


The Role of Geometry in Cobalt–Polypyridine Complexes in the Electrochemical Reduction of CO2 Using UV-Vis Spectroelectrochemistry

 ,
,  , and
, and 
Abstract

1. Introduction
2. Results and Discussion
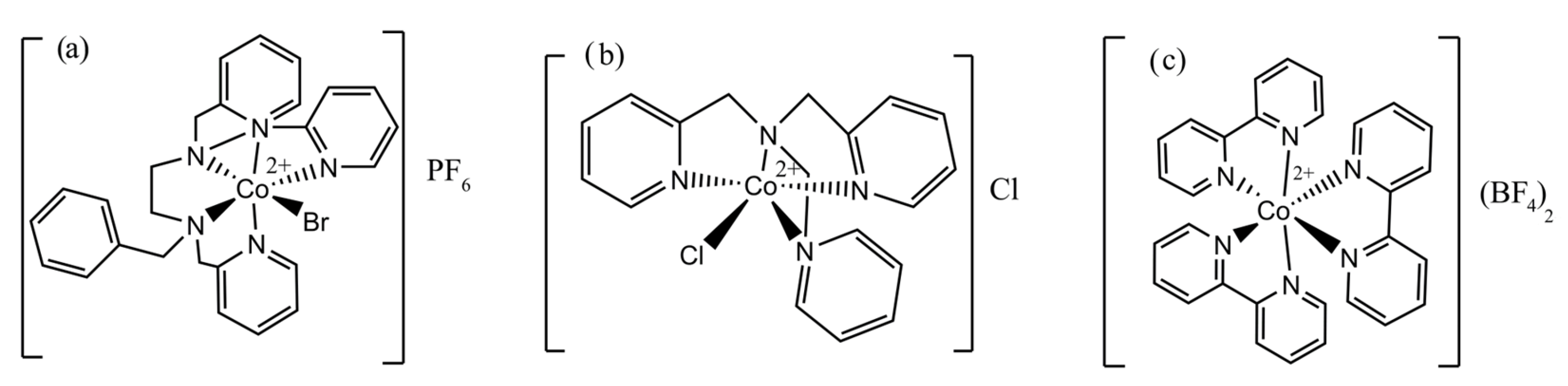
2.1. Complexes’ Characterization
2.1.1. IR Spectroscopy
2.1.2. UV-Vis Spectroscopy
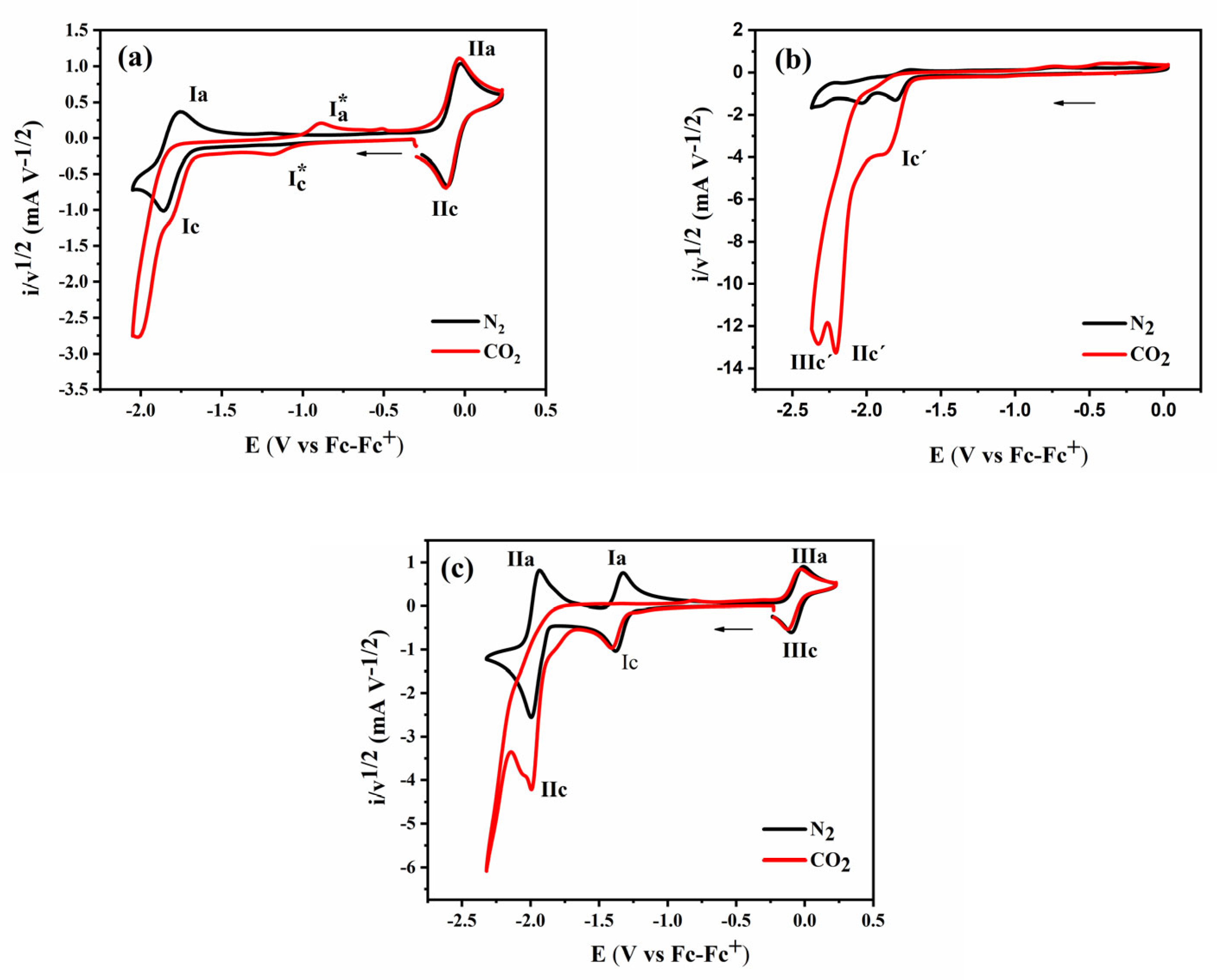
2.2. Cyclic Voltammetry Response of Metal Complexes
2.2.1. Cyclic Voltammetry of [CoII(bztpen)Br]PF6
2.2.2. Cyclic Voltammetry of [CoII (tpa)Cl]Cl
2.2.3. Cyclic Voltammetry of [CoII(bpy)3](BF4)2
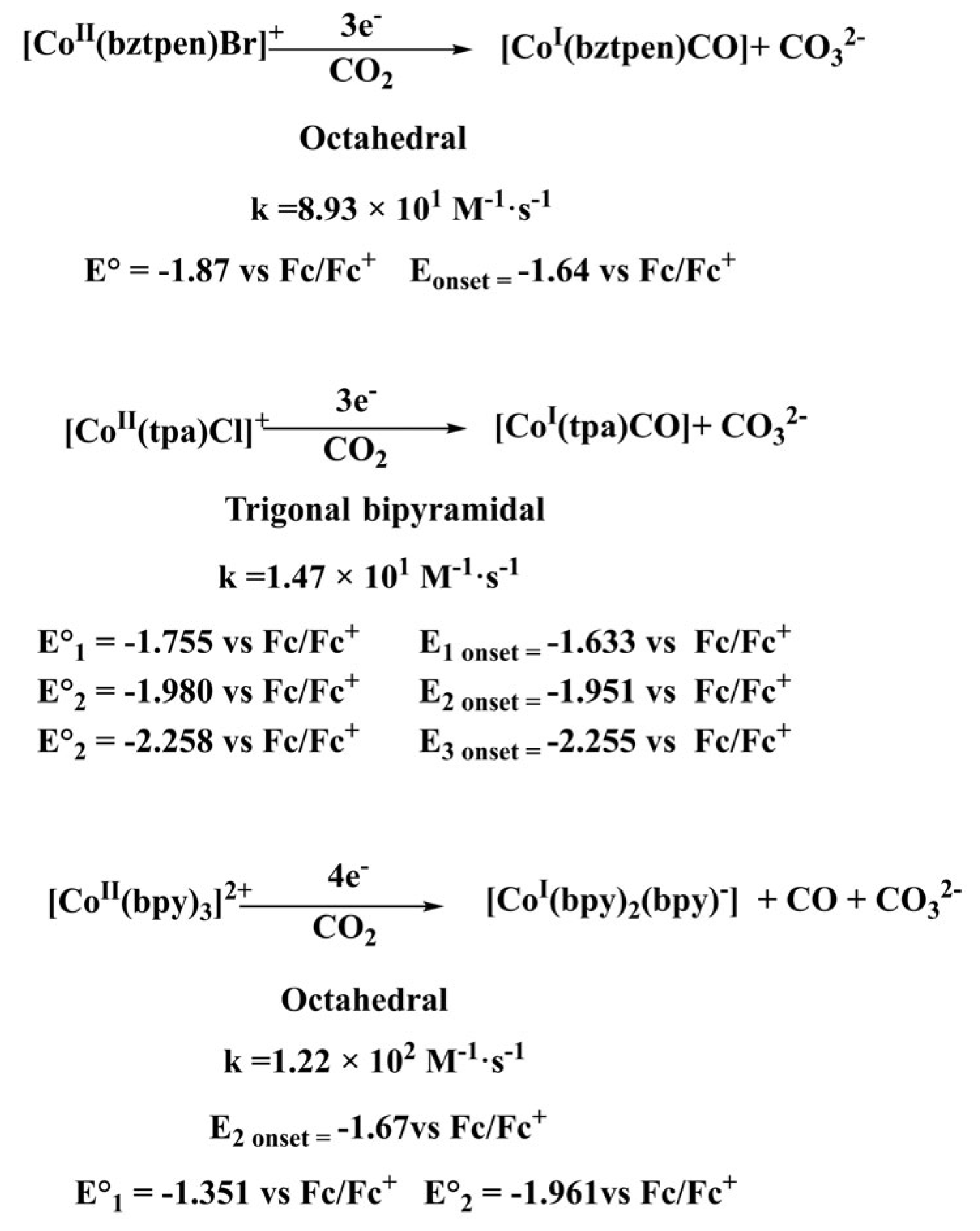
2.3. Cyclic Voltammetry Response of Complexes in the Presence of CO2
2.3.1. Cyclic Voltammetry of [CoII(bztpen)Br]PF6 in the presence of CO2
2.3.2. Cyclic Voltammetry of [CoII (tpa)Cl]Cl in the presence of CO2
2.3.3. Cyclic Voltammetry of [CoII (bpy)3(BF4)2] in the presence of CO2
2.3.4. FOWA
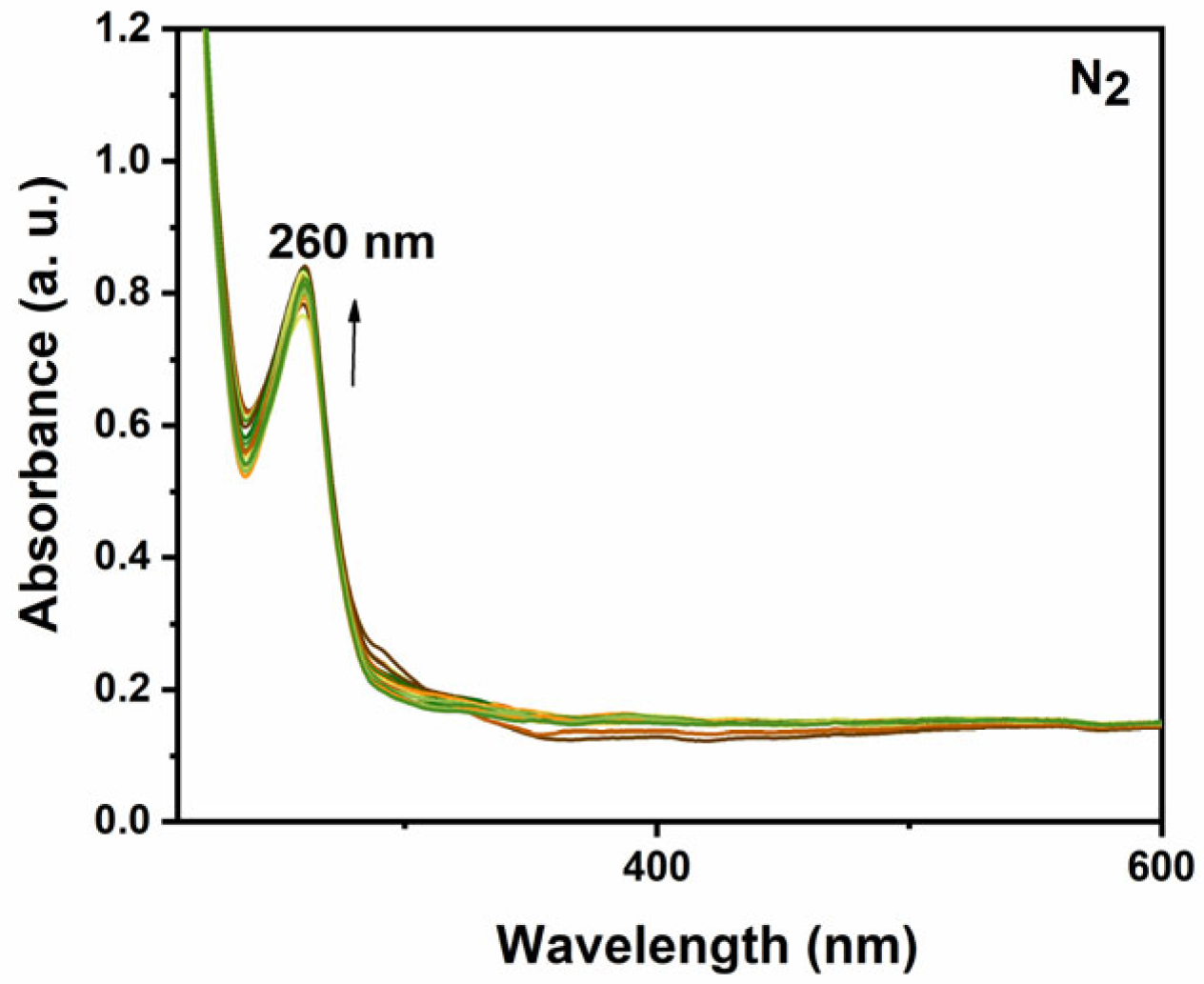
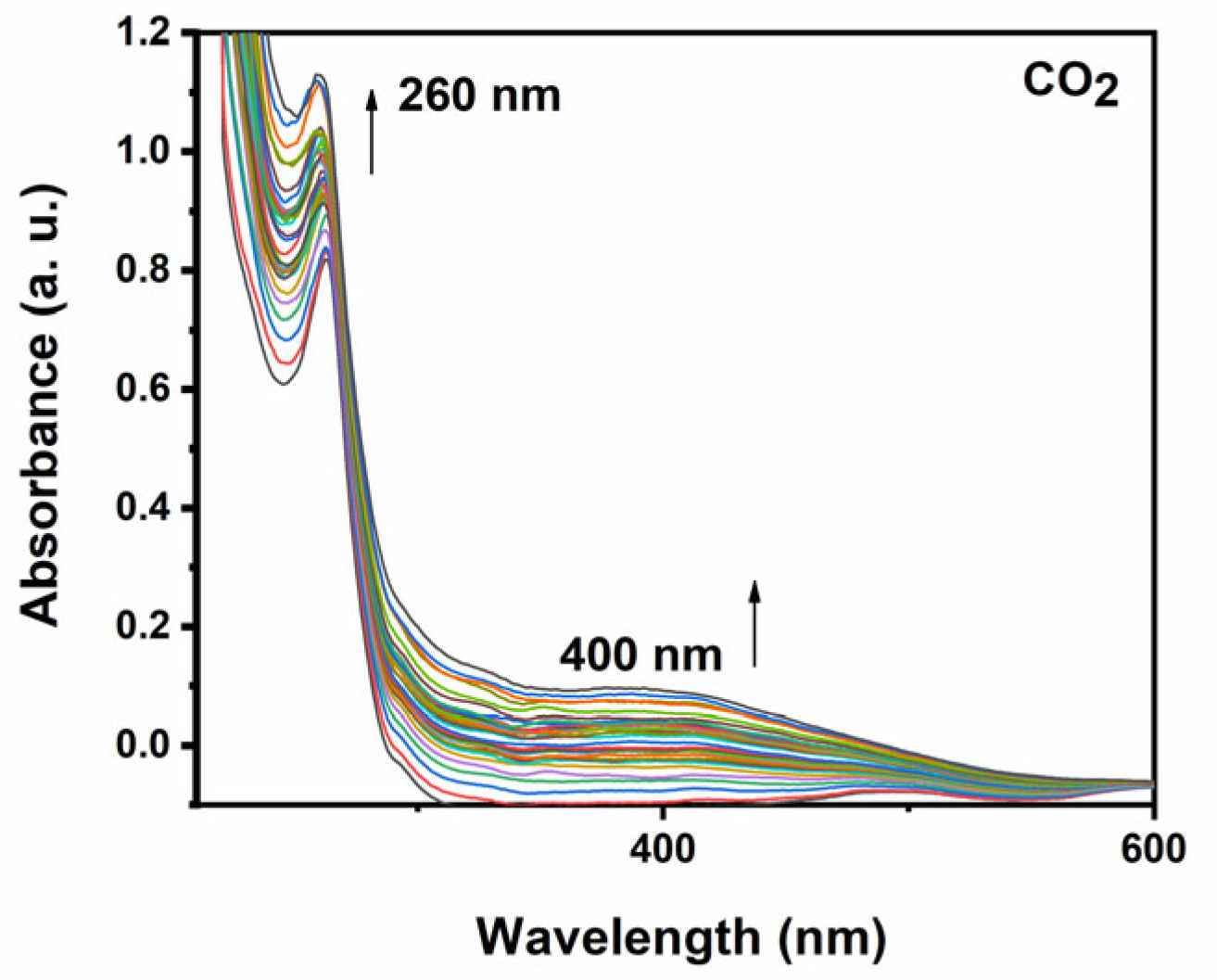
2.4. UV-Vis Spectroelectrochemical Response of the Complexes in the Presence and in the Absence of CO2
2.4.1. UV-Vis Spectroelectrochemical Response of [CoII(Bztpen)Br]PF6
2.4.2. UV-Vis Spectroelectrochemical Response of [CoII(tpa)Cl]Cl
2.4.3. UV-Vis Spectroelectrochemical Response of [CoII(bpy)3](BF4)2
2.4.4. Kinetics of the Formation of Intermediate Species
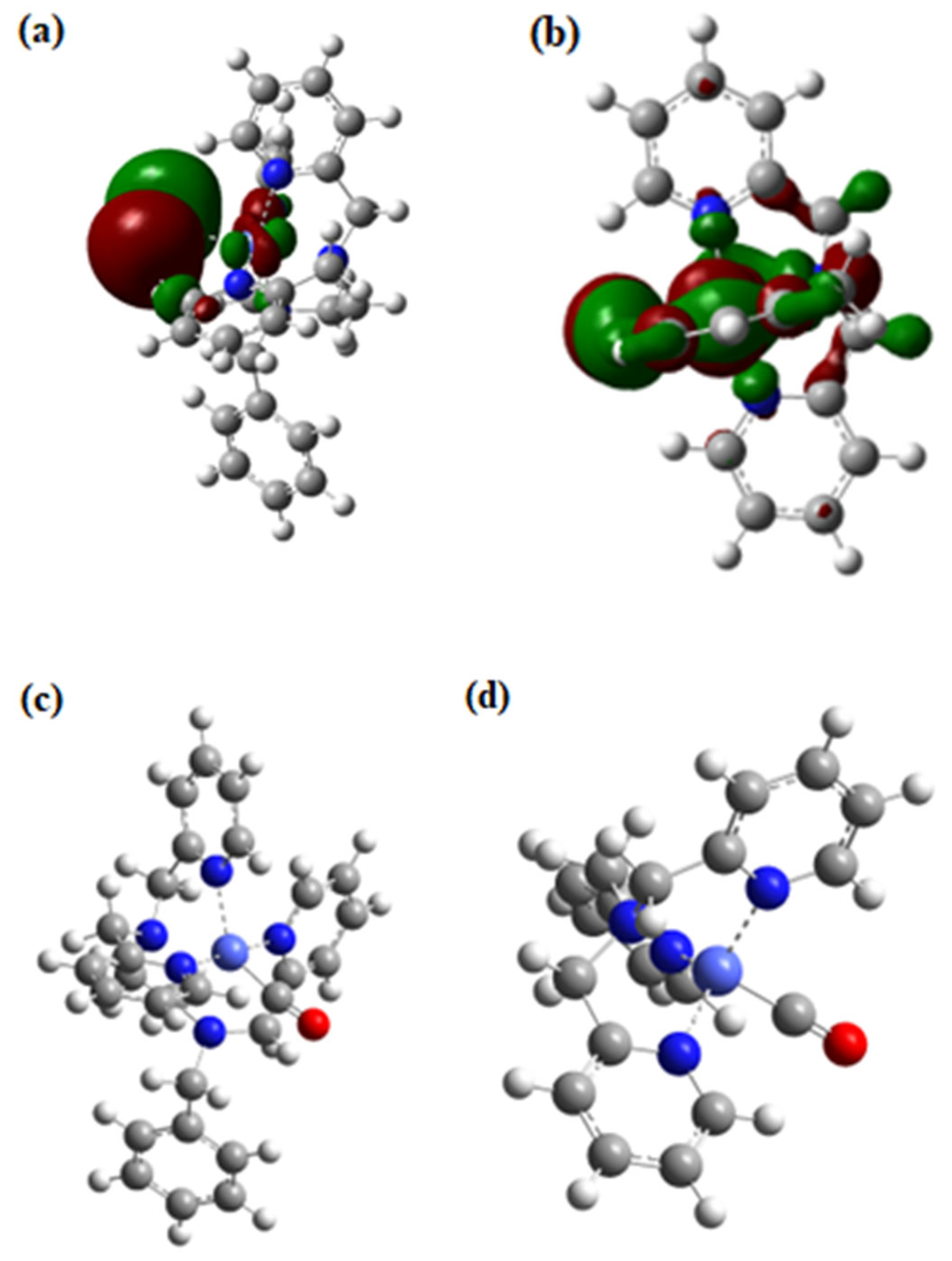
2.5. DFT Calculations
3. Materials and Methods
3.1. Reagents
3.2. FTIR-IR and UV-Vis Measurements
3.3. Synthesis of Complexes
3.3.1. [Co(bztpen)Br]PF6
3.3.2. [Co(tpa)Cl]Cl
3.3.3. [Co(bpy)3](BF4)2
3.4. Electrochemical Studies in the Absence and in the Presence of CO2
3.5. UV-Vis Spectroelectrochemical Studies in the Absence and in the Presence of CO2
3.6. DFT Theoretical Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Core Writing Team; Lee, H.; Romero, J. (Eds.) Summary for Policymakers. In Climate Change 2023: Synthesis Report. Contribution of Working Groups I, II and III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2023; pp. 1–34. [Google Scholar] [CrossRef]
- Ritchie, H.; Roser, M. CO2 Emissions. Published Online at OurWorldinData.org. [Online Resource]. 2020. Available online: https://ourworldindata.org/co2-emissions (accessed on 26 June 2025).
- Hogue, C. CO Levels Climb Ever Higher. Chem. Eng. News 2015, 93, 6. [Google Scholar] [CrossRef]
- Albero, J.S.; Albero, A.S.; Casco, M.E.; Reinoso, F.R. Retos actuales para la captura y almacenamiento de CO2. Real Sociedad Española de Química. An. Quim. 2014, 110, 30–34. [Google Scholar]
- Bui, A.T.; Hartley, N.A.; Thom, A.J.W.; Forse, A.C. Trade-Off between Redox Potential and the Strength of Electrochemical CO2 Capture in Quinone. J. Phys. Chem. C 2022, 126, 14163–14172. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Maji, S.; Ott, S. Mechanistic insights into electrocatalytic CO2 reduction within [RuII(tpy)(NN)X]n+ architectures. Dalton Trans. 2014, 43, 15028–15037. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.K.; Kumari, N.; Pachfule, P.; Banerjee, R.; Mishra, L. Metal [Zn(II), Cd(II)], 1,10-Phenanthroline Containing Coordination Polymers Constructed on the Skeleton of Polycarboxylates: Synthesis, Characterization, Microstructural, and CO2 Gas Adsorption Studies. Cryst. Growth Des. 2012, 12, 5311–5319. [Google Scholar] [CrossRef]
- Thoi, V.S.; Sun, Y.; Long, J.R.; Chang, C.J. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 2013, 42, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Dempsey, Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 998–10002. [Google Scholar] [CrossRef]
- Garba, M.D.; Usman, M.; Khan, S.; Shehzad, F.; Galadima, A.; Ehsan, M.F.; Ghanem, A.S.; Humayun, M. CO2 towards fuels: A review of catalytic conversion of carbon dioxide to hydrocarbons. J. Environ. Chem. Eng. 2021, 9, 104756. [Google Scholar] [CrossRef]
- Schrodt, A.; Neubrand, A.; van Eldik, R. Fixation of CO2 by Zinc(II) Chelates in Alcoholic Medium. X-ray Structures of {[Zn(cyclen)]3(μ3-CO3)}(ClO4)4 and [Zn(cyclen)EtOH](ClO4)2. Inorg. Chem. 1997, 36, 4579–4584. [Google Scholar] [CrossRef]
- Chiericato, G.; Arana, C.; Casado, C.; Cuadrado, I.; Abruña, H. Electrocatalytic reduction of carbon dioxide mediated by transition metal complexes with terdentate ligands derived from diacetylpyridine. Inorg. Chim. Acta 2000, 300, 32–42. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef]
- Berardi, S.; Drouet, S.; Francàs, L.; Gimbert-Suriñach, C.; Guttentag, M.; Richmond, C.; Stoll, T.; Llobet, A. Molecular artificial photosynthesis. Chem. Soc. Rev. 2014, 43, 7501–7519. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Bai, X.; Xu, X.; Husile, A.; Zhang, S.; Qi, L.; Guan, J. Advances and challenges in the electrochemical reduction of carbon dioxide. Chem. Sci. 2024, 15, 7870–7907. [Google Scholar] [CrossRef]
- Bonin, J.; Robert, M.; Routier, M. Selective and Efficient Photocatalytic CO2 Reduction to CO Using Visible Light and an Iron-Based Homogeneous Catalyst. J. Am. Chem. Soc. 2014, 136, 16768–16771. [Google Scholar] [CrossRef]
- Simpson, T.C.; Durand, R.R. Ligand participation in the reduction of CO2 catalyzed by complexes of 1,10 o-phenanthroline. Electrochim. Acta. 1998, 33, 581–583. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) polypyridine complexes: Photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Usman, M.; Humayun, M.; Garba, M.D.; Ullah, L.; Zeb, Z.; Helal, A.; Suliman, M.H.; Alfaifi, B.Y.; Iqbal, N.; Abdinejad, M.; et al. Electrochemical reduction of CO2: A review of cobalt based catalyst for carbon dioxide conversion to fuels. Nanomaterials 2021, 11, 2029. [Google Scholar] [CrossRef]
- Intrator, J.A.; Velazquez, D.A.; Fan, S.; Mastrobattista, E.; Yu, C.J.; Marinescu, S.C. A Electrocatalytic CO2 reduction to formate by a cobalt phosphino–thiolate complex. Chem. Sci. 2024, 15, 6385–6396. [Google Scholar] [CrossRef]
- Rebolledo-Chávez, J.P.F.; Toral, G.T.; Ramírez-Delgado, V.; Reyes-Vidal, Y.; Jiménez-González, M.L.; Cruz-Ramírez, M.; Mendoza, A.; Ortiz-Frade, L. The Role of Redox Potential and Molecular Structure of Co(II)-Polypyridine Complexes on the Molecular Catalysis of CO2 Reduction. Catalysts 2021, 11, 948. [Google Scholar] [CrossRef]
- Reimann, C.W. Electron absorption spectrum of cobalt (II)-doped trisphenanthrolinezinc nitrate dihydrate. J. Res. Nat. Stand. A Phys. Chem. 1966, 70, 417–419. [Google Scholar] [CrossRef]
- Louise, I.S.Y.; Nabila, S.; Sugiyarto, K.H. Complex of Tris (Phenan- throline) Cobalt (II) Trifluoroacetate: Characterization and Powder XRD Analysis. Orient. J. Chem. 2019, 35, 1500–1507. [Google Scholar] [CrossRef]
- Chan, S.L.-F.; Lam, T.L.; Yang, C.; Lai, J.; Cao, B.; Zhou, Z.; Zhu, Q. Cobalt(II) tris(2-pyridylmethyl)amine complexes [Co(TPA)X] + bearing co- ordinating anion (X = Cl−, Br−, I− and NCS−): Synthesis and application for carbon dioxide reduction. Polyhedron 2017, 125, 156–163. [Google Scholar] [CrossRef]
- Massoud, S.S.; Broussard, K.T.; Mautner, F.A.; Vicente, R.; Saha, M.K.; Bernal, I. Five-coordinate cobalt (II) complexes of tris(2-pyridylmethyl) amine (TPA): Synthesis, structural and magnetic characterization of a terephthalato-bridged dinuclear cobalt (II) complex. Inor. Chim. Acta 2008, 361, 123–131. [Google Scholar] [CrossRef]
- Blackman, A.G. Tripodal tetraamine ligands containing three pyridine units: The other polypyridyl ligands. Eur. J. Inorg. Chem. 2008, 17, 2633–2647. [Google Scholar] [CrossRef]
- Blackman, A.G. The coordination chemistry of tripodal tetraamine ligands. Polyhedron 2005, 24, 1–39. [Google Scholar] [CrossRef]
- England, J.; Bill, E.; Weyhermüller, T.; Neese, F.; Atanasov, M.; Wieghardt, K. Molecular and Electronic Structures of Homoleptic Six-Coordinate Cobalt(I) Complexes of 2,2:6,2-Terpyridine, 2,2-Bpyridine, and 1,10-Phenanthroline. An Experimental and Computational Study. Inorg. Chem. 2015, 54, 12002–12018. [Google Scholar] [CrossRef]
- Kurtz, D.A.; Zhang, J.; Sookezian, A.; Kallick, J.; Hill, M.G.; Hunter, B.M. A cobalt phosphine complex in five oxidation states. Inorg. Chem. 2021, 60, 17445–17449. [Google Scholar] [CrossRef]
- Isaacs, M.; Canales, J.; Aguirre, M.; Estiú, G.; Caruso, F.; Ferraudi, G.; Costamagna, J. Electrocatalytic reduction of CO2 by aza-macrocyclic complexo f Ni (II), Co (II), and Cu (II). Theoretical contribution to probable mechanisms. Inorg. Chim. Acta 2002, 339, 224. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, M.; Yang, Y.; Yao, T.; Sun, L. A molecular copper catalyst for electrochemical water reduction with a large hydrogen-generation rate constant in aqueous solution. Angew. Chem. Int. Ed. 2014, 126, 14023–14027. [Google Scholar] [CrossRef]
- Chan, S.L.-F.; Lam, T.L.; Yang, C.; Yan, S.-C.; Cheng, N.M. A robust and efficient cobalt molecular catalyst for CO2 reduction. Chem. Commun. 2015, 51, 7799. [Google Scholar] [CrossRef] [PubMed]
- Keene, F.R.; Creutz, C.; Sutin, N. Reduction of carbon dioxide by tris(2,2′-bpyridine)cobalt(I), Coord. Chem. Rev. 1985, 64, 247–260. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Passard, G.; Robert, M.; Savéant, J.-M. Proton-Coupled Electron Transfer Cleavage of Heavy-Atom Bonds in Electrocatalytic Processes. Cleavage of a CO Bond in the Catalyzed Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2003, 135, 9023–9031. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. A Study of Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- McSkimming, A.; Bhadbhade, M.; Colbran, S.B. Hydride ion-carrier ability in Rh(i) complexes of a nicotinamide-functionalised N-heterocyclic carbene ligand. Dalton Trans. 2010, 39, 10581–10584. [Google Scholar] [CrossRef]
- Záliš, S.; Consani, C.; El Nahhas, A.; Cannizzo, A.; Chergui, M.; Hartl, F.; Vlček, A., Jr. Origin of electronic absorption spectra of MLCT-excited and one-electron reduced 2,2’ -bpyridine and 1,10-phenanthroline complexes. Inorg. Chim. Acta. 2011, 374, 578–585. [Google Scholar] [CrossRef]
- Keszei, E. Kinetics of Composite Reactions. Reaction Kinetics; Springer: Cham, Switzerland, 2021; pp. 71–116. [Google Scholar] [CrossRef]
- Trasatti, S. The Absolute Electrode Potential: An Explanatory Note (Recommendations 1986). Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 7, 1912–1919. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 399, 51–57. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., Ed.; Plenum: New York, NY, USA, 1977; Volume 3, pp. 1–28. [Google Scholar]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Redox Potential (V vs. Fc/Fc+) | Onset Potential (V vs. Fc/Fc+) |
|---|---|---|
| [CoII(bztpen)Br]PF6 | ||
| −1.807 | −1.640 | |
| −0.0655 | n.o. | |
| [CoII(tpa)Cl]Cl | ||
| −1.755 | −1.633 | |
| −1.980 | −1.951 | |
| −2.258 | −2.255 | |
| [CoII(bpy)3](BF4)2 | ||
| −1.351 | n.o. | |
| −1.961 | −1.667 |
| Compound | Potential Applied (V vs. Fc/Fc+) | Wavelength (nm) | k (s−1) |
|---|---|---|---|
| [CoII(bztpen)Br]PF6 | −1.95 | 400 | 0.026 ± 0.001 |
| [CoII(tpa)Cl]Cl | −2.06 | 400 | 0.037 ± 0.001 |
| [CoII(tpa)Cl]Cl | −2.24 | 400 | 0.015 ± 0.001 |
| [CoII(bpy)3](BF4)2 | −1.55 | 380 | 0.008 ± 0.002 |
| [CoII(bpy)3](BF4)2 | −2.25 | 380 | 0.0001 ± 0.002 |
| [CoII(bztpen)Br]PF6 | −1.95 | 260 | n.o. |
| [CoII(tpa)Cl]Cl | −2.06 | 260 | 0.031 ± 0.002 |
| [CoII(tpa)Cl]Cl | −2.24 | 260 | 0.021 ± 0.002 |
| [CoII(bpy)3](BF4)2 | −1.55 | 245 | 0.020 ± 0.002 |
| [CoII(bpy)3](BF4)2 | −2.25 | 245 | n.o. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha-Ortiz, G.; Barrios-Velasco, A.; Monsalvo Zúñiga, O.; Cruz-Ramírez, M.; Mendoza, A.; Ramírez-Palma, L.G.; Rebolledo-Chávez, J.P.F.; Ortiz-Frade, L. The Role of Geometry in Cobalt–Polypyridine Complexes in the Electrochemical Reduction of CO2 Using UV-Vis Spectroelectrochemistry. Catalysts 2025, 15, 641. https://doi.org/10.3390/catal15070641
Rocha-Ortiz G, Barrios-Velasco A, Monsalvo Zúñiga O, Cruz-Ramírez M, Mendoza A, Ramírez-Palma LG, Rebolledo-Chávez JPF, Ortiz-Frade L. The Role of Geometry in Cobalt–Polypyridine Complexes in the Electrochemical Reduction of CO2 Using UV-Vis Spectroelectrochemistry. Catalysts. 2025; 15(7):641. https://doi.org/10.3390/catal15070641
Chicago/Turabian StyleRocha-Ortiz, Gilberto, Anahí Barrios-Velasco, Omar Monsalvo Zúñiga, Marisela Cruz-Ramírez, Angel Mendoza, Lillian G. Ramírez-Palma, Juan Pablo F. Rebolledo-Chávez, and Luis Ortiz-Frade. 2025. "The Role of Geometry in Cobalt–Polypyridine Complexes in the Electrochemical Reduction of CO2 Using UV-Vis Spectroelectrochemistry" Catalysts 15, no. 7: 641. https://doi.org/10.3390/catal15070641
APA StyleRocha-Ortiz, G., Barrios-Velasco, A., Monsalvo Zúñiga, O., Cruz-Ramírez, M., Mendoza, A., Ramírez-Palma, L. G., Rebolledo-Chávez, J. P. F., & Ortiz-Frade, L. (2025). The Role of Geometry in Cobalt–Polypyridine Complexes in the Electrochemical Reduction of CO2 Using UV-Vis Spectroelectrochemistry. Catalysts, 15(7), 641. https://doi.org/10.3390/catal15070641