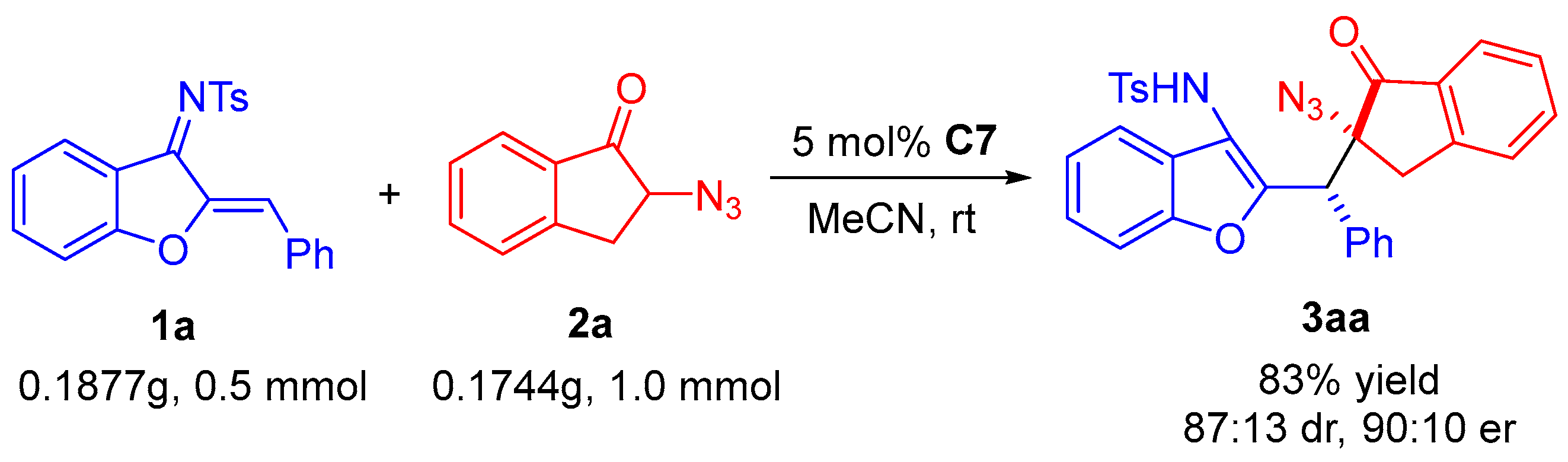
3.4. Procedure for the Synthesis of Chiral Compounds 3
To a dried small bottle were added 1 (0.1 mmol), 2 (0.2 mmol), chiral organocatalyst C7 (5 mol%), and CH3CN (1.0 mL). The mixture was stirred at room temperature, and after completion, the reaction mixture was concentrated and the crude product was directly purified by silica gel column chromatography using eluent (ethyl acetate/petroleum ether = 1:5) to afford the desired product 3.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofuran-3-yl)-4-methyl-benzenesulfonamide (3aa). White solid, 43.4 mg (79% yield), m.p. 94–96 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 85:15, flow rate 1.0 mL/min, detection at 254 nm): tR = 54.7 min (minor), tR = 59.5 min (major); 90:10 er. [α]D20 = −74.5 (c 1.1, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.2 Hz, 1H, ArH), 7.54–7.49 (m, 4H, ArH), 7.42 (d, J = 8.0 Hz, 1H, ArH), 7.34–7.28 (m, 3H, ArH), 7.21 (t, J = 7.2 Hz, 1H, ArH), 7.14–7.11 (m, 2H, ArH), 7.08–7.06 (m, 3H, ArH), 6.97 (d, J = 8.0 Hz, 2H, ArH), 6.49 (s, 1H, NH), 4.86 (s, 1H, CH), 3.75 (d, J = 18.0 Hz, 1H, CH2), 3.11 (d, J = 18.0 Hz, 1H, CH2), 2.26 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 200.8, 153.5, 152.4, 151.9, 143.7, 136.1, 136.0, 134.7, 134.0, 129.5, 129.4, 128.2, 128.0, 127.4, 127.3, 126.2, 125.7, 125.0, 124.9, 123.5, 120.0, 116.2, 111.5, 71.4, 44.9, 36.5, 21.5 ppm. HRMS (ESI): m/z calculated for C31H28N5O4S [M + NH4]+ 566.1857, found 566.1852.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(4-bromophenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ba). White solid, 31.2 mg (50% yield), m.p. 103–105 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 10.2 min (major), tR = 12.6 min (minor); 68:32 er. [α]D20 = −21.5 (c 1.1, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 7.6 Hz, 1H, ArH), 7.56 (td, J1 = 0.8 Hz, J2 = 7.2 Hz, 1H, ArH), 7.52 (d, J = 8.0 Hz, 1H, ArH), 7.48 (d, J = 8.0 Hz, 2H, ArH), 7.36−7.31 (m, 4H, ArH), 7.22–7.18 (m, 3H, ArH), 7.05 (d, J = 8.4 Hz, 2H, ArH), 6.98 (d, J = 8.4 Hz, 2H, ArH), 6.38 (s, 1H, NH), 4.89 (s, 1H, CH), 3.67 (d, J = 18.0 Hz, 1H, CH2), 3.10 (d, J = 18.4 Hz, 1H, CH2), 2.30 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 200.5, 153.5, 152.0, 151.7, 144.0, 136.4, 135.9, 133.9, 133.8, 131.4, 131.2, 129.5, 128.2, 127.3, 126.3, 125.5, 125.2, 125.1, 123.6, 121.8, 119.9, 116.4, 111.5, 71.1, 44.1, 36.4, 21.6 ppm. HRMS (ESI): m/z calculated for C31H2379BrN4NaO4S [M + Na]+ 649.0516, found 649.0519; calculated for C31H2381BrN4NaO4S [M + Na]+ 651.0496, found 651.0482.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(p-tolyl)methyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ca). White solid, 37.6 mg (67% yield), m.p. 91–93 °C. HPLC (Daicel Chiralpak IC, n-hexane/ethyl acetate = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 10.5 min (minor), tR = 13.3 min (major); 76:24 er. [α]D20 = −37.9 (c 1.4, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.6 Hz, 1H, ArH), 7.54–7.49 (m, 4H, ArH), 7.40 (d, J = 7.6 Hz, 1H, ArH), 7.33–7.29 (m, 3H, ArH), 7.19 (t, J = 7.6 Hz, 1H, ArH), 7.02–6.97 (m, 4H, ArH), 6.87 (d, J = 8.0 Hz, 2H, ArH), 6.39 (s, 1H, NH), 4.81 (s, 1H, CH), 3.73 (d, J = 18.0 Hz, 1H, CH2), 3.09 (d, J = 18.0 Hz, 1H, CH2), 2.27 (s, 3H, CH3), 2.19 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.7, 153.5, 152.7, 151.9, 143.8, 137.1, 136.04, 135.96, 133.9, 131.6, 129.5, 129.3, 129.0, 127.9, 127.3, 126.2, 125.8, 125.0, 124.9, 123.4, 119.9, 116.0, 111.5, 71.4, 44.4, 36.5, 21.5, 21.0 ppm. HRMS (ESI): m/z calculated for C32H30N5O4S [M + NH4]+ 580.2013, found 580.2017.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(4-fluorophenyl)methyl)benzo-furan-3-yl)-4-methylbenzenesulfonamide (3da). White solid, 46.6 mg (82% yield), m.p. 95–97 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 85:15, flow rate 1.0 mL/min, detection at 254 nm): tR = 28.3 min (major), tR = 37.8 min (minor); 90:10 er. [α]D20 = −72.9 (c 0.83, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 7.6 Hz, 1H, ArH), 7.57–7.48 (m, 4H, ArH), 7.35–7.31(m, 4H, ArH), 7.21–7.14 (m, 3H, ArH), 7.00 (d, J = 8.0 Hz, 2H, ArH), 6.79–6.75 (m, 2H, ArH), 6.41 (s, 1H, NH), 4.90 (s, 1H, CH), 3.72 (d, J = 18.0 Hz, 1H, CH2), 3.10 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.7, 161.9 (d, 1JC–F = 245.0 Hz), 153.5, 152.3, 151.8, 143.9, 136.3, 136.0, 133.9, 131.3 (d, 3JC–F = 8.0 Hz), 130.6 (d, 4JC–F = 3.1 Hz), 129.5, 128.1, 127.3, 126.2, 125.6, 125.1, 125.0, 123.6, 119.9, 116.2, 115.2 (d, 2JC–F = 21.2 Hz), 111.5, 71.3, 44.1, 36.4, 21.5 ppm; HRMS (ESI): m/z calculated for C31H23FN4NaO4S [M + Na]+ 589.1316, found 589.1303.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(4-chlorophenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ea). White solid, 38.3 mg (66% yield), m.p. 93–95 °C. HPLC (Daicel Chiralpak IC, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.0 min (major), tR = 7.7 min (minor); 56:44 er. [α]D20 = −6.4 (c 1.36, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 7.6 Hz, 1H, ArH), 7.56 (td, J1 = 0.8 Hz, J2 = 7.6 Hz, 1H, ArH), 7.52 (d, J = 8.4 Hz, 1H, ArH), 7.47 (d, J = 8.4 Hz, 2H, ArH), 7.37–7.31 (m, 4H, ArH), 7.19 (t, J = 7.6 Hz, 1H, ArH), 7.12 (d, J = 8.8 Hz, 2H, ArH), 7.05 (d, J = 8.8 Hz, 2H, ArH), 6.97 (d, J = 8.0 Hz, 2H, ArH), 6.53 (s, 1H, NH), 4.93 (s, 1H, CH), 3.69 (d, J = 18.0 Hz, 1H, CH2), 3.11 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.6, 153.5, 152.0, 151.8, 143.9, 136.4, 135.9, 133.8, 133.5, 133.4, 130.8, 129.4, 128.4, 128.2, 127.3, 126.3, 125.5, 125.15, 125.05, 123.6, 119.9, 116.4, 111.5, 71.2, 44.1, 36.4, 21.5 ppm. HRMS (ESI): m/z calculated for C31H27ClN5O4S [M + NH4]+ 600.1467, found 600.1480.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(4-(trifluoromethyl)phenyl)methyl)benzo-furan-3-yl)-4-methylbenzenesulfonamide (3fa). White solid, 40.4 mg (66% yield), m.p. 109–111 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 5.6 min (major), tR = 6.8 min (minor); 85:15 er. [α]D20 = −56.3 (c 1.06, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 7.6 Hz, 1H, ArH), 7.56 (td, J1 = 1.2 Hz, J2 = 7.6 Hz, 1H, ArH), 7.52 (d, J = 8.4 Hz, 1H, ArH), 7.48 (d, J = 8.0 Hz, 2H, ArH), 7.37–7.30 (m, 7H, ArH), 7.27–7.24 (m, 1H, ArH), 7.19–7.15 (m, 1H, ArH), 6.98 (d, J = 8.4 Hz, 2H, ArH), 6.50 (s, 1H, NH), 5.03 (s, 1H, CH), 3.65 (d, J = 18.0 Hz, 1H, CH2), 3.12 (d, J = 18.0 Hz, 1H, CH2), 2.27 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.3, 153.6, 151.7, 151.5, 143.9, 139.0, 136.4, 136.0, 133.7, 129.9, 129.5, 128.3, 127.3, 126.3, 125.4, 125.3, 125.14 (d, JC–F = 3.6 Hz), 125.13, 123.6, 122.5, 119.7, 116.6, 111.6, 71.2, 44.6, 36.7, 21.4 ppm. HRMS (ESI): m/z calculated for C32H23F3N4NaO4S [M + Na]+ 639.1284, found 639.1272.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(4-(oxo-l6-methyl)phenyl)methyl)benzo-furan-3-yl)-4-methylbenzenesulfonamide (3ga). White solid, 30.6 mg (53% yield), m.p. 91–93 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 77:23, flow rate 1.0 mL/min, detection at 254 nm): tR = 30.5 min (major), tR = 35.0 min (minor); 82:18 er. [α]D20 = −34.6 (c 0.34, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.6 Hz, 1H, ArH), 7.55–7.50 (m, 4H, ArH), 7.41 (d, J = 7.6 Hz, 1H, ArH), 7.34–7.30 (m, 3H, ArH), 7.23–7.19 (m, 1H, ArH), 7.05 (d, J = 8.8 Hz, 2H, ArH), 7.01 (d, J = 8.0 Hz, 2H, ArH), 6.62–6.58 (m, 2H, ArH), 6.31 (s, 1H, NH), 4.80 (s, 1H, CH), 3.75 (d, J = 18.0 Hz, 1H, CH2), 3.69 (s, 3H, CH3), 3.09 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.8, 158.7, 153.5, 152.8, 152.0, 143.8, 136.1, 136.0, 134.0, 130.6, 129.5, 128.0, 127.3, 126.6, 126.2, 125.8, 124.9, 123.5, 119.9, 115.9, 113.6, 111.5, 71.4, 55.0, 44.0, 36.3, 21.5 ppm. HRMS (ESI): m/z calculated for C32H30N5O5S [M + NH4]+ 596.1962, found 596.1940.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(m-tolyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ha). White solid, 29.8 mg (53% yield), m.p. 123–125 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.6 min (major), tR = 8.0 min (minor); 58:42 er. [α]D20 = −22.2 (c 1.12, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.0 Hz, 1H, ArH), 7.53–7.50 (m, 4H, ArH), 7.44 (d, J = 7.6 Hz, 1H, ArH), 7.34–7.29 (m, 3H, ArH), 7.21 (t, J = 7.6 Hz, 1H, ArH), 7.00 (d, J = 8.0 Hz, 2H, ArH), 6.95–6.87 (m, 4H, ArH), 6.36 (s, 1H, NH), 4.81 (s, 1H, CH), 3.73 (d, J = 18.0 Hz, 1H, CH2), 3.10 (d, J = 18.0 Hz, 1H, CH2), 2.27 (s, 3H, CH3), 2.17 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.7, 153.5, 152.5, 151.9, 143.7, 137.8, 136.1, 136.0, 134.6, 134.0, 130.0, 129.5, 128.2, 128.1, 128.0, 127.3, 126.5, 126.2, 125.8, 124.93, 124.90, 123.5, 120.0, 116.1, 111.5, 71.4, 44.8, 36.5, 21.5, 21.4 ppm. HRMS (ESI): m/z calculated forC32H30N5O4S [M + NH4]+ 580.2013, found 580.2015.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(3-bromophenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ia). White solid, 27.7 mg (44% yield), m.p. 133–135 °C. HPLC (Daicel Chiralpak IC, n-hexane/ethyl acetate = 85:15, flow rate 1.0 mL/min, detection at 254 nm): tR = 14.0 min (major), tR = 17.3 min (minor); 68:32 er. [α]D20 = −30.8 (c 0.50, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 7.6 Hz, 1H, ArH), 7.58–7.48 (m, 4H, ArH), 7.40–7.32 (m, 4H, ArH), 7.28 (d, J = 1.6 Hz, 1H, ArH), 7.24–7.19 (m, 2H, ArH), 7.13 (d, J = 7.6 Hz, 1H, ArH), 7.01 (d, J = 8.0 Hz, 2H, ArH), 6.96 (t, J = 8.0 Hz, 1H, ArH), 6.38 (s, 1H, NH), 4.85 (s, 1H, CH), 3.67 (d, J = 18.0 Hz, 1H, CH2), 3.10 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.5, 153.6, 151.7, 151.6, 143.9, 137.0, 136.3, 135.8, 133.9, 132.2, 130.7, 129.8, 129.5, 128.24, 128.19, 127.3, 126.3, 125.5, 125.2, 125.1, 123.6, 122.3, 120.0, 116.6, 111.6, 71.3, 44.4, 36.5, 21.6 ppm. HRMS (ESI): m/z calculated for C31H2779BrN5O4S [M + NH4]+ 644.0962, found 644.0983; calculated for C31H2781BrN5O4S [M + NH4]+ 646.0941, found 646.0965.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(naphthalen-2-yl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ja). White solid, 27.3 mg (46% yield), m.p. 109–111 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 28.9 min (major), tR = 33.8 min (minor); 57:43 er. [α]D20 = −111.3 (c 0.68, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.72–7.65 (m, 3H, ArH), 7.59–7.53 (m, 4H, ArH), 7.49–7.35 (m, 6H, ArH), 7.28–7.24 (m, 4H, ArH), 6.75 (d, J = 8.4 Hz, 2H, ArH), 6.37 (s, 1H, NH), 5.06 (s, 1H, CH), 3.75 (d, J = 18.0 Hz, 1H, CH2), 3.14 (d, J = 18.0 Hz, 1H, CH2), 1.91 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.7, 153.7, 152.4, 152.0, 143.8, 136.2, 135.6, 133.8, 133.0, 132.4, 132.3, 129.3, 128.8, 128.1, 128.0, 127.9, 127.4, 127.2, 126. 9, 126.3, 126.2, 125.8, 125.1, 125.0, 123.6, 120.3, 116.4, 111. 6, 71.4, 44.5, 36.4, 21.1 ppm. HRMS (ESI): m/z calculated for C35H30N5O4S [M + NH4]+ 616.2013, found 616.2026.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(thiophen-2-yl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ka). White solid, 36.7 mg (66% yield), m.p. 119–121 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 18.4 min (major), tR = 28.0 min (minor); 61:39 er. [α]D20 = −19.4 (c 1.53, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 7.6 Hz, 1H, ArH), 7.58–7.50 (m, 4H, ArH), 7.43 (d, J = 7.6 Hz, 1H, ArH), 7.37–7.31 (m, 3H, ArH), 7.22 (t, J = 7.6 Hz, 1H, ArH), 7.03–7.01 (m, 3H, ArH), 6.72–6.70 (m, 1H, ArH), 6.63 (d, J = 3.2 Hz, 1H, ArH), 6.48 (s, 1H, NH), 5.08 (s, 1H, CH), 3.85 (d, J = 18.0 Hz, 1H, CH2), 3.07 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.3, 153.4, 152.1, 151.3, 143.9, 136.2, 135.9, 135.8, 133.9, 129.5, 128.4, 128.1, 127.3, 126.34, 126.28, 125.6, 125.5, 125.2, 125.0, 123.6, 120.0, 116.2, 111.5, 71.5, 40.7, 36.4, 21.5 ppm. HRMS (ESI): m/z calculated for C29H26N5O4S2 [M + NH4]+ 572.1421, found 572.1424.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(3,5-dichlorophenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3la). White solid, 41.5 mg (67% yield), m.p. 103–105 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 9.1 min (major), tR = 10.0 min (minor); 72:28 er. [α]D20 = −12.6 (c 1.58, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.4 Hz, 1H, ArH), 7.77 (d, J = 7.6 Hz, 1H, ArH), 7.59–7.56 (m, 3H, ArH), 7.40–7.36 (m, 2H, ArH), 7.33 (d, J =2.4 Hz, 1H, ArH), 7.30 (d, J = 8.0 Hz, 1H, ArH), 7.24–7.19 (m, 2H, ArH), 7.11 (d, J = 8.4 Hz, 1H, ArH), 7.04–7.01 (m, 1H, ArH), 6.93–6.90 (m, 1H, ArH), 6.37 (s, 1H, NH), 5.50 (s, 1H, CH), 3.40 (d, J = 17.6 Hz, 1H, CH2), 2.99 (d, J = 17.6 Hz, 1H, CH2), 2.34 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 199.3, 153.3, 151.1, 150.1, 144.0, 136.6, 136.1, 135.2, 134.4, 134.2, 132.8, 132.0, 129.6, 129.4, 128.3, 127.5, 127.0, 126.1, 125.1, 125.0, 125.0, 123.3, 119.4, 115.9, 111.6, 71.0, 41.6, 39.2, 21.5 ppm. HRMS (ESI): m/z calculated for C31H22Cl2N4NaO4S [M + Na]+ 639.0631, found 639.0638.
N-(2-((2-azido-1-oxo-2,3-dihydro-1H-inden-2-yl(2-chloropheny)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ma). White solid, 29.2 mg (50% yield), m.p. 92–95 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 8.8 min (major), tR = 10.7 min (minor); 71:29 er. [α]D20 = −22.4 (c 0.81, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.96 (dd, J1 = 1.8 Hz, J2 = 7.4 Hz, 1H, ArH), 7.77 (d, J = 7.6 Hz, 1H, ArH), 7.62 (d, J = 8.4 Hz, 2H, ArH), 7.57 (td, J1 = 0.8 Hz, J2 = 7.6 Hz, 1H, ArH), 7.40 (d, J = 8.4 Hz, 1H, ArH), 7.37 (t, J = 7.6 Hz, 1H, ArH), 7.32–7.29 (m, 2H, ArH), 7.24–7.17 (m, 3H, ArH), 7.12 (d, J = 8.0 Hz, 2H, ArH), 7.05 (t, J = 7.6 Hz, 1H, ArH), 7.00 (d, J = 8.0 Hz, 1H, ArH), 6.27(s, 1H, NH), 5.51 (s, 1H, CH), 3.44 (d, J = 17.6 Hz, 1H, CH2), 2.97 (d, J = 17.6 Hz, 1H, CH2), 2.33 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 199.4, 153.3, 151.5, 150.2, 143.8, 136.6, 136.0, 134.4, 134.3, 133.3, 132.0, 129.7, 129.6, 129.1, 128.1, 127.5, 126.7, 126.1, 125.2, 125.1, 124.9, 123.2, 119.5, 115.7, 111.6, 71.2, 42.0, 39.1, 21.5 ppm. HRMS (ESI): m/z calculated for C31H27ClN5O4S [M + NH4]+ 600.1467, found 600.1471.
N-(2-((2-azido-6-fluoro-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ab). White solid, 25.8 mg (46% yield), m.p. 85–87 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 15.5 min (major), tR =22.0 min (minor); 53:47 er. [α]D20 = −3.8 (c 0.64, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.51 (t, J = 7.8 Hz, 3H, ArH), 7.37–7.27 (m, 4H, ArH), 7.25–7.18 (m, 2H, ArH), 7.14–7.07 (m, 5H, ArH), 6.99 (d, J = 8.0 Hz, 2H, ArH), 6.28 (s, 1H, NH), 4.87 (s, 1H, CH), 3.74 (d, J = 18.0 Hz, 1H, CH2), 3.06 (d, J = 18.0 Hz, 1H, CH2), 2.28 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.1, 162.3 (d, 1JC–F = 248.0 Hz), 153.5, 152.4, 147.4, 143.9, 135.9, 135.6 (d, 3JC–F = 7.7 Hz), 134.5, 129.6, 129.4, 128.3, 127.7 (d, 3JC–F = 8.0 Hz), 127.6, 127.3, 125.7, 125.1, 123.9 (d, 2JC–F = 23.5 Hz), 123.5, 119.9, 116.2, 111.5, 110.6 (d, 2JC–F = 22.0 Hz), 72.0, 44.9, 36.0, 21.5 ppm. HRMS (ESI): m/z calculated for C31H23FN4O4SNa [M + Na]+ 589.1316, found 589.1302.
N-(2-((2-azido-6-methyl-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide) (3ac). White solid, 22.2 mg (39% yield), m.p. 105–107 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 12.2 min (major), tR = 19.4 min (minor); 73:27 er. [α]D20 = −46.8 (c 0.8, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.53–7.49 (m, 4H, ArH), 7.43 (d, J = 7.6 Hz, 1H, ArH), 7.35–7.30 (m, 2H, ArH), 7.23–7.17 (m, 2H, ArH), 7.13–7.06 (m, 5H, ArH), 6.97 (d, J = 8.0 Hz, 2H, ArH), 6.38 (s, 1H, NH), 4.83 (s, 1H, CH), 3.69 (d, J = 18.0 Hz, 1H, CH2), 3.05 (d, J = 18.0 Hz, 1H, CH2), 2.33 (s, 3H, CH3), 2.27 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 200.6, 153.5, 152.5, 149.3, 143.7, 138.1, 137.5, 135.9, 134.7, 134.1, 129.5, 129.4, 128.2, 127.4, 127.3, 125.9, 125.8, 125.0, 124.8, 123.5, 120.0, 116.1, 111.5, 71.7, 44.7, 36.1, 21.5, 21.0 ppm. HRMS (ESI): m/z calculated for C32H30N5O4S [M + NH4]+ 580.2013, found 580.2007.
N-(2-((2-azido-5,6-Dimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofu-ran-3-yl)-4-methylbenzenesulfonamide (3ad). White solid, 45.9 mg (75% yield), m.p. 107–109 °C. HPLC (Daicel Chiralpak IC, n-hexane/ethyl acetate = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 43.3 min (major), tR = 48.8 min (minor); 81:19 er. [α]D20 = −71.0 (c 1.6, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.52–7.47 (m, 4H, ArH), 7.33 (td, J1 = 1.2 Hz, J2 = 7.2 Hz, 1H, ArH), 7.24–7.20 (m, 1H, ArH),7.12–7.06 (m, 6H, ArH), 6.96 (d, J = 8.0 Hz, 2H, ArH), 6.69 (s, 1H, ArH), 6.46 (s, 1H, NH), 4.83 (s, 1H, CH), 3.89 (s, 3H, CH3), 3.86 (s, 3H, CH3), 3.69 (d, J = 18.0 Hz, 1H, CH2), 3.01 (d, J = 18.0 Hz, 1H, CH2), 2.26 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 199.1, 156.7, 153.5, 152.4, 149.8, 147.8, 143.6, 135.9, 134.8, 129.4, 129.3, 128.1, 127.3, 127.2, 126.6, 126.4, 125.8, 124.9, 123.4, 120.1, 116.2, 111.4, 106.9, 104.9, 71.9, 56.2, 56.0, 44.8, 36.1, 21.5 ppm. HRMS (ESI): m/z calculated for C33H32N5O6S [M + NH4]+ 626.2068, found 626.2074.
N-(2-((2-azido-5-fluoro-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3ae) White solid, 33.6 mg (59% yield), m.p. 98–100 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 75:25, flow rate 1.0 mL/min, detection at 254 nm): tR = 24.0 min (major), tR =27.5 min (minor); 83:17 er. [α]D20 = −51.2 (c 1.3, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.71 (dd, J1 = 5.2 Hz, J2 = 8.4 Hz, 1H, ArH), 7.52 (d, J = 8.0 Hz, 1H, ArH), 7.49 (d, J= 8.0 Hz, 2H, ArH), 7.37 (d, J = 8.0 Hz, 1H, ArH), 7.34–7.30 (m, 1H, ArH), 7.20 (t, J = 7.6 Hz, 1H, ArH), 7.15–7.08 (m, 5H, ArH), 7.02–6.95 (m, 4H, ArH), 6.36 (s, 1H, NH), 4.90 (s, 1H, CH), 3.76 (d, J = 18.4 Hz, 1H, CH2), 3.08 (d, J = 18.0 Hz, 1H, CH2), 2.27 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 199.0, 167.8 (d, 1JC–F = 257.5 Hz), 154.9 (d, 3JC–F = 10.4 Hz), 153.5, 152.4, 143.8, 135.9, 134.6, 130.4, 129.5, 129.4, 128.3, 127.6, 127.4 (d, 3JC-F = 10.7 Hz), 127.3, 125.7, 125.1, 123.5, 119.9, 116.5 (d, 2JC–F = 23.7 Hz), 116.2, 113.0 (d, 2JC–F = 22.5 Hz), 111.5, 71.5, 44.8, 36.5, 21.5 ppm. HRMS (ESI): m/z calculated for C31H23FN4NaO4S [M + Na]+ 589.1316, found 589.1296.
N-(2-((2-azido-5-bromo-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofuran-3-yl)-4-methylbenzenesulfonamide (3af). White solid, 20.2 mg (32% yield), m.p. 98–100 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.9 min (major), tR = 8.7 min (minor); 74:26 er. [α]D20 = −63.1 (c 0.86, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 8.4 Hz, 1H, ArH), 7.52 (d, J = 8.0 Hz, 1H, ArH), 7.49 (d, J = 8.0 Hz, 3H, ArH), 7.44 (d, J = 8.0 Hz, 1H, ArH), 7.35 (d, J = 8.0 Hz, 1H, ArH), 7.31 (dd, J1 = 1.2 Hz, J2 = 8.4 Hz, 1H, ArH), 7.22–7.17 (m, 1H, ArH) 7.14–7.07 (m, 5H, ArH), 6.98 (d, J = 8.0 Hz, 2H, ArH), 6.34 (s, 1H, NH), 4.90 (s, 1H, CH), 3.75 (d, J = 18.4 Hz, 1H, CH2), 3.07 (d, J = 18.0 Hz, 1H, CH2), 2.27 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 199.6, 153.5, 153.4, 152.4, 143.8, 135.9, 134.5, 132.7, 131.7, 129.5, 129.4, 128.4, 127.6, 127.3, 126.0, 125.6, 125.1, 123.5, 119.8, 116.2, 111.5, 71.3, 44.8, 36.2, 21.5 ppm. HRMS (ESI): m/z calculated for C31H2379BrN4O4SNa [M + Na]+ 649.0516, found 649.0524; calculated. for C31H2381BrN4O4SNa [M + Na]+ 651.0495, found 651.0484.
N-(2-((2-azido-5-(oxo-l6-methyl)-1-oxo-2,3-dihydro-1H-inden-2-yl(phenyl)methyl)benzofu-ran-3-yl)-4-methylbenzenesulfonamide (3ag). White solid, 23.5 mg (41% yield), m.p. 108–110 °C. HPLC (Daicel Chiralpak IA, n-hexane/ethyl acetate = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 7.9 min (major), tR = 11.2 min (minor); 56:44 er. [α]D20 = −3.4 (c 0.48, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 8.4 Hz, 1H, ArH), 7.52–7.47 (m, 4H, ArH), 7.35–7.31 (m, 1H, ArH), 7.23 (t, J = 7.6 Hz, 1H, ArH), 7.11–7.04 (m, 5H, ArH), 6.97 (d, J = 8.0 Hz, 2H, ArH), 6.83 (dd, J1 = 2.0 Hz, J2 = 8.4 Hz, 1H, ArH), 6.71 (d, J = 1.6 Hz, 1H, ArH), 6.36 (s, 1H, NH), 4.80 (s, 1H, CH), 3.82 (s, 3H, CH3), 3.69 (d, J = 18.0 Hz, 1H, CH2), 3.04 (d, J = 18.0 Hz, 1H, CH2), 2.26 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 198.5, 166.4, 155.0, 153.6, 152.5, 143.7, 135.9, 134.8, 129.5, 129.4, 128.2, 127.4, 127.3, 127.1, 126.8, 125.9, 125.0, 123.5, 120.1, 116.2, 111.4, 109.3, 71.8, 55.7, 44.7, 36.4, 21.6 ppm; HRMS (ESI): m/z calculated for C32H30N5O5S [M + NH4]+ 596.1962, found 596.1978.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



