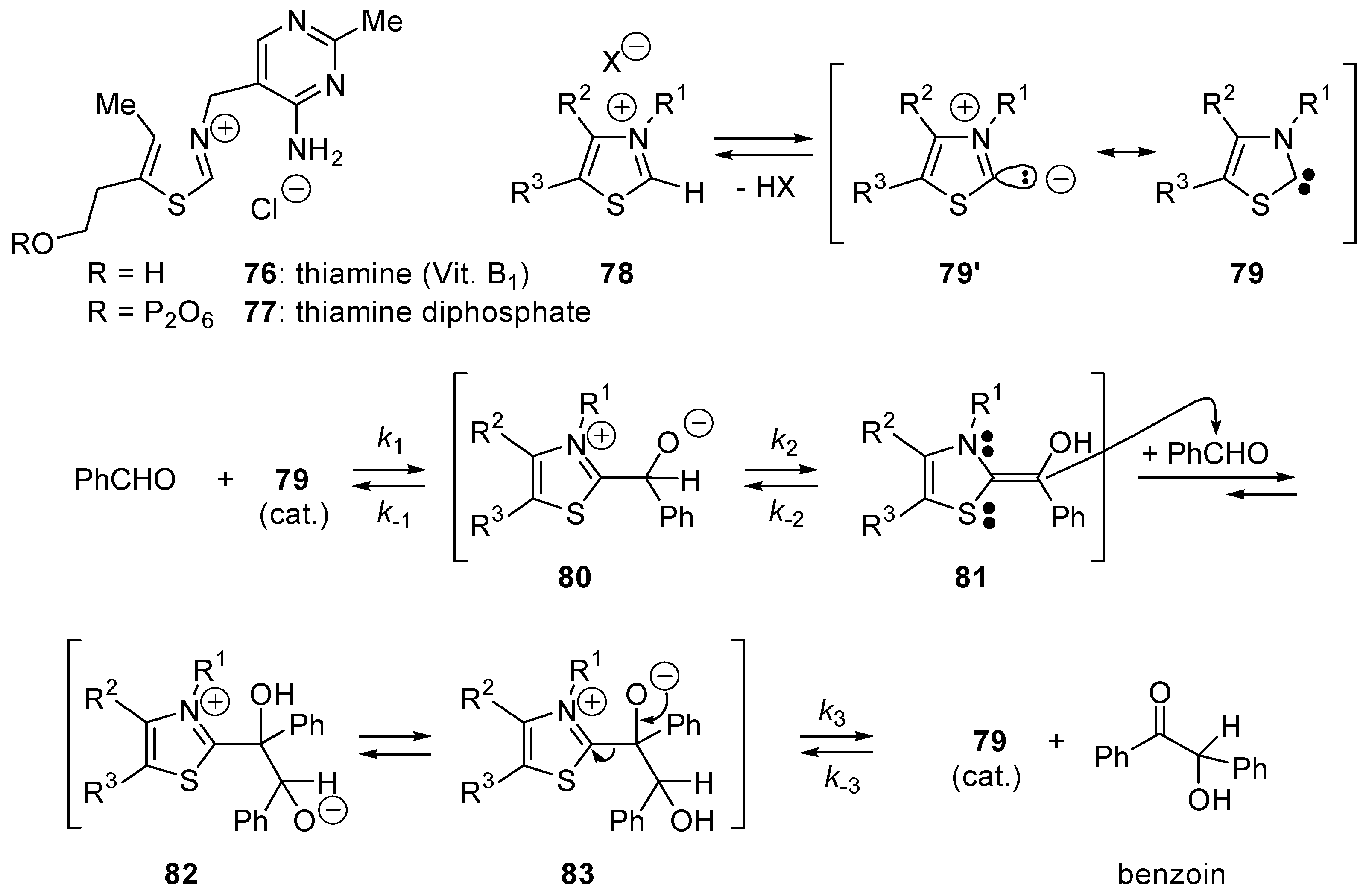
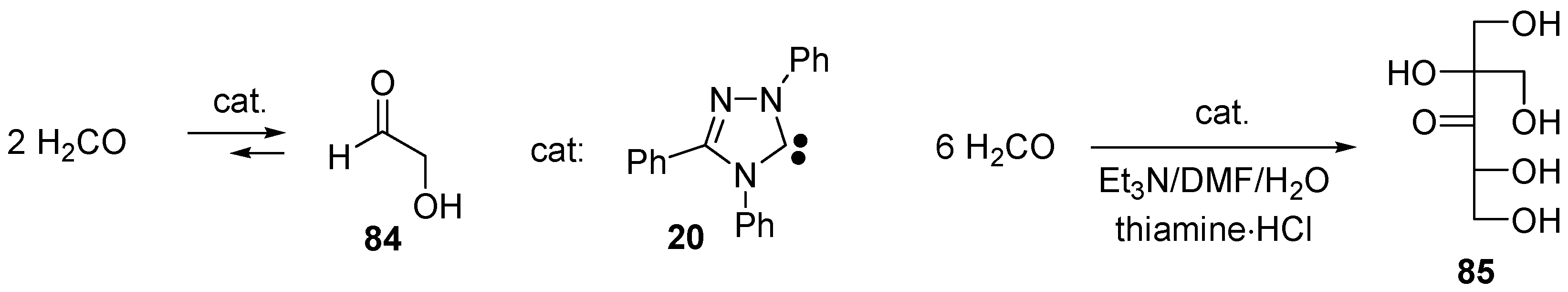
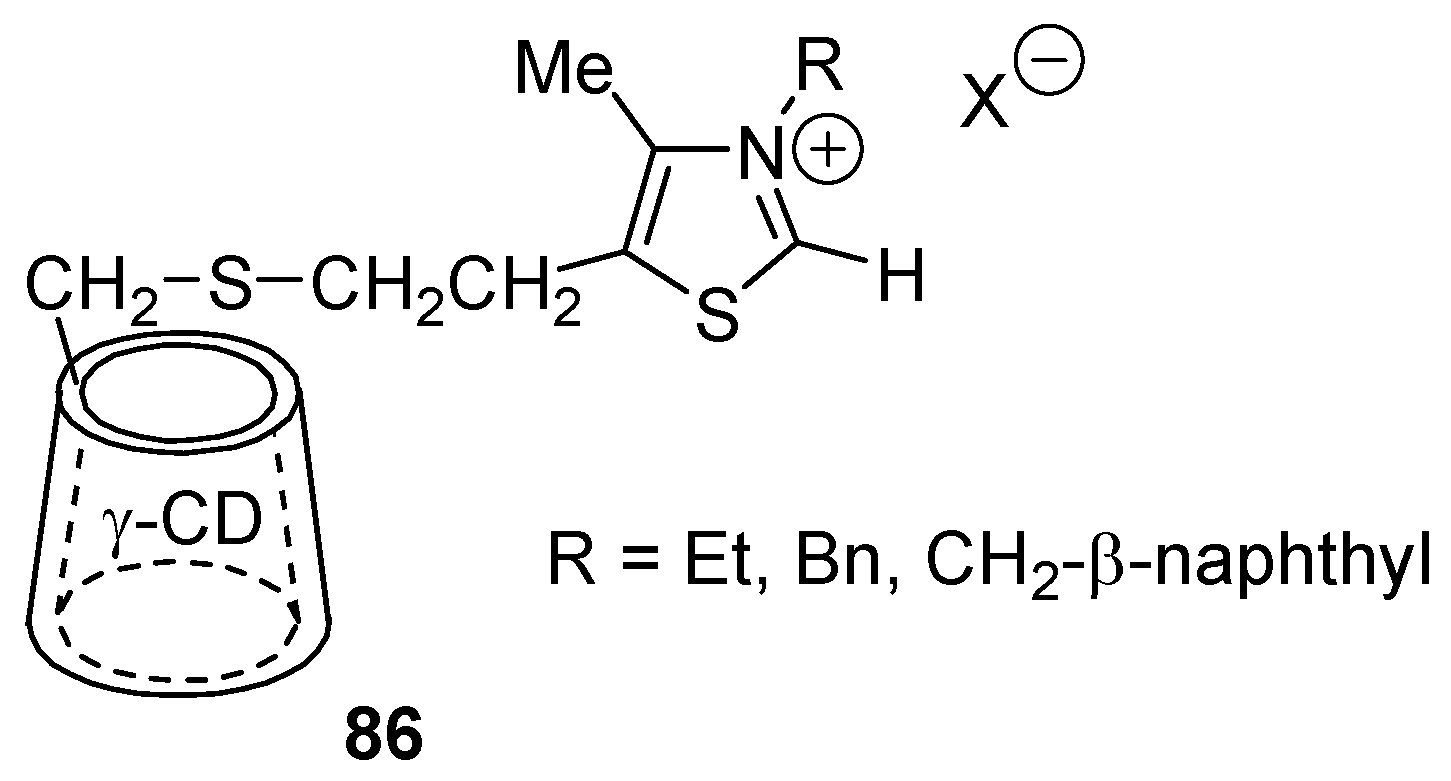
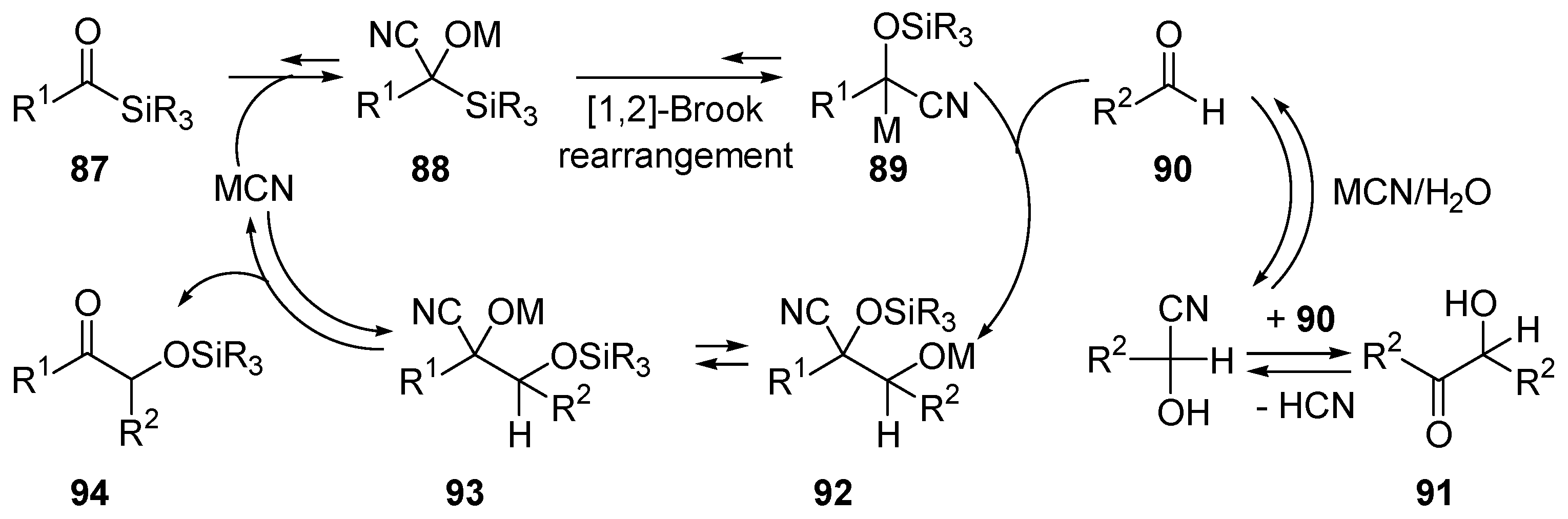
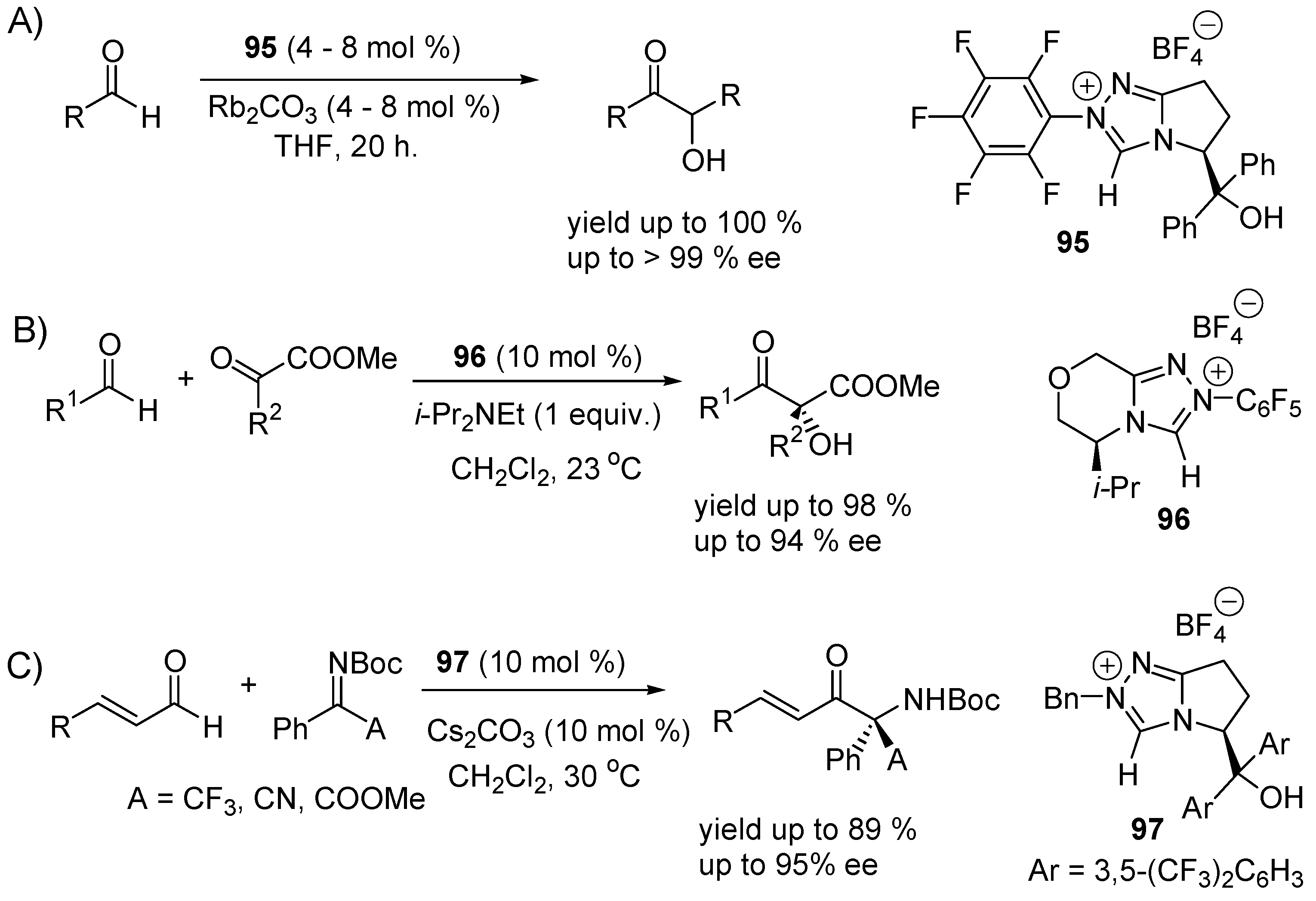
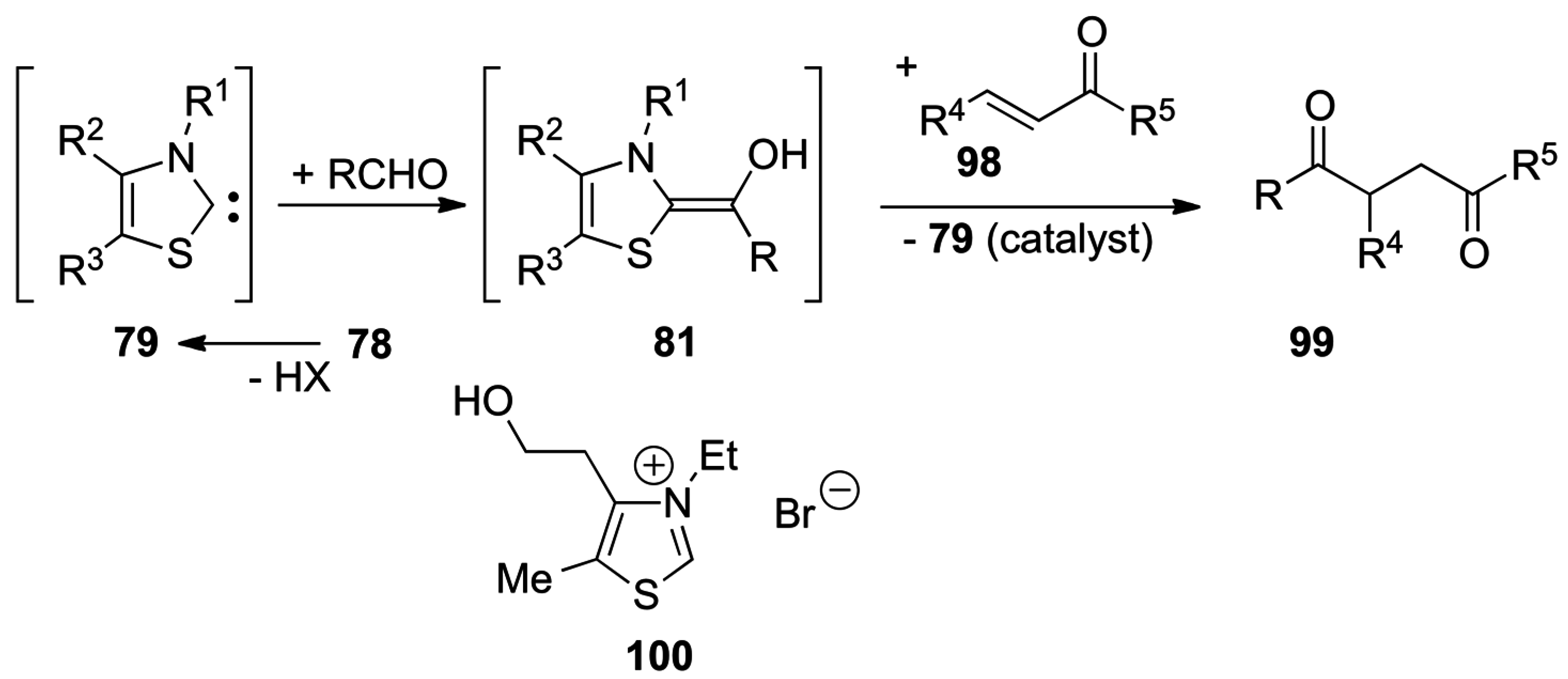
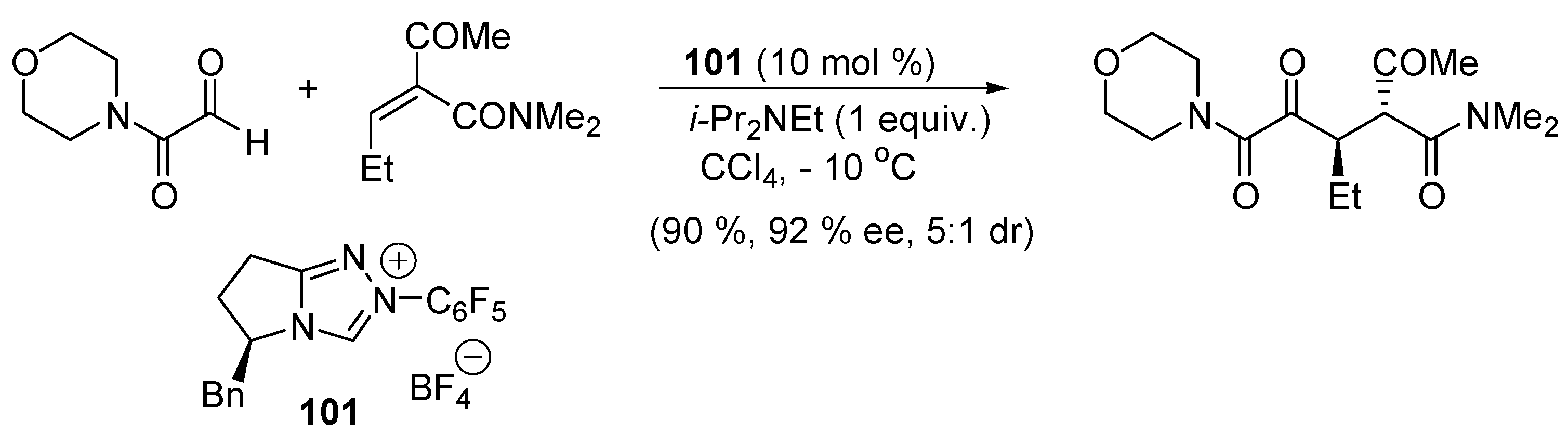
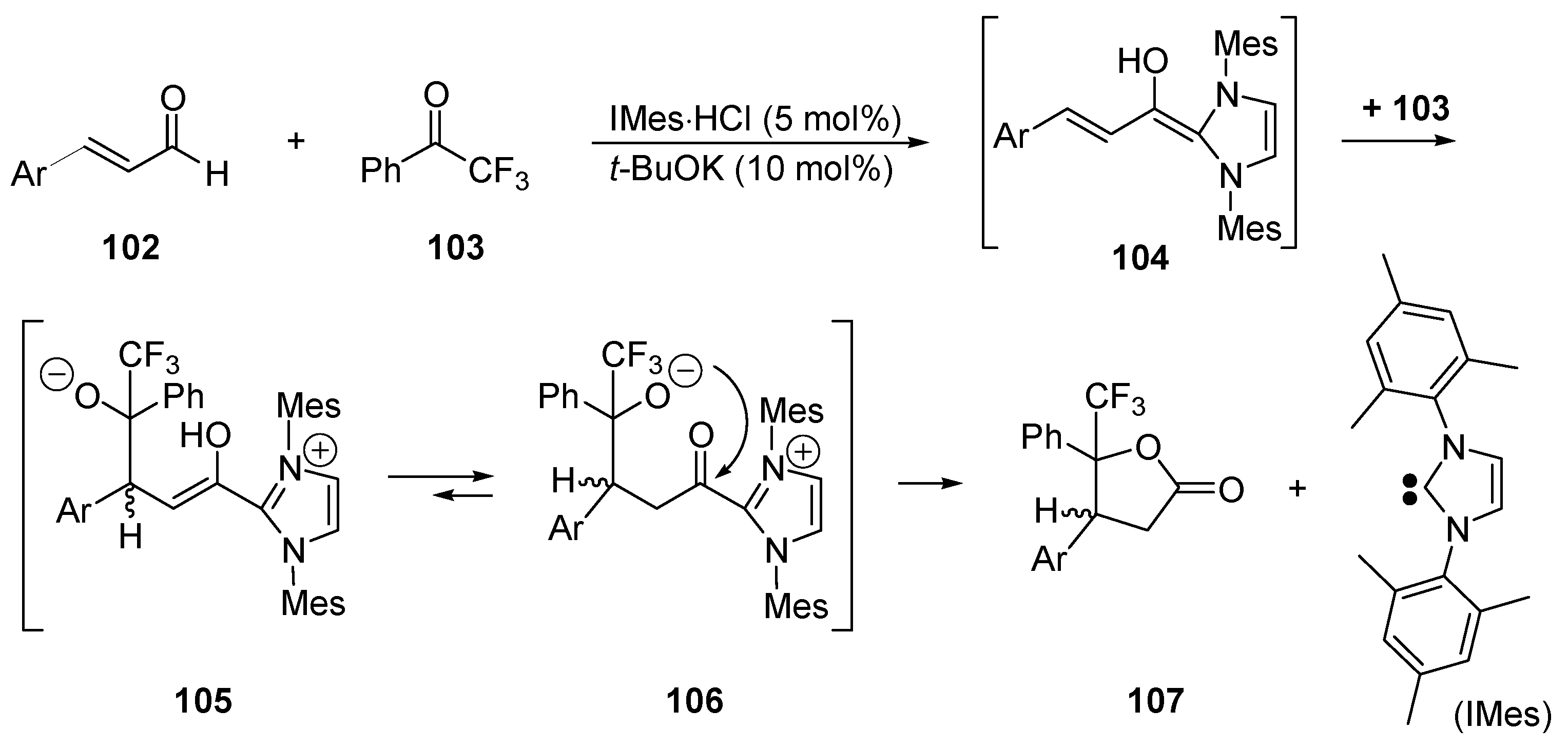
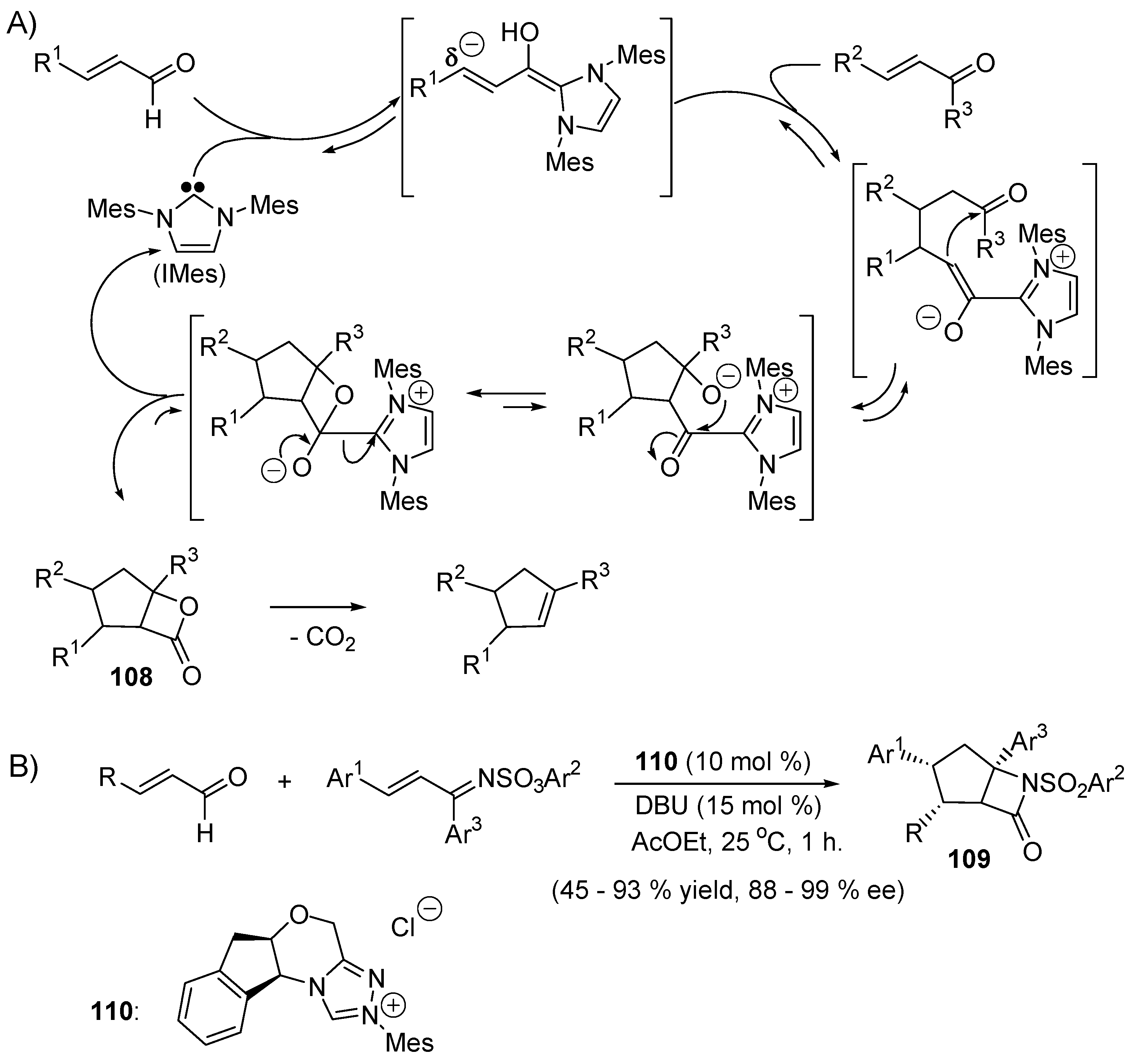
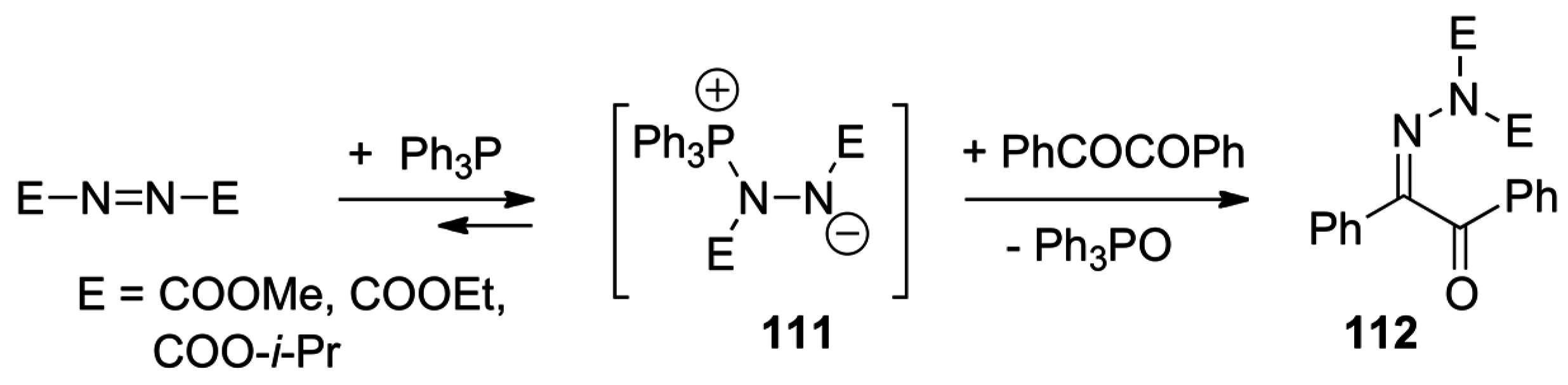
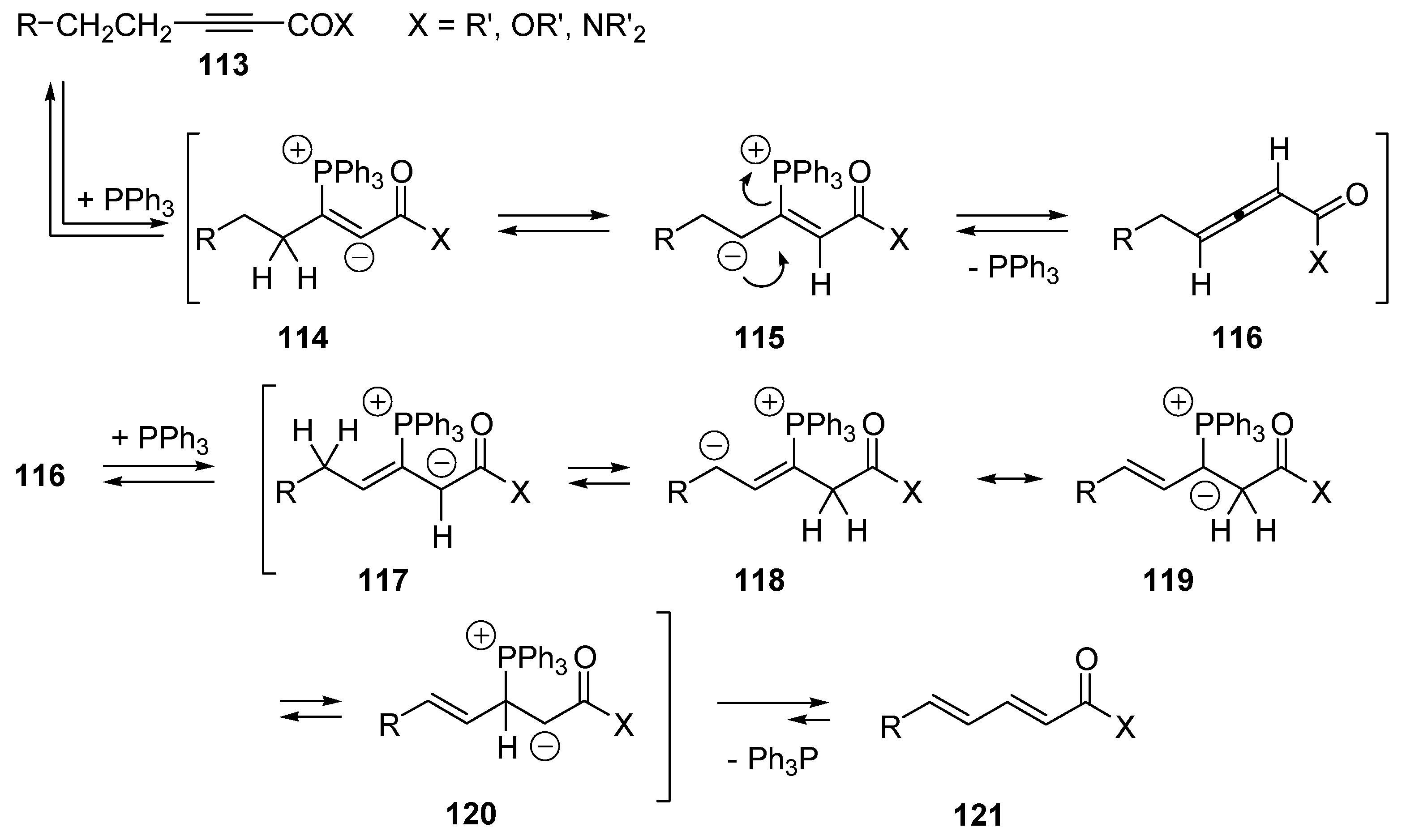
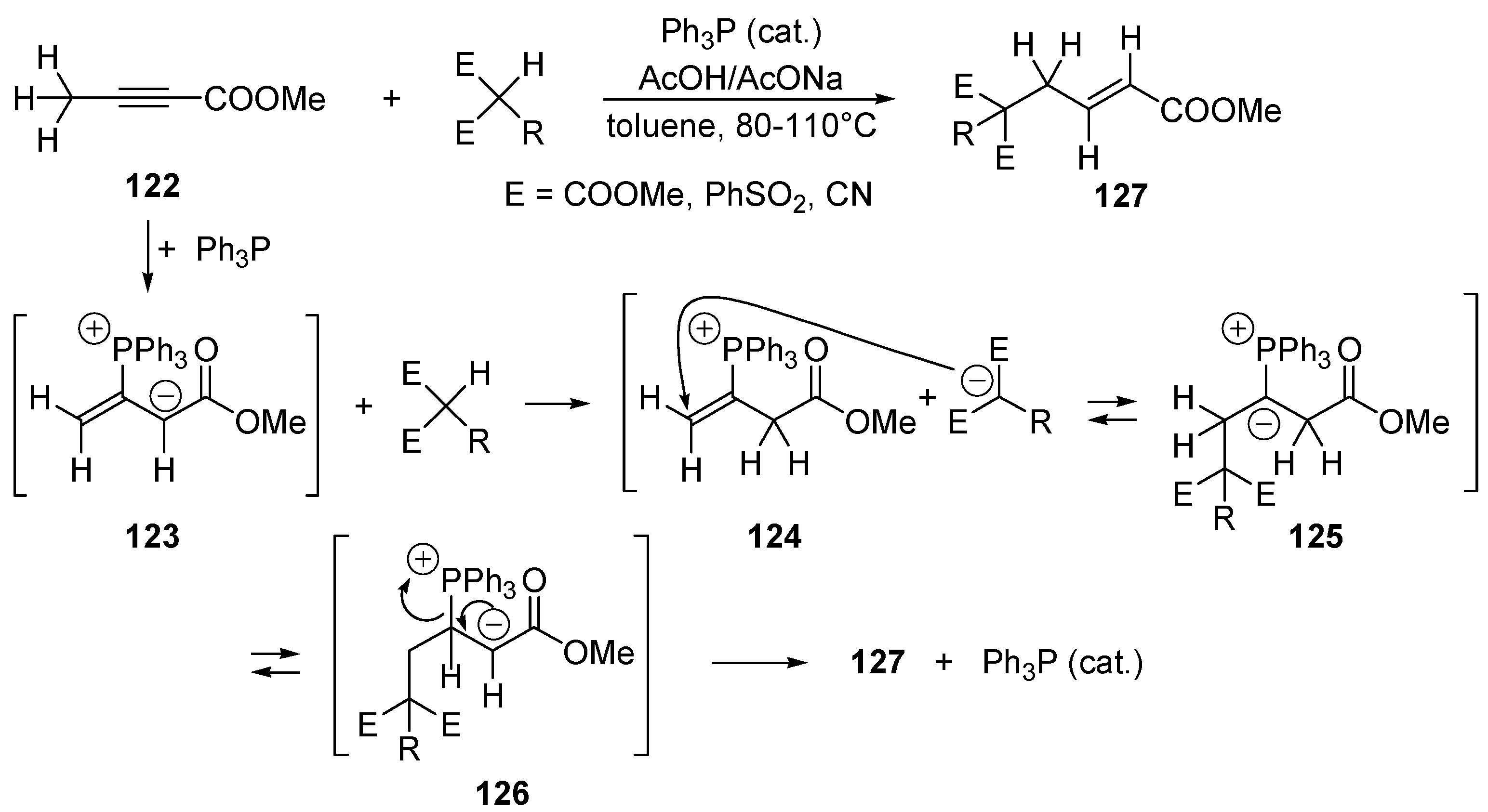
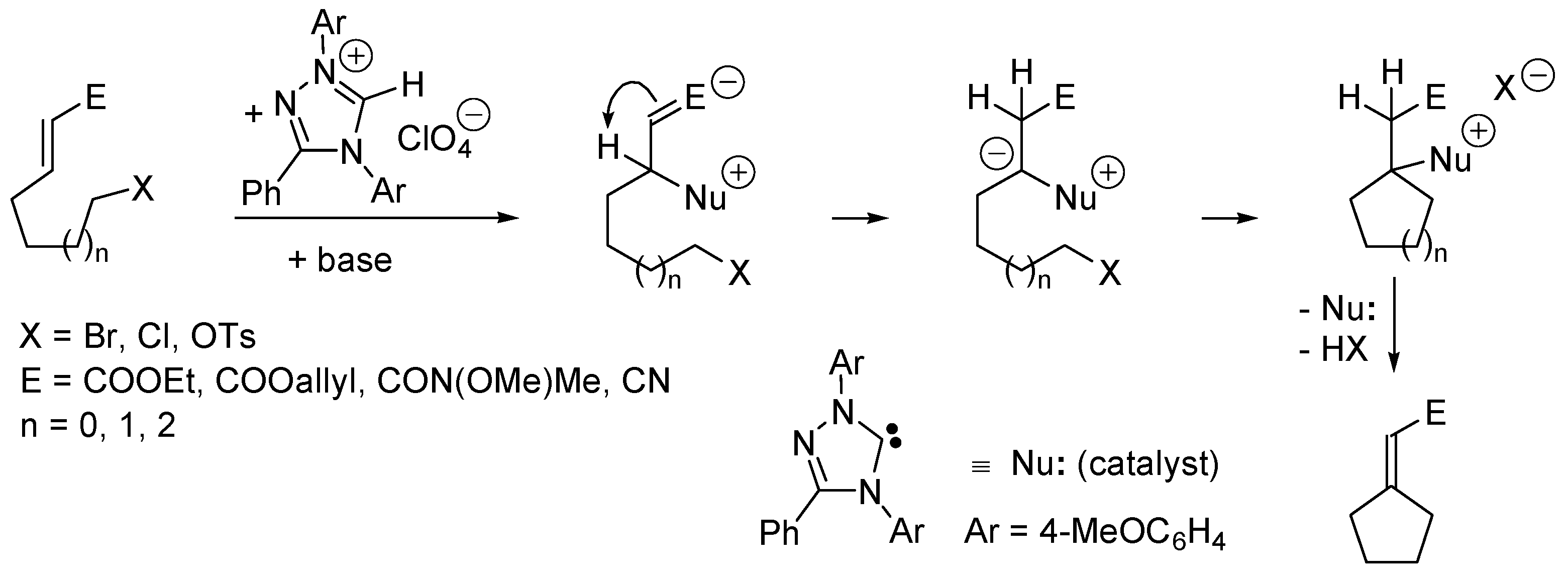
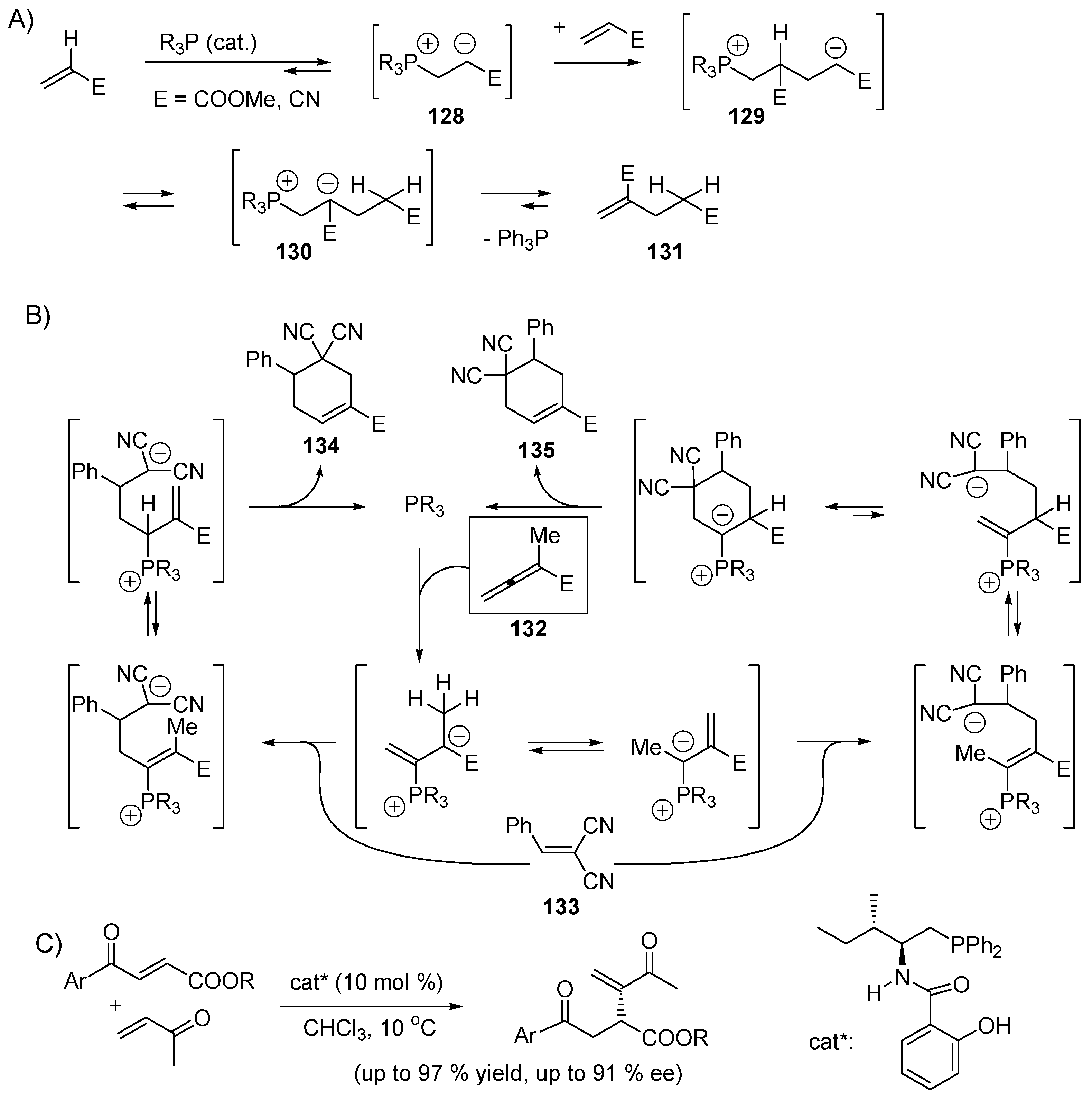
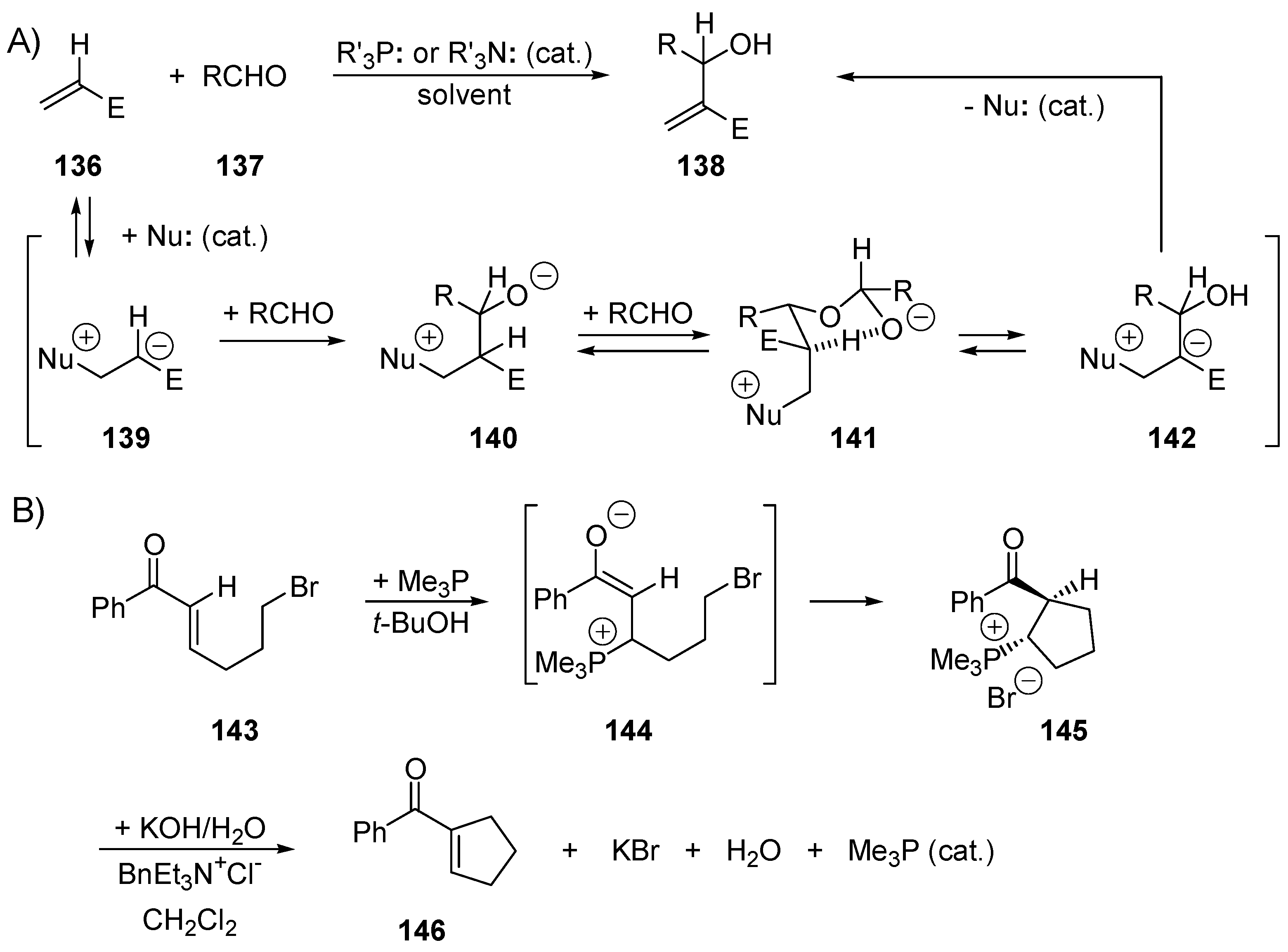
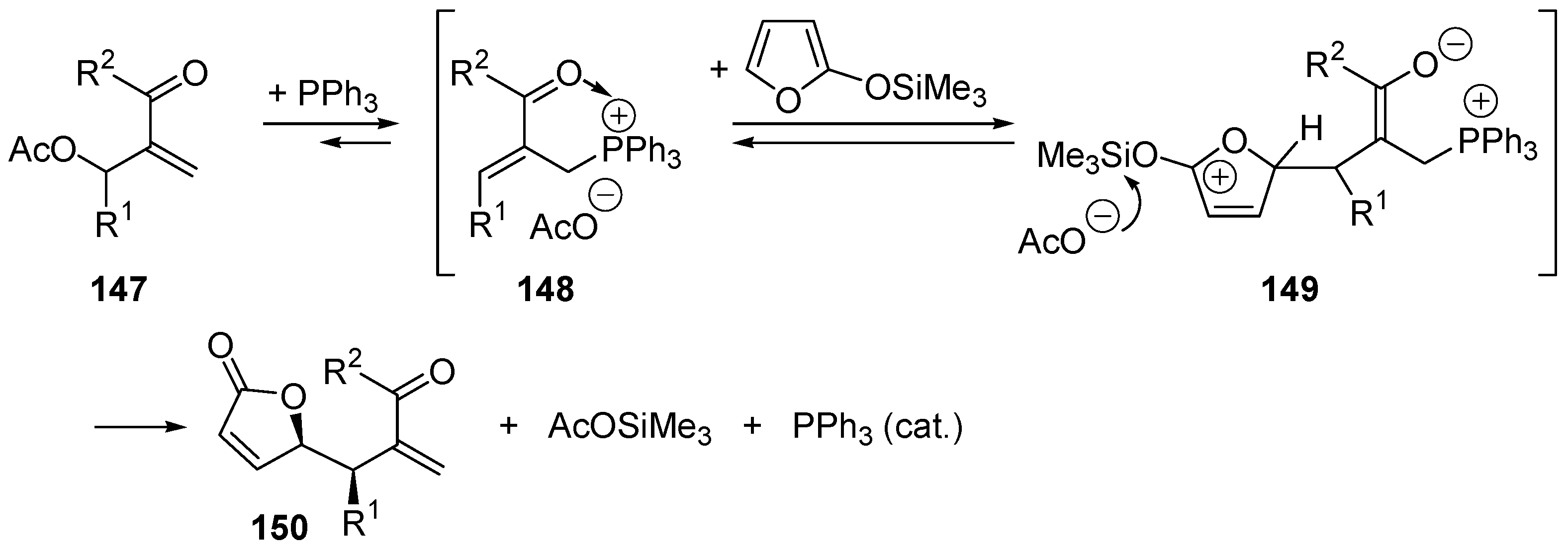

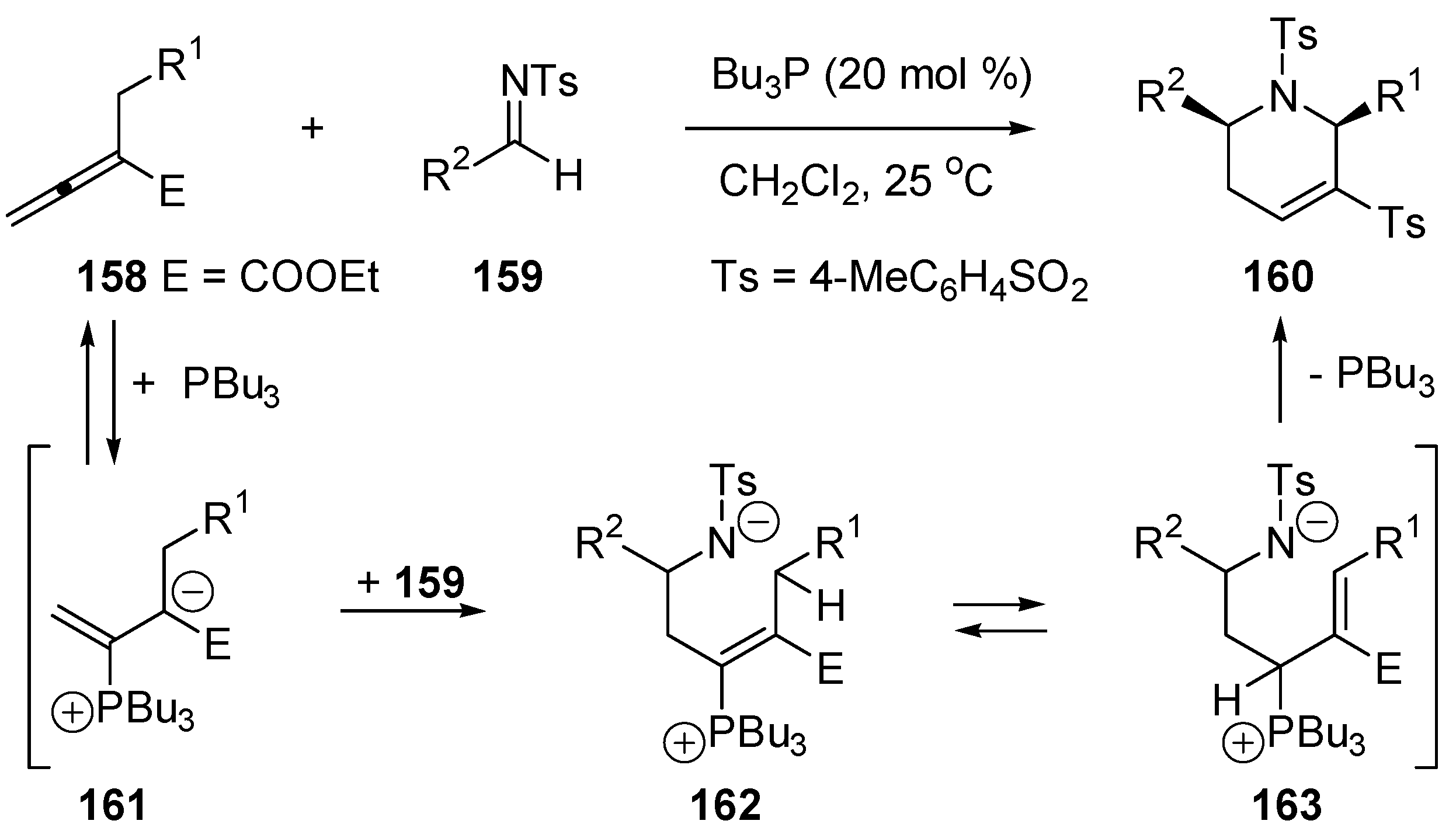
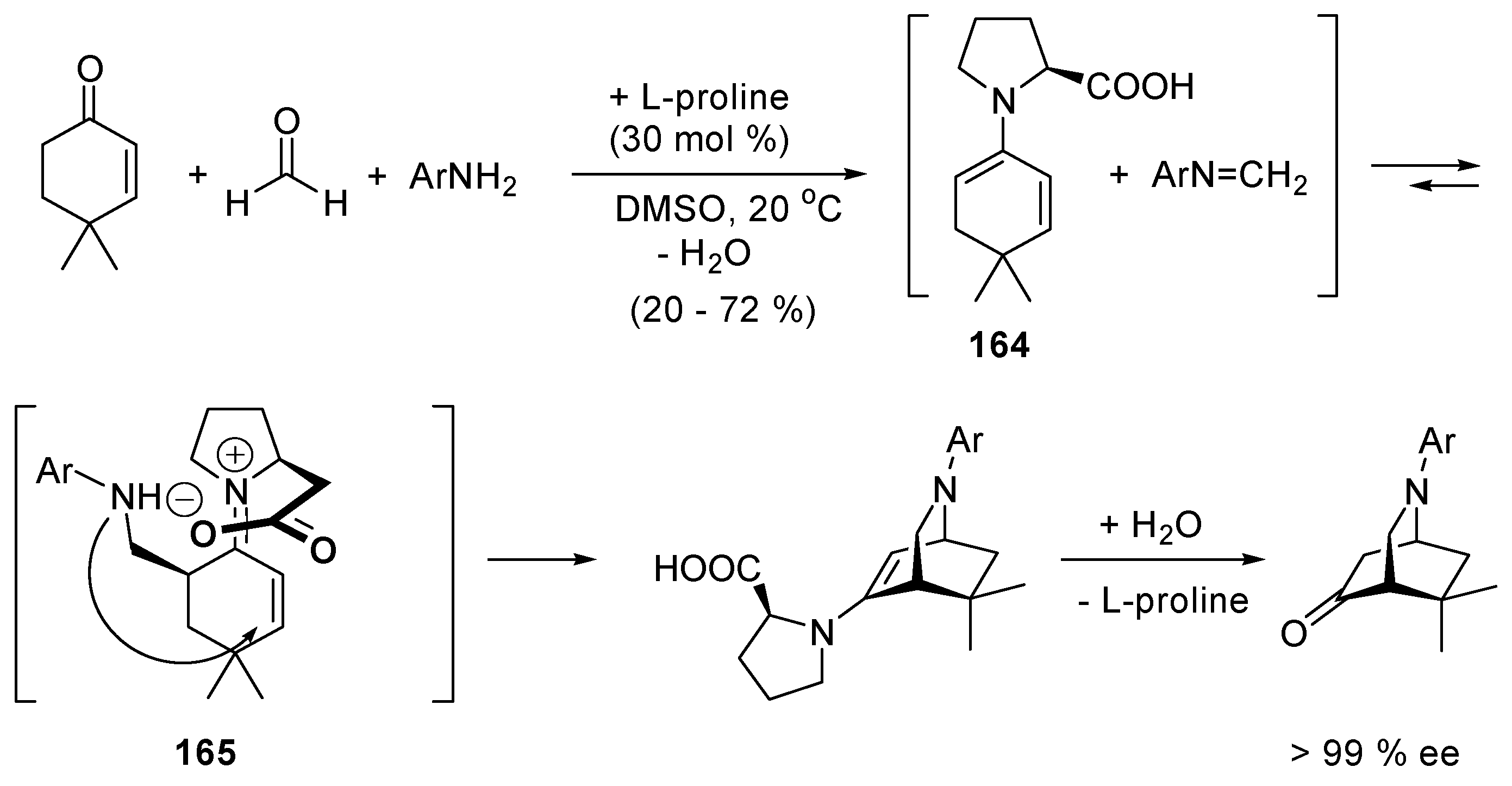
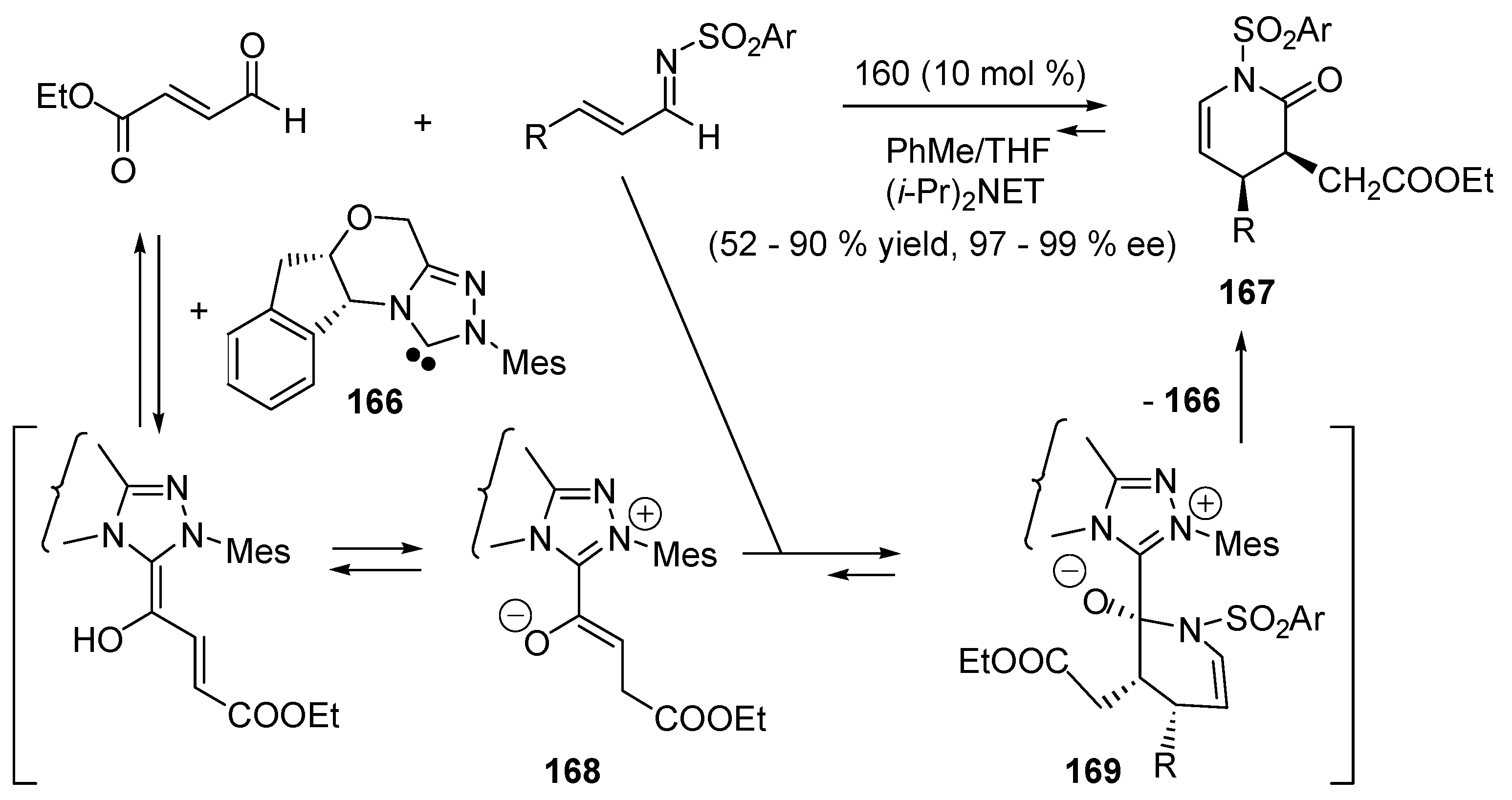
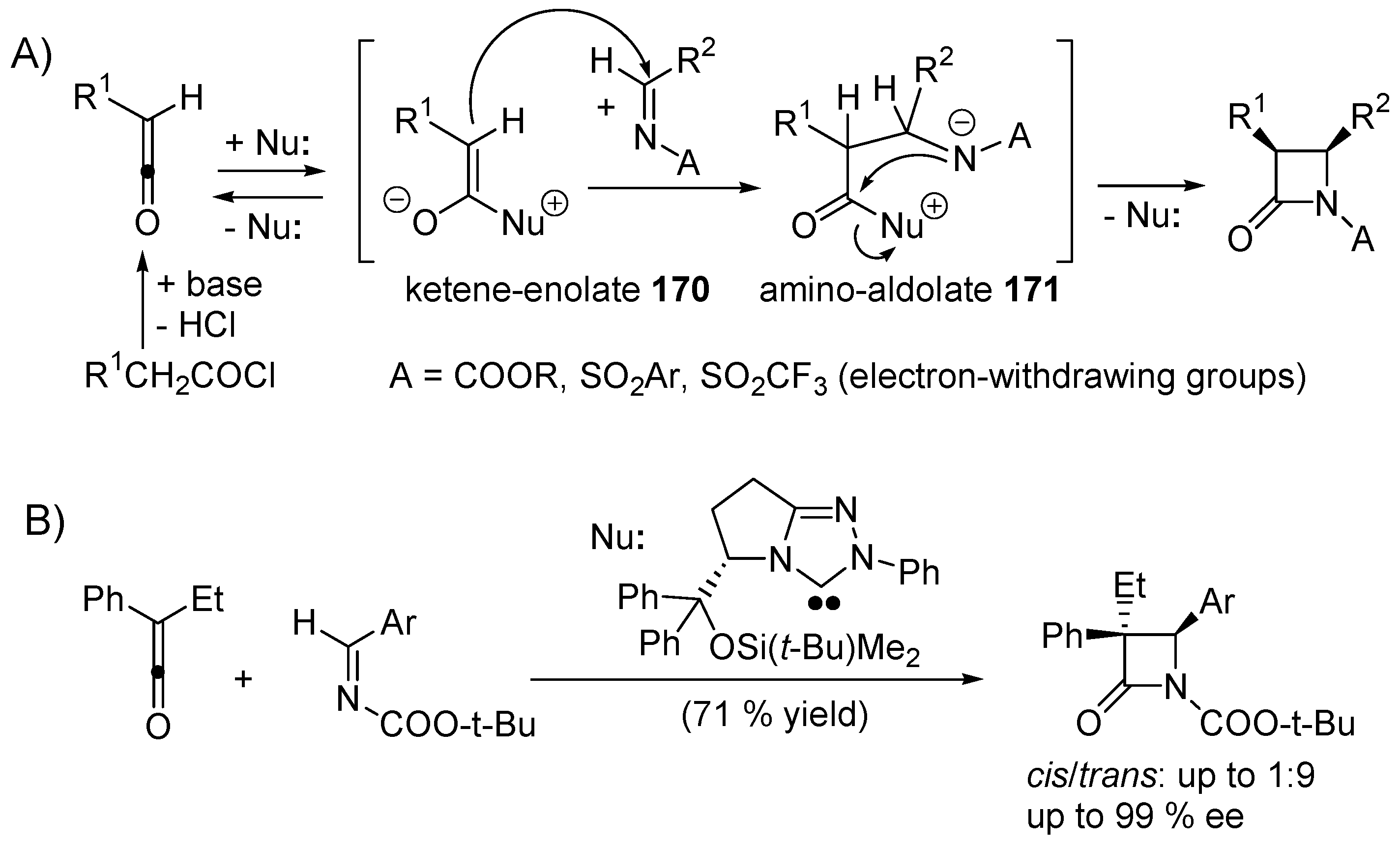
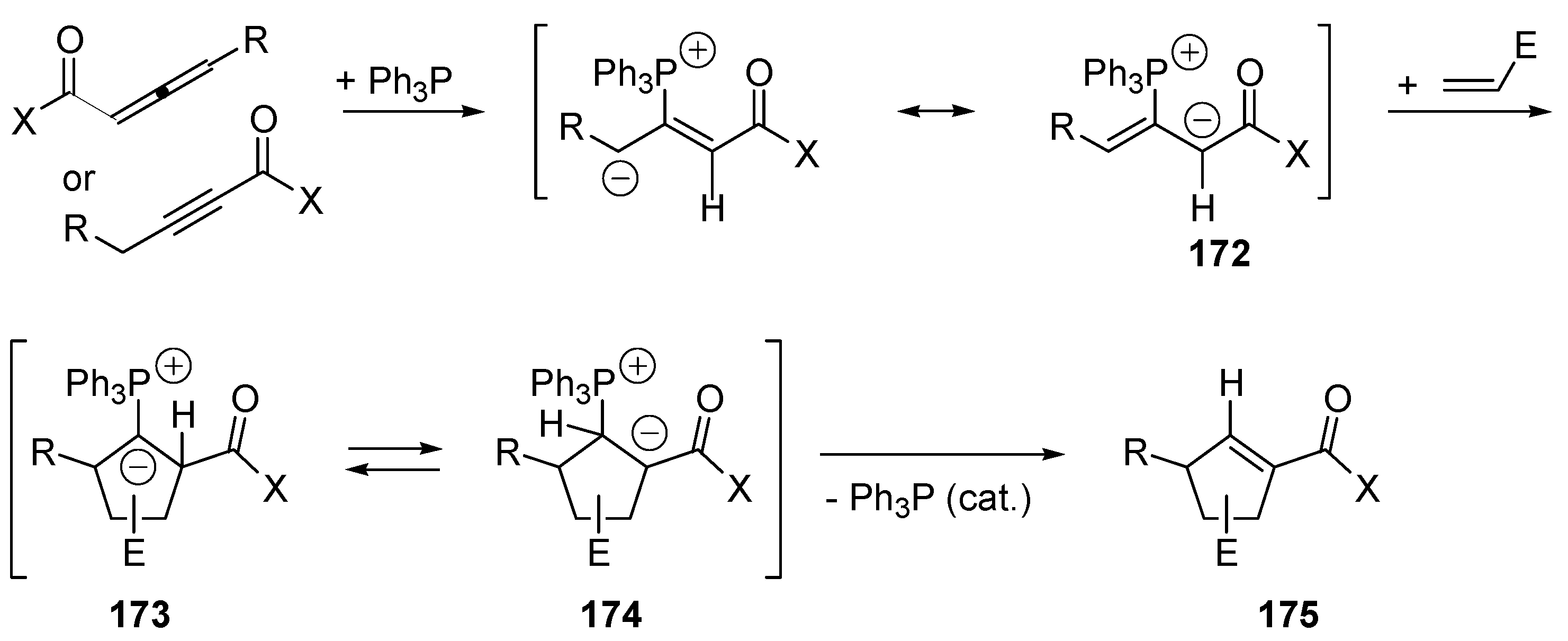
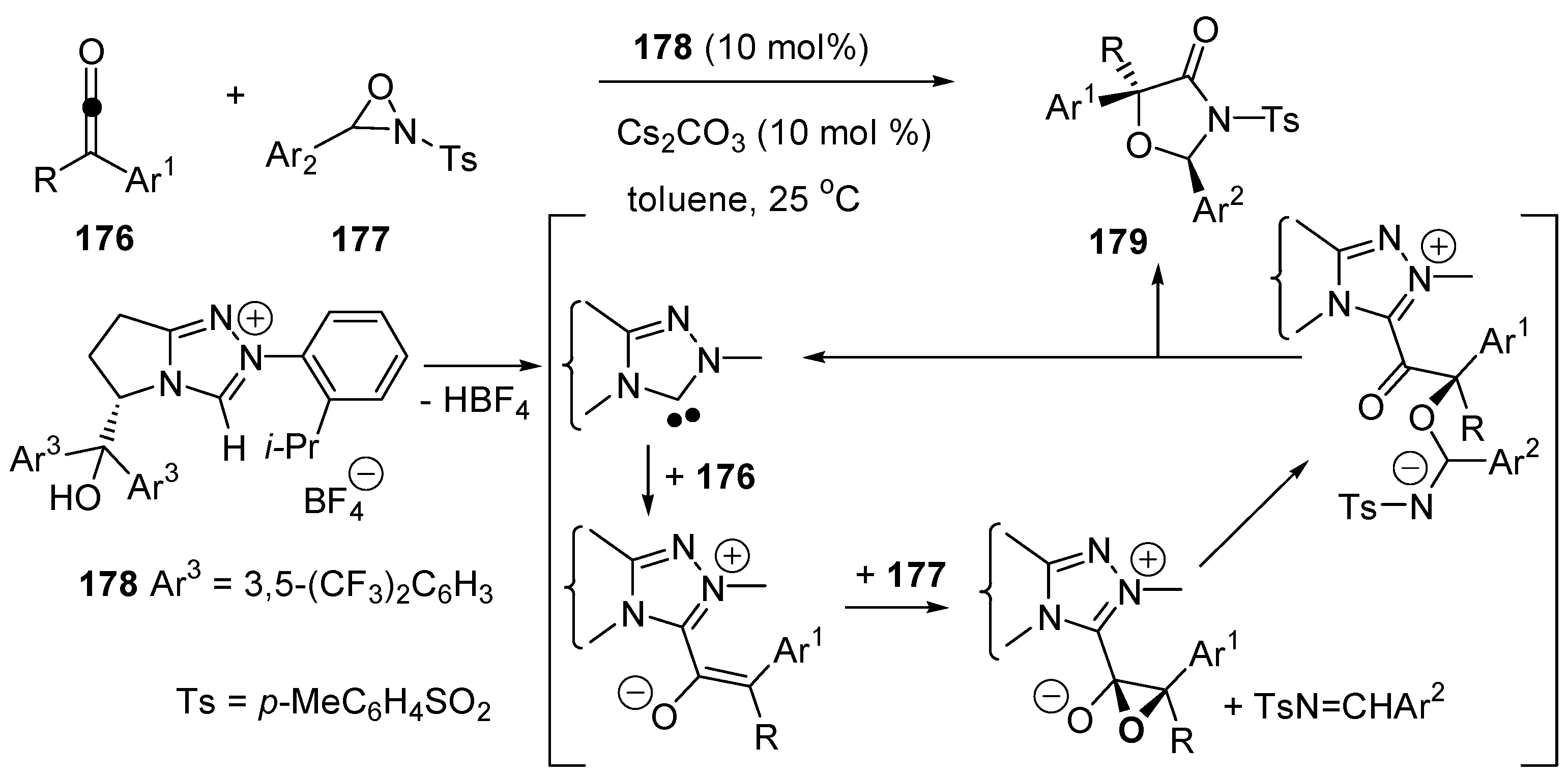

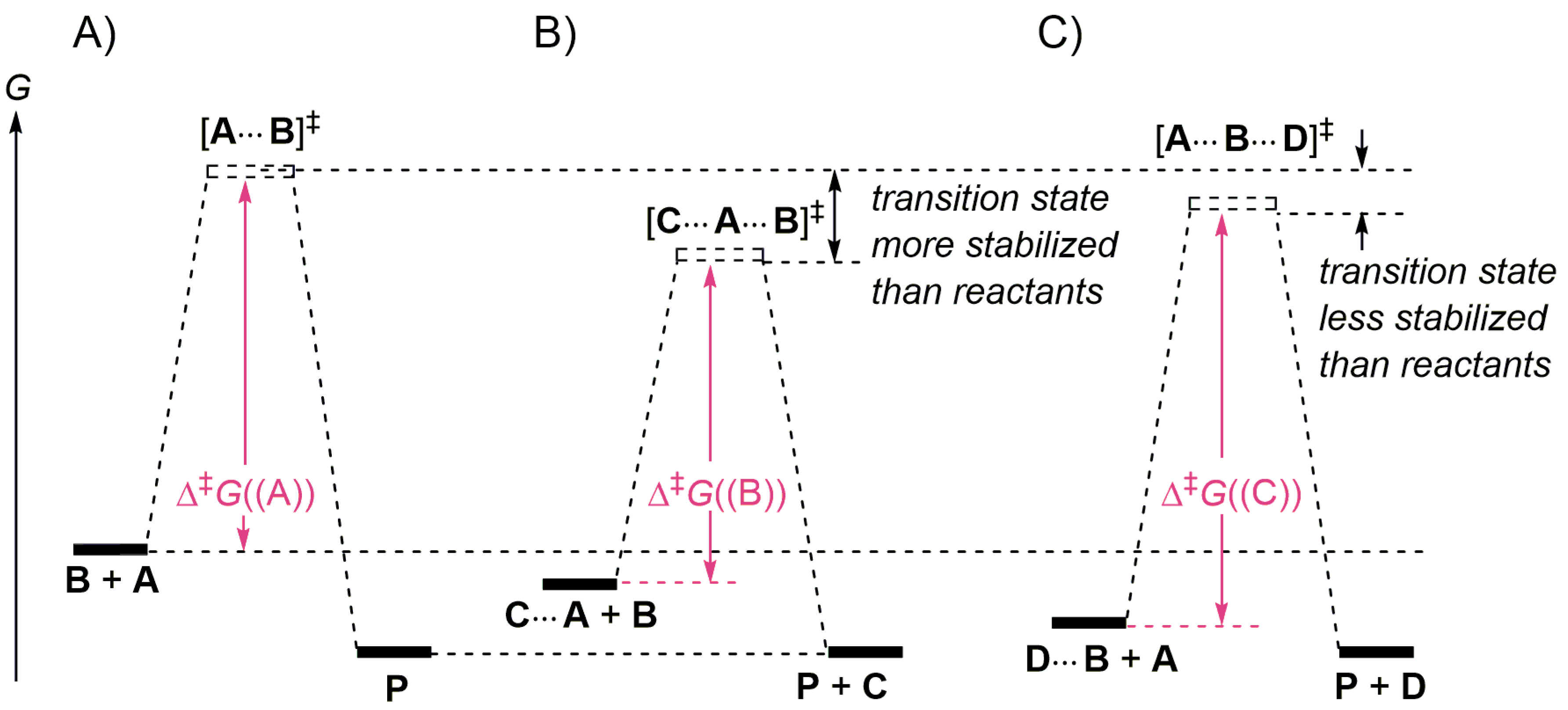{kind=link}
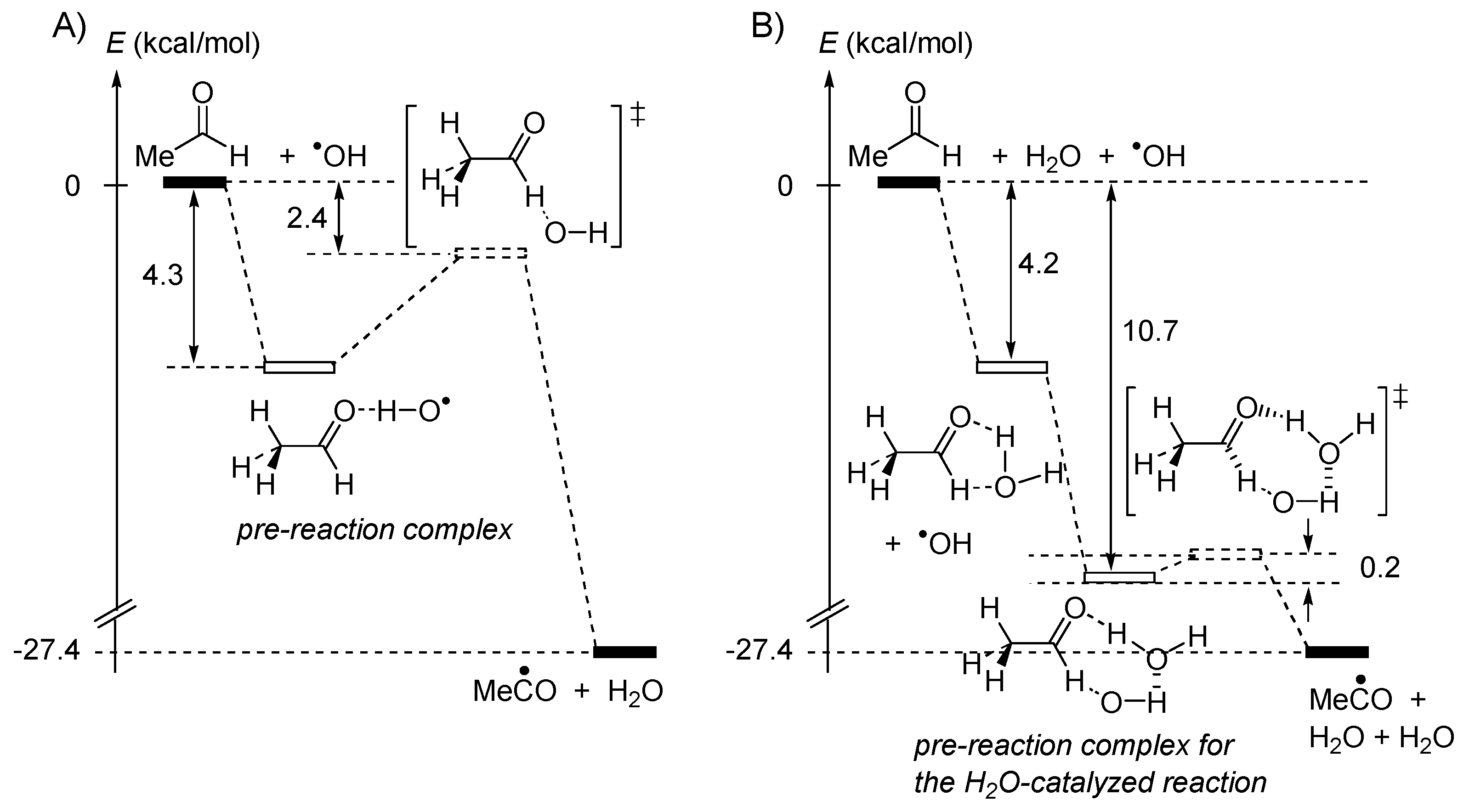{kind=link}
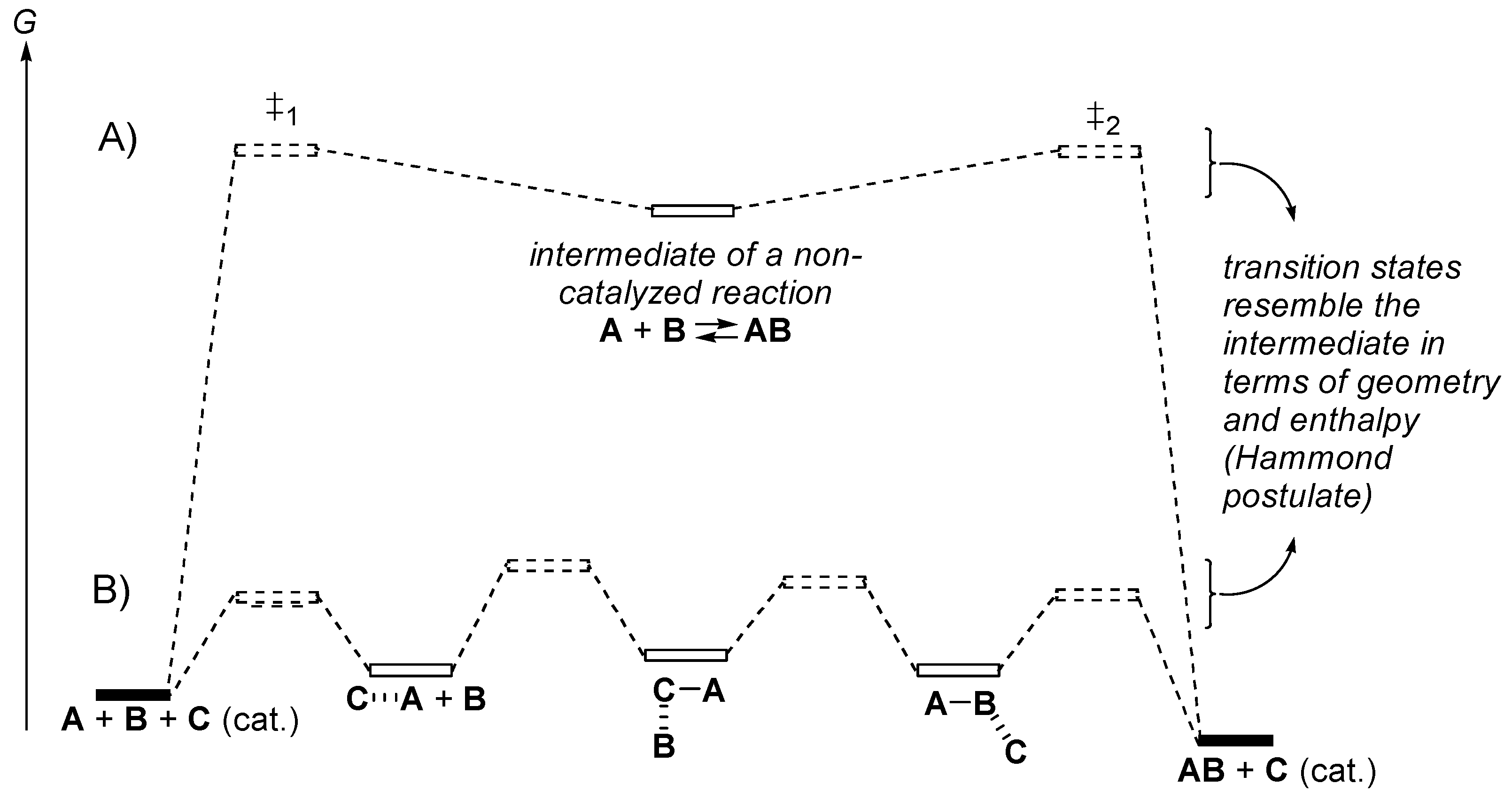{kind=link}
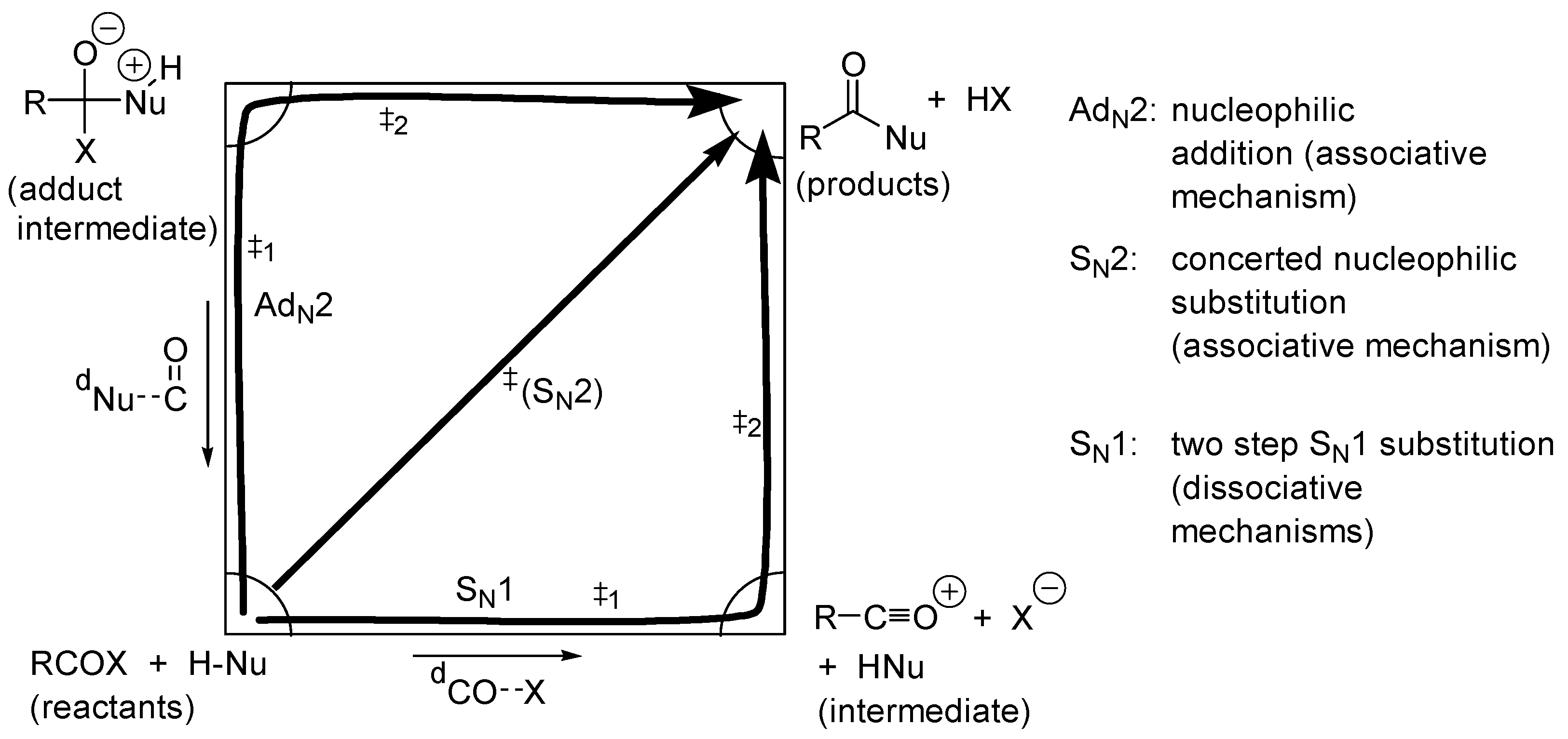{kind=link}
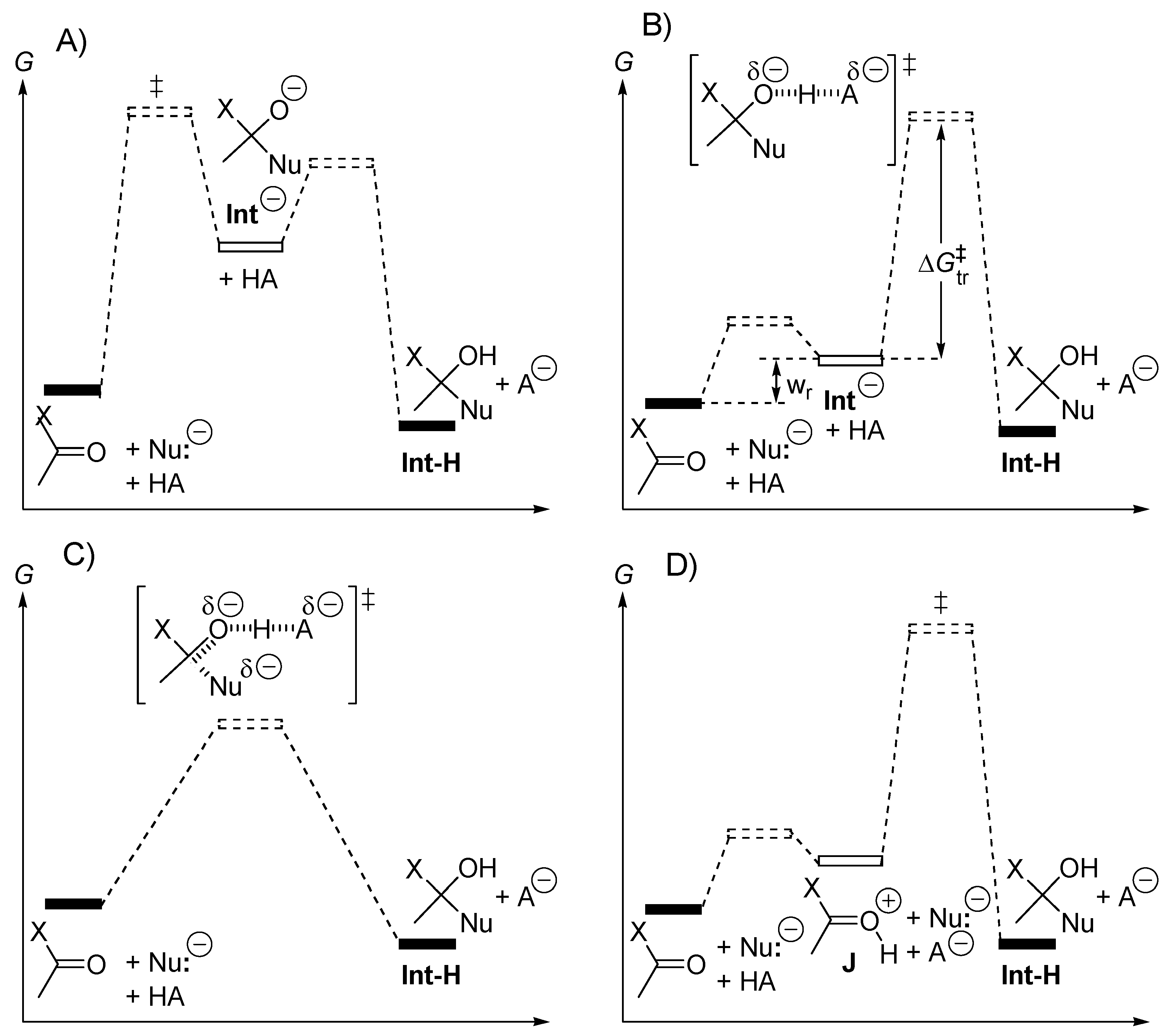{kind=link}
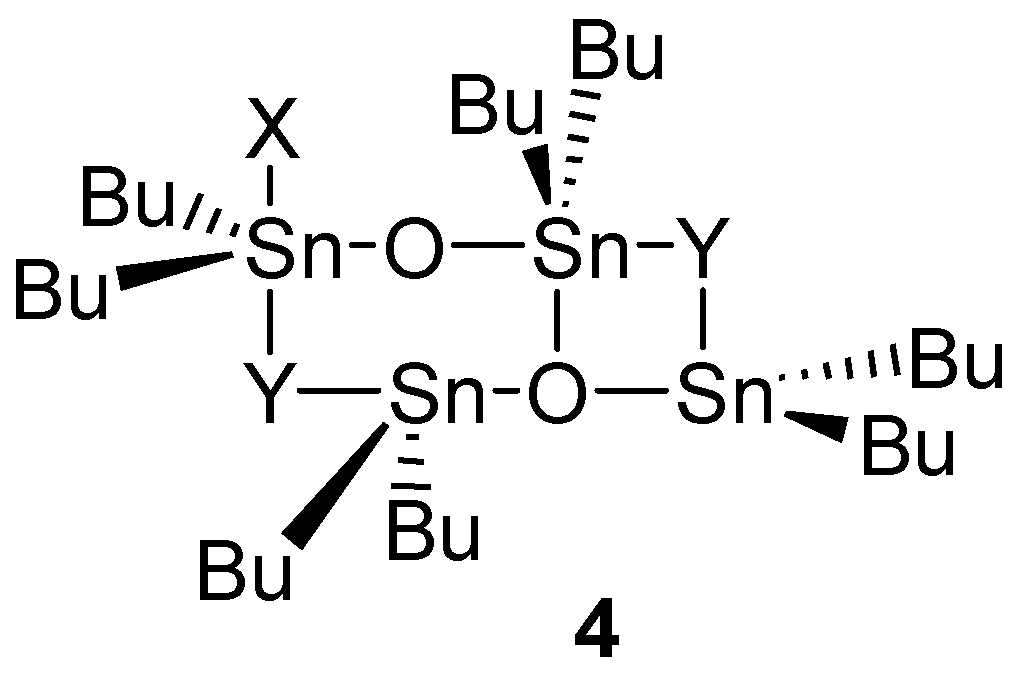{kind=link}
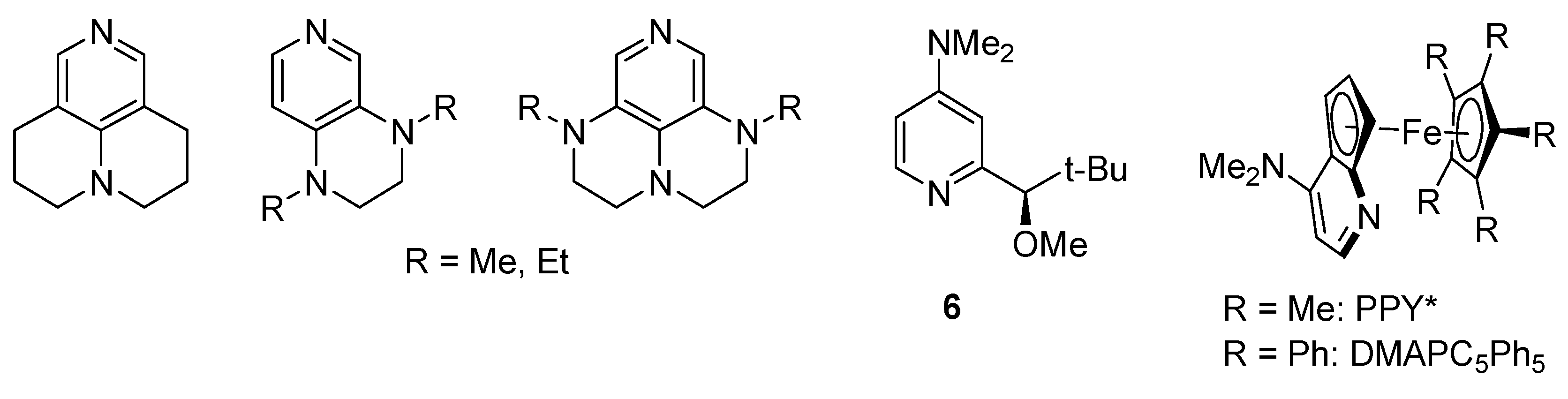{kind=link}
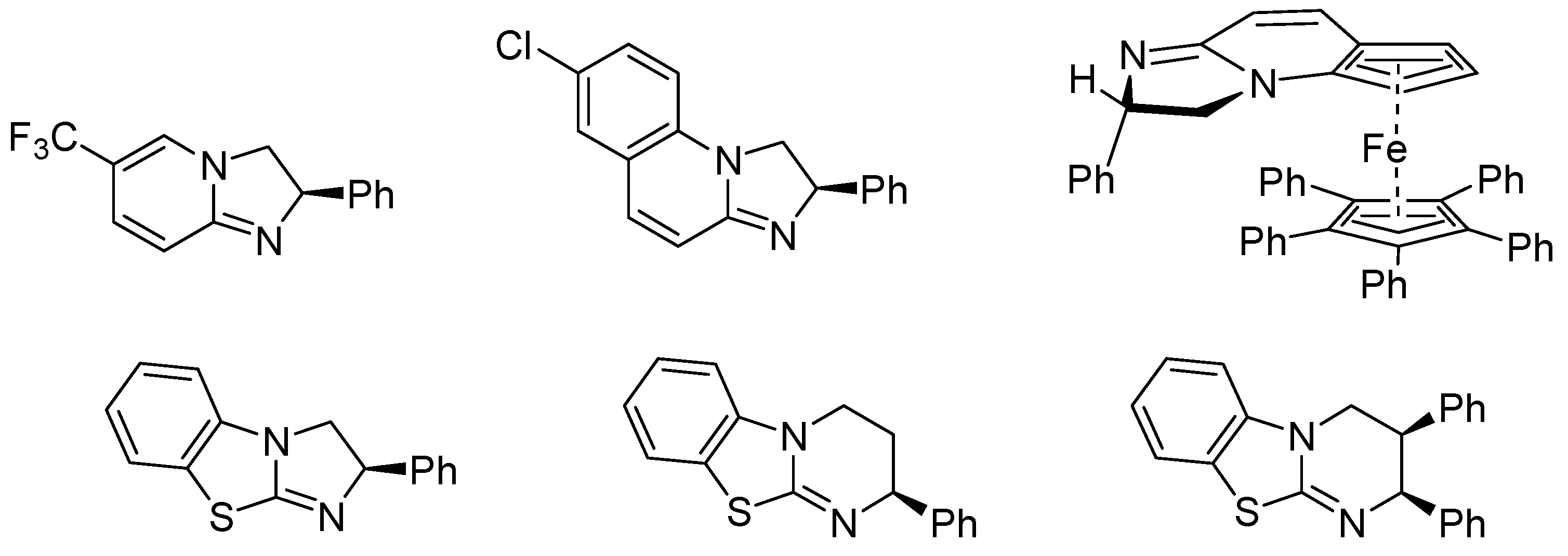{kind=link}
{kind=link}
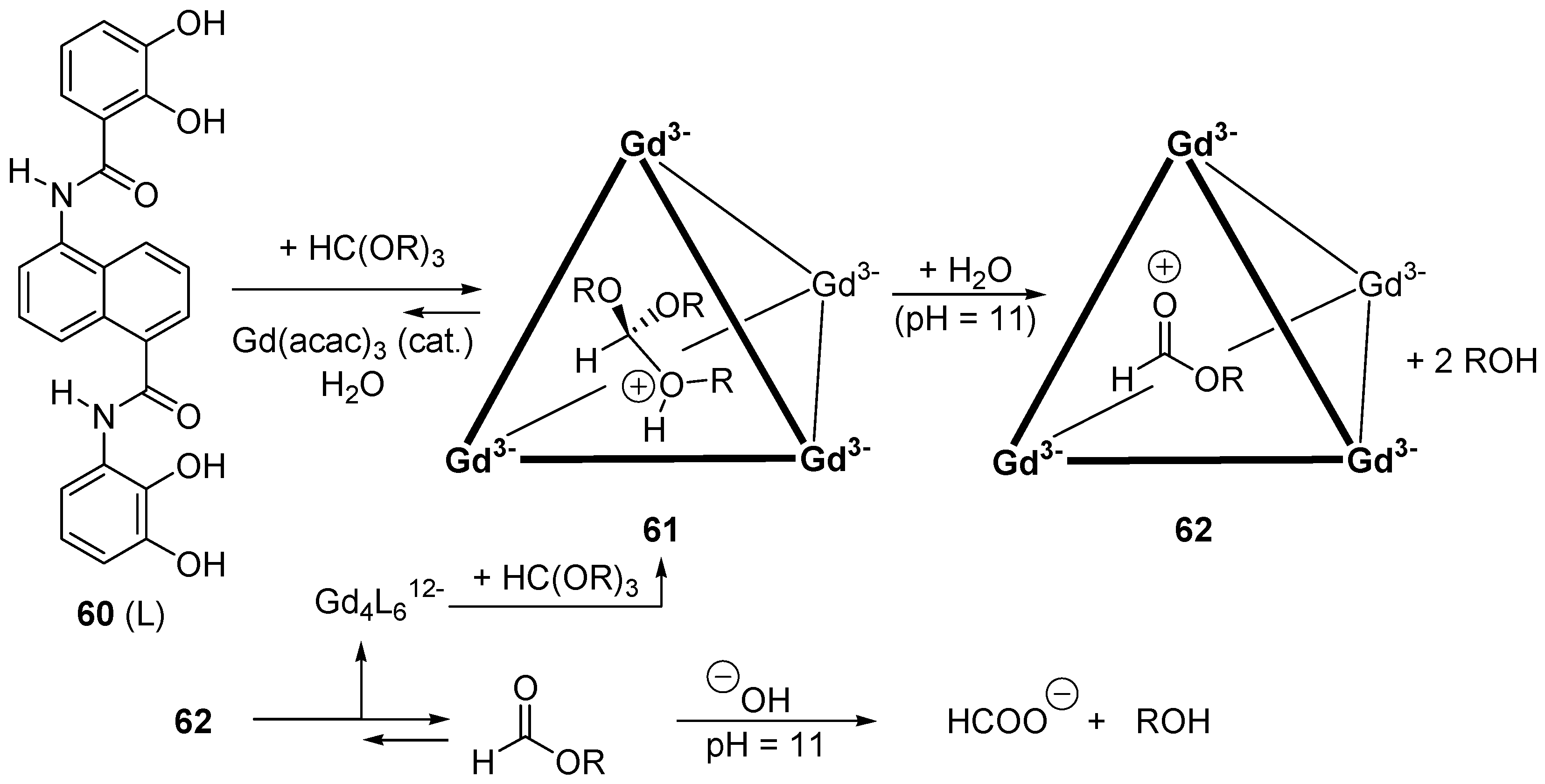{kind=link}
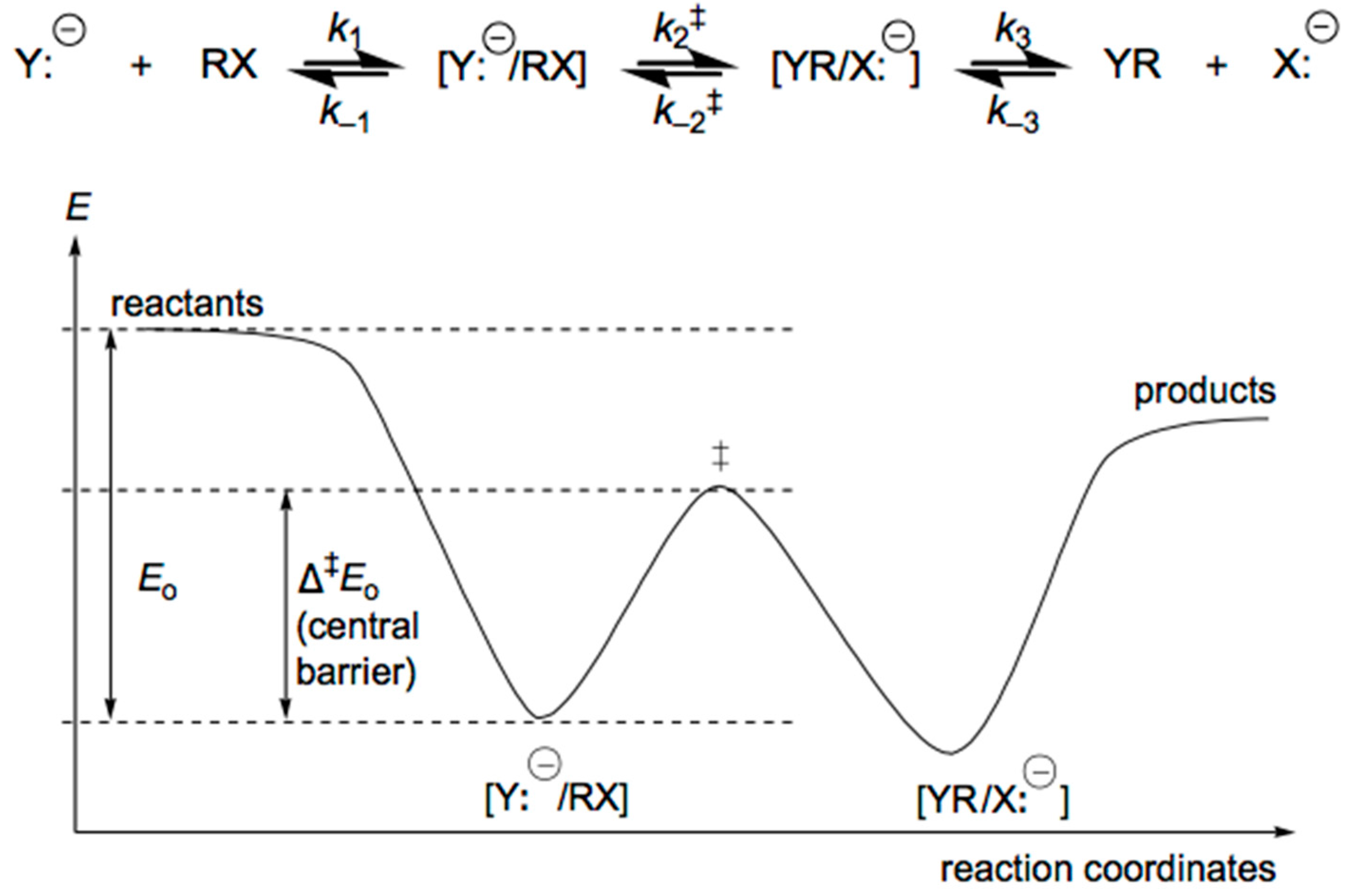{kind=link}
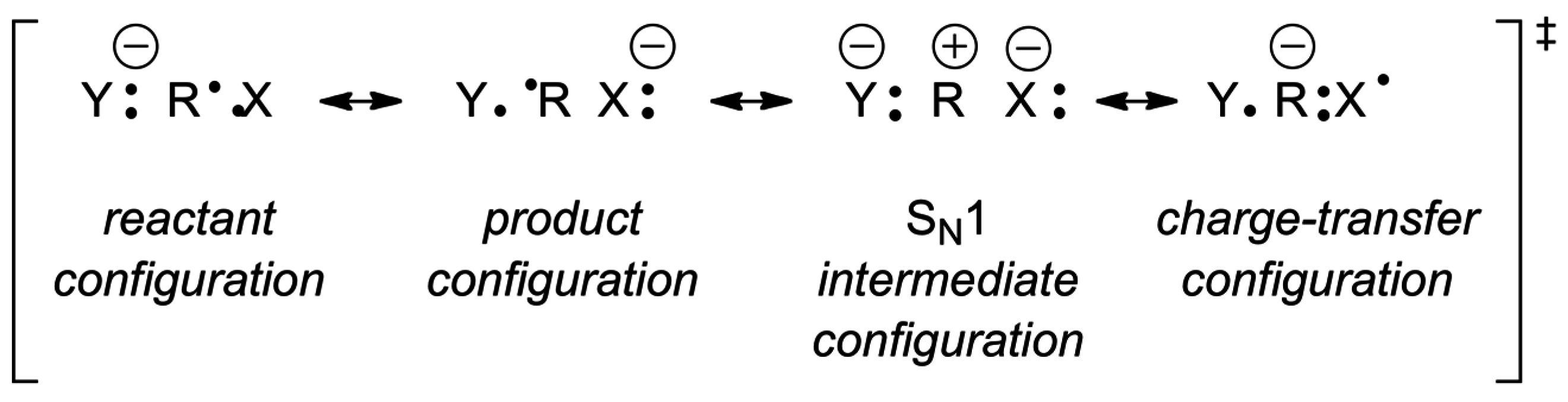{kind=link}
{kind=link}
{kind=link}
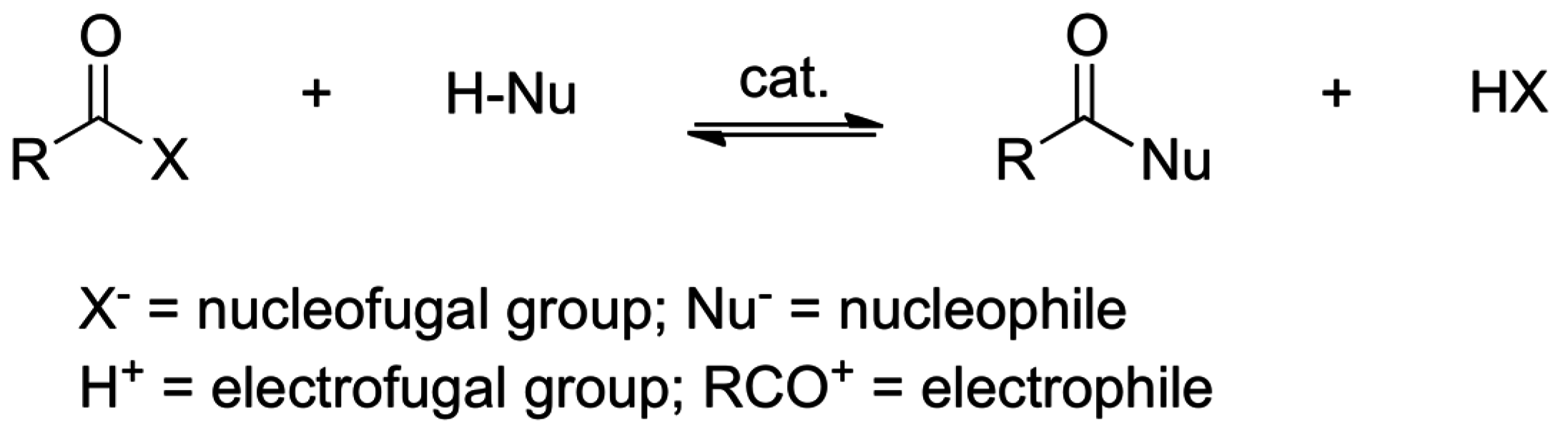{kind=link}
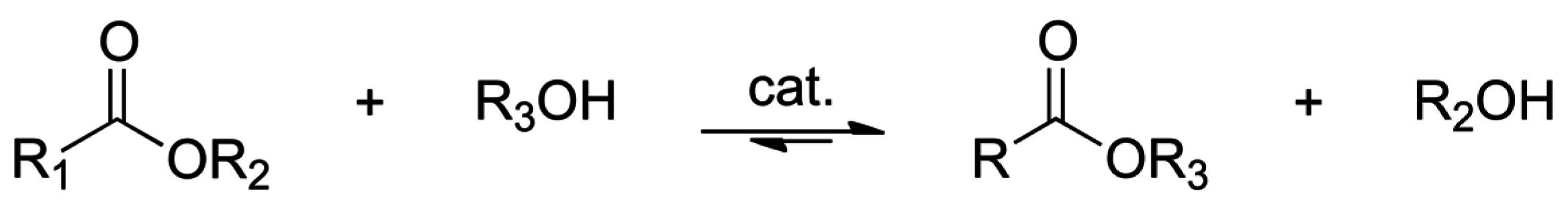{kind=link}
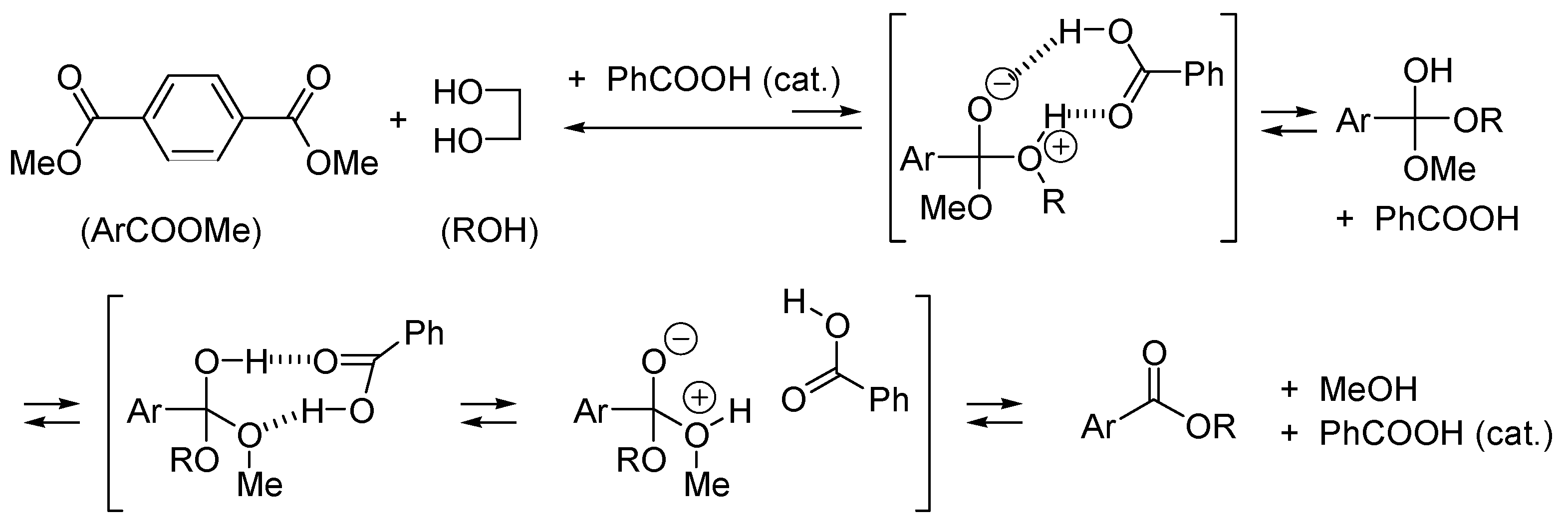{kind=link}
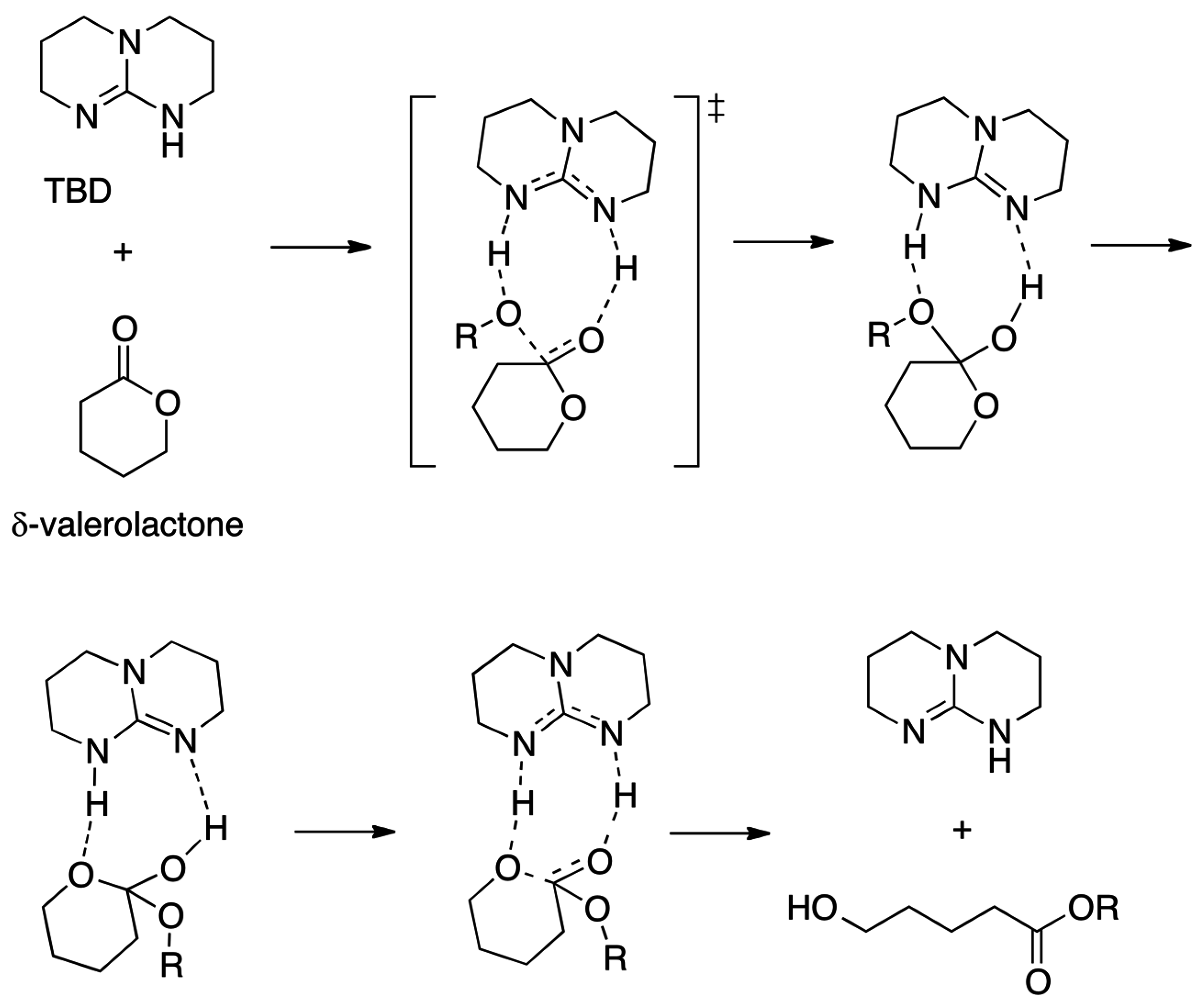{kind=link}
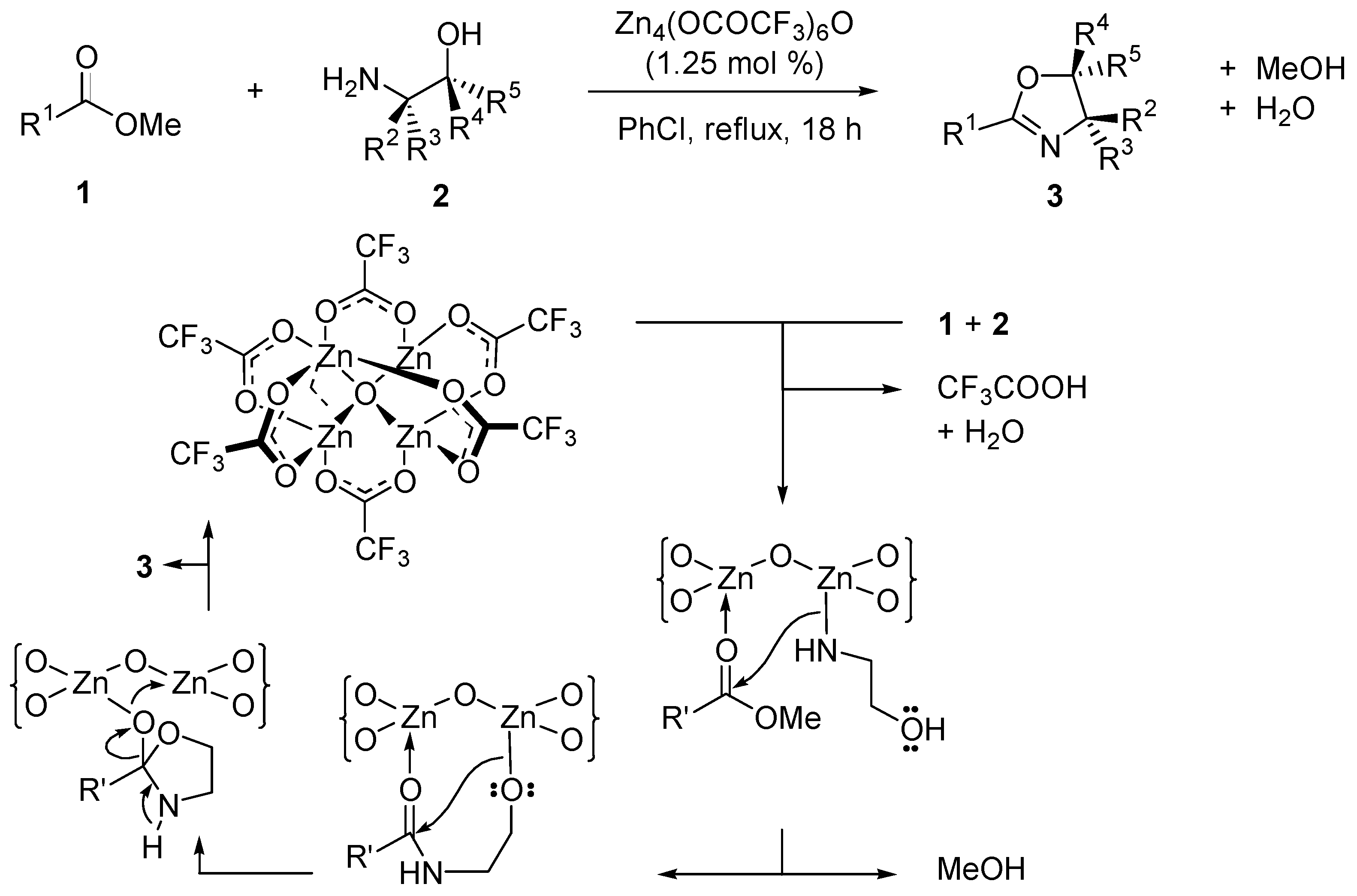{kind=link}
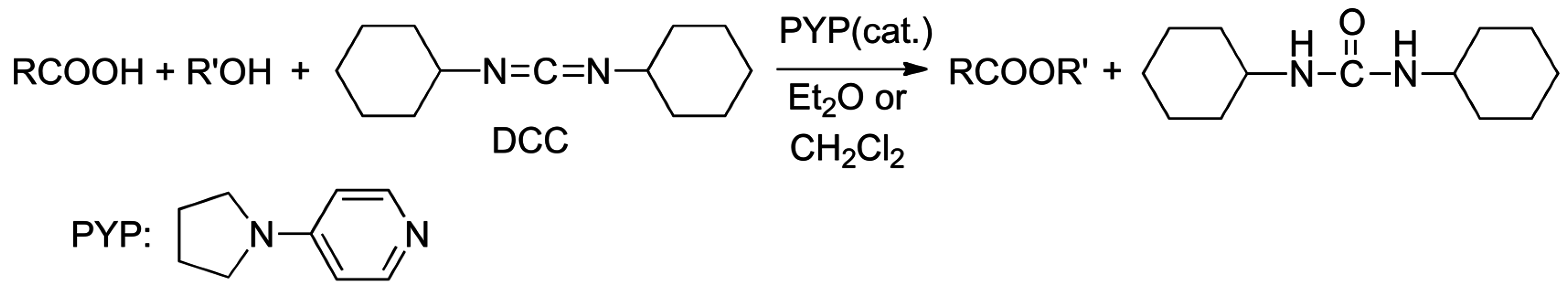{kind=link}
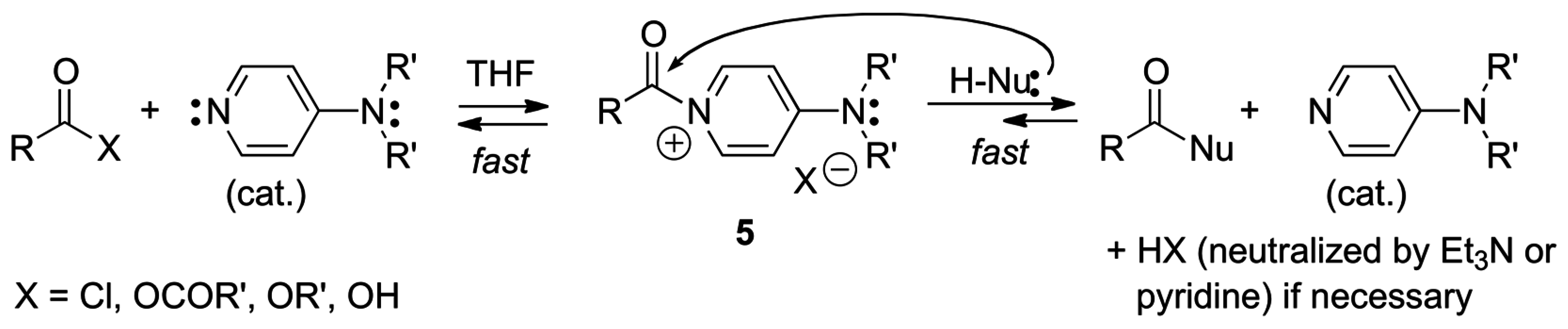{kind=link}
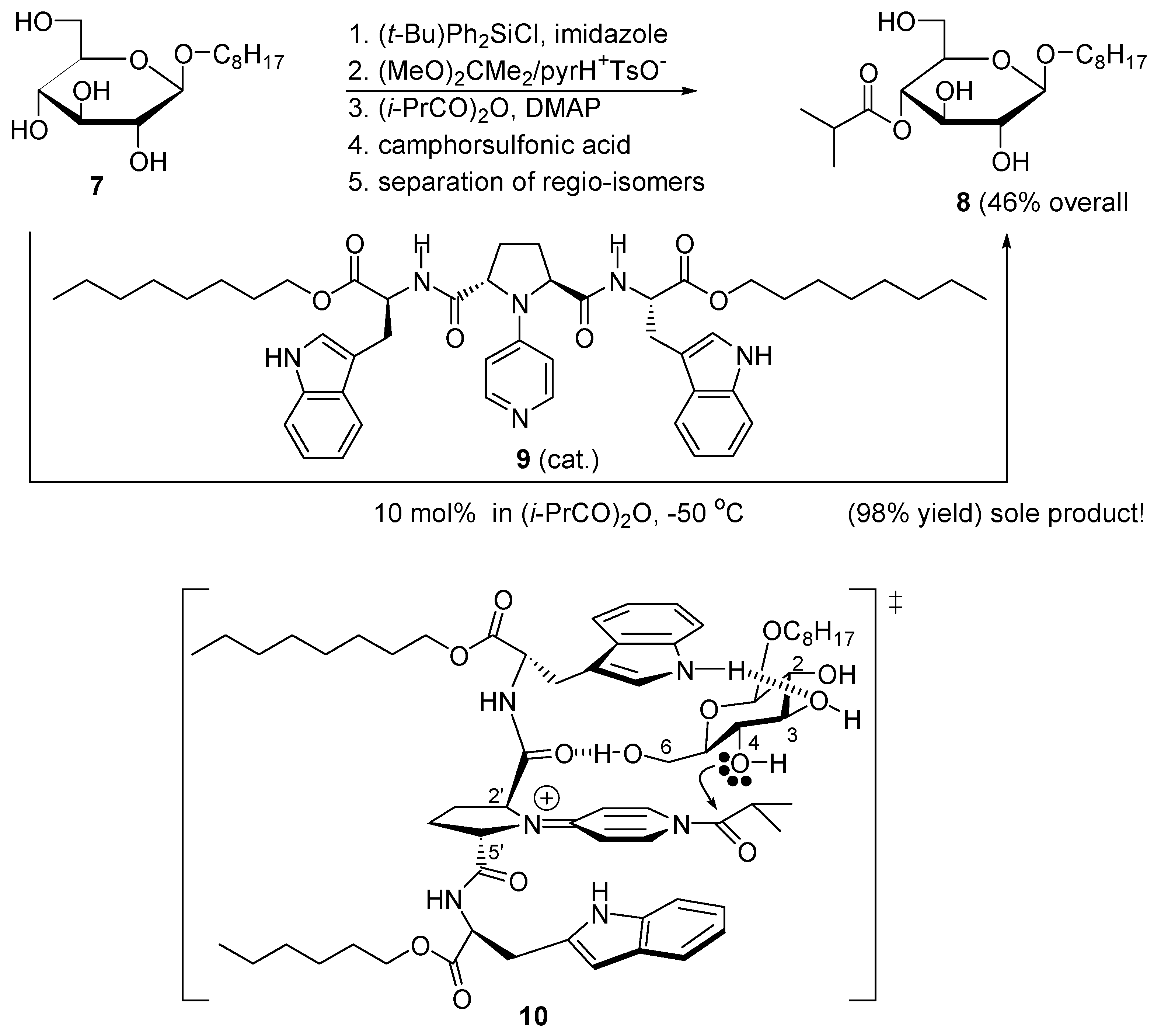{kind=link}
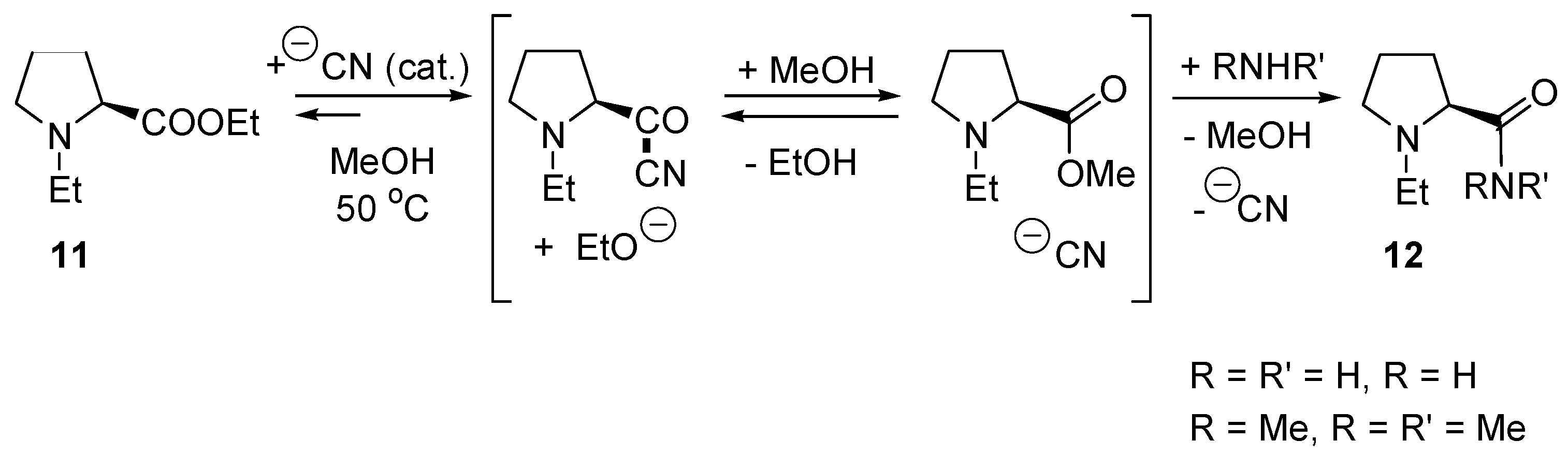{kind=link}
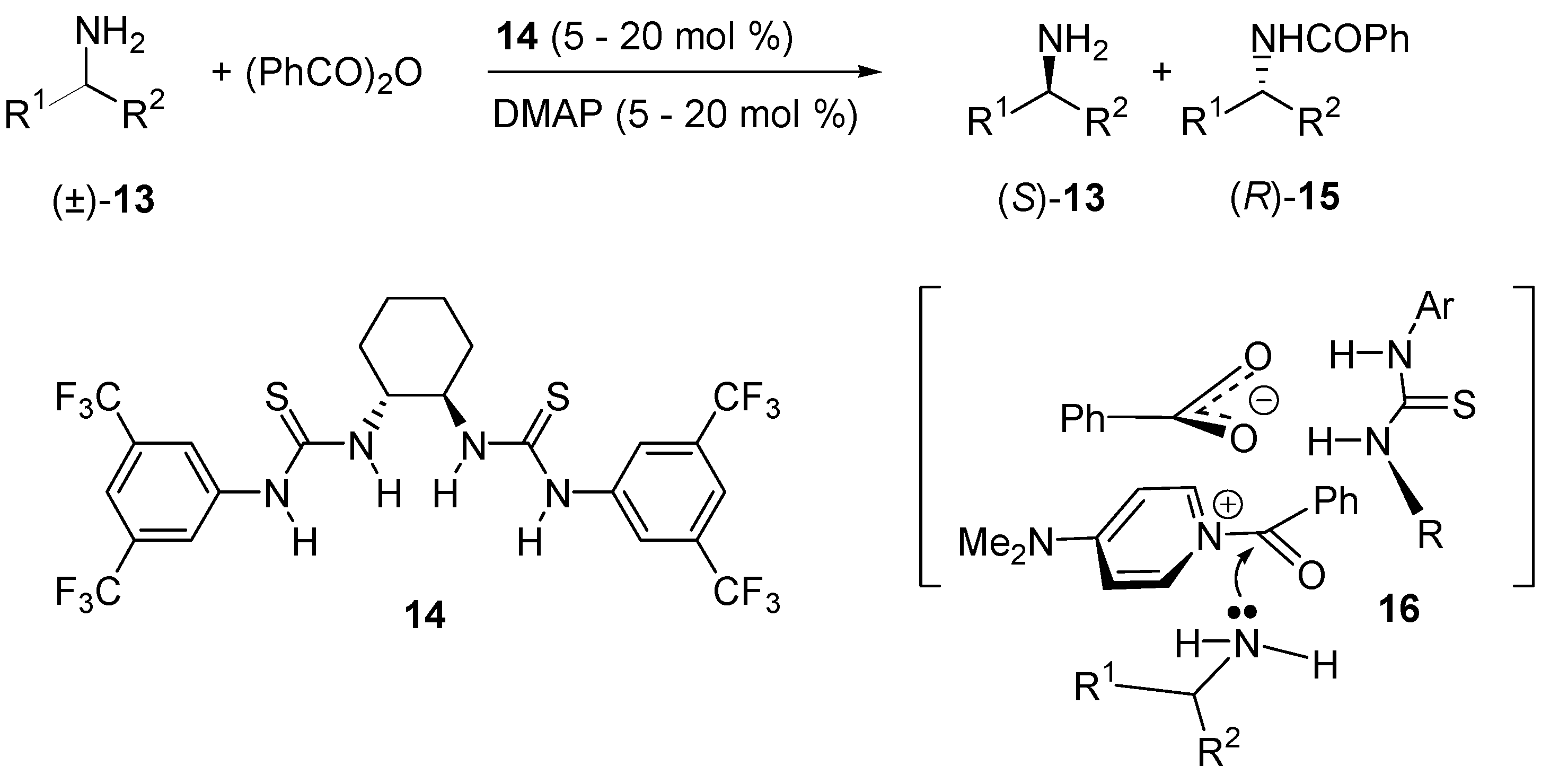{kind=link}
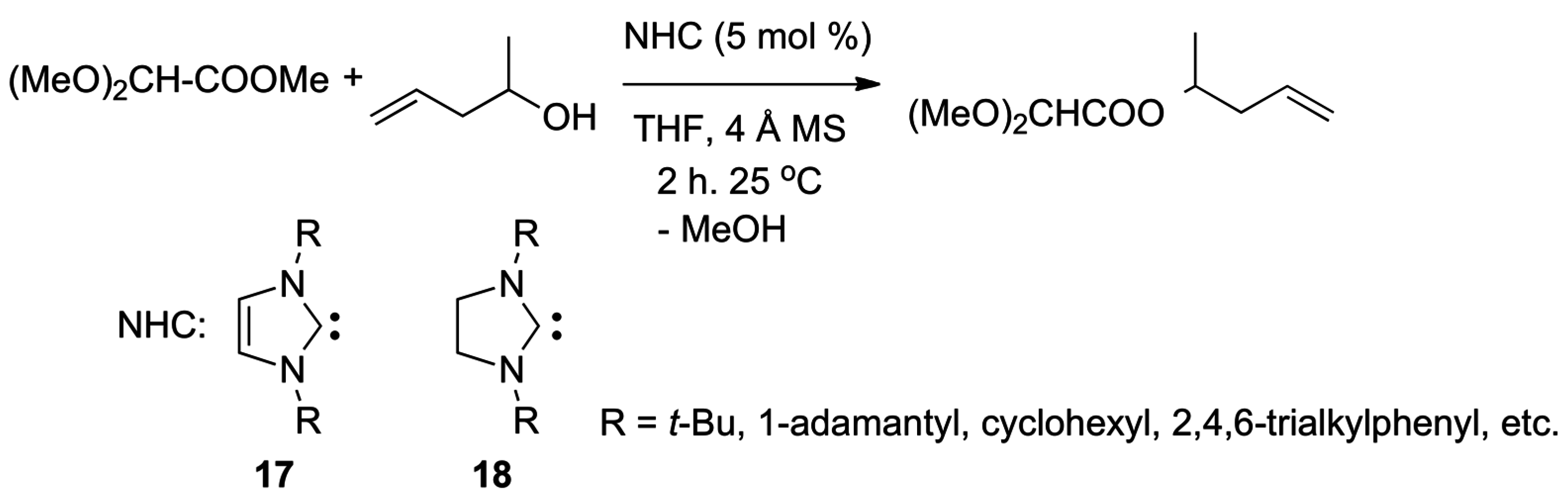{kind=link}
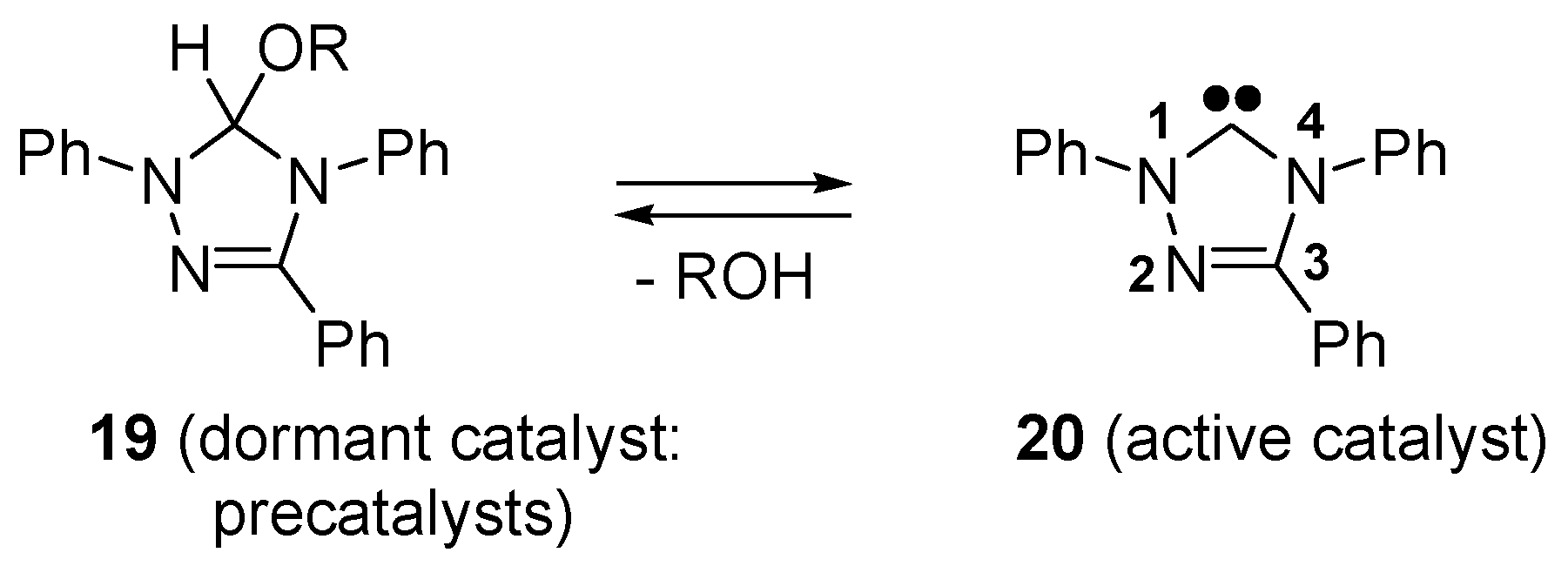{kind=link}
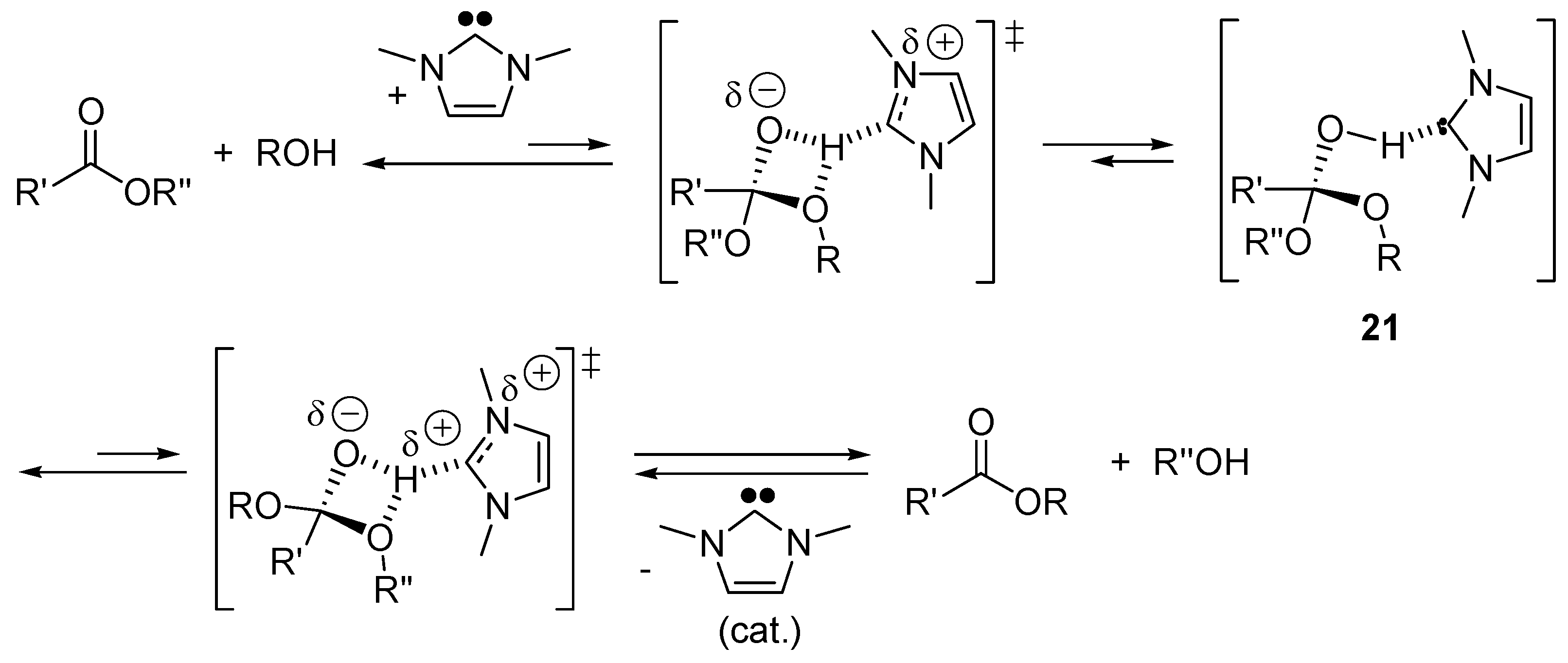{kind=link}
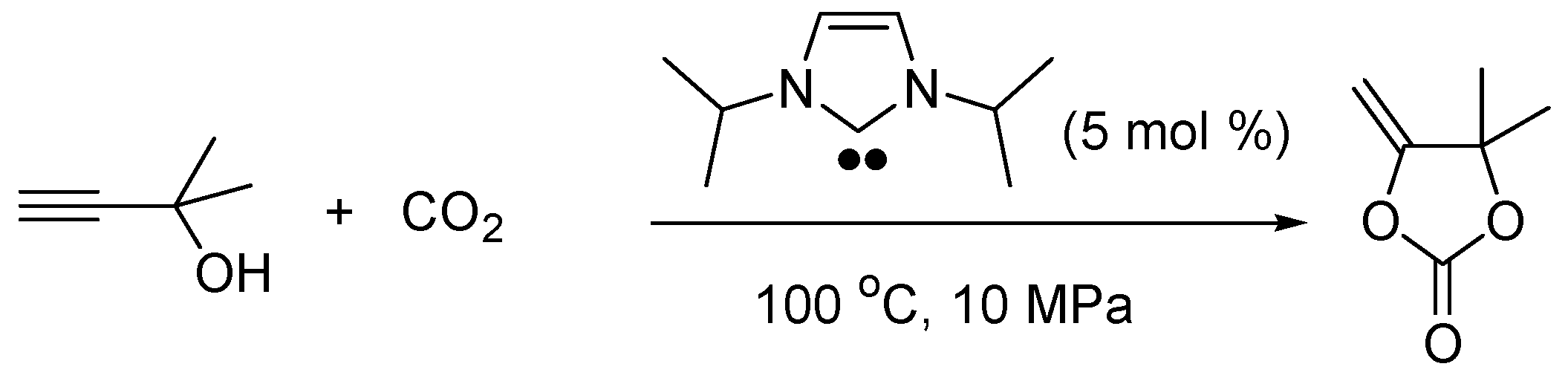{kind=link}
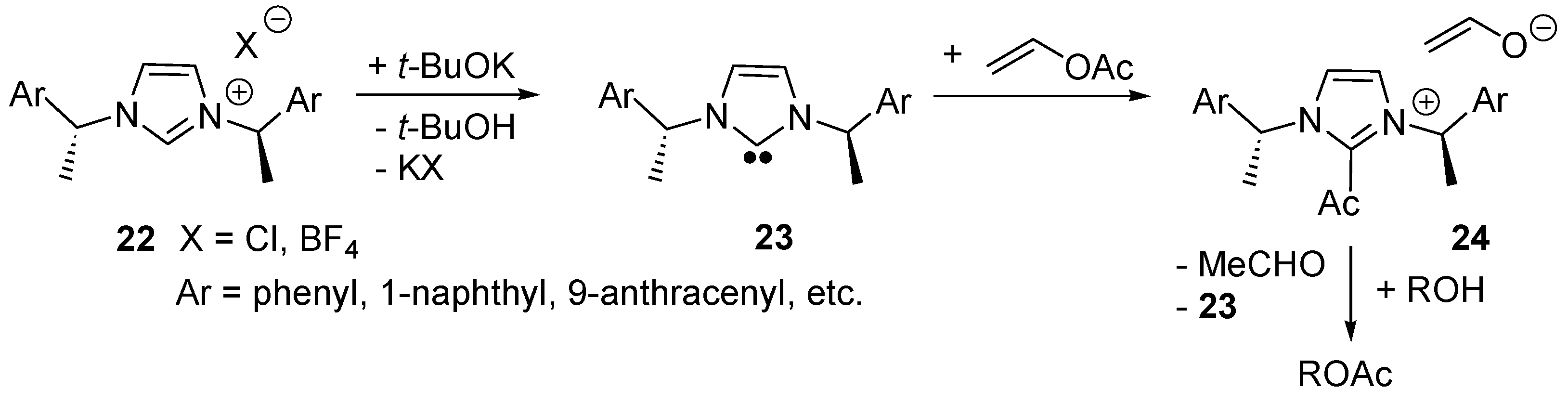{kind=link}
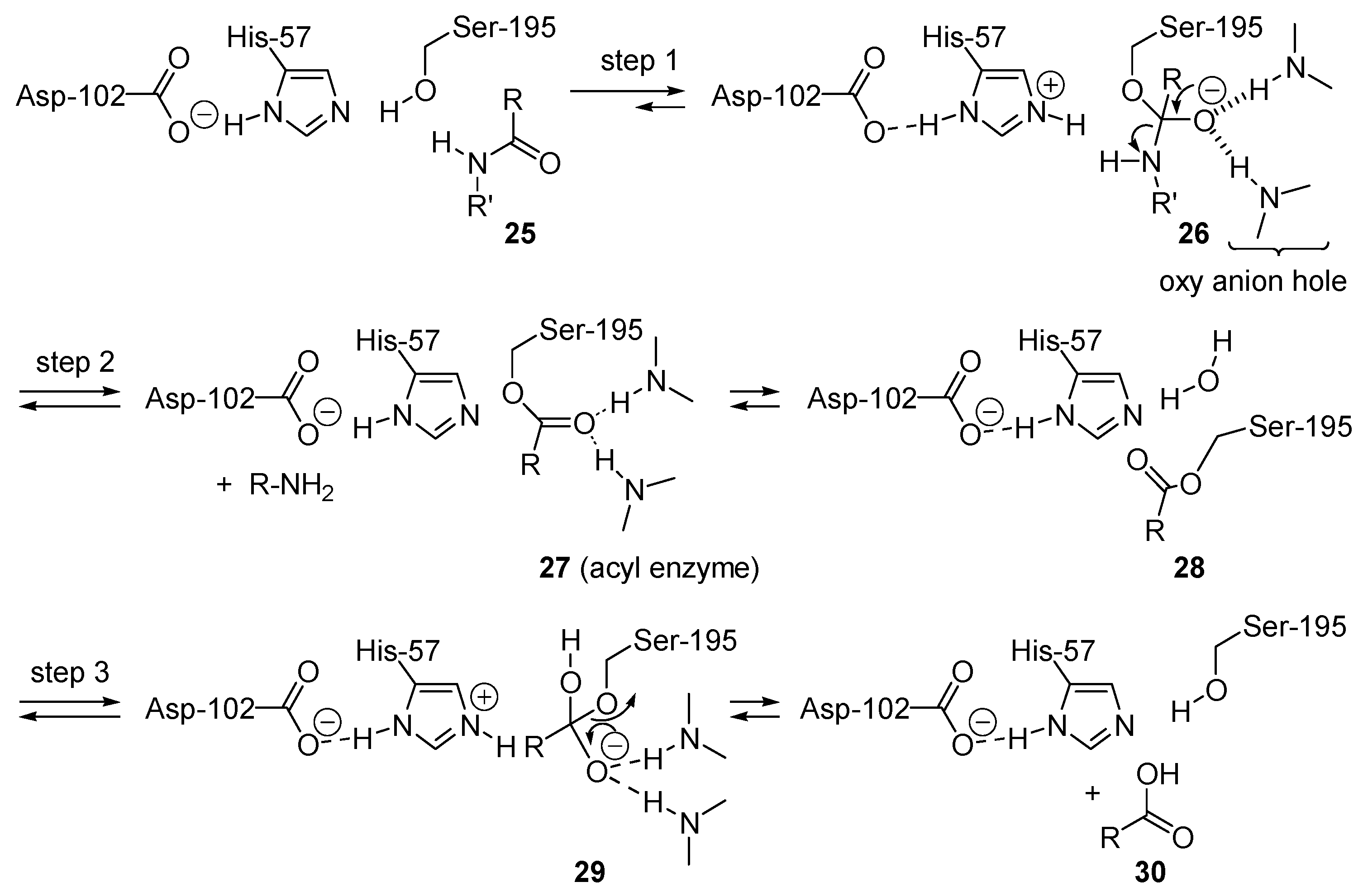{kind=link}
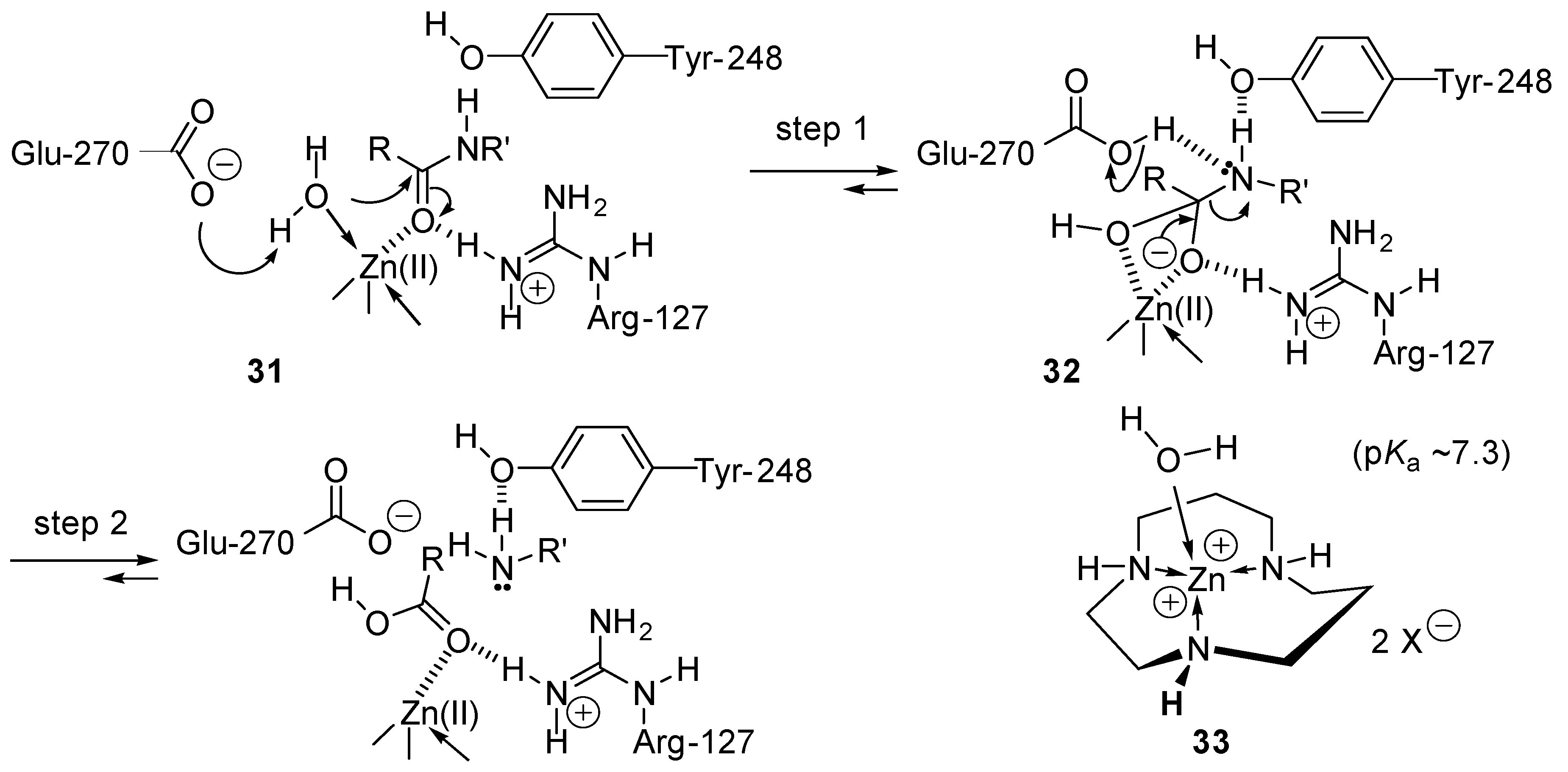{kind=link}
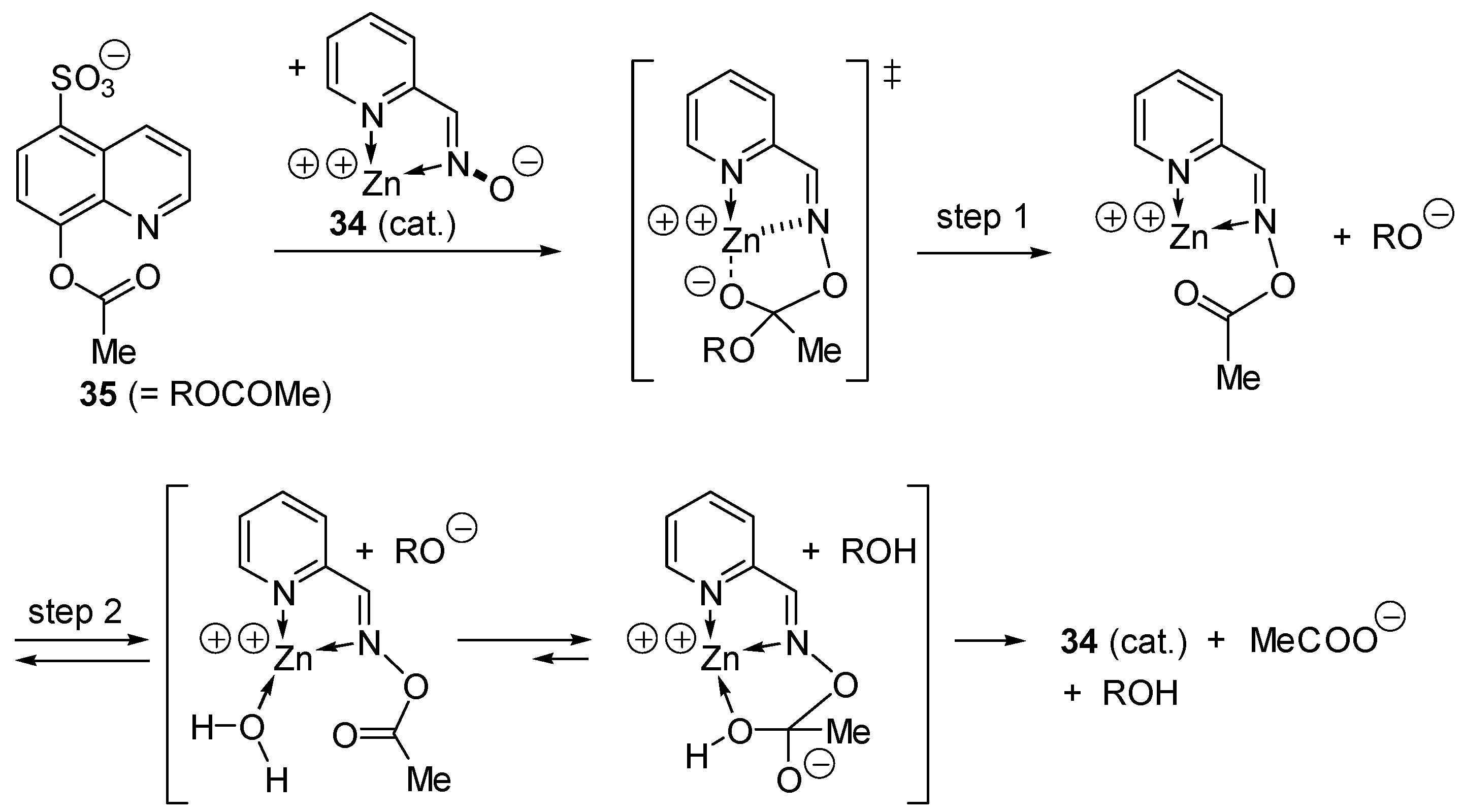{kind=link}
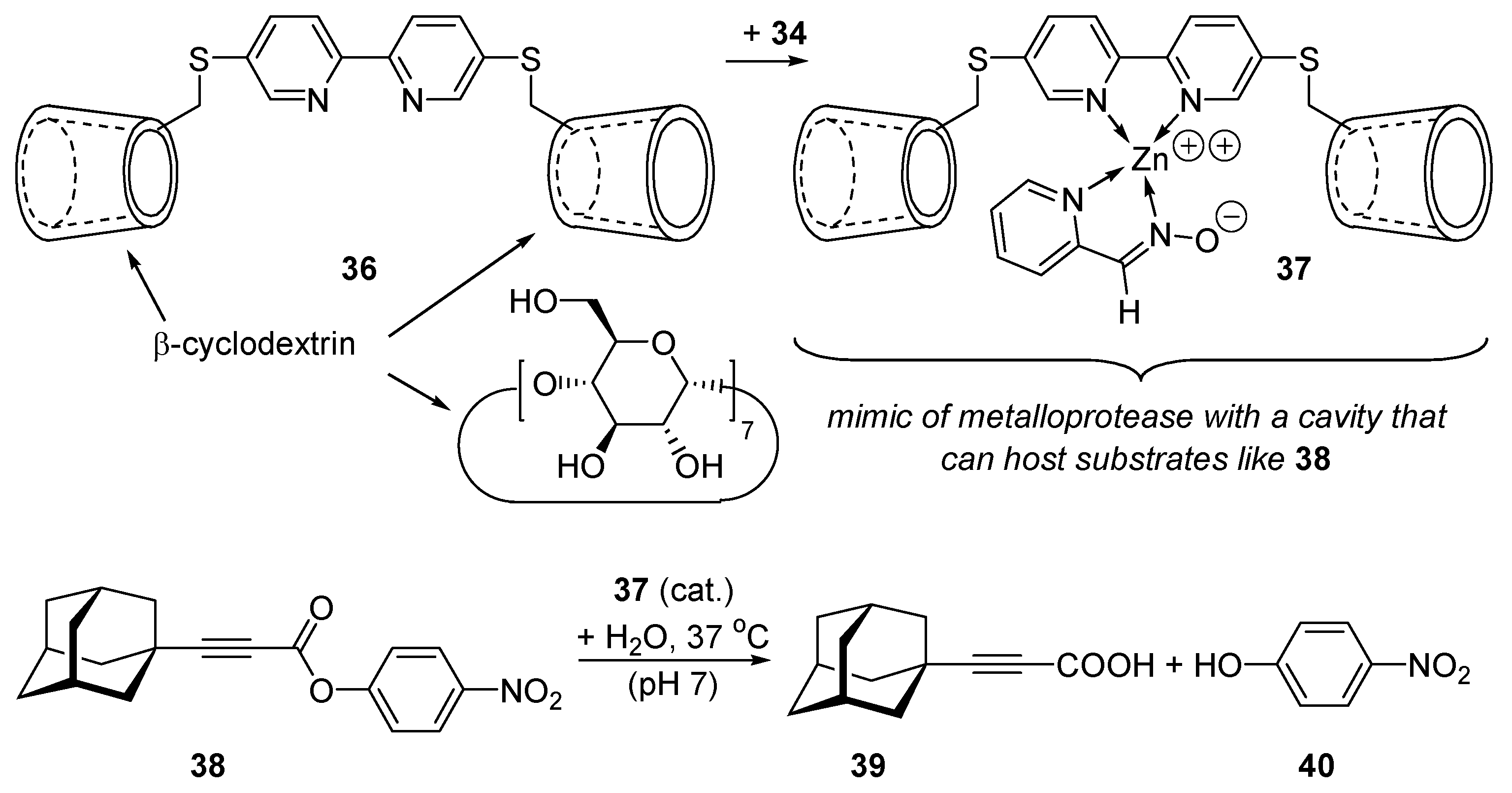{kind=link}
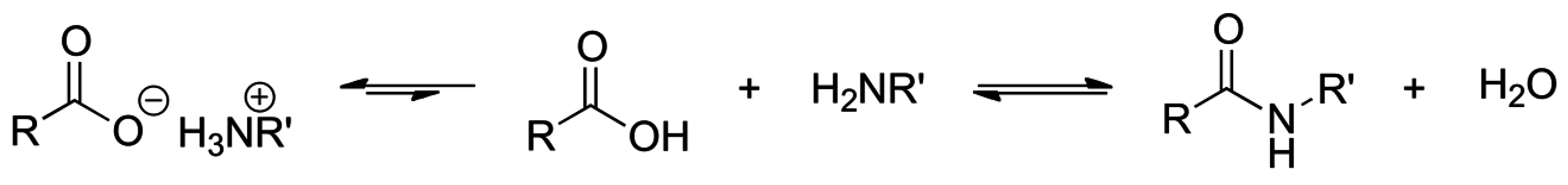{kind=link}
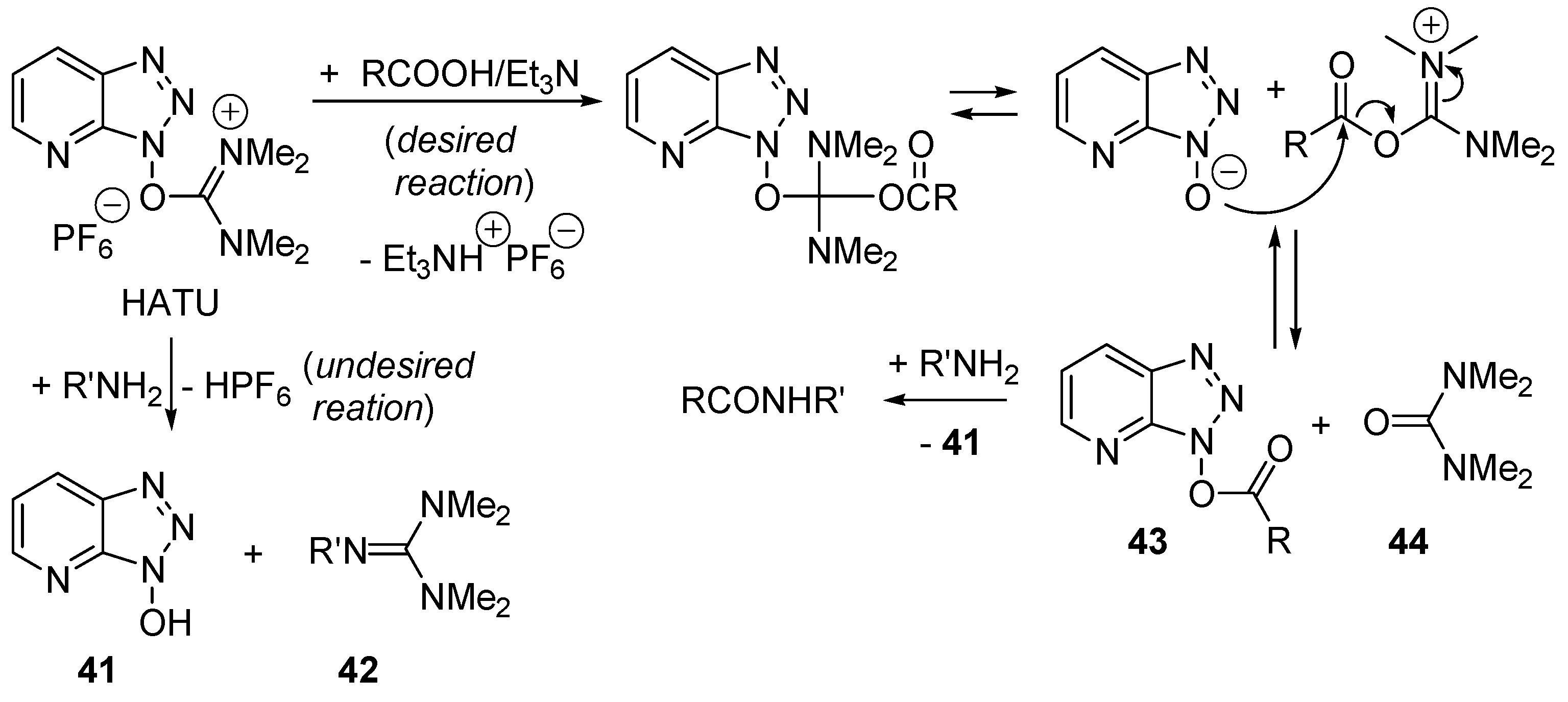{kind=link}
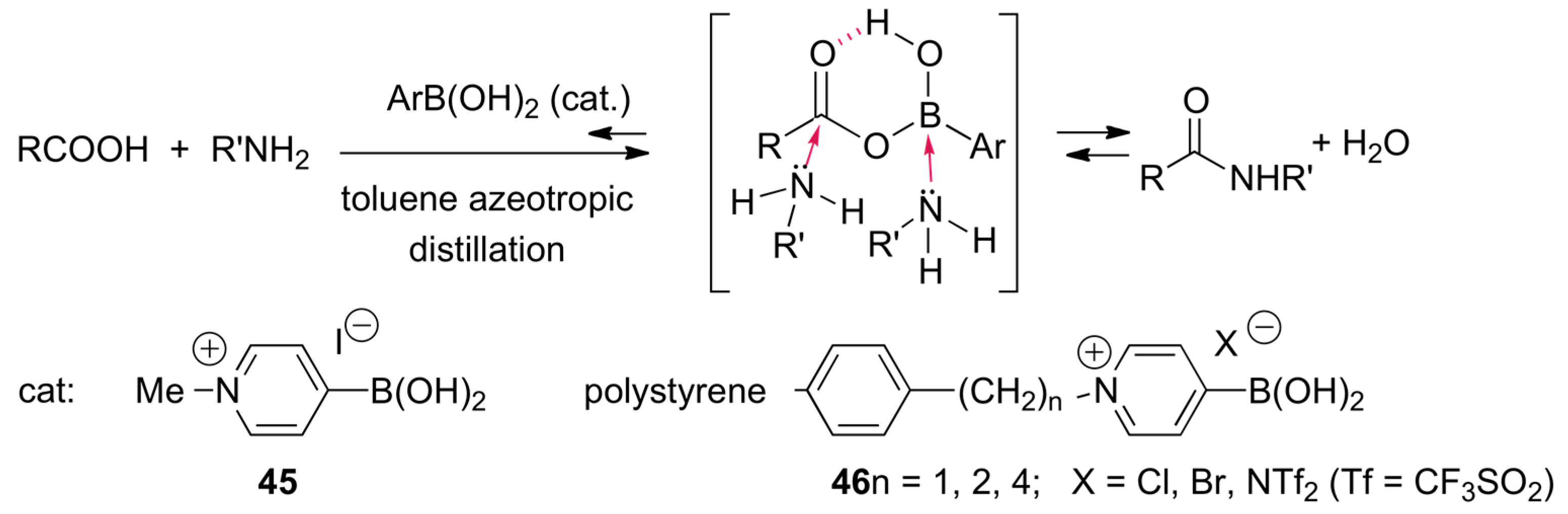{kind=link}
{kind=link}
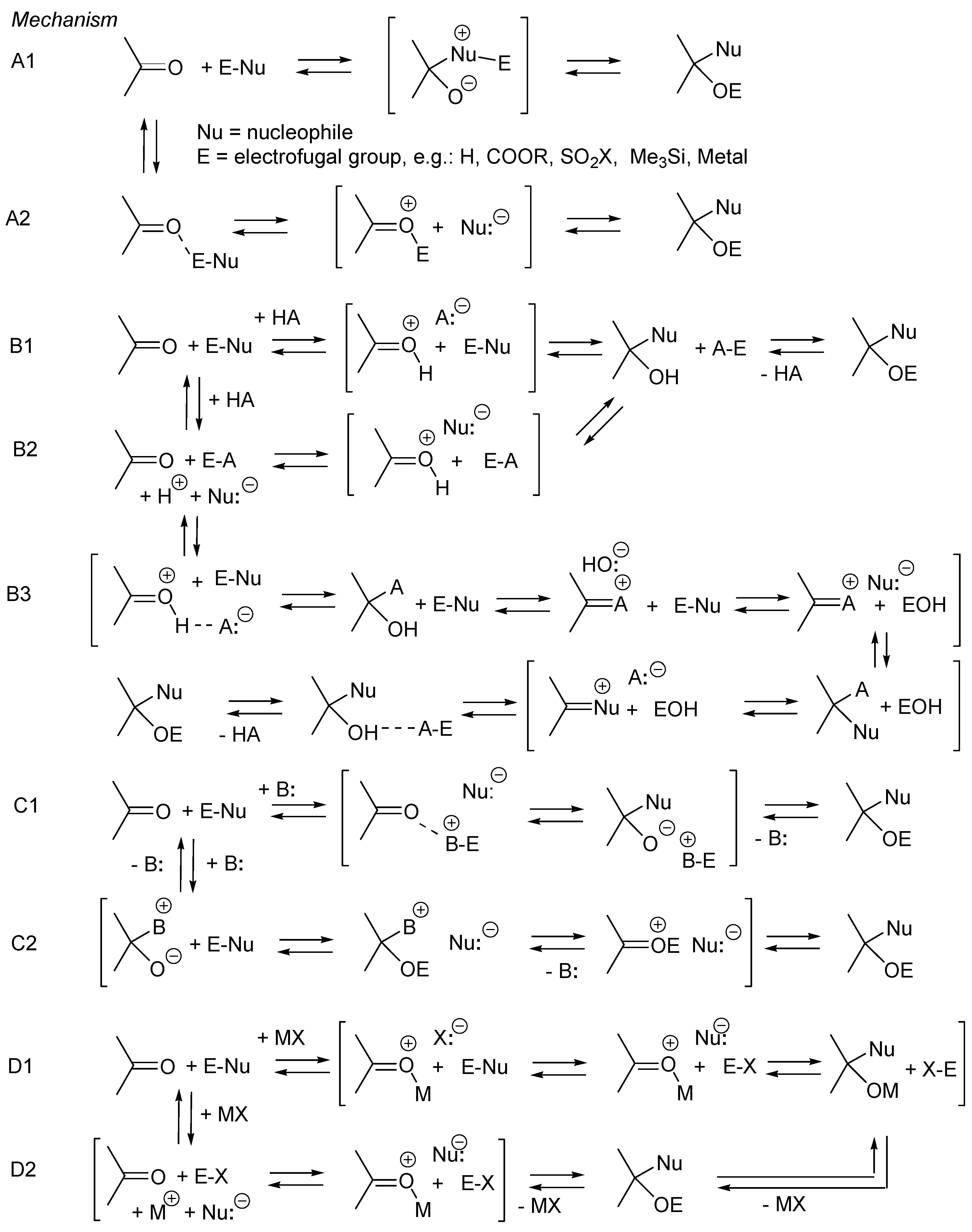{kind=link}
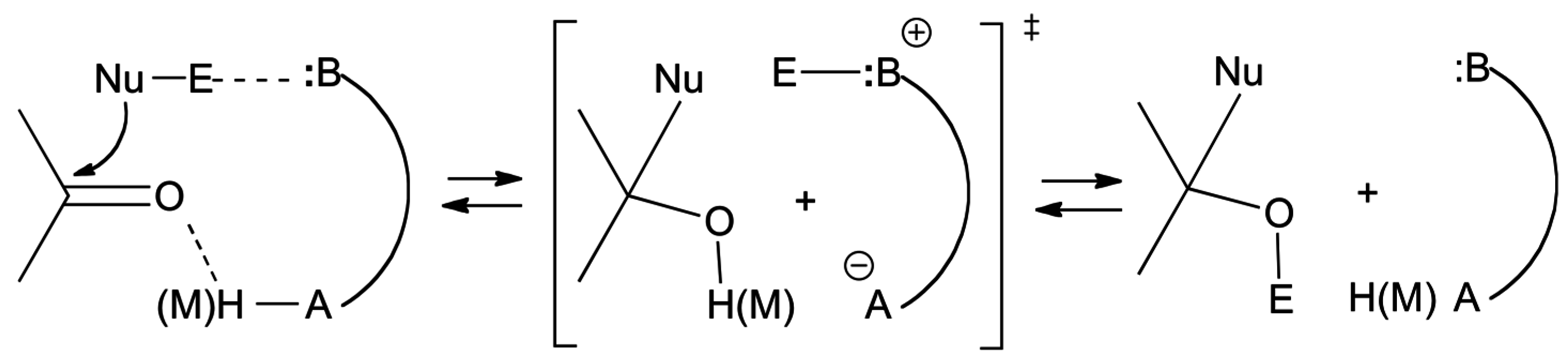{kind=link}
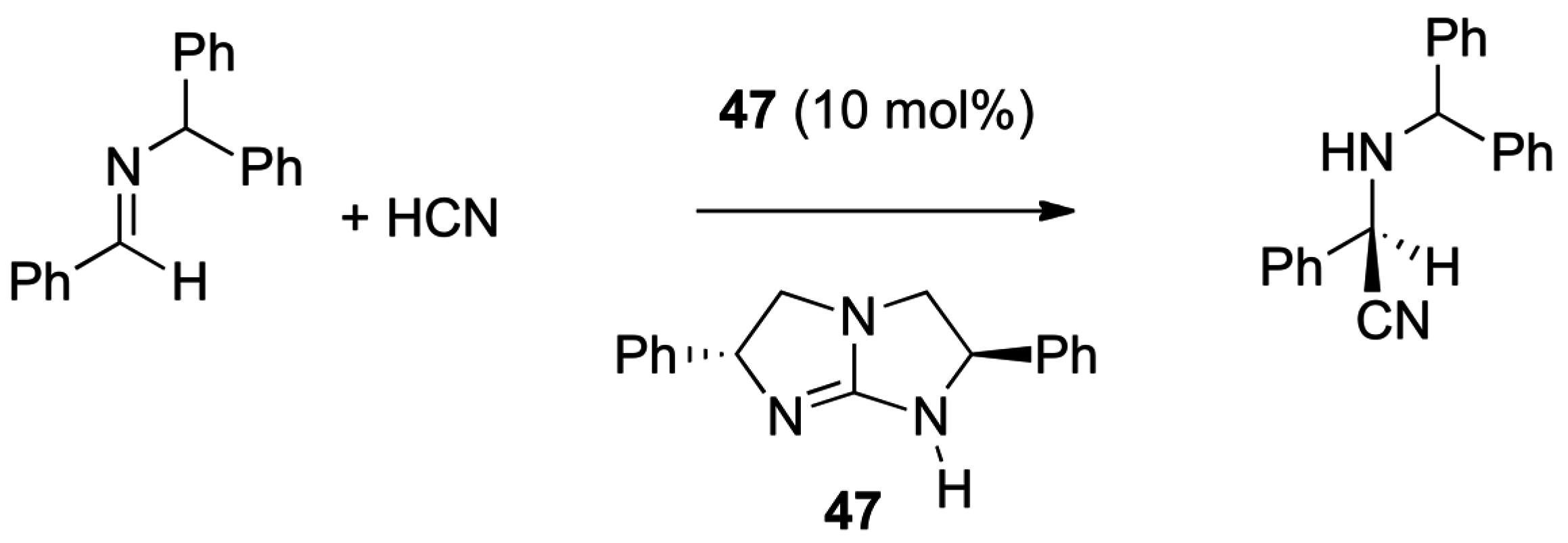{kind=link}
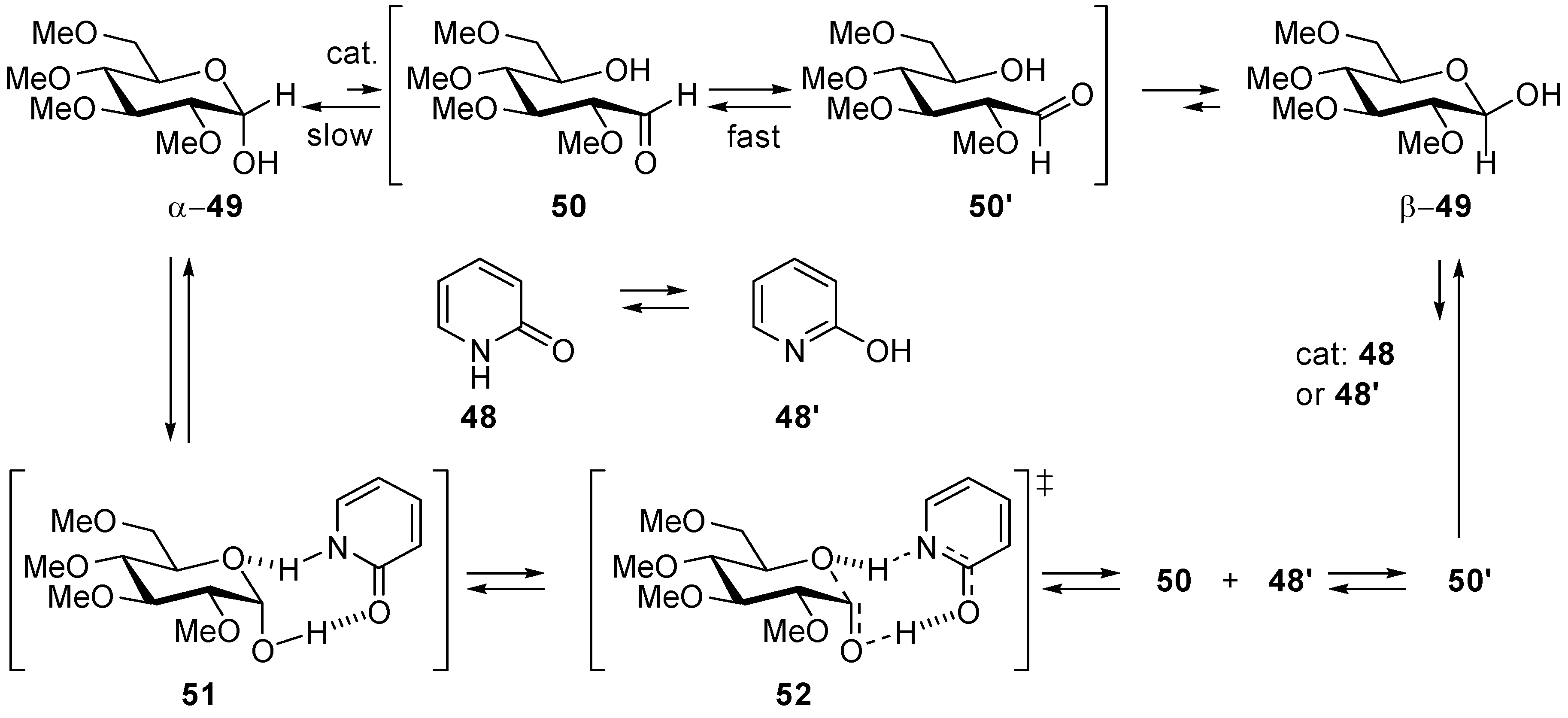{kind=link}
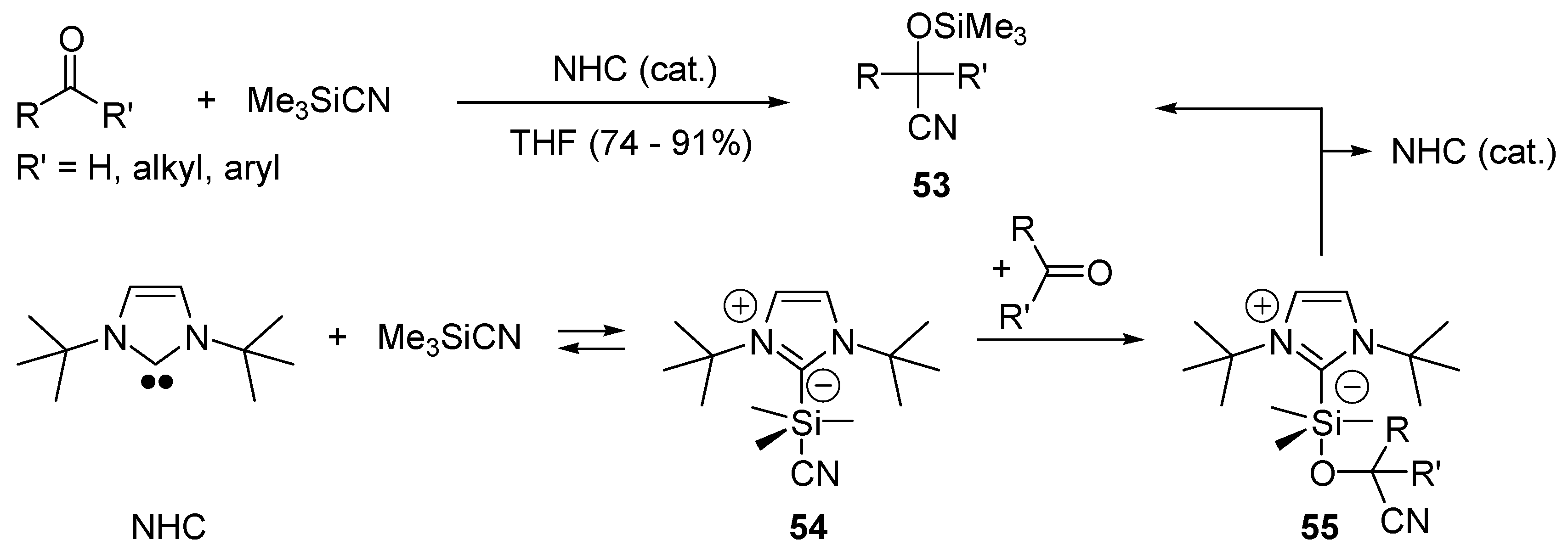{kind=link}
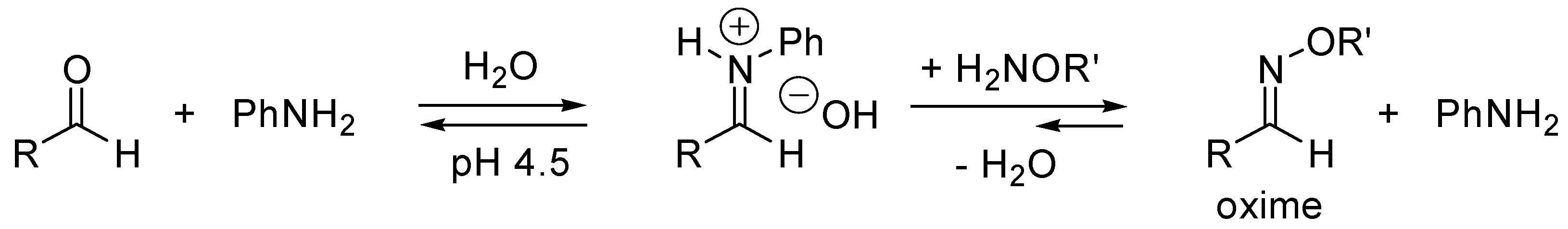{kind=link}
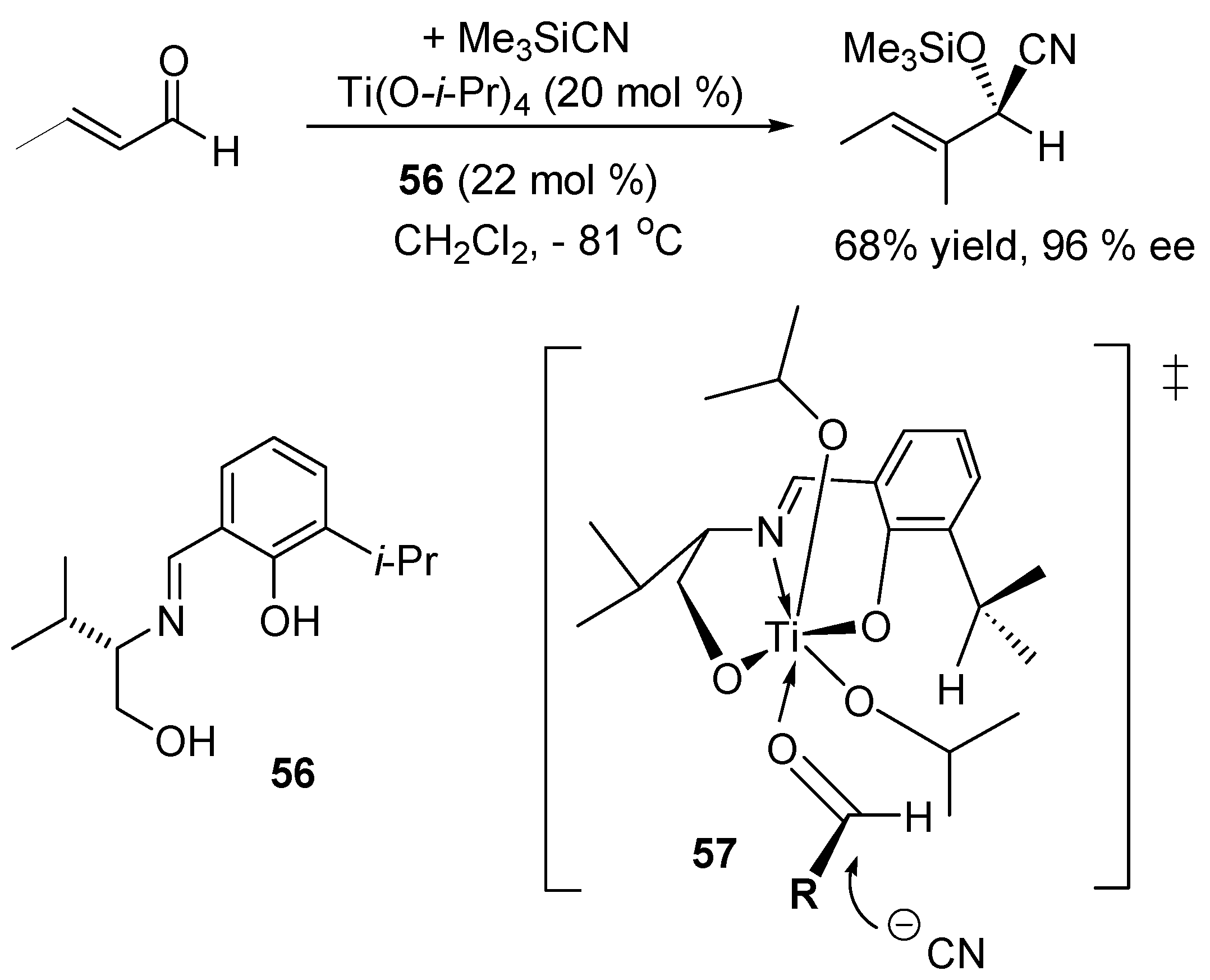{kind=link}
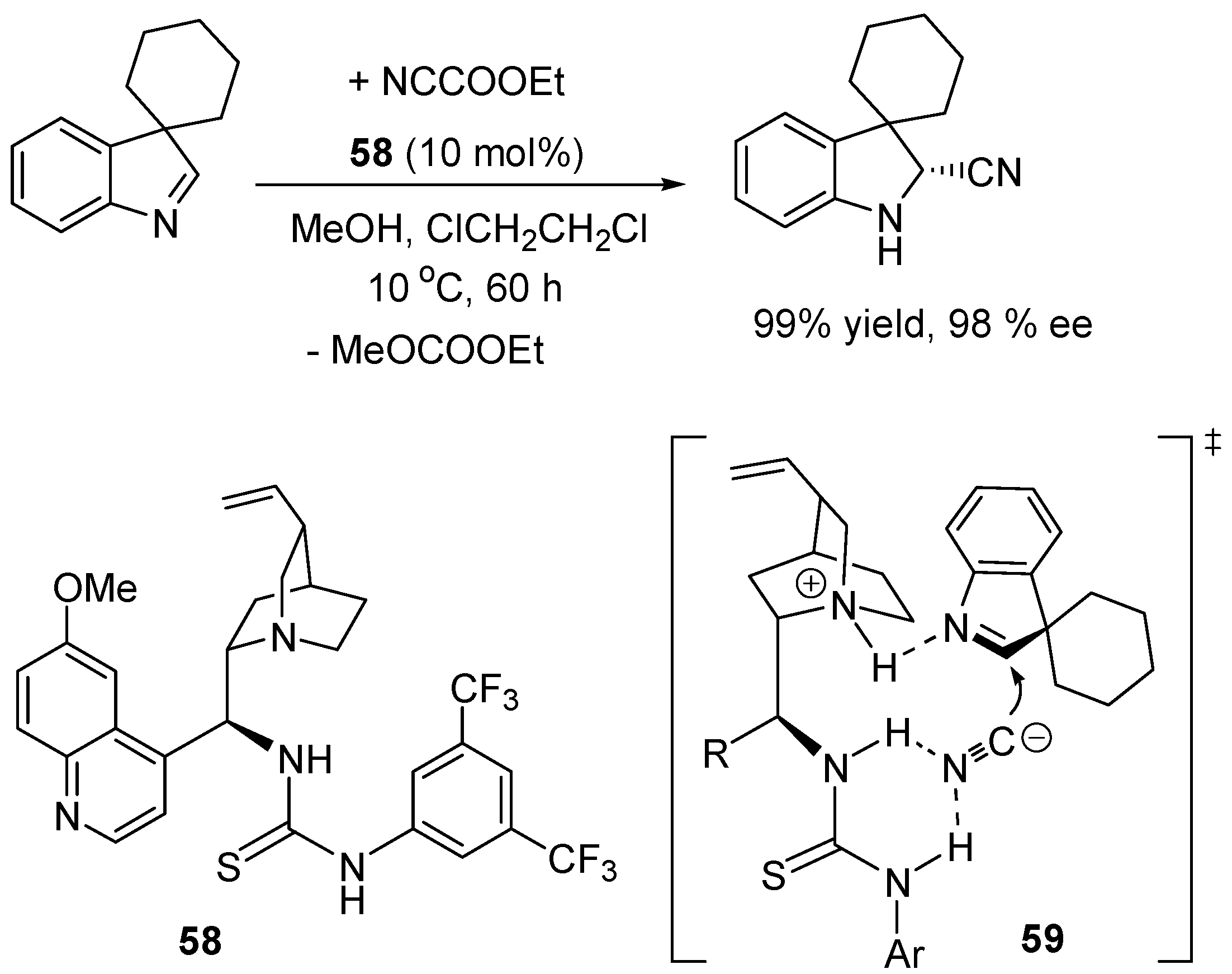{kind=link}
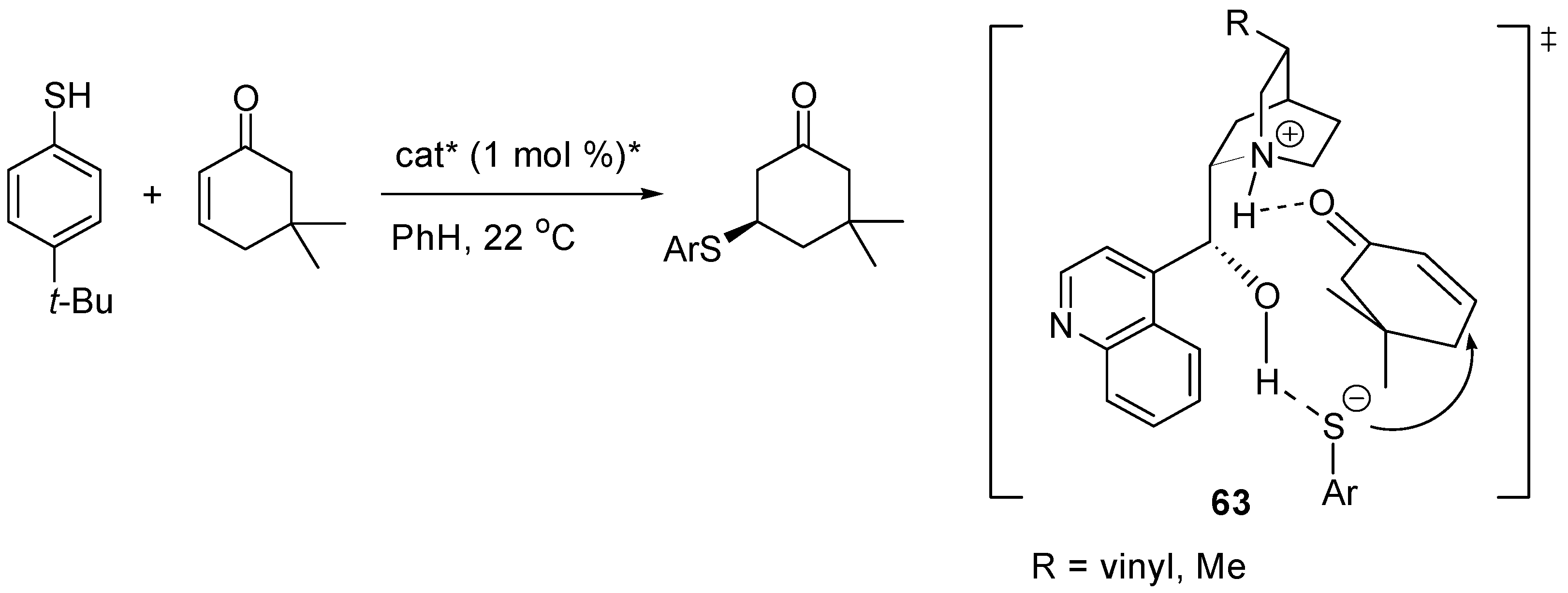{kind=link}
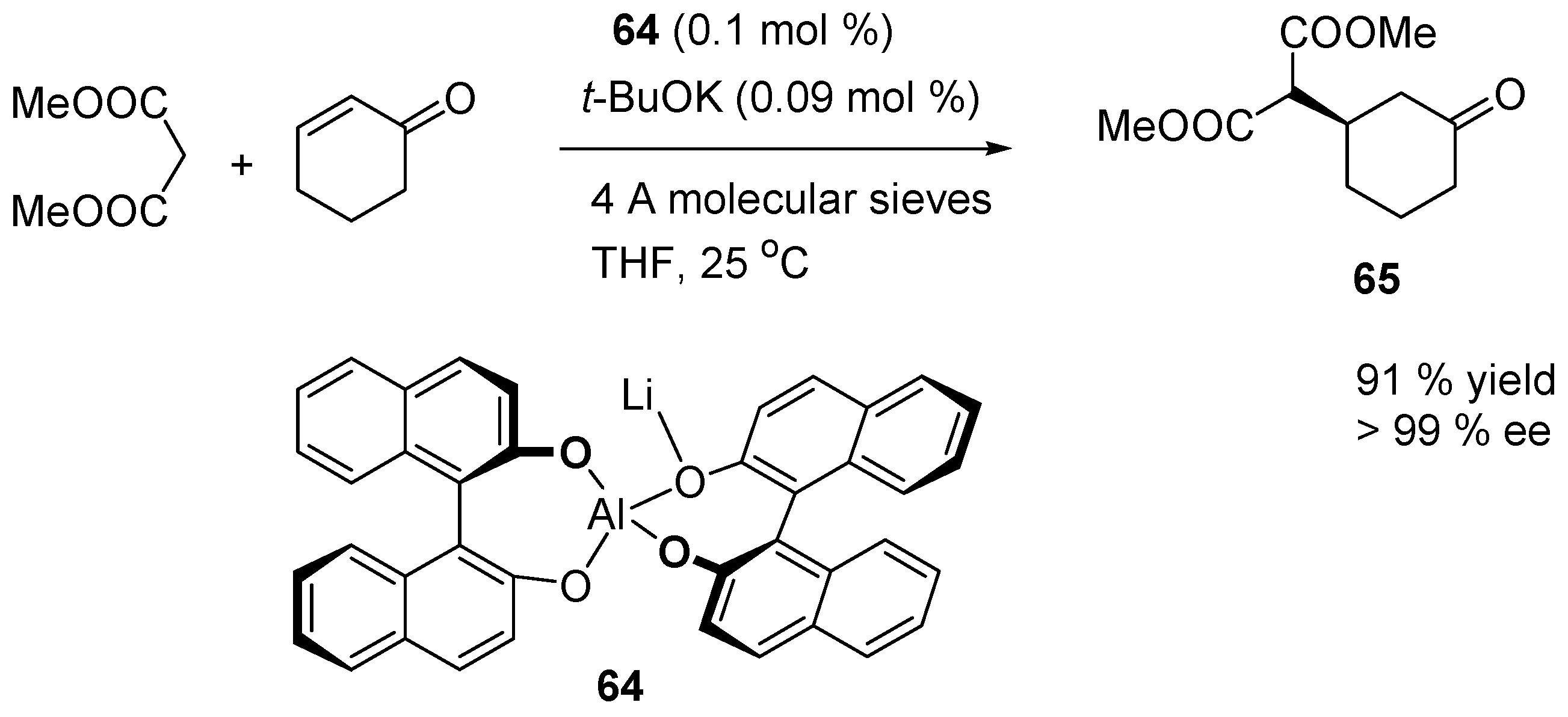{kind=link}
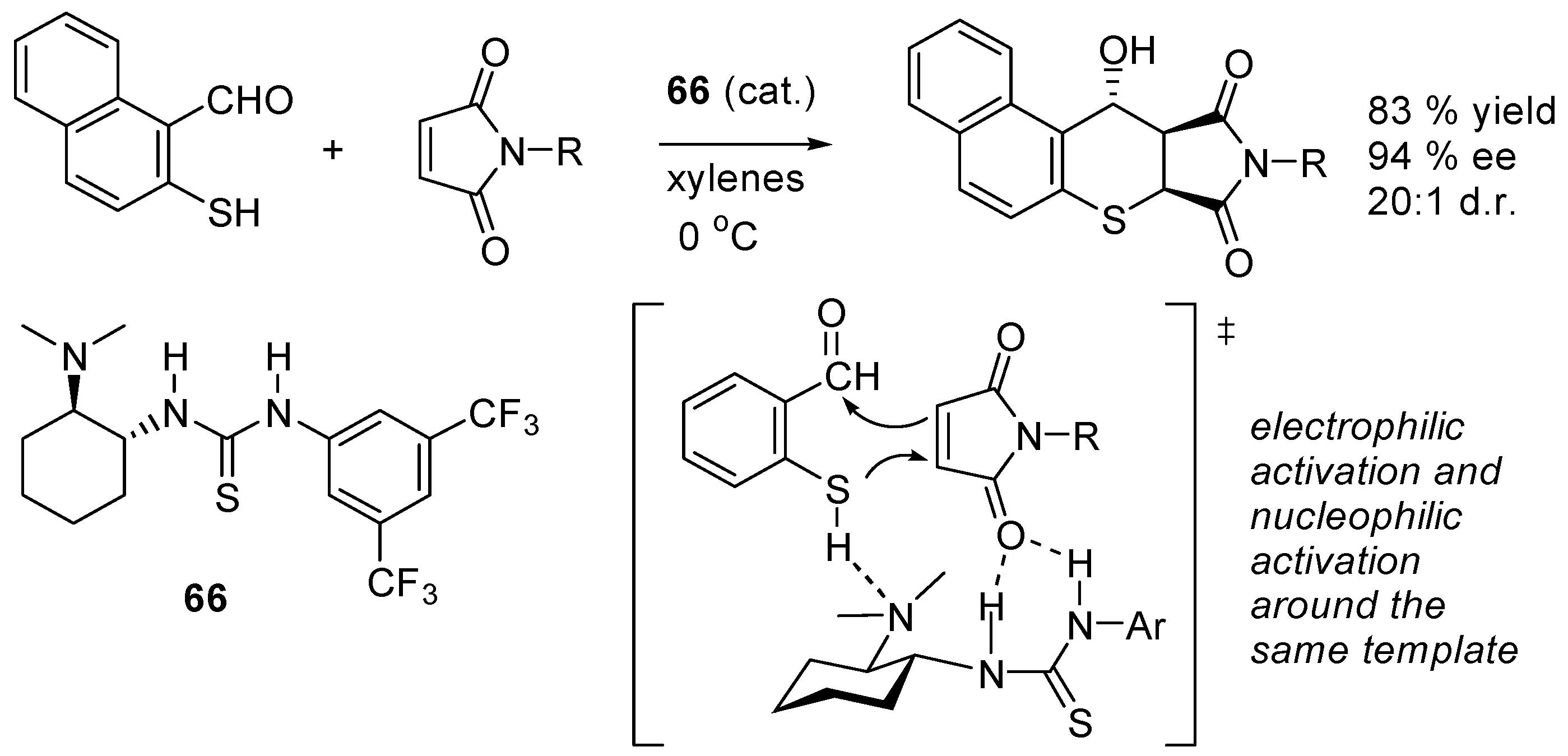{kind=link}
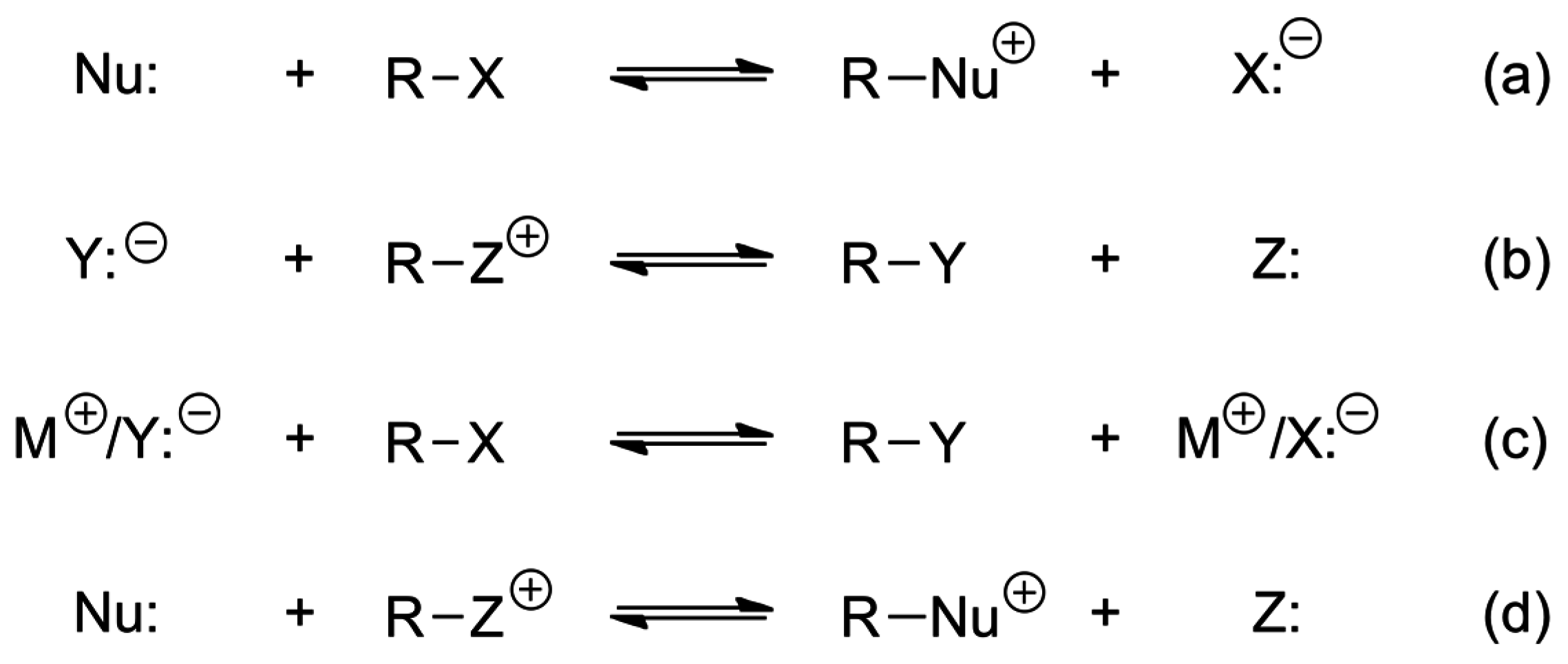{kind=link}
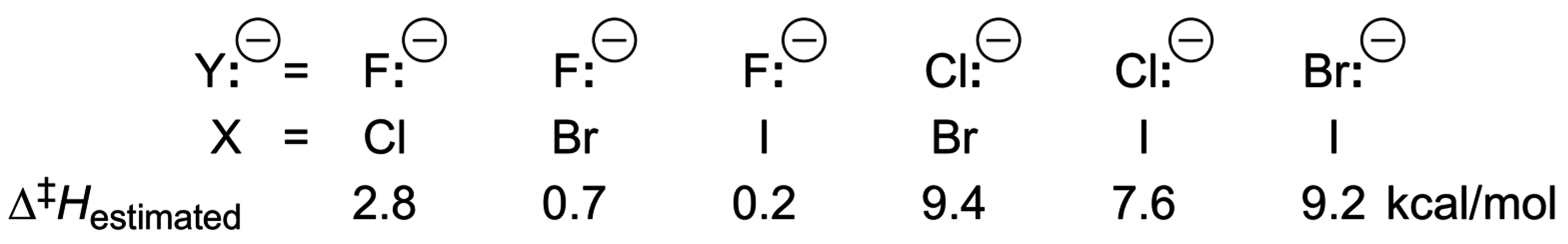{kind=link}
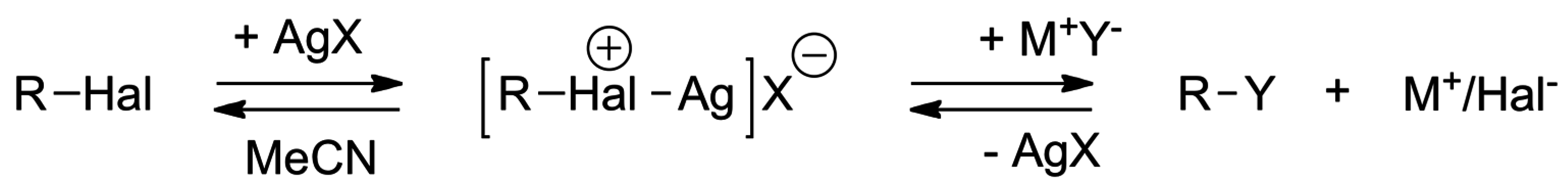{kind=link}
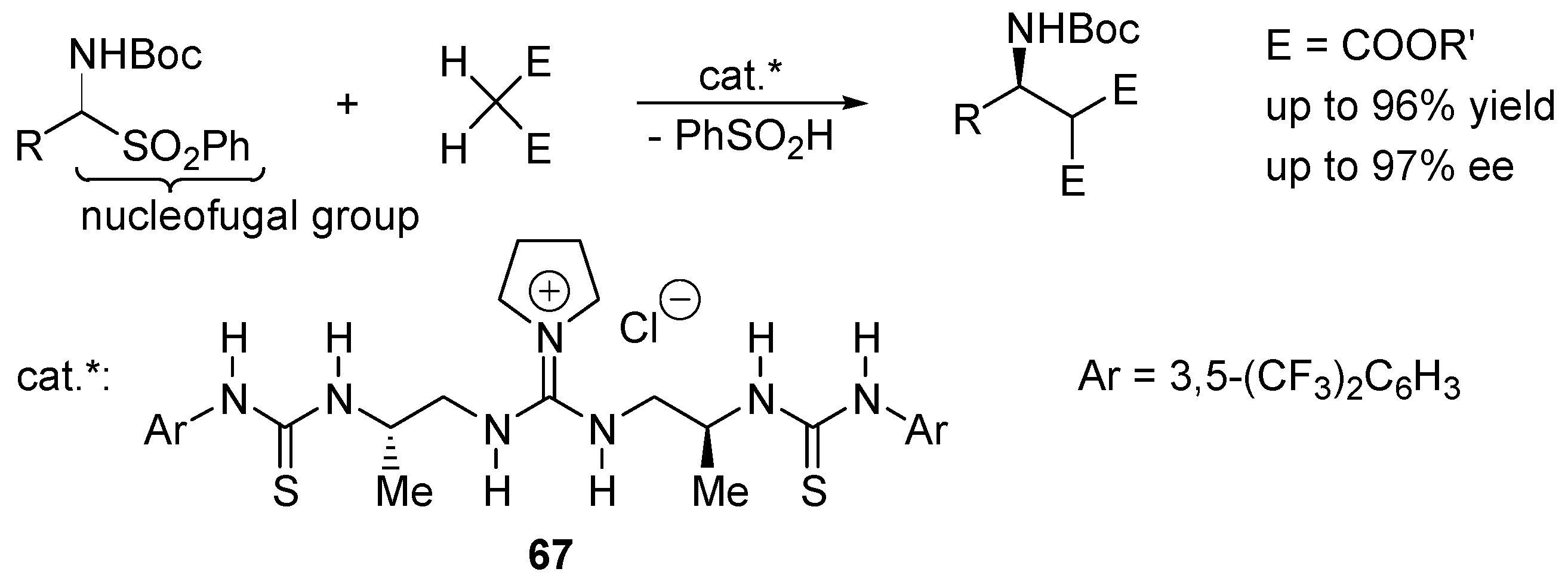{kind=link}
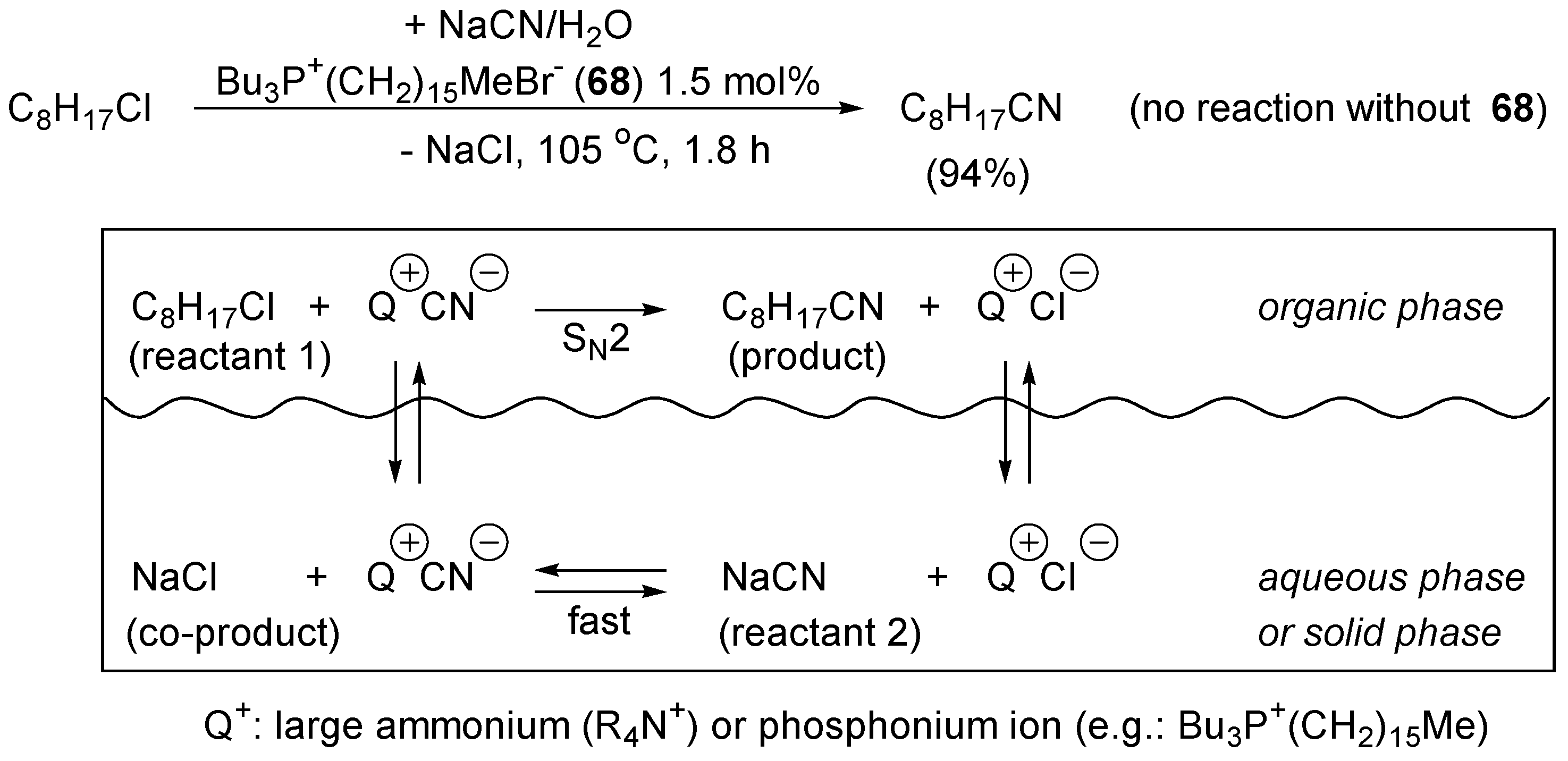{kind=link}
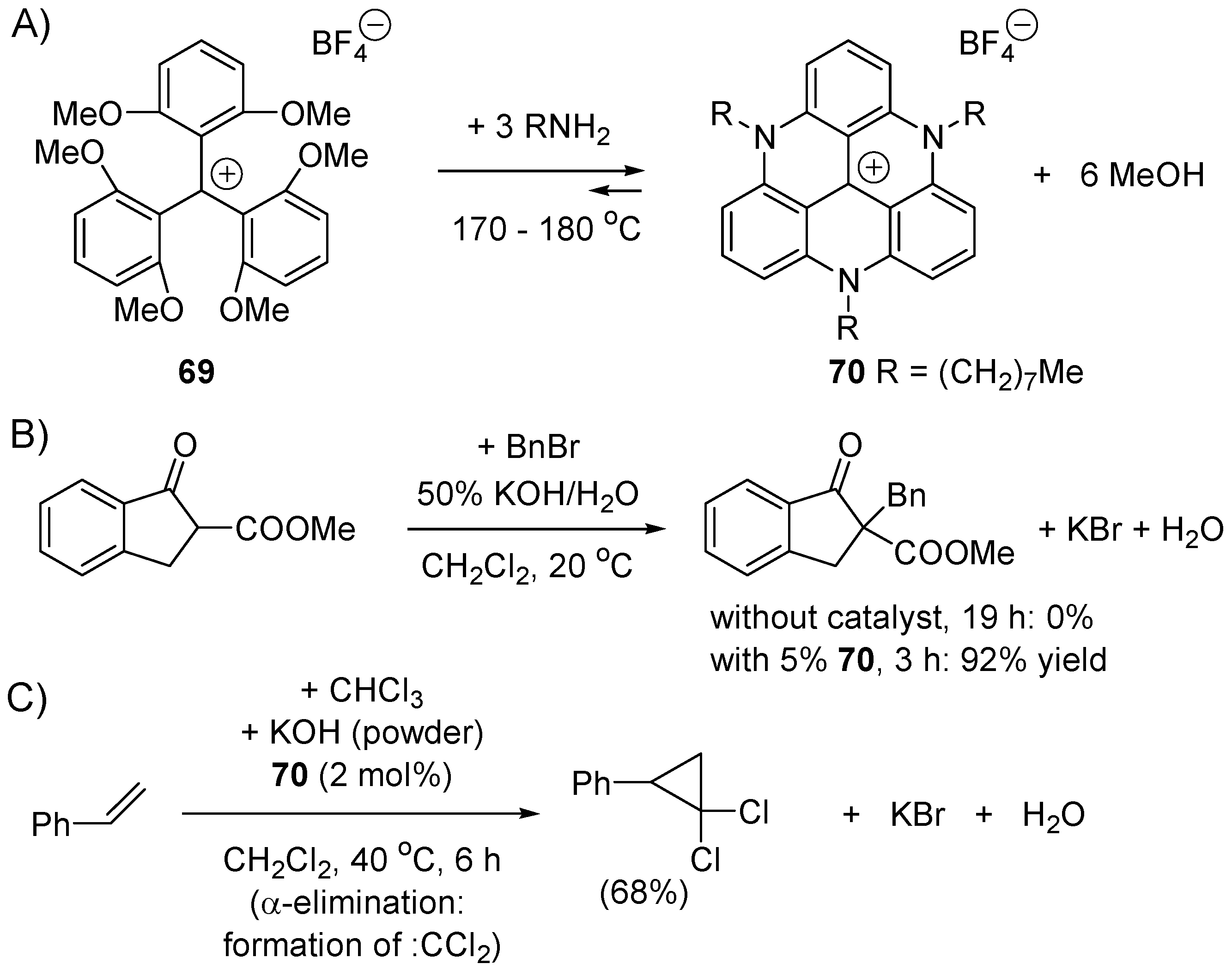{kind=link}
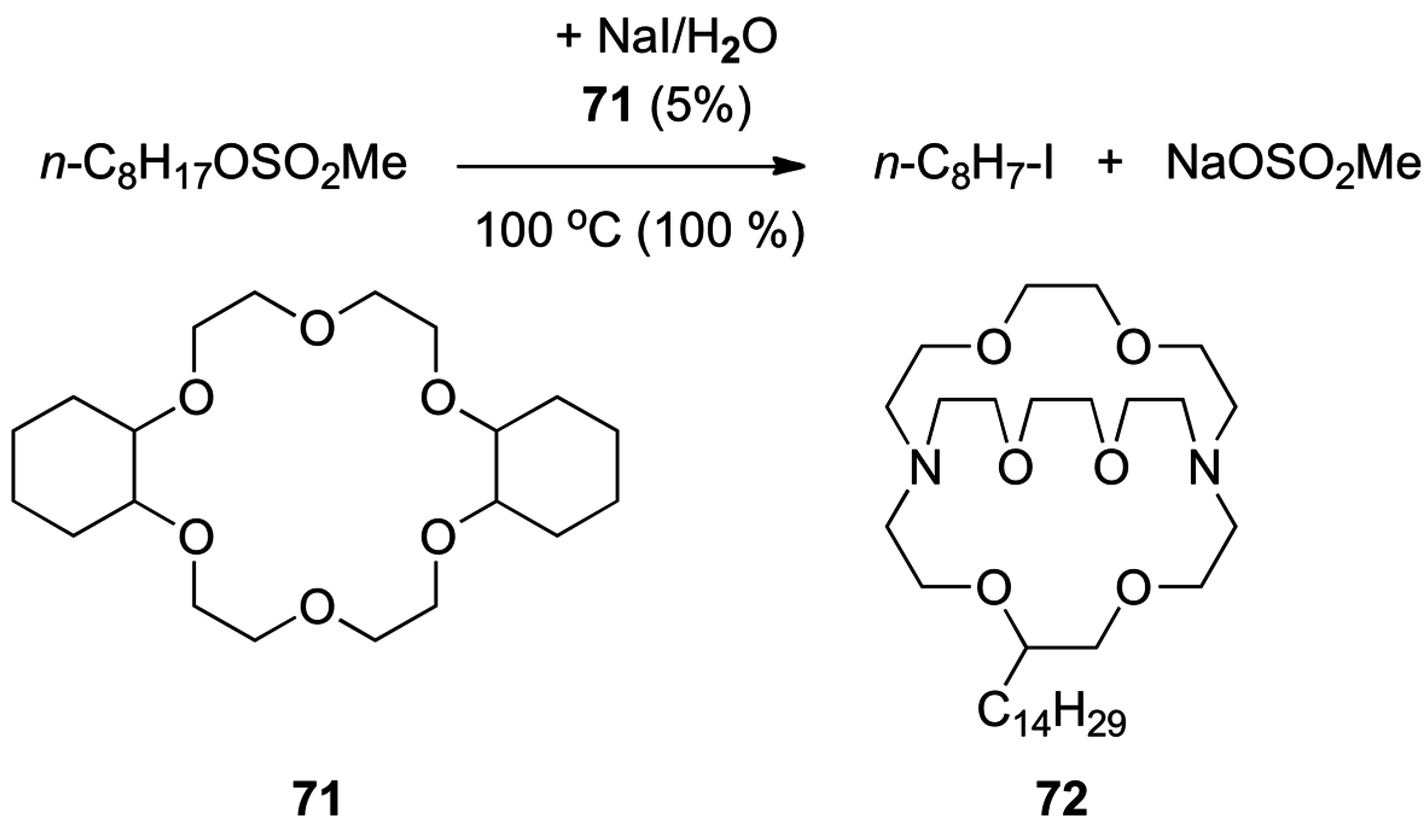{kind=link}
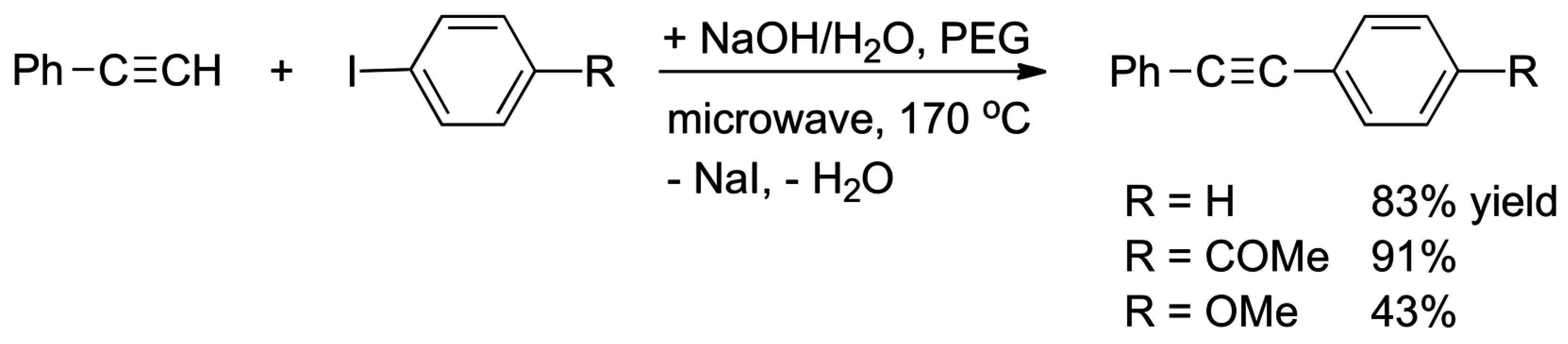{kind=link}
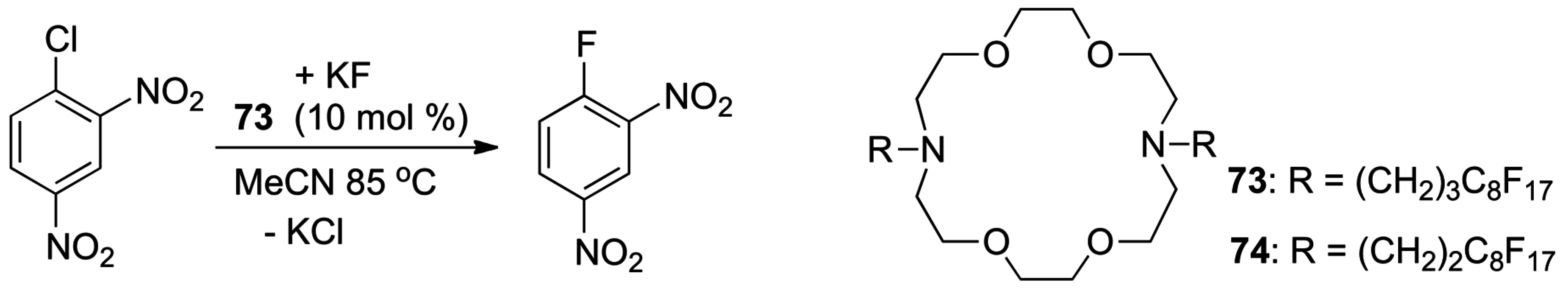{kind=link}
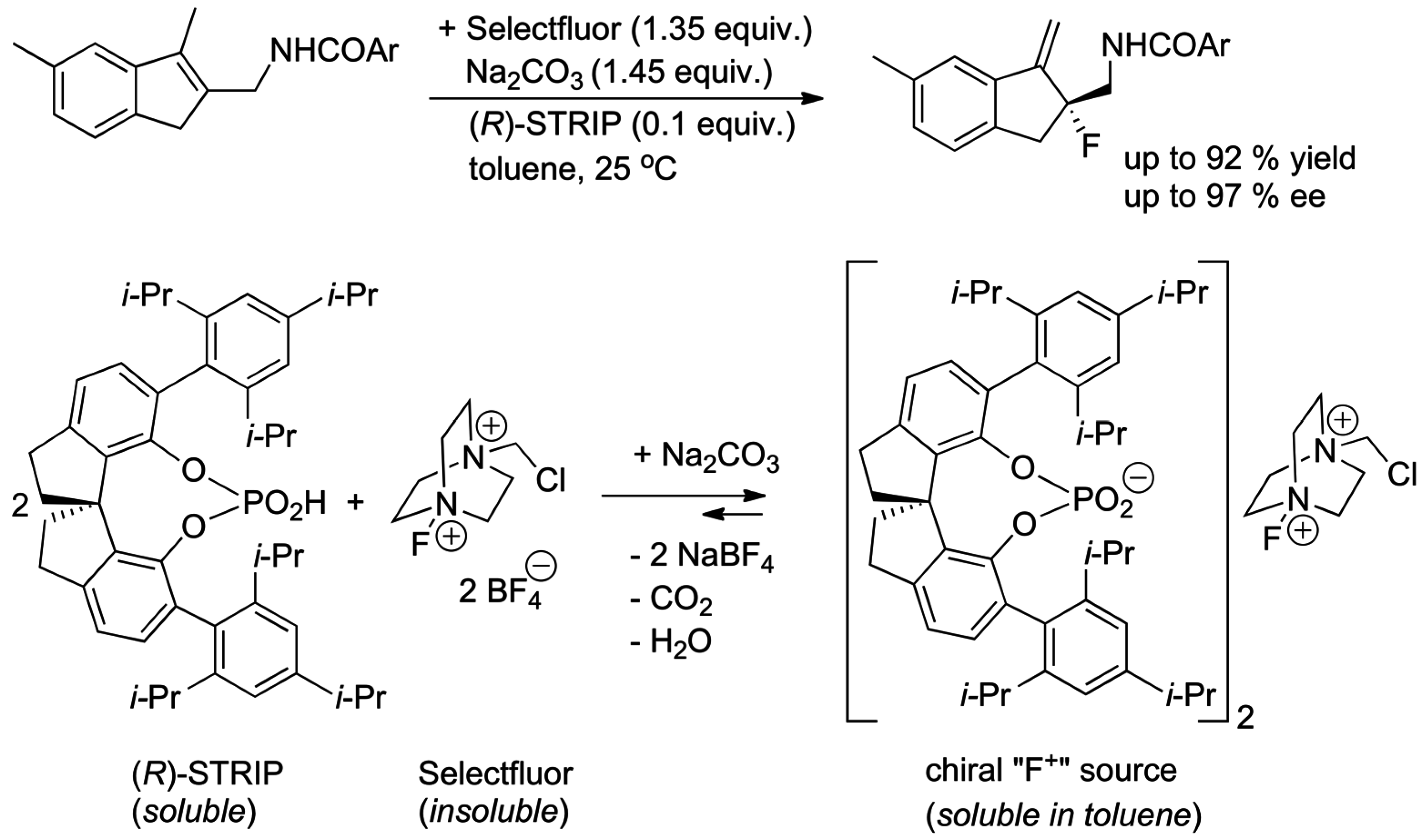{kind=link}
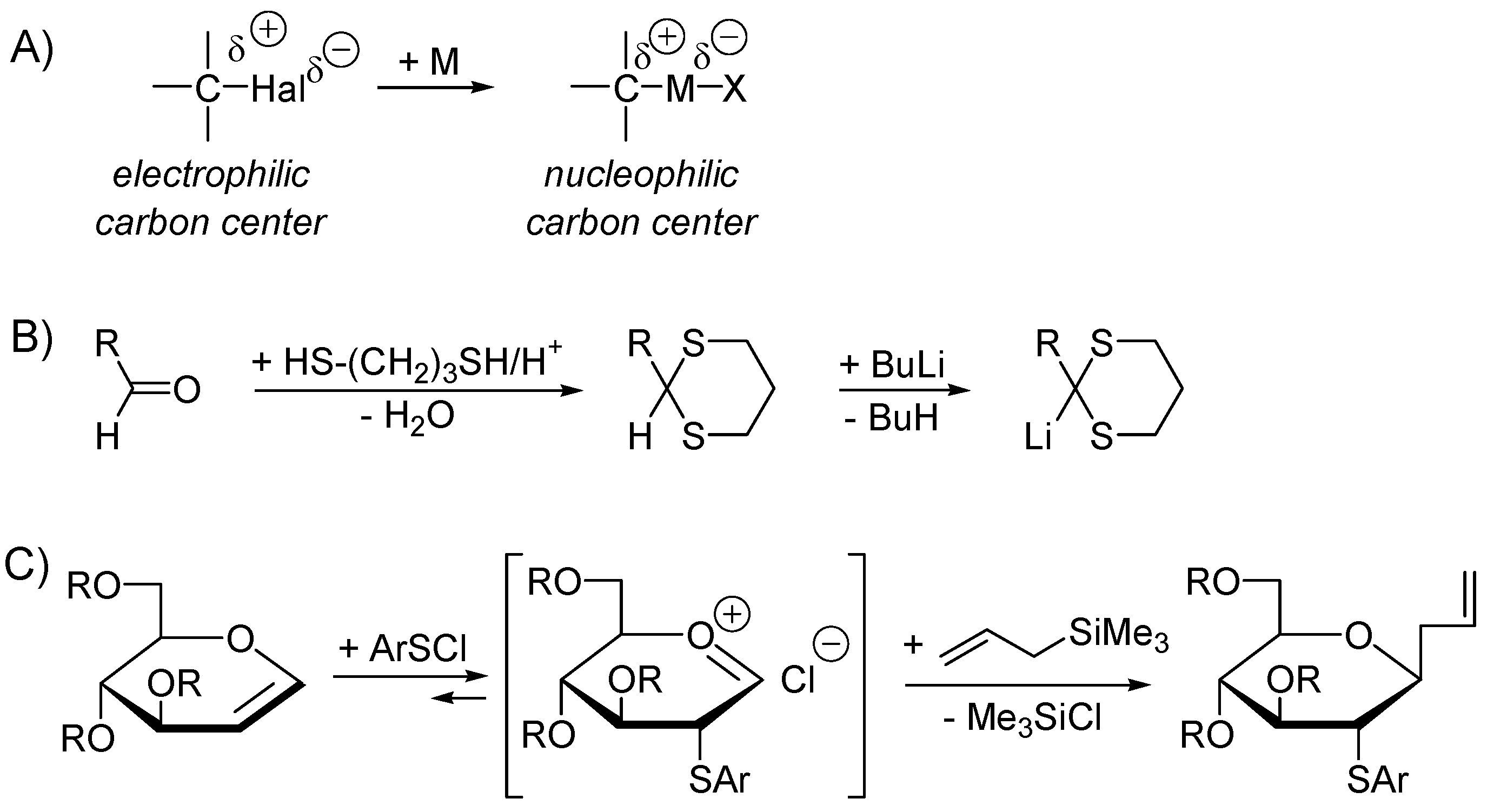{kind=link}
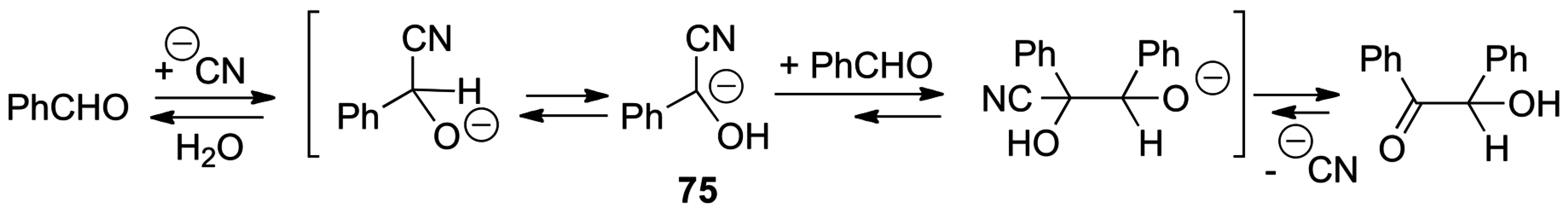{kind=link}
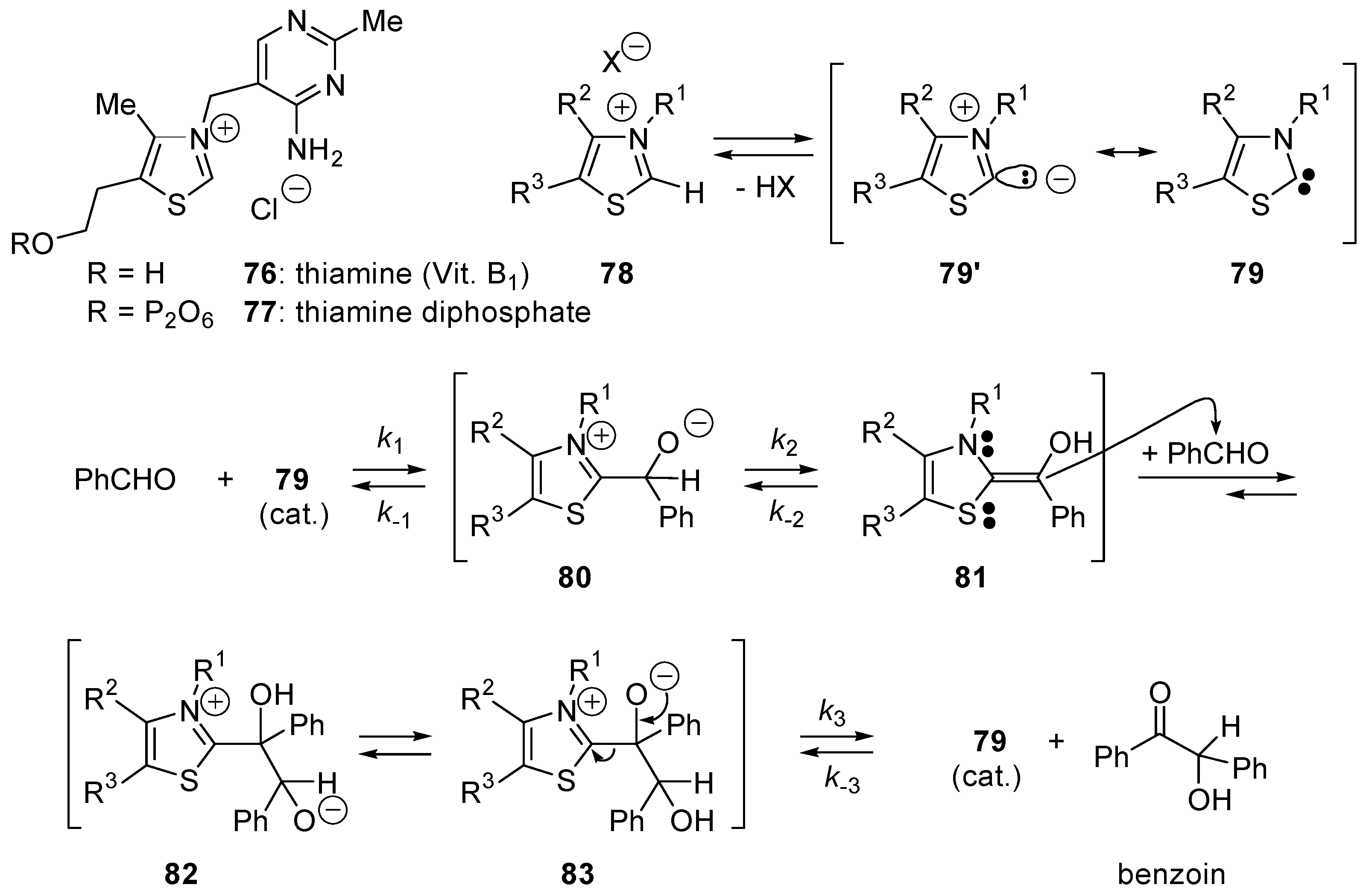{kind=link}
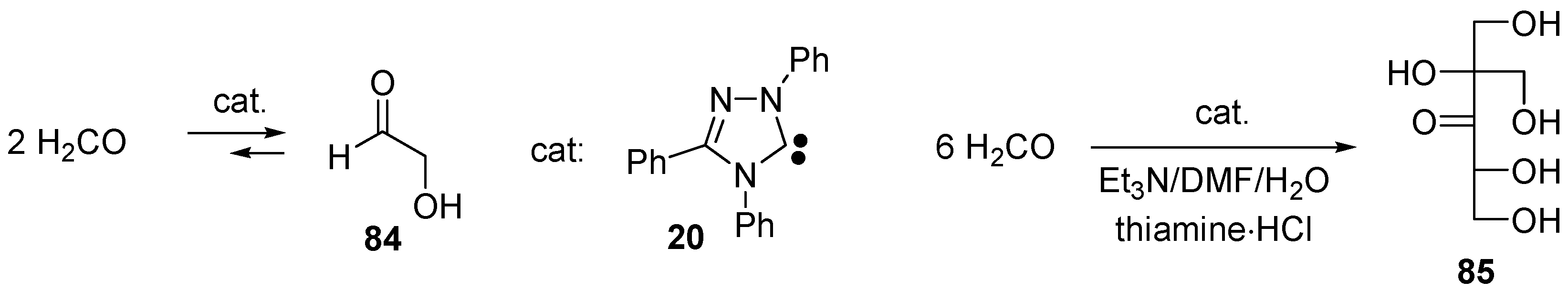{kind=link}
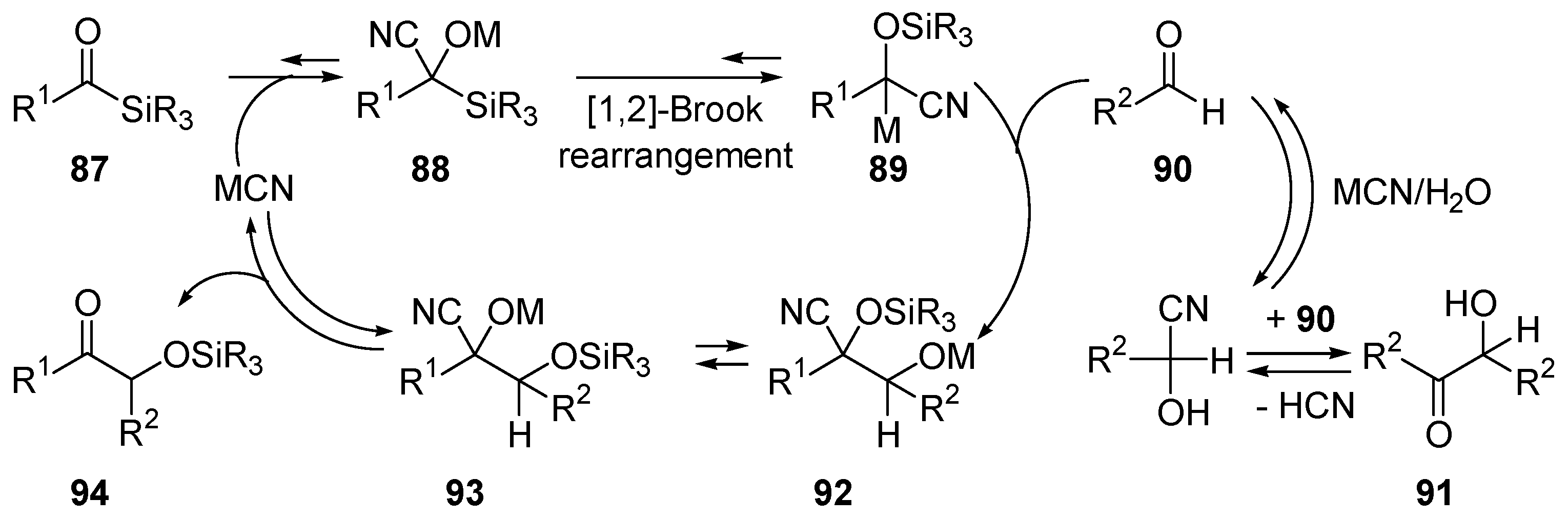{kind=link}
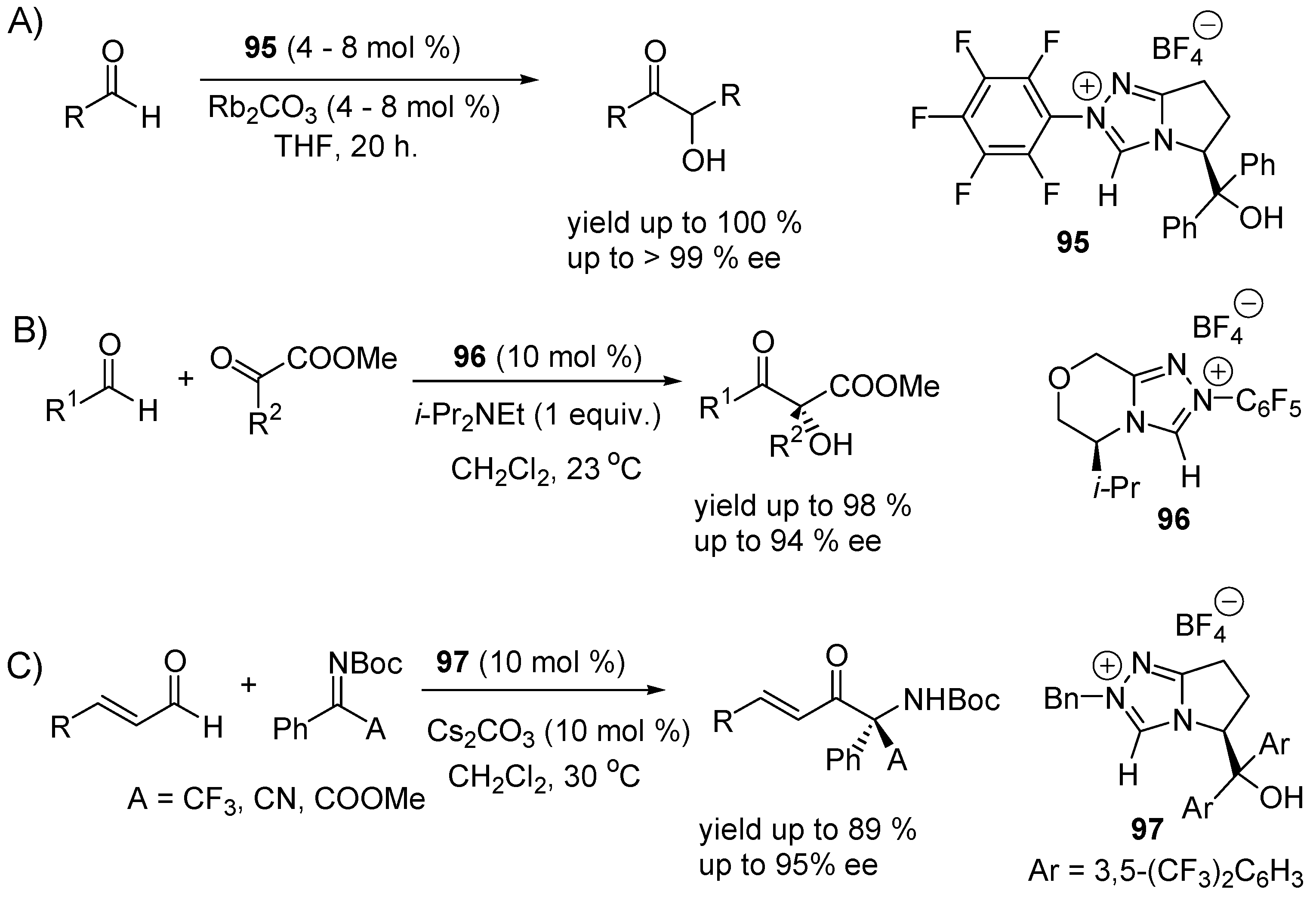{kind=link}
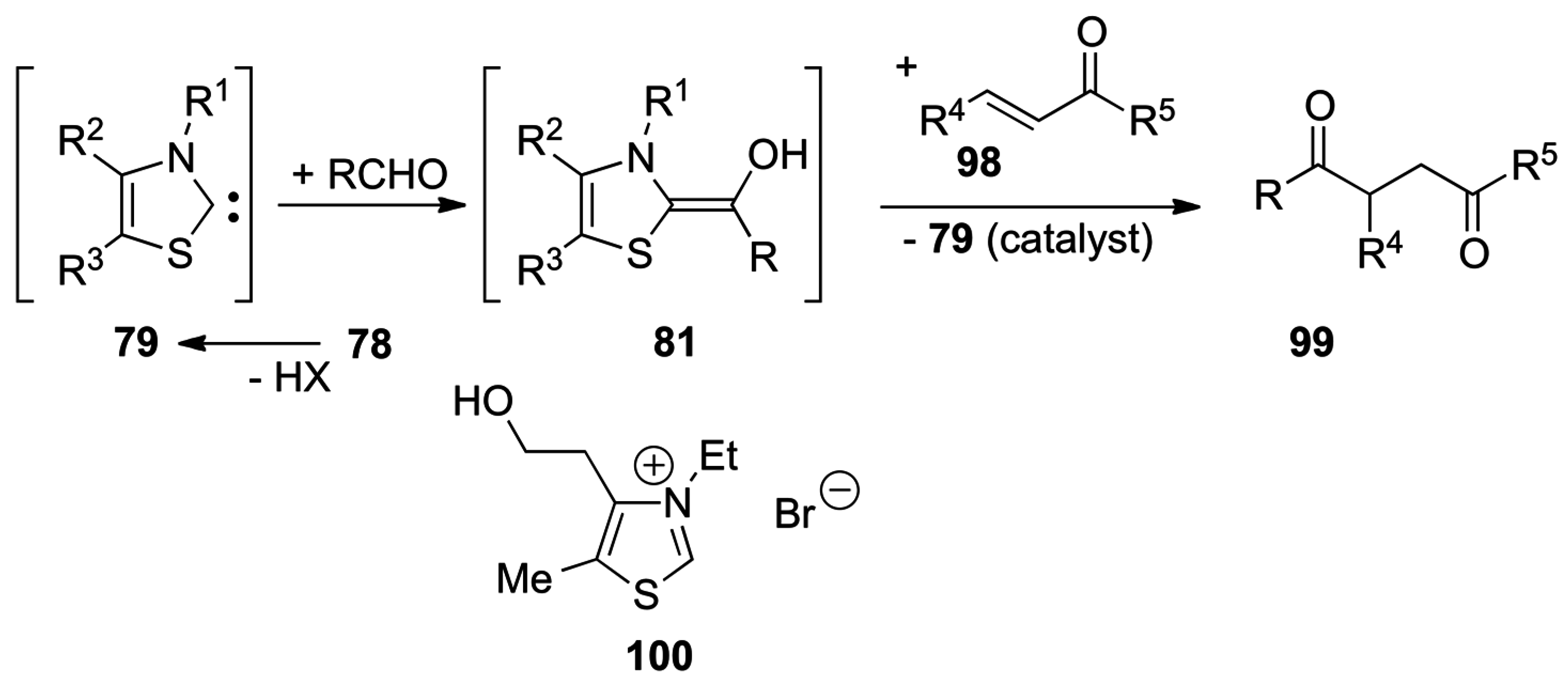{kind=link}
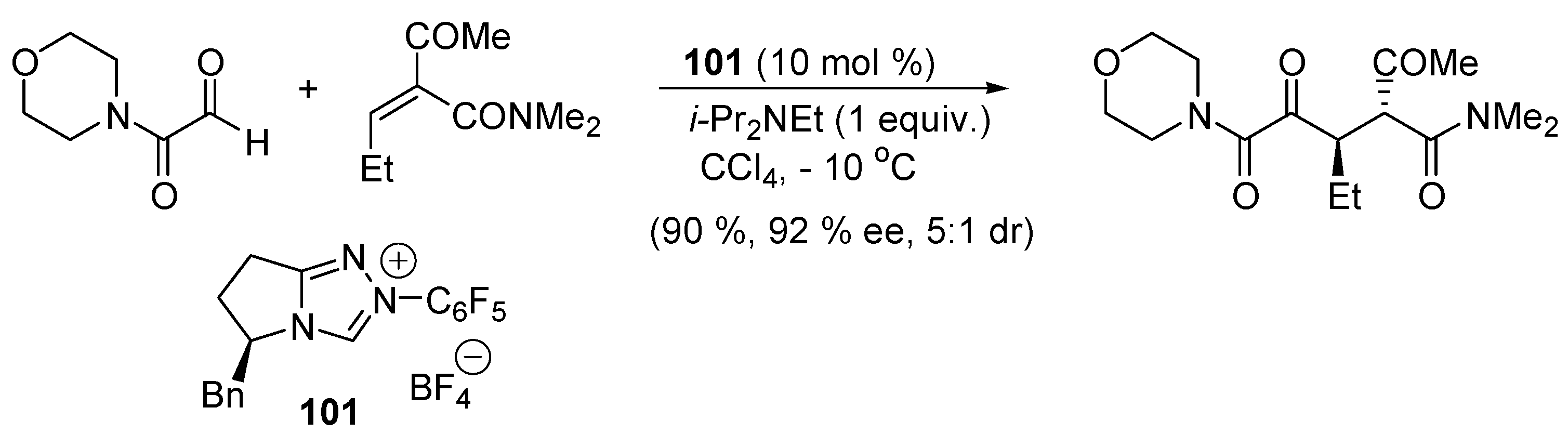{kind=link}
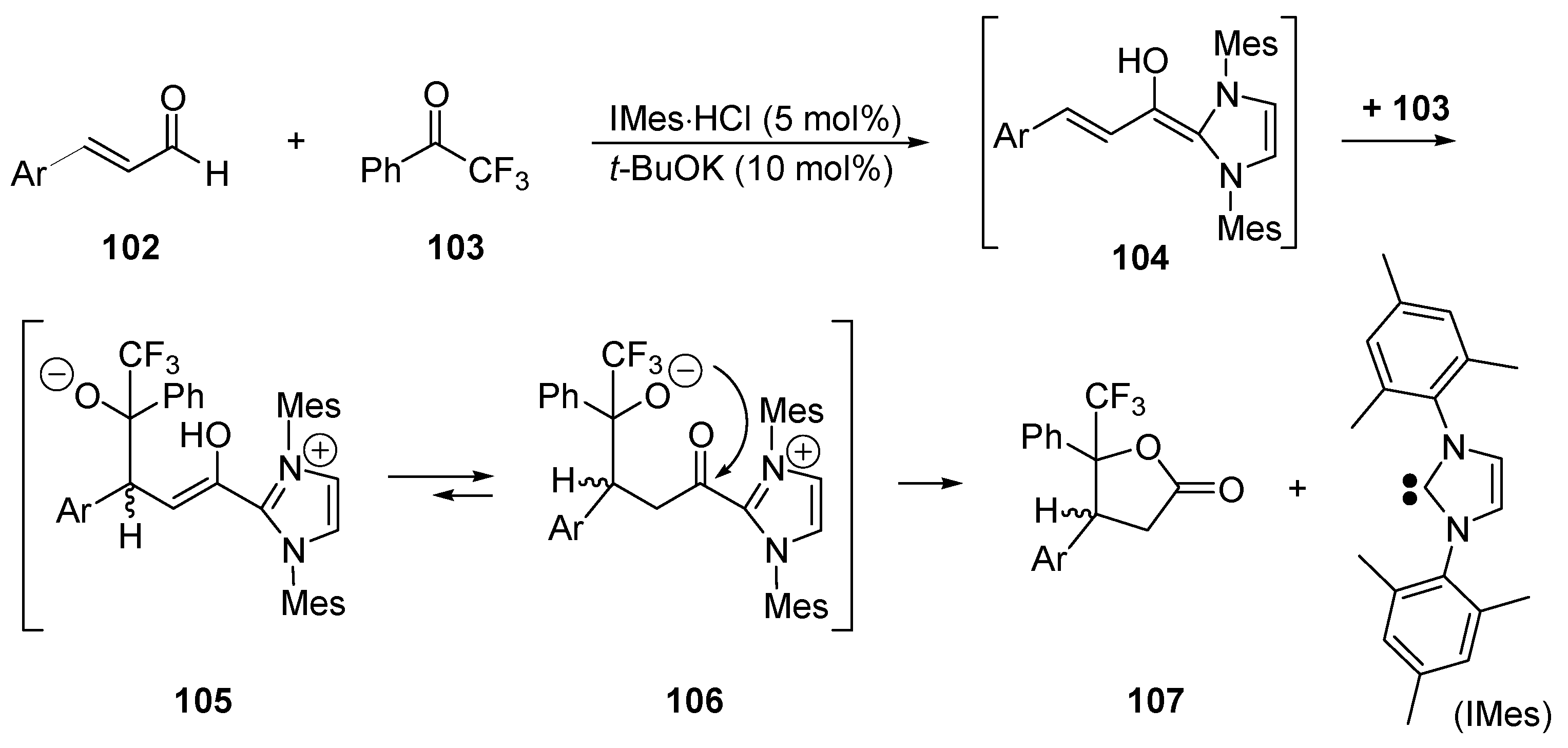{kind=link}
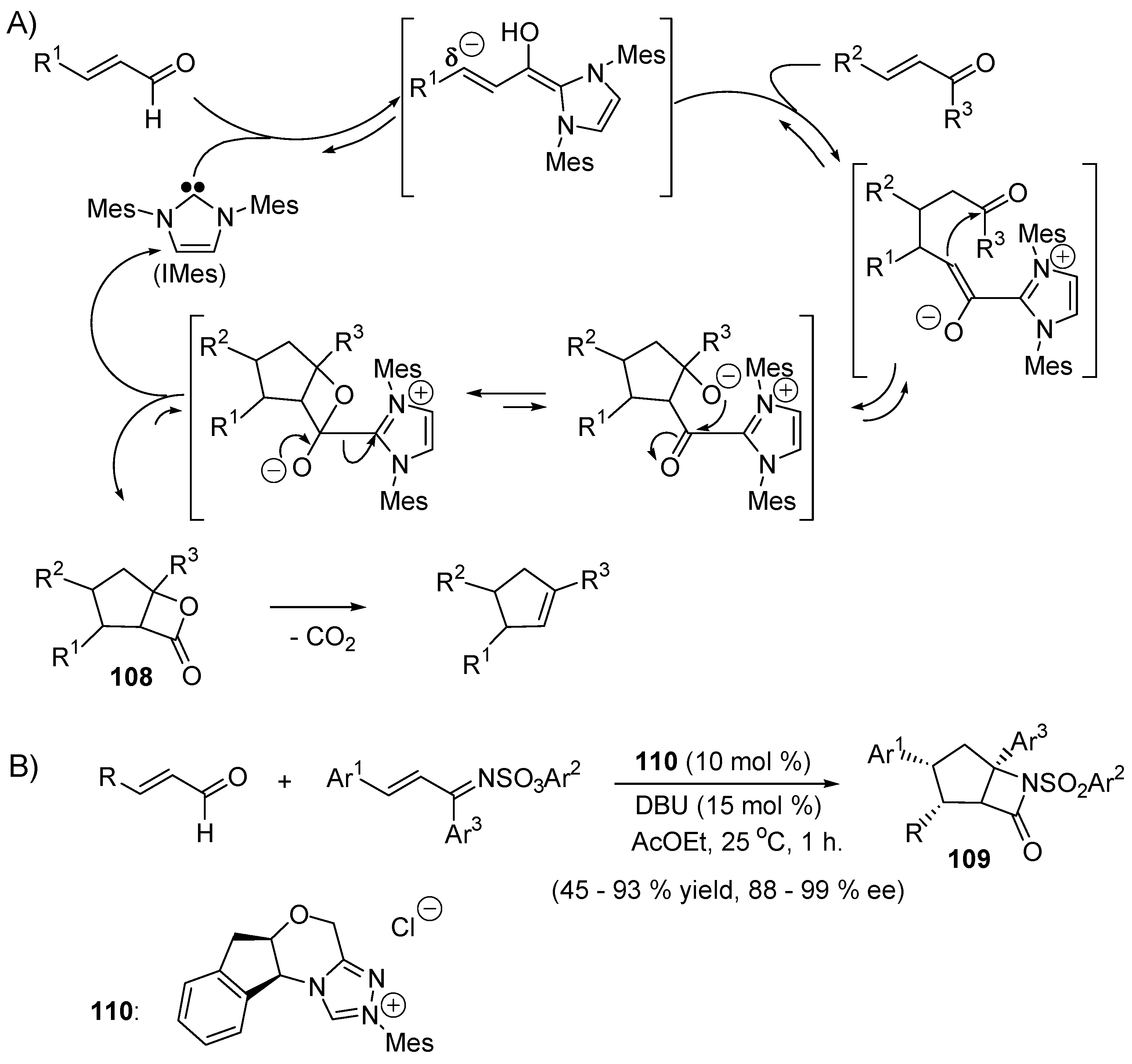{kind=link}
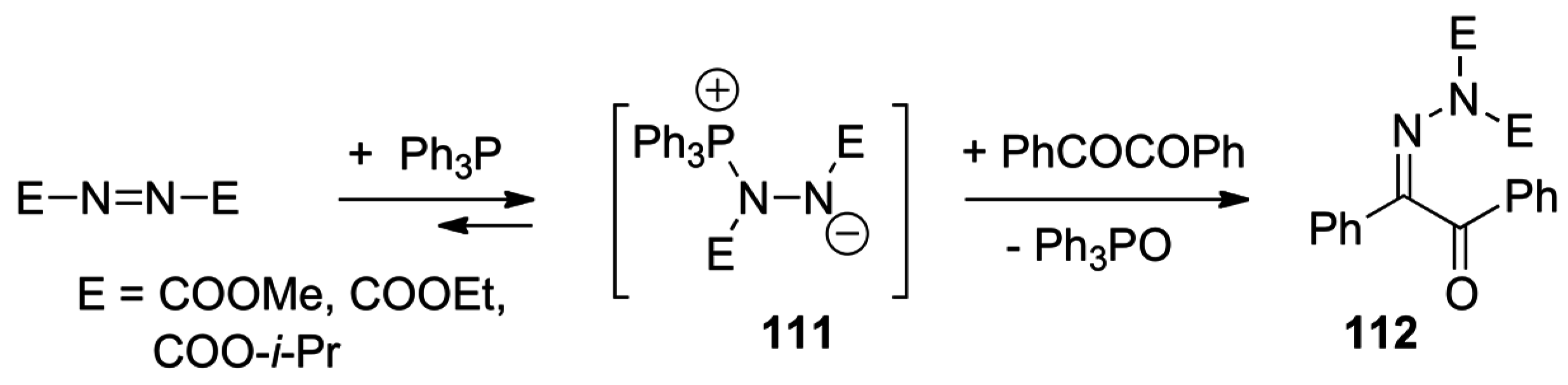{kind=link}
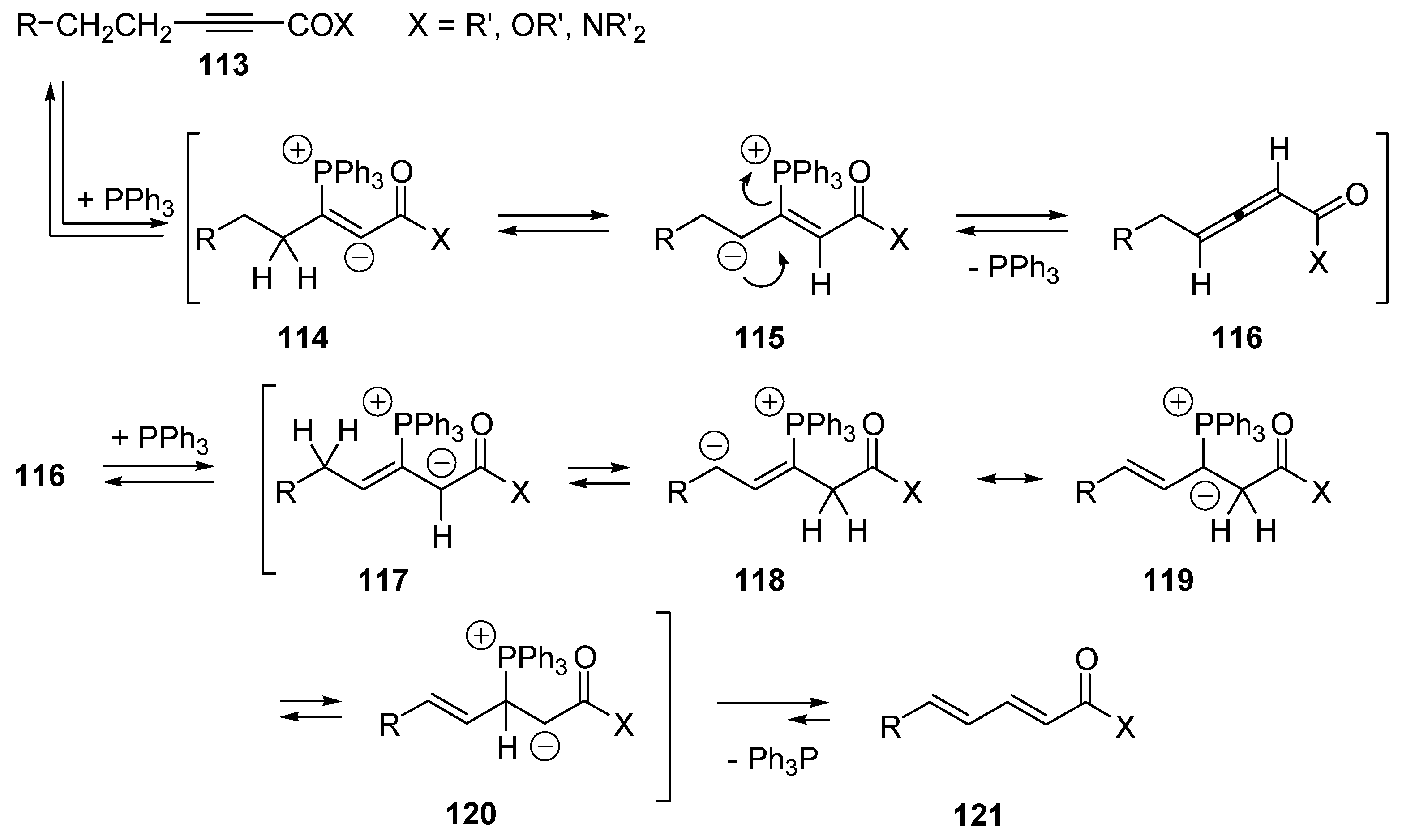{kind=link}
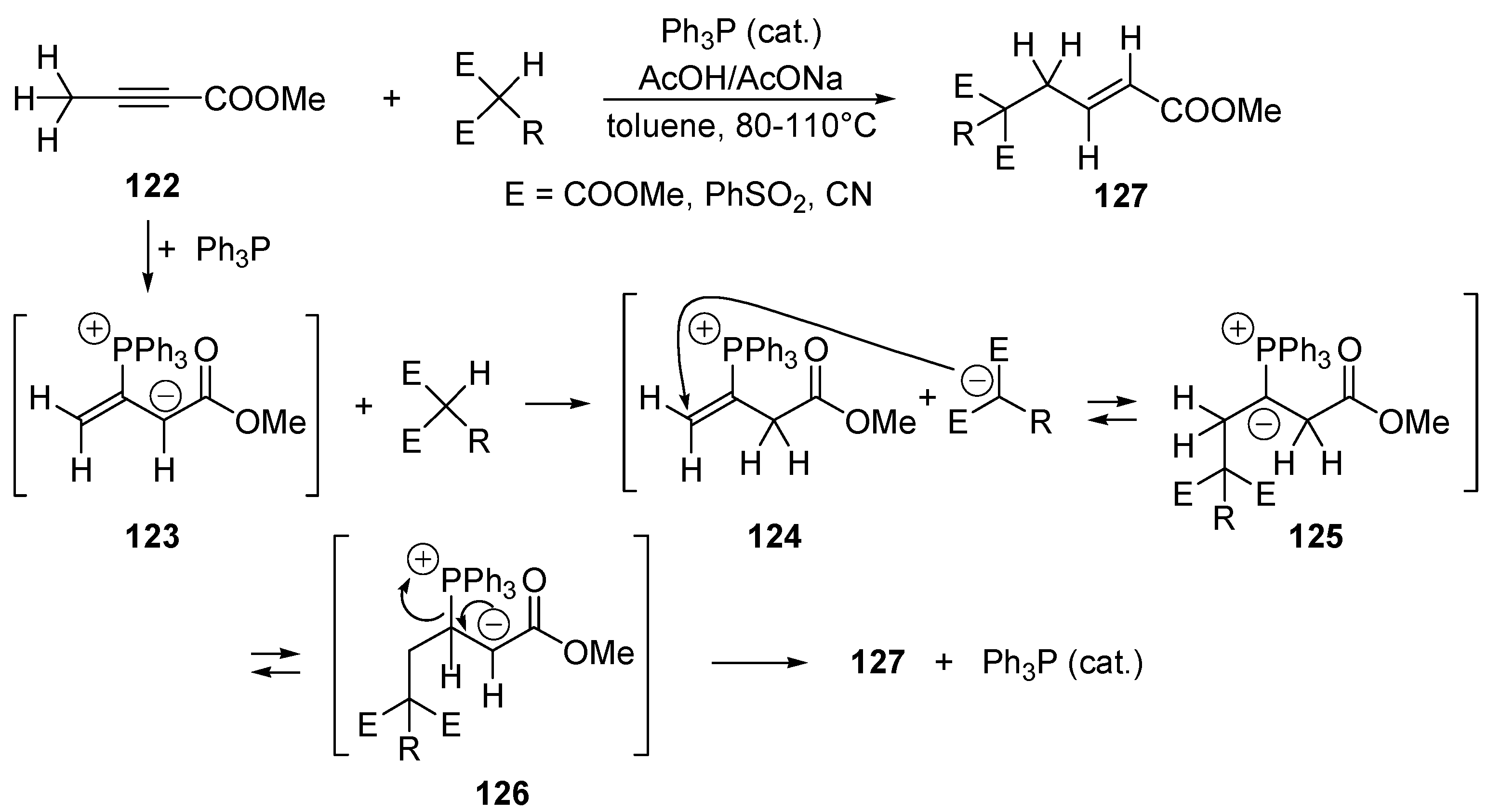{kind=link}
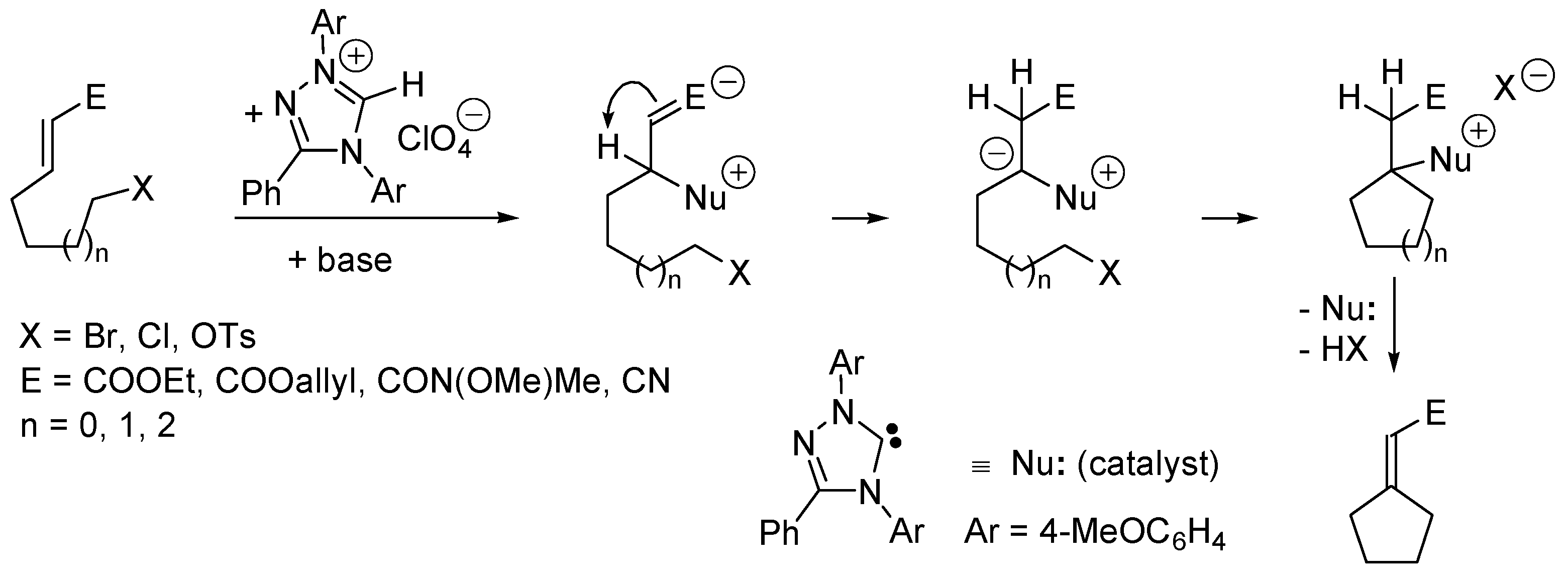{kind=link}
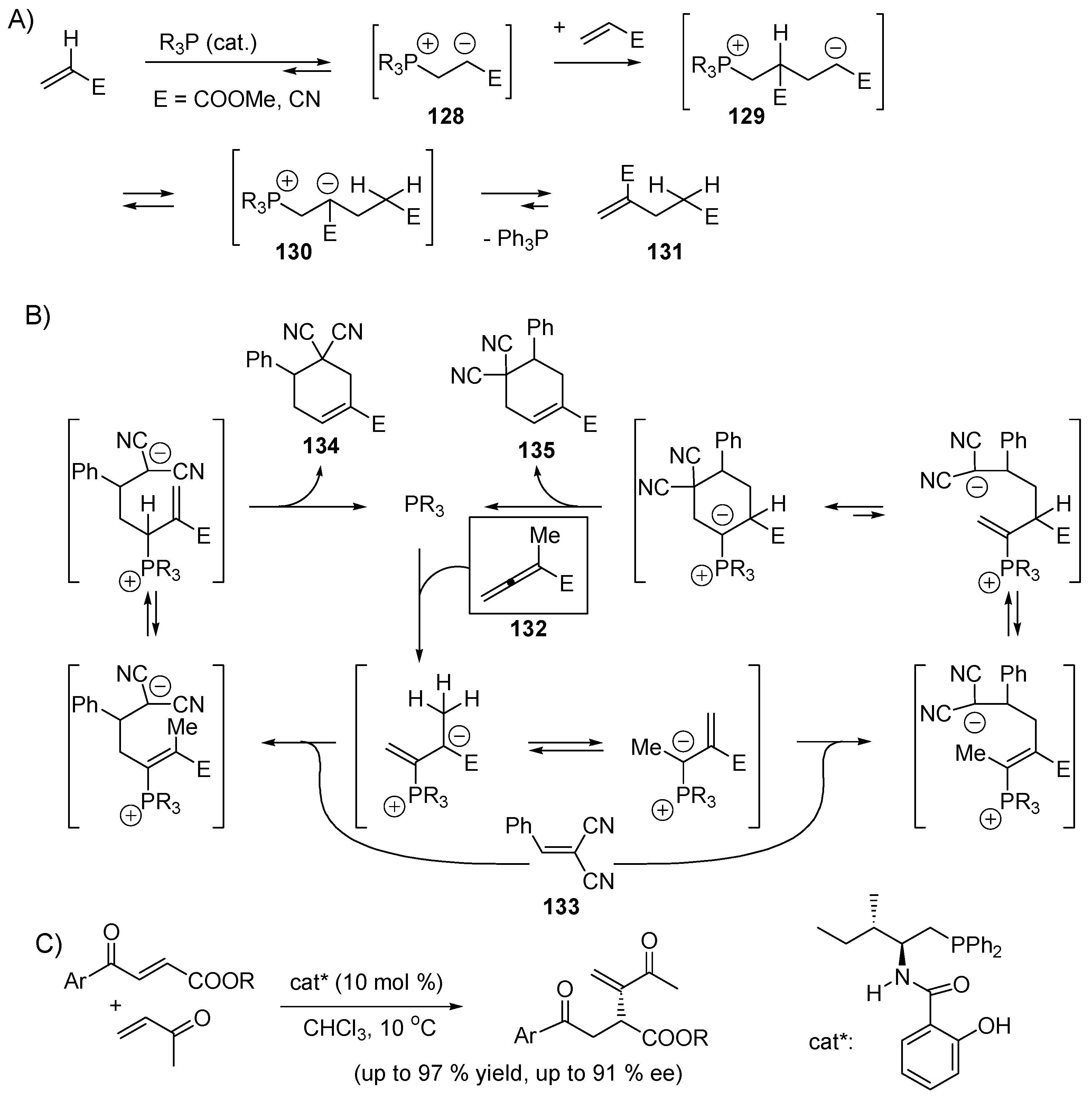{kind=link}
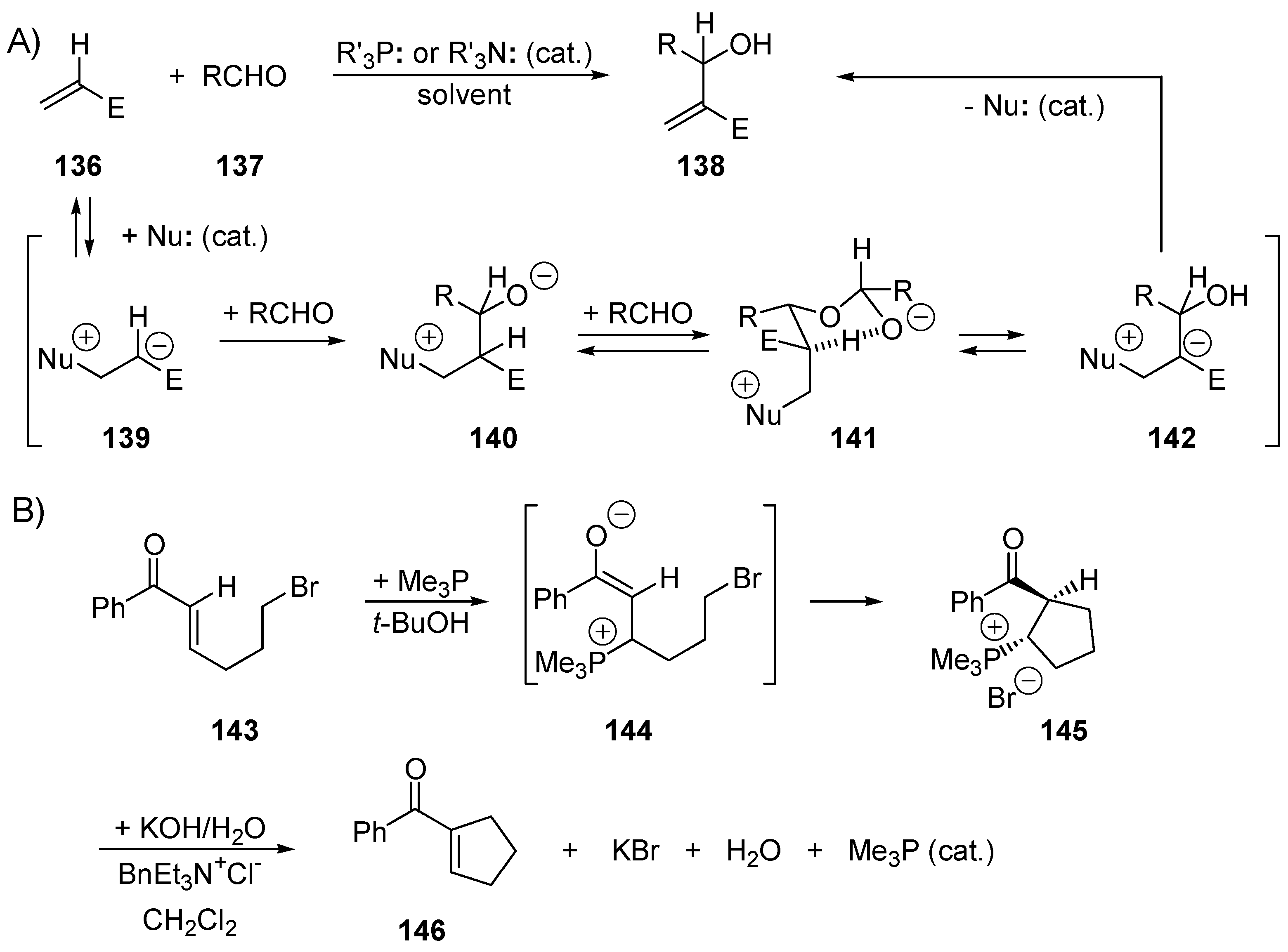{kind=link}
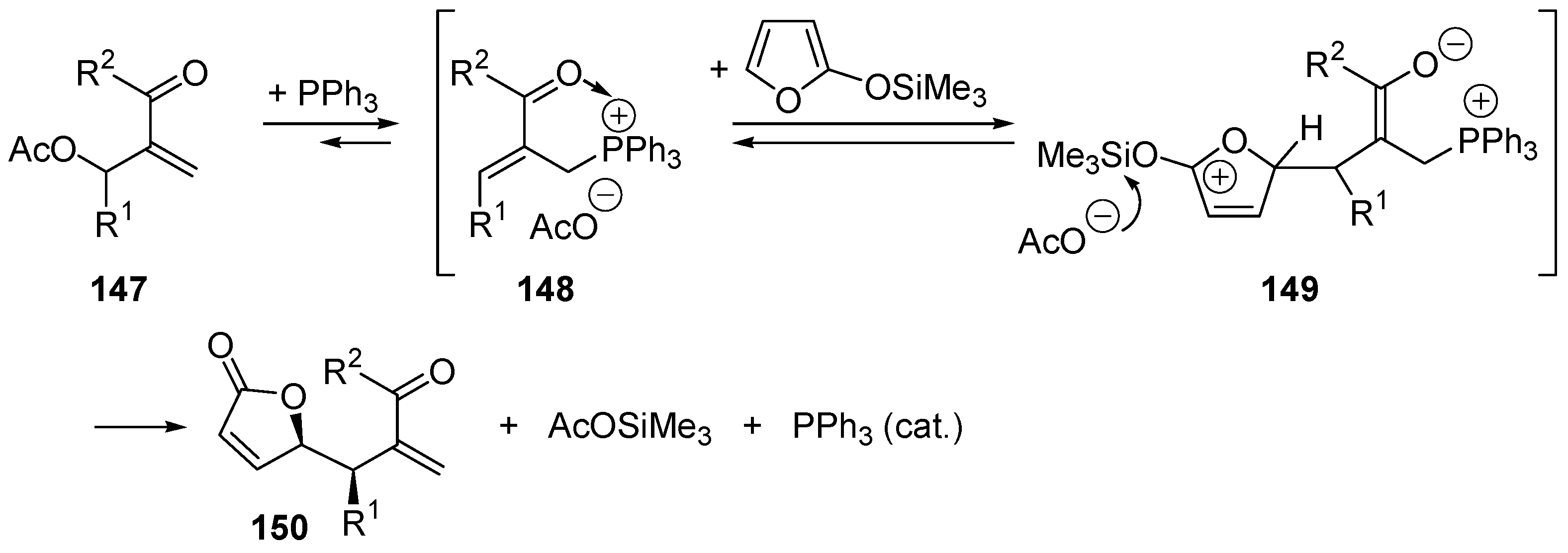{kind=link}
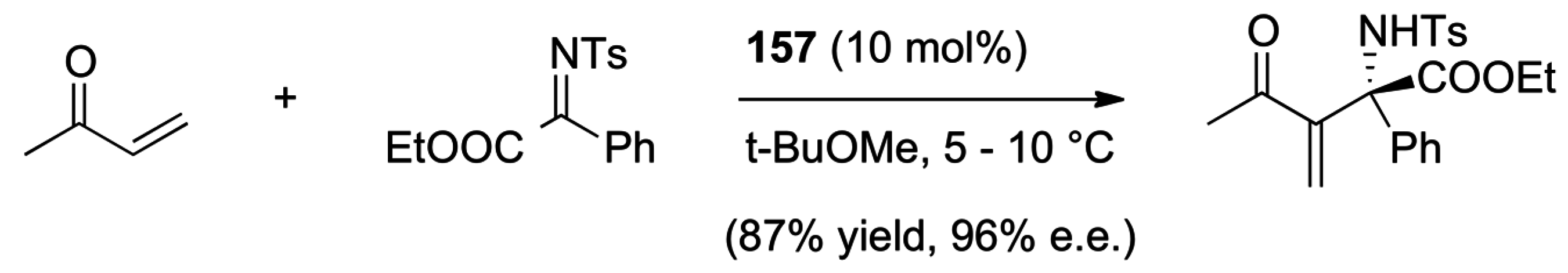{kind=link}
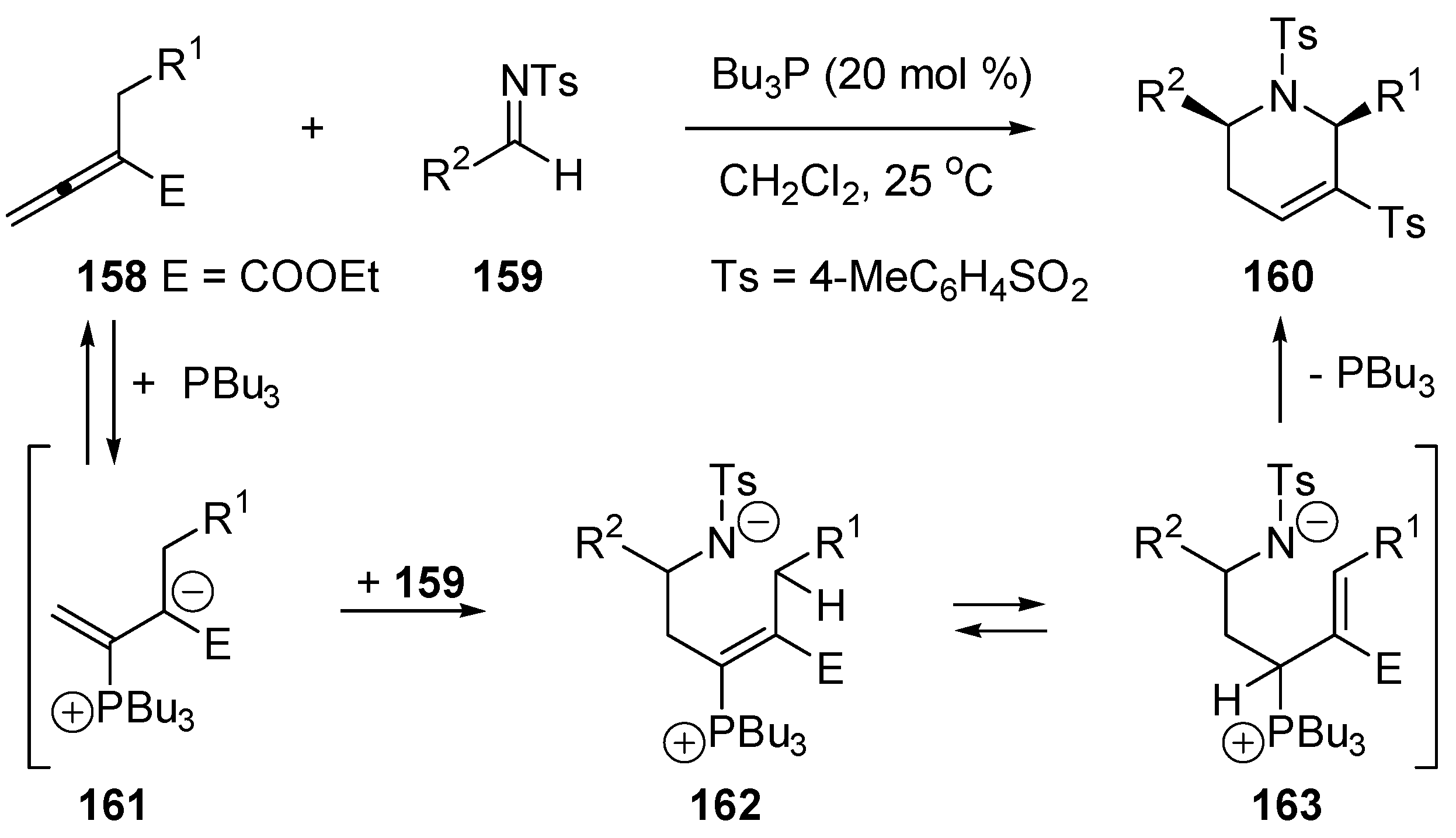{kind=link}
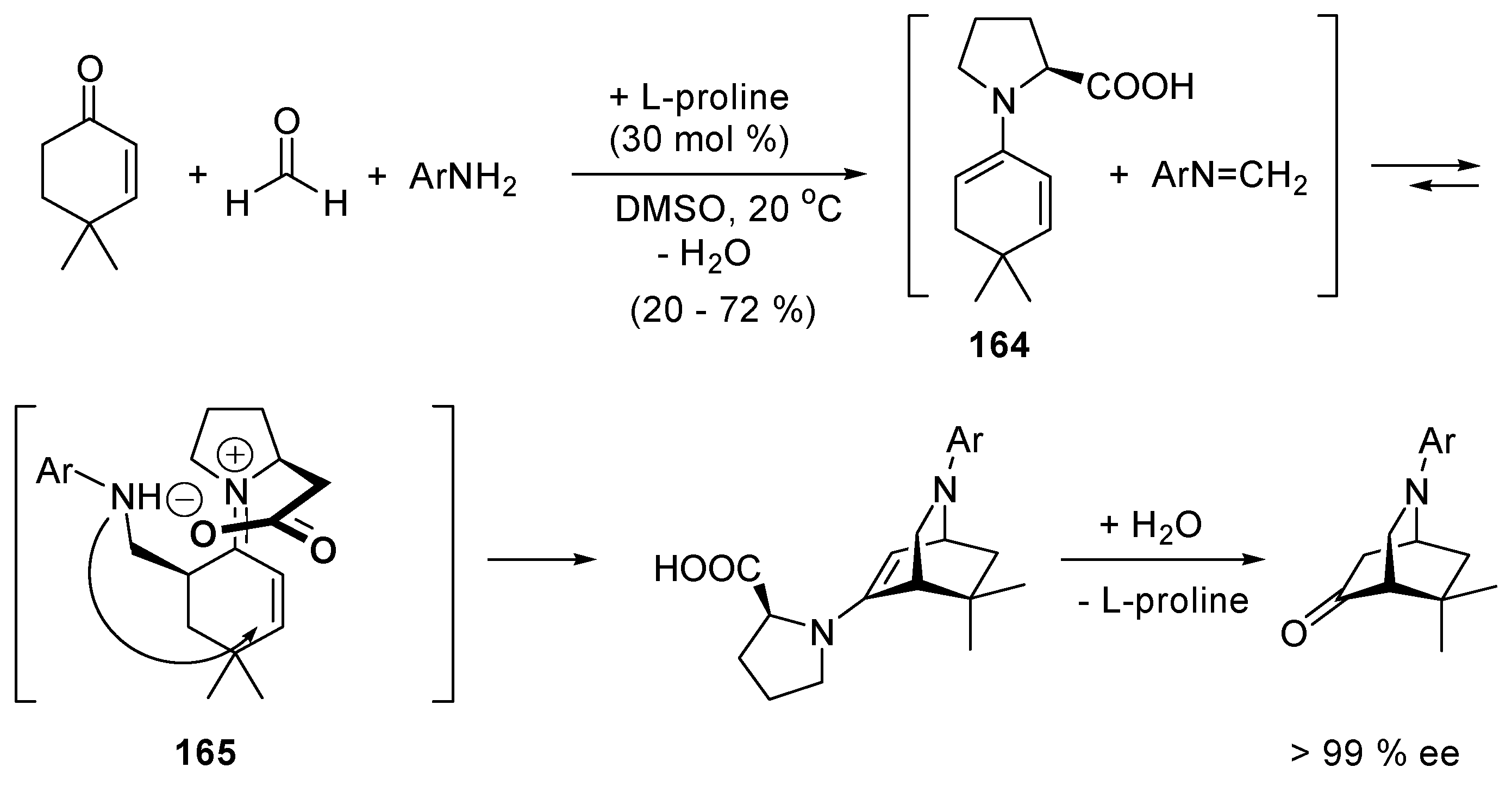{kind=link}
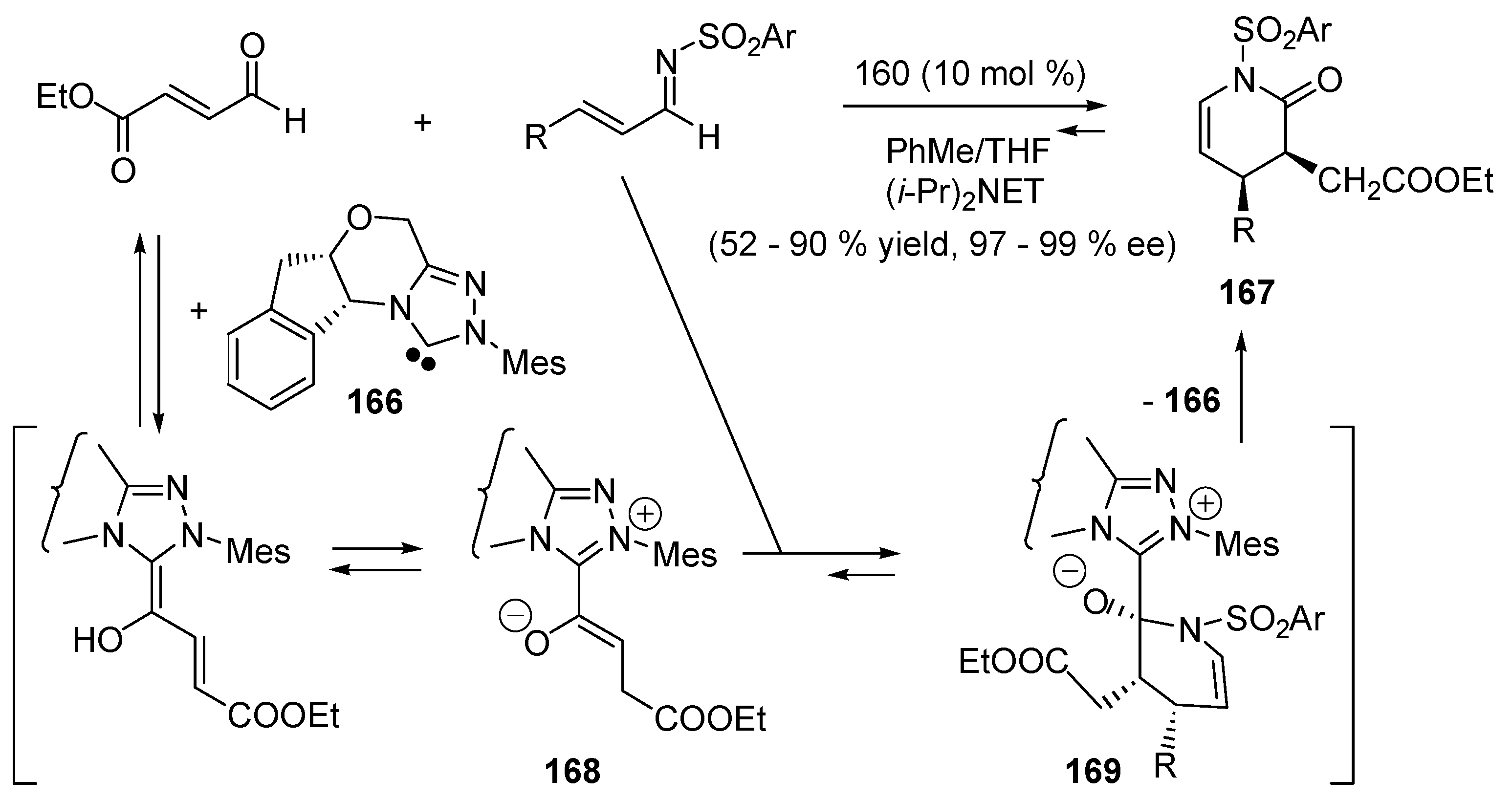{kind=link}
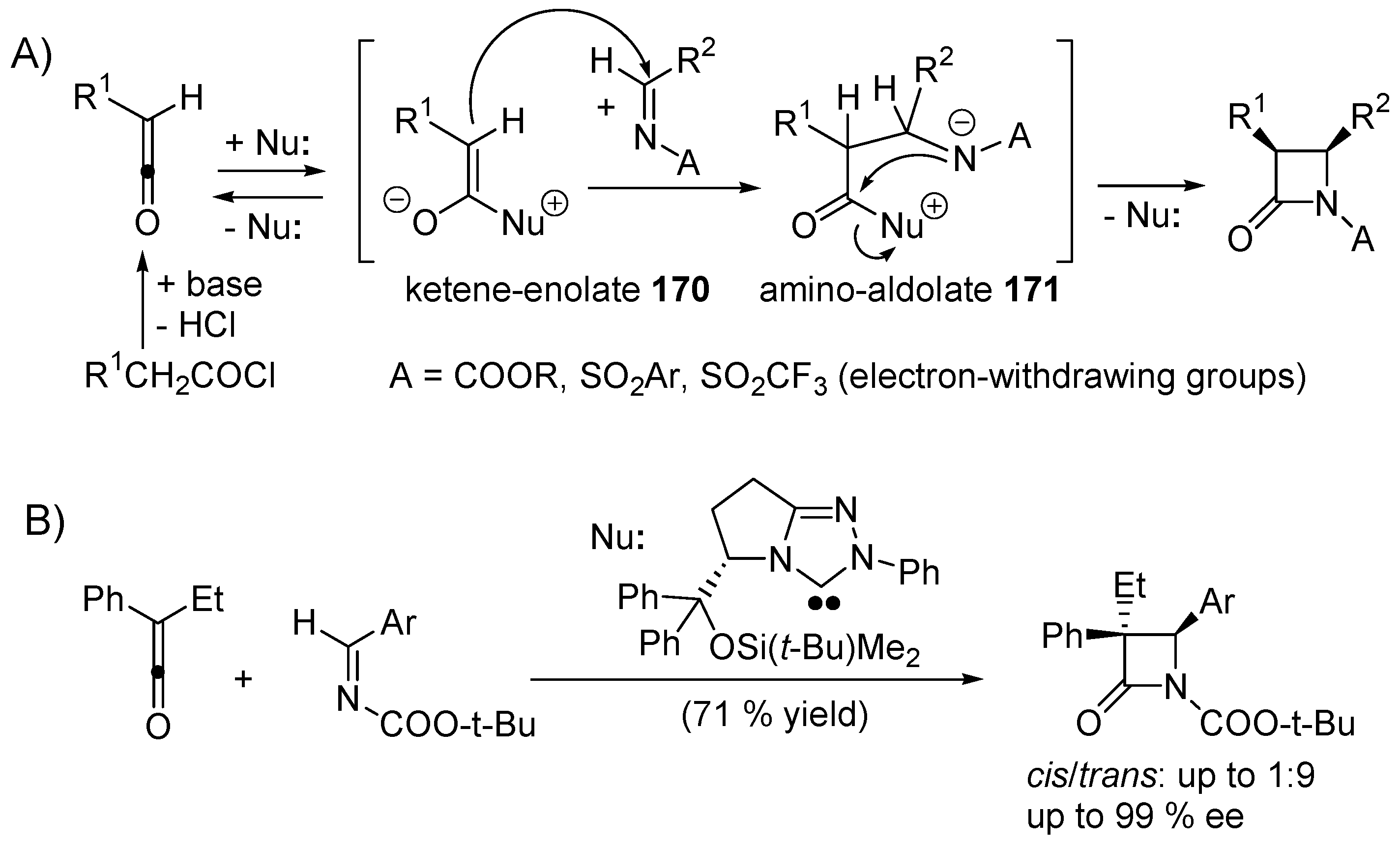{kind=link}
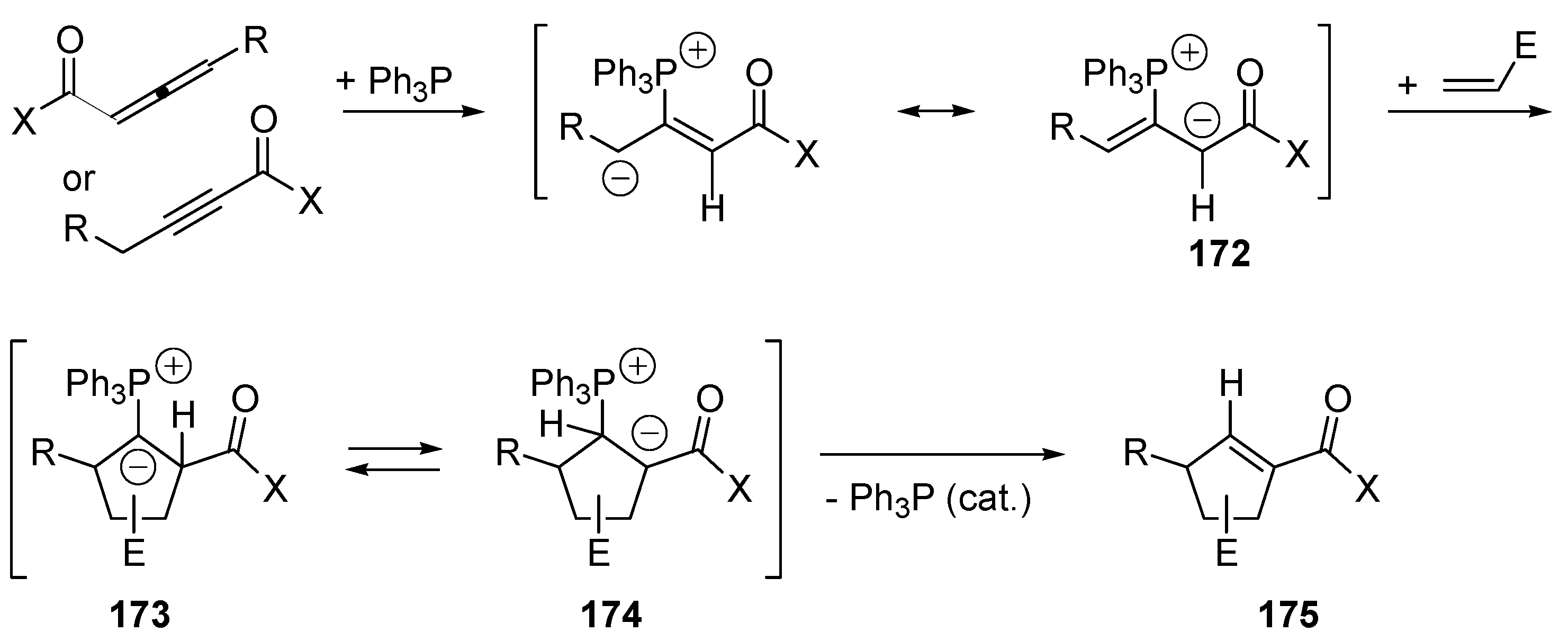{kind=link}
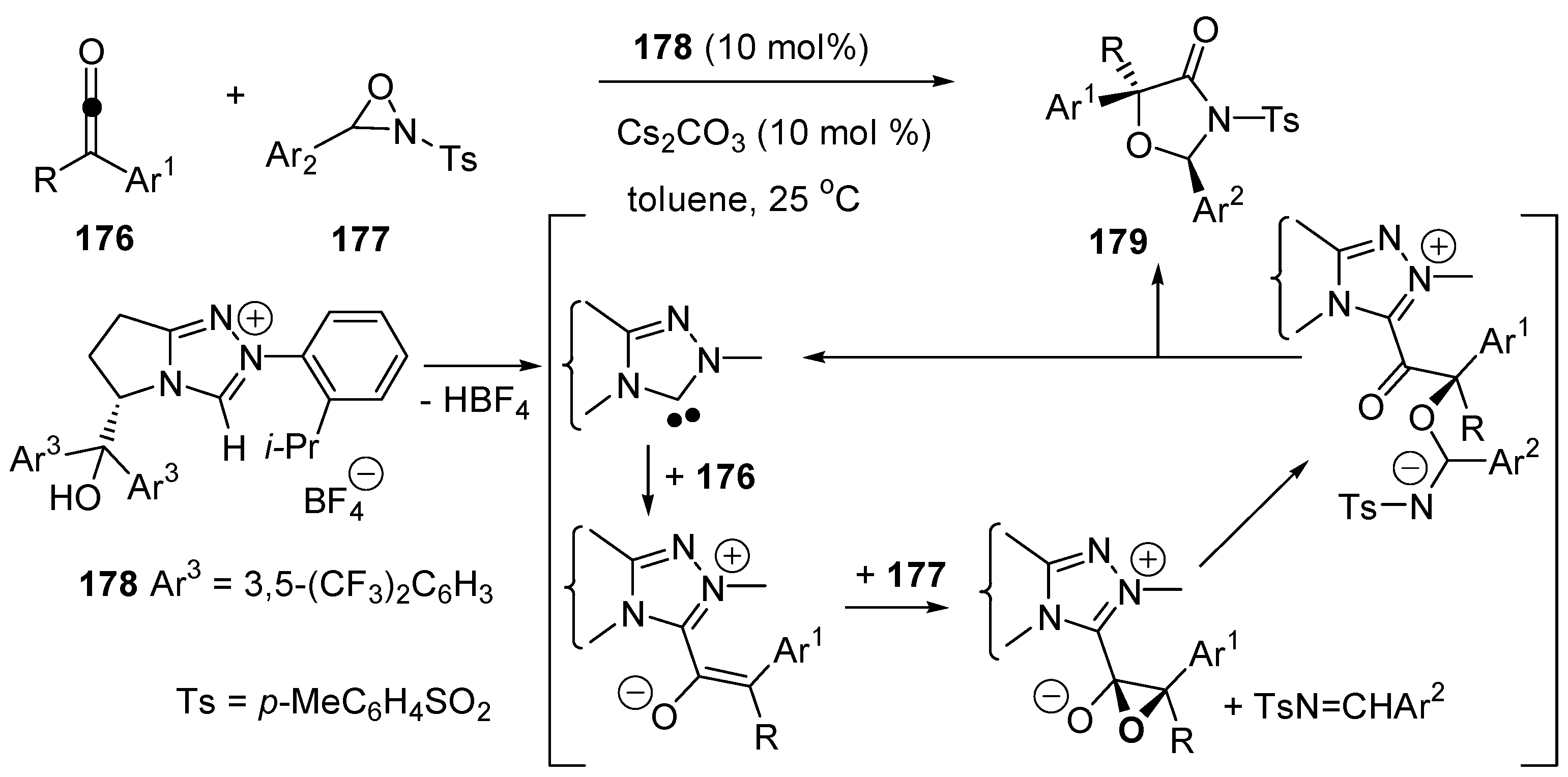{kind=link}
| 1 | Acid catalyzed acyl cleavage AAC1 (IUPAC: AN+DN+AN+DN) (SN1 like at the acyl group) RCOOR' + H3O+ + H2O ⇄ RCO(O+H)R'·· (H2O)2 ⇄ [RCO+] + R'OH + 2 H2O (the slowest step) ⇄ RCO(O+H2) + R'OH + H2O ⇄ RC+(OH)2 + R'OH + H2O ⇄ RCOOH + R'OH + H3O+ |
| 2 | Acid-catalyzed acyl cleavage AAC2 (Ah+AN+AhDh+Dh) (add./elim: adduct intermediate) RCOOR' + H3O+ + H2O ⇄ RC+(OR')OH·· OH2 + H2O ⇄ RC(OR')(OH)(O+H2 ··OH2) (slow) ⇄ RC(OH)2(O+HR') ··OH2 ⇄ RC+(OH)2·· OH2 + R'OH (slow) ⇄ RCOOH + R'OH + H3O+ |
| 3 | Acid-catalyzed alkyl cleavage AAL1 (Ah+AN+AN+Dh) (SN1 like at the alkyl group) RCOOR' + H3O+ + H2O ⇄ RC+(OH)(OR') ··(H2O)2 ⇄ RCOOH + [R'+] + 2 H2O (the slowest step) ⇄ RCOOH + R'O+H2·· OH2 ⇄ RCOOH + R'OH + H3O+ |
| 4 | Acid-catalyzed alkyl cleavage AAL2 (Ah+ANDN+DN) (SN2 like at the alkyl group) RCOOR' + H3O+ + H2O ⇄ RCO(O+HR') ··(OH2)2 ⇄ RCOOH + R'O+H2·· OH2 (slow) ⇄ RCOOH + R'OH + H3O+ |
| 5 | Base-catalyzed acyl cleavage BAC1 (DN+AN+AxhDh) (SN1 like at the acyl group) RCOOR' + HO− ⇄ [RCO+] + R'O−+ HO− (slow) ⇄ RCOOH + R'O− ⇄ RCOO− + R'OH |
| 6 | Base-catalyzed acyl cleavage BAC2 (AN+DN+AxhDh) (adduct intermediate) RCOOR' + HO− ⇄ R(OH)(OR')(O−) (slow) ⇄ RCOOH + [R'O−] ⇄ RCOO− + R'OH |
| 7 | Base-catalyzed alkyl cleavage BAL1 (DN+AN+AxhDh) (SN1 like at the alkyl group) RCOOR' + HO− ⇄ RCOO− + [R'+]/HO− (slow) ⇄ RCOO− + R'OH |
| 8 | Base-catalyzed alkyl cleavage BAL2 (ANDN) (SN2 like at the alkyl group) RCOOR' + HO− ⇄ RCOO− + R'OH |
| 9 | Ketene formation (Dh+DN+AN+Ah) (Elim./add.) RCH2COOR' + HO− ⇄ RCH=C=O + R'O− + H2O ⇄ RCH=C=O + R'OH + HO− ⇄ RCH=C(OH)O− + R'OH ⇄ RCH2COO− + R'OH |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogel, P.; Lam, Y.-h.; Simon, A.; Houk, K.N. Organocatalysis: Fundamentals and Comparisons to Metal and Enzyme Catalysis. Catalysts 2016, 6, 128. https://doi.org/10.3390/catal6090128
Vogel P, Lam Y-h, Simon A, Houk KN. Organocatalysis: Fundamentals and Comparisons to Metal and Enzyme Catalysis. Catalysts. 2016; 6(9):128. https://doi.org/10.3390/catal6090128
Chicago/Turabian StyleVogel, Pierre, Yu-hong Lam, Adam Simon, and Kendall N. Houk. 2016. "Organocatalysis: Fundamentals and Comparisons to Metal and Enzyme Catalysis" Catalysts 6, no. 9: 128. https://doi.org/10.3390/catal6090128
APA StyleVogel, P., Lam, Y.-h., Simon, A., & Houk, K. N. (2016). Organocatalysis: Fundamentals and Comparisons to Metal and Enzyme Catalysis. Catalysts, 6(9), 128. https://doi.org/10.3390/catal6090128