
Constructing Hydrazone-Linked Chiral Covalent Organic Frameworks with Different Pore Sizes for Asymmetric Catalysis
 and
and
Abstract

1. Introduction
2. Results and Discussion
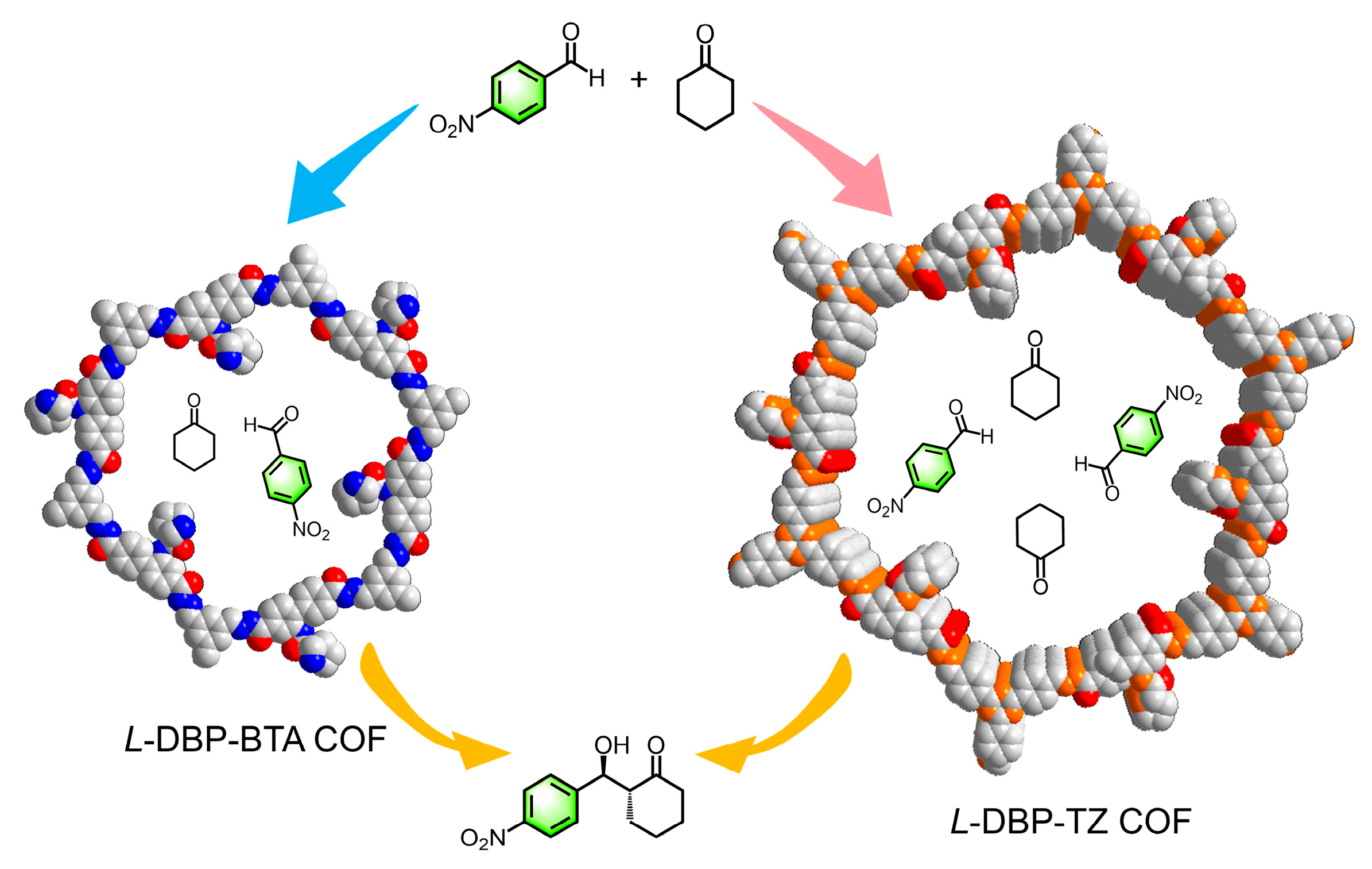
2.1. Facile Synthesis of Pyrrolidine-Functionalized Chiral COFs
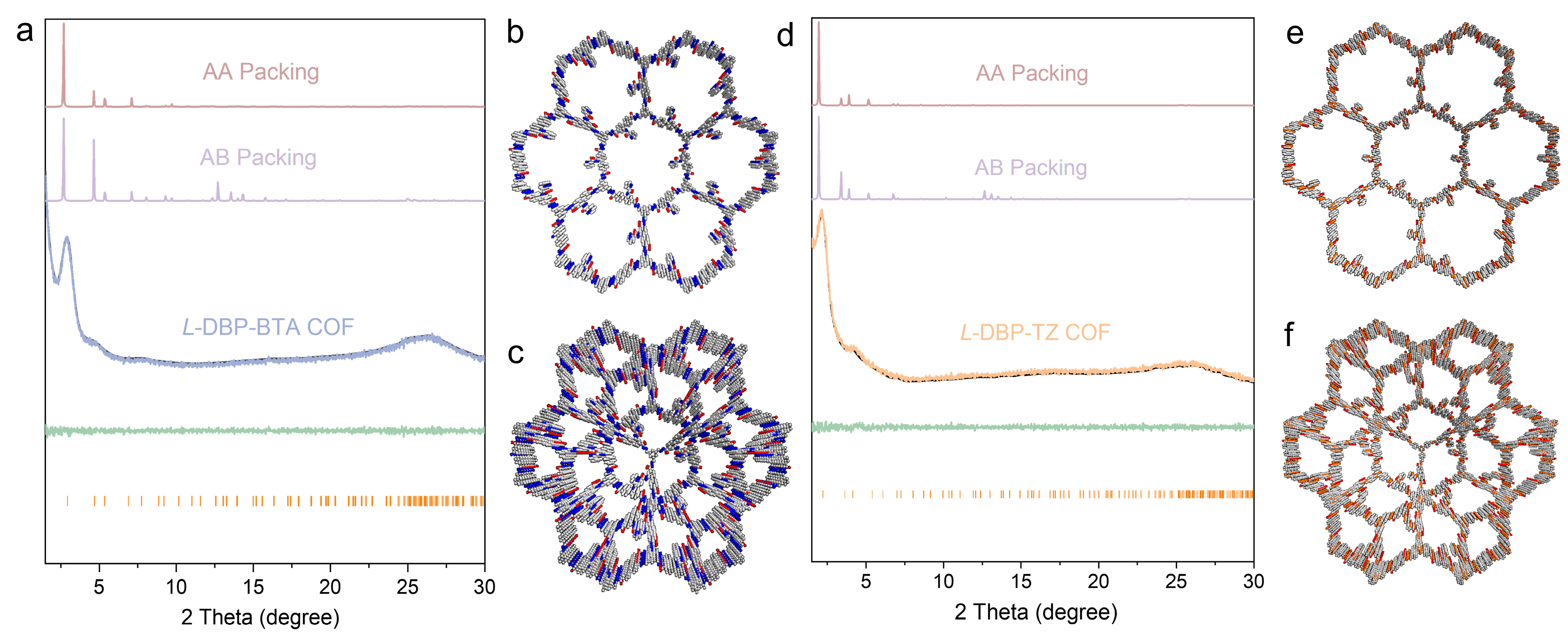
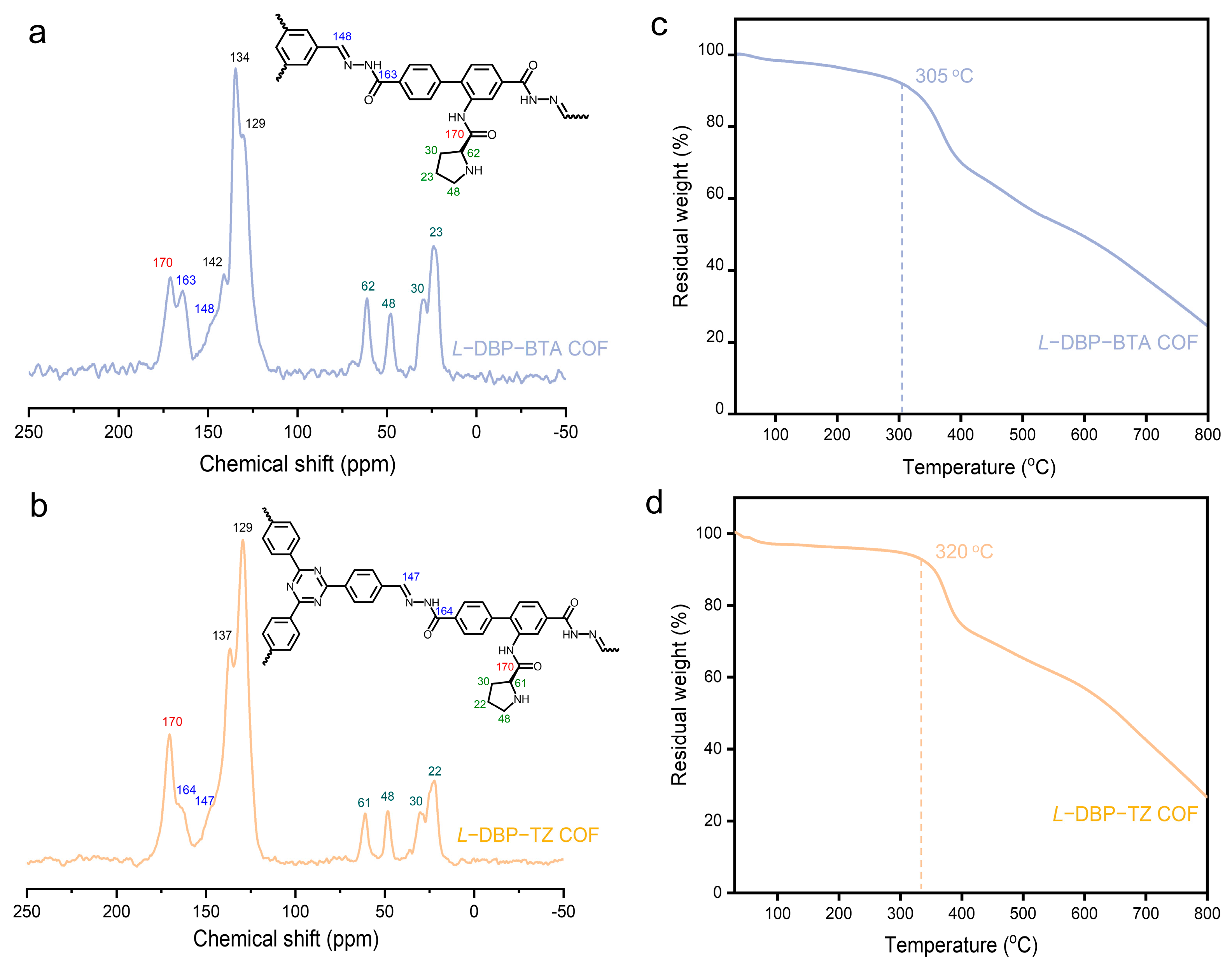
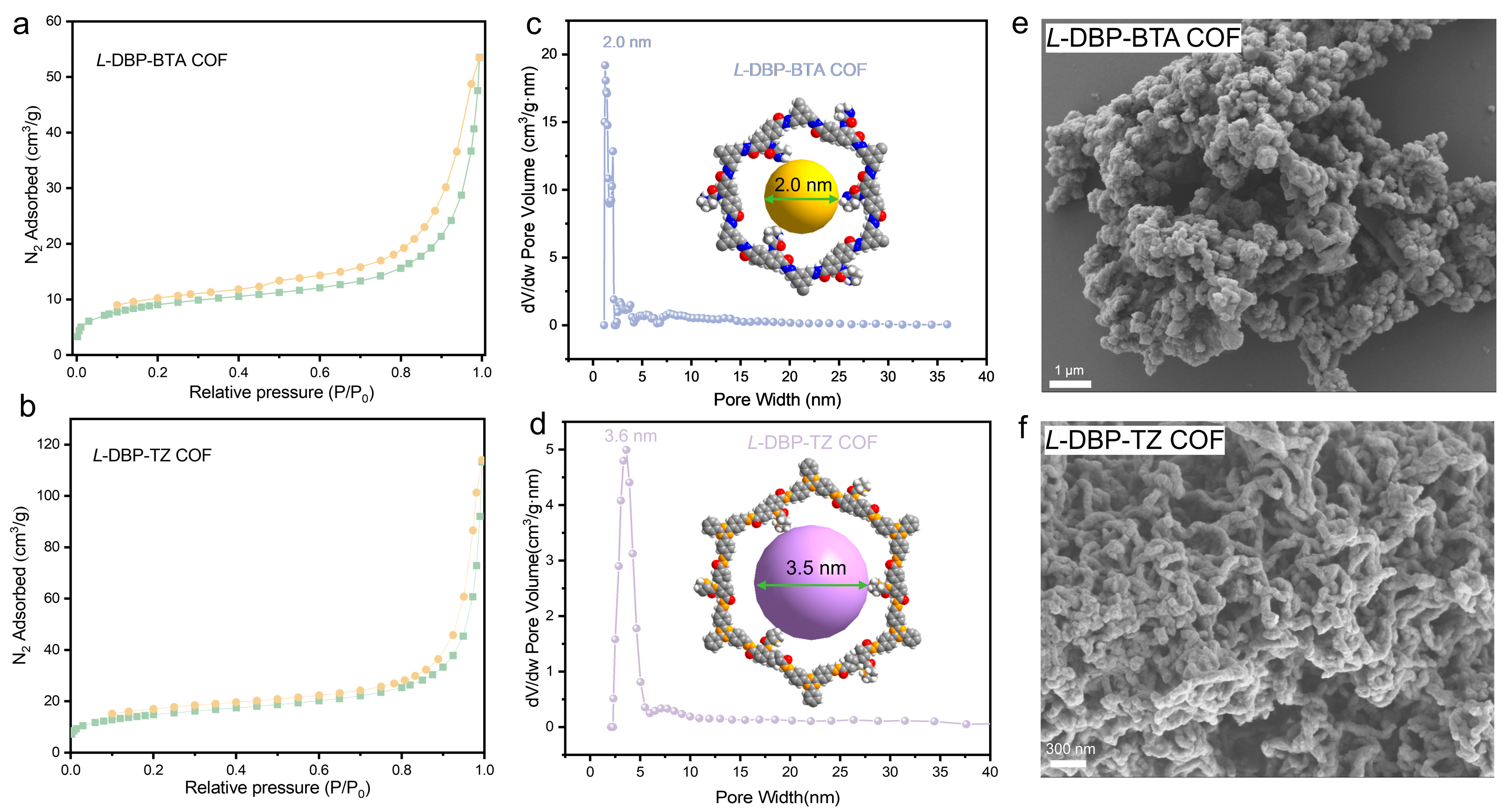
2.2. Structural Characterization of Chiral COFs
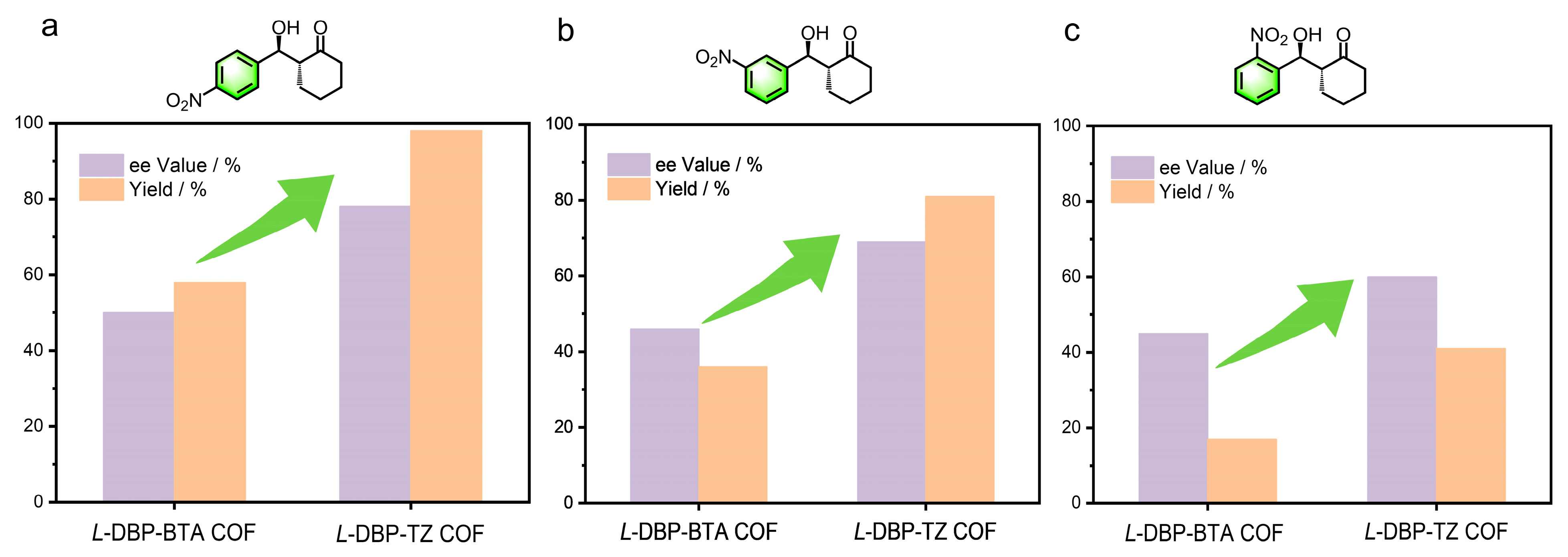
2.3. Impact of Pore Size on Asymmetric Catalytic Performance in Chiral COFs
3. Materials and Methods
3.1. Materials and Instruments
3.2. Synthesis of the Precursors
3.2.1. Dimethyl 2-Amino-[1,1′-biphenyl]-4,4′-dicarboxylate (1)
3.2.2. Dimethyl(S)-2-(1-(tert-butoxycarbonyl)pyrrolidine-2-carboxamido)-[1,1′-biphenyl]-4,4′-dicarboxylate (2)
3.2.3. L-DPD
3.2.4. L-DBP-Boc
3.3. Synthesis of the Chiral COFs
3.3.1. L-DBP-BTA COF
3.3.2. L-DBP-TZ COF
3.4. Experimental Procedure for Asymmetric Aldol Reactions
3.5. The Procedure for Recycled Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Han, X.; Yuan, C.; Hou, B.; Liu, L.; Li, H.; Liu, Y.; Cui, Y. Chiral covalent organic frameworks: Design, synthesis and property. Chem. Soc. Rev. 2020, 49, 6248–6272. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Stephens, E.R.; Spector-Watts, B.M.; Li, Z.; Liu, Y.; Liu, L.; Cui, Y. Challenges and opportunities for chiral covalent organic frameworks. Chem. Sci. 2022, 13, 9811–9832. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sheng, J.; Zhao, Y. Chiral covalent organic frameworks for asymmetric catalysis and chiral separation. Sci. China Chem. 2017, 60, 1015–1022. [Google Scholar] [CrossRef]
- Wu, X.; Han, X.; Zhang, J.; Jiang, H.; Hou, B.; Liu, Y.; Cui, Y. Metal–and covalent organic frameworks threaded with chiral polymers for heterogeneous asymmetric catalysis. Organometallics 2019, 38, 3474–3479. [Google Scholar] [CrossRef]
- Hu, H.; Yan, Q.; Ge, R.; Gao, Y. Covalent organic frameworks as heterogeneous catalysts. Chin. J. Catal. 2018, 39, 1167–1179. [Google Scholar] [CrossRef]
- Dong, Y.; Yin, C.F.; Kan, J.L.; Chang, C.R.; Ma, J.P.; Liu, J.B.; Dong, Y.B. Au-Acyclic Diaminocarbene-Linked Homochiral Covalent Organic Frameworks: Synthesis and Asymmetric Catalytic Application. Chem. Mater. 2024, 37, 473–479. [Google Scholar] [CrossRef]
- Kang, X.; Cheng, C.; Chen, X.; Dong, J.; Liu, Y.; Cui, Y. Three-Dimensional Homochiral Covalent Organic Frameworks with Intrinsic Chiral qzd Topology. J. Am. Chem. Soc. 2024, 146, 8407–8416. [Google Scholar] [CrossRef]
- Li, J.; Zhang, K.; Tang, X.; Yang, X.; Chen, H.; Zheng, S.; Fan, J.; Xie, M.; Zhang, W.; Li, X.; et al. Primary Amine-Functionalized Chiral Covalent Organic Framework Enables High-Efficiency Asymmetric Catalysis in Water. ACS Appl. Mater. Interfaces 2024, 16, 59379–59387. [Google Scholar] [CrossRef]
- Yu, J.; Yang, G.; Gao, M.L.; Wang, H.; Jiang, H.L. Chiral Ligand-Decorated Rhodium Nanoparticles Incorporated in Covalent Organic Framework for Asymmetric Catalysis. Angew. Chem. Int. Ed. 2024, 63, e202412643. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Wu, X.; Liu, Y.; Cui, Y. Multivariate Chiral Covalent Organic Frameworks with Controlled Crystallinity and Stability for Asymmetric Catalysis. J. Am. Chem. Soc. 2017, 139, 8277–8285. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, Y.; Chen, H.; Cai, D.; Tang, X.; Zheng, S.; Cai, S. Dimensionality control of chiral covalent organic frameworks for enhanced asymmetric catalysis. Chem. Eng. J. 2025, 515, 163613. [Google Scholar] [CrossRef]
- Liu, P.; Dai, W.; Shen, X.; Shen, X.; Zhao, Y.; Liu, J.J. Recent Advances in the Utilization of Chiral Covalent Organic Frameworks for Asymmetric Photocatalysis. Molecules 2024, 29, 5006. [Google Scholar] [CrossRef]
- Han, X.; Zhang, J.; Huang, J.; Wu, X.; Yuan, D.; Liu, Y.; Cui, Y. Chiral induction in covalent organic frameworks. Nat. Commun. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Han, X.; Xu, Q.; Liu, Y.; Yuan, C.; Yang, S.; Liu, Y.; Jiang, J.; Cui, Y. Chiral BINOL-Based Covalent Organic Frameworks for Enantioselective Sensing. J. Am. Chem. Soc. 2019, 141, 7081–7089. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Fu, S.; Yang, K.; Hou, B.; Liu, Y.; Jiang, J.; Cui, Y. Crystalline C-C and C=C Bond-Linked Chiral Covalent Organic Frameworks. J. Am. Chem. Soc. 2021, 143, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.Y.; Song, L.P.; Shi, Y.H.; Zhang, L.; Pan, Z.X.; Yang, P.; Tang, B. Chiral ionic covalent organic framework as an enantioselective fluorescent sensor for phenylalaninol determination. Anal. Chem. 2023, 95, 11078–11084. [Google Scholar] [CrossRef]
- Yuan, L.; Tang, X.; Zhang, K.; Chen, H.; Yang, X.; Fan, J.; Cai, S. Construction of a Defective Chiral Covalent Organic Framework for Fluorescence Recognition of Amino Acids. Chem.-Asian J. 2024, 19, e202400753. [Google Scholar] [CrossRef]
- Song, Q.; Yang, J.; Zheng, K.; Zhang, T.; Yuan, C.; Yuan, L.M.; Hou, X. Chiral Memory in Dynamic Transformation from Porous Organic Cages to Covalent Organic Frameworks for Enantiorecognition Analysis. J. Am. Chem. Soc. 2024, 146, 7594–7604. [Google Scholar] [CrossRef]
- Zhang, K.; Cai, S.L.; Yan, Y.L.; He, Z.-H.; Lin, H.M.; Huang, X.L.; Zheng, S.R.; Fan, J.; Zhang, W.G. Construction of a hydrazone-linked chiral covalent organic framework-silica composite as the stationary phase for high performance liquid chromatography. J. Chromatogr. A 2017, 1519, 100–109. [Google Scholar] [CrossRef]
- Zhang, S.; Zheng, Y.; An, H.; Aguila, B.; Yang, C.X.; Dong, Y.; Xie, W.; Cheng, P.; Zhang, Z.; Chen, Y.; et al. Covalent Organic Frameworks with Chirality Enriched by Biomolecules for Efficient Chiral Separation. Angew. Chem. Int. Ed. 2018, 57, 16754–16759. [Google Scholar] [CrossRef]
- Xu, J.; Feng, G.; Ao, D.; Li, X.; Li, M.; Lei, S.; Wang, Y. Functional Covalent Organic Frameworks Microspheres Synthesized by Self-Limited Dynamic Linker Exchange for Stationary Phases. Adv. Mater. 2024, 36, 2406256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, Y.; An, H.; Aguila, B.; Yang, C.X.; Dong, Y.; Ma, S. A Chiral COFs Membrane for Enantioselective Amino Acid Separation. Angew. Chem. Int. Ed. 2025, 64, e202417088. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, B.; Li, Z.; Fu, S.; Liu, S.; Jia, L.; Cui, Y. Highly Enantioselective Transportation Across Liquid Membranes Mediated by Porous Covalent Organic Frameworks. Angew. Chem. Int. Ed. 2025, 64, e202419916. [Google Scholar] [CrossRef]
- Fu, S.; Qin, G.; Dong, J.; Yuan, C.; Liu, Y.; Yuan, L.M.; Cui, Y. Construction of chiral crown ethers in robust covalent organic frameworks for electrochromatographic enantioseparation. Natl. Sci. Rev. 2024, 11, 256. [Google Scholar] [CrossRef]
- Yuan, C.; Wang, Z.; Xiong, W.; Huang, Z.; Lai, Y.; Fu, S.; Dong, J.; Duan, A.; Hou, X.; Yuan, L.M.; et al. Cyclodextrin Incorporation into Covalent Organic Frameworks Enables Extensive Liquid and Gas Chromatographic Enantioseparations. J. Am. Chem. Soc. 2023, 145, 8956–18967. [Google Scholar] [CrossRef]
- Chen, H.; Gu, Z.G.; Zhang, J. Chiral-Induced Ultrathin Covalent Organic Frameworks Nanosheets with Tunable Circularly Polarized Luminescence. J. Am. Chem. Soc. 2022, 144, 7245–7252. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liao, X.; Cai, X.; Wu, J.; Wu, X.; Zhang, Q.; Yan, Y.; Zheng, S.; Jiang, H.; Fan, J.; et al. Self-Assembly of Helical Nanofibrous Chiral Covalent Organic Frameworks. Angew. Chem. Int. Ed. 2023, 62, e202216310. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, K.; Xue, R.; Zheng, Y.; Chen, S.; Zheng, S.; Fan, J.; Zhang, Y.; Ye, W.; Zhang, W.; et al. Self-Standing Chiral Covalent Organic Framework Thin Films with Full-Color Tunable Guest-Induced Circularly Polarized Luminescence. Angew. Chem. Int. Ed. 2024, 63, e202413171. [Google Scholar] [CrossRef]
- Gu, Q.; Zha, J.; Chen, C.; Wang, X.; Yao, W.; Liu, J.; Zhang, Q. Constructing chiral covalent-organic frameworks for circularly polarized light detection. Adv. Mater. 2024, 36, 2306414. [Google Scholar] [CrossRef]
- Tang, X.; Zha, J.; Wu, X.; Tong, J.; Gu, Q.; Zhang, K.; Zhang, Y.; Zheng, S.; Fan, J.; Zhang, W.; et al. Construction of Chiral Covalent Organic Frameworks Through a Linker Decomposition Chiral Induction Strategy for Circularly Polarized Light Detection. Angew. Chem. Int. Ed. 2025, 64, e202413675. [Google Scholar] [CrossRef]
- Jing, S.M.; Gu, Z.G.; Zhang, J. Chiral Cross-Linked Covalent Organic Framework Films for Highly Sensitive Circularly Polarized Luminescence Probing. J. Am. Chem. Soc. 2025, 147, 8948–8958. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Zhou, J.J.; Lan, Y.B.; Ding, S.Y.; Yu, W.; Wang, W. Divergent synthesis of chiral covalent organic frameworks. Angew. Chem. Int. Ed. 2019, 131, 9543–9547. [Google Scholar] [CrossRef]
- Liu, S.; Li, W.L.; Zhang, J.P. Revealing the potential application of chiral covalent organic frameworks in CO2 adsorption and separation. New J. Chem. 2020, 44, 95–101. [Google Scholar] [CrossRef]
- Zhuo, S.; Zhang, X.; Luo, H.; Wang, X.; Ji, Y. The application of covalent organic frameworks for chiral chemistry. Macromol. Rapid. Comm. 2020, 41, 2000404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Z.; Sun, J.; Lan, Y.; Hu, J.; Xiao, Y.; Gao, D. Recent Advance in the Synthesis and Applications of Chiral Covalent Organic Frameworks. J. Sep. Sci. 2025, 48, e70101. [Google Scholar] [CrossRef]
- Weng, W.; Guo, J. Chiral Covalent Organic Framework Films with Enhanced Photoelectrical Performances. J. Am. Chem. Soc. 2024, 146, 13201–13209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Wu, X.; Liu, Y.; Cui, Y. Chiral DHIP- and Pyrrolidine-Based Covalent Organic Frameworks for Asymmetric Catalysis. ACS Sustain. Chem. Eng. 2019, 7, 5065–5071. [Google Scholar] [CrossRef]
- Zhang, K.; Tang, X.; Yang, X.; Wu, J.; Guo, B.; Xiao, R.; Xie, Y.; Zheng, S.; Jiang, H.; Fan, J.; et al. Raising the Asymmetric Catalytic Efficiency of Chiral Covalent Organic Frameworks by Tuning the Pore Environment. ACS Appl. Mater. Interfaces 2024, 16, 10661–10670. [Google Scholar] [CrossRef]
- Xu, H.S.; Ding, S.Y.; An, W.K.; Wu, H.; Wang, W. Constructing Crystalline Covalent Organic Frameworks from Chiral Building Blocks. J. Am. Chem. Soc. 2016, 138, 11489–11492. [Google Scholar] [CrossRef]
- He, T.; Liu, R.; Wang, S.; On, I.K.W.; Wu, Y.; Zhao, Y. Bottom–Up Design of Photoactive Chiral Covalent Organic Frameworks for Visible-Light-Driven Asymmetric Catalysis. J. Am. Chem. Soc. 2023, 145, 18015–18021. [Google Scholar] [CrossRef]
- Lun, D.J.; Waterhouse, G.I.N.; Telfer, S.G. A General Thermolabile Protecting Group Strategy for Organocatalytic Metal−Organic Frameworks. J. Am. Chem. Soc. 2011, 133, 5806–5809. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Y.; Chen, H.; Chen, X.; Zhang, K.; Liang, W.; Cai, S. Construction of a spherical chiral covalent organic framework for efficient asymmetric aldol catalysis. J. Solid State Chem. 2025, 348, 125350. [Google Scholar] [CrossRef]
- Chen, G.; Lan, H.H.; Cai, S.L.; Sun, B.; Li, X.L.; He, Z.H.; Zhang, W.G. Stable hydrazone-linked covalent organic frameworks containing O,N,O′-chelating sites for Fe (III) detection in water. ACS Appl. Mater. Interfaces 2019, 11, 12830–12837. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [PubMed]
- Meso, G.; Gregorich, D.; Waite, A.J.; Smith, S.; Liu, L.; Cortez, J.; Liu, Y. Organocatalytic Assembly Based on Aromatic Donor–Acceptor Interactions for the Asymmetric Aldol Reaction on Water. ChemCatChem 2023, 15, e202300012. [Google Scholar] [CrossRef]
- Newar, R.; Kalita, R.; Akhtar, N.; Antil, N.; Chauhan, M.; Manna, K. N-Formylation of amines utilizing CO2 by a heterogeneous metal–organic framework supported single-site cobalt catalyst. Catal. Sci. Technol. 2022, 12, 6795. [Google Scholar] [CrossRef]
- Shan, H.; Zhang, Z.; Jiang, Y.; Cai, D.; Qin, P.; Baeyens, J. Two-dimensional porphyrin-based covalent organic frameworks for heterogeneous photocatalysis: Influence of pore size on photocatalytic performance. Sep. Purif. Technol. 2025, 363, 132102. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
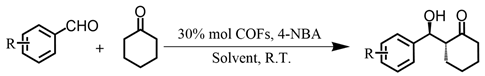 | |||||||
|---|---|---|---|---|---|---|---|
| Entry a | Catalyst | R | Solvent | t/d | Yield/% b | ee/% c | Syn/Anti d |
| 1 e | L-DBP-TZ COF | 4-NO2 | DCM:H2O (10:1) | 4 | 55 | 60 | 1:3.4 |
| 2 e | L-DBP-TZ COF | 4-NO2 | ACN:H2O (10:1) | 4 | 43 | 60 | 1:3.0 |
| 3 e | L-DBP-TZ COF | 4-NO2 | TBME:H2O (10:1) | 4 | 60 | 63 | 1:3.2 |
| 4 e | L-DBP-TZ COF | 4-NO2 | EtOH:H2O (10:1) | 4 | 43 | 53 | 1:1.4 |
| 5 e | L-DBP-TZ COF | 4-NO2 | DMF:H2O (10:1) | 4 | 65 | 62 | 1:2.0 |
| 6 f | L-DBP-TZ COF | 4-NO2 | DMF:H2O (10:3) | 4 | 78 | 66 | 1:3.3 |
| 7 g | L-DBP-TZ COF | 4-NO2 | DMF:H2O (3:10) | 4 | 87 | 68 | 1:3.3 |
| 8 h | L-DBP-TZ COF | 4-NO2 | DMF:H2O (1:10) | 4 | 90 | 72 | 1:4.2 |
| 9 | L-DBP-TZ COF | 4-NO2 | H2O | 3 | 98 | 78 | 1:5.2 |
| 10 i | L-DPD | 4-NO2 | H2O | 3 | 97 | 89 | 1:6.4 |
| 11 | L-DBP-TZ COF | 3-NO2 | H2O | 3 | 81 | 69 | 1:3.8 |
| 12 | L-DBP-TZ COF | 2-NO2 | H2O | 3 | 41 | 60 | 1:2.0 |
| 13 | L-DBP-BTA COF | 4-NO2 | H2O | 3 | 58 | 50 | 1:5.1 |
| 14 | L-DBP-BTA COF | 3-NO2 | H2O | 3 | 36 | 46 | 1:2.2 |
| 15 | L-DBP-BTA COF | 2-NO2 | H2O | 3 | 17 | 45 | 1:2.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Zhang, K.; Zheng, Y.; Chen, H.; Cai, D.; Zheng, S.; Fan, J.; Cai, S. Constructing Hydrazone-Linked Chiral Covalent Organic Frameworks with Different Pore Sizes for Asymmetric Catalysis. Catalysts 2025, 15, 640. https://doi.org/10.3390/catal15070640
Huang H, Zhang K, Zheng Y, Chen H, Cai D, Zheng S, Fan J, Cai S. Constructing Hydrazone-Linked Chiral Covalent Organic Frameworks with Different Pore Sizes for Asymmetric Catalysis. Catalysts. 2025; 15(7):640. https://doi.org/10.3390/catal15070640
Chicago/Turabian StyleHuang, Haichen, Kai Zhang, Yuexin Zheng, Hong Chen, Dexuan Cai, Shengrun Zheng, Jun Fan, and Songliang Cai. 2025. "Constructing Hydrazone-Linked Chiral Covalent Organic Frameworks with Different Pore Sizes for Asymmetric Catalysis" Catalysts 15, no. 7: 640. https://doi.org/10.3390/catal15070640
APA StyleHuang, H., Zhang, K., Zheng, Y., Chen, H., Cai, D., Zheng, S., Fan, J., & Cai, S. (2025). Constructing Hydrazone-Linked Chiral Covalent Organic Frameworks with Different Pore Sizes for Asymmetric Catalysis. Catalysts, 15(7), 640. https://doi.org/10.3390/catal15070640