
Promotion of Carbon Dioxide Biofixation through Metabolic and Enzyme Engineering


Abstract
:1. Introduction
2. Comparative Analysis of Pathways for Carbon Dioxide Biofixation
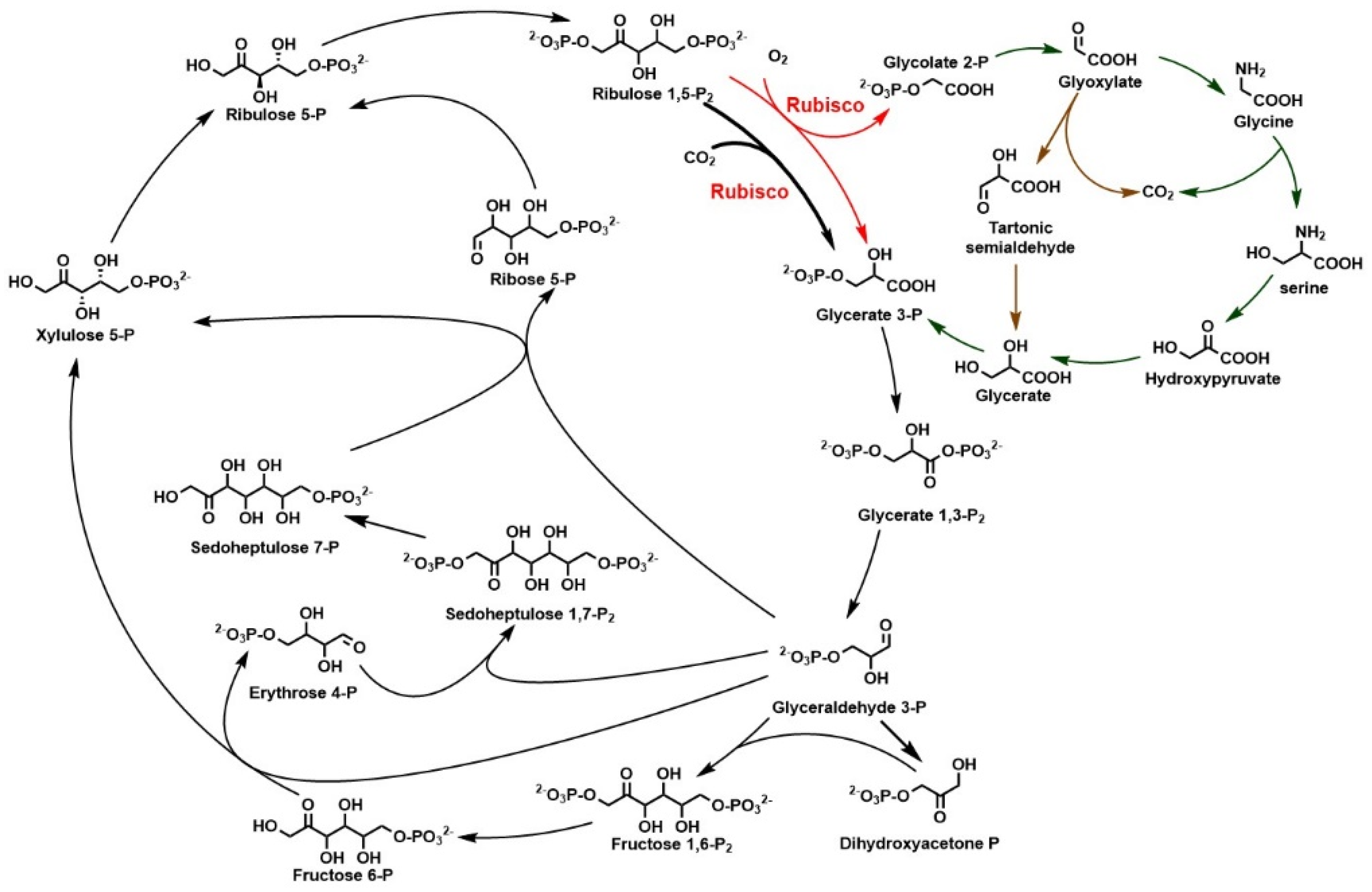
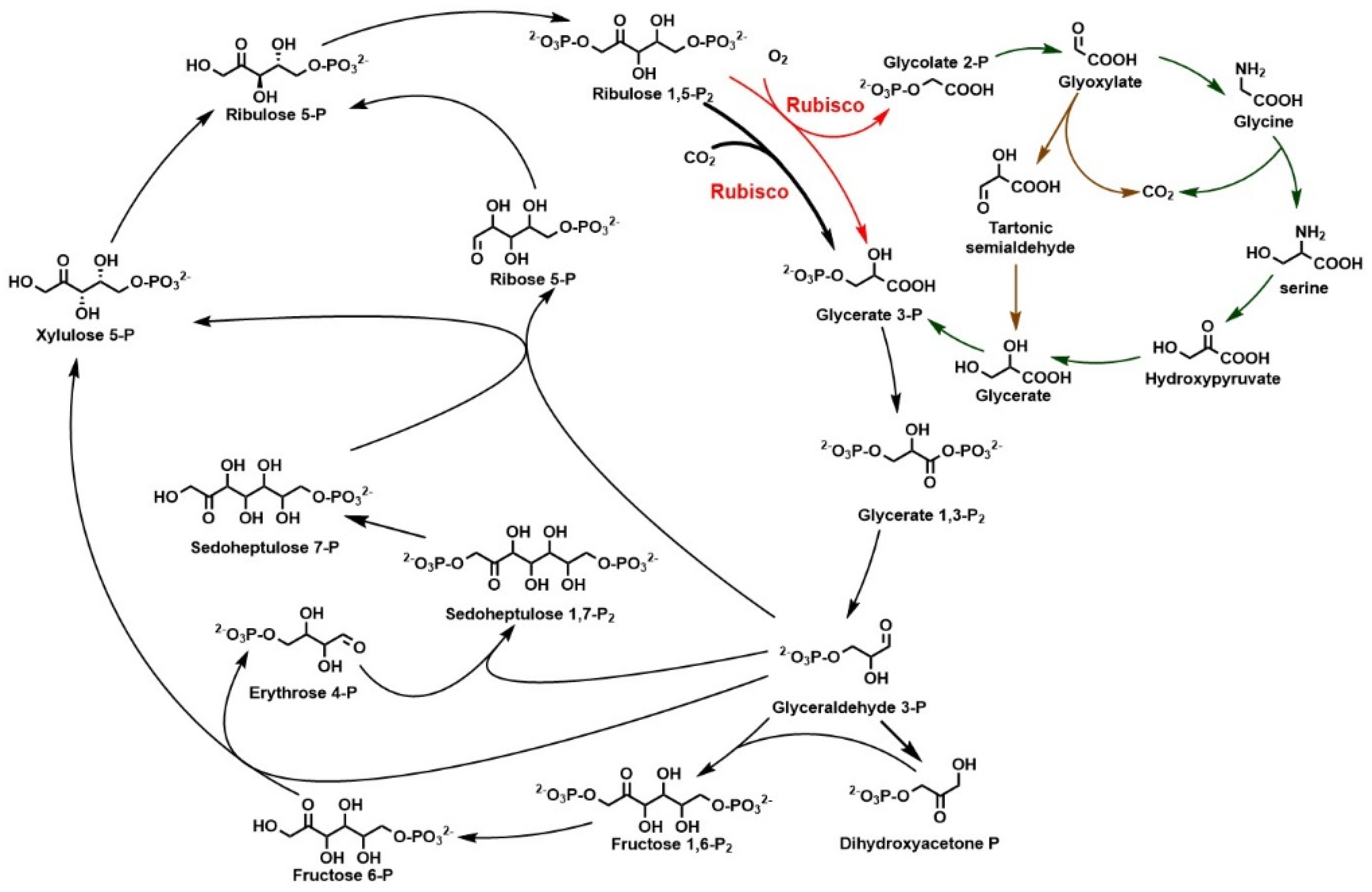
2.1. Calvin–Benson–Basham Cycle and Rubisco
2.2. The Reductive Tricarboxylic Cycle and Its Variant roTCA Cycle
2.3. The Reductive Acetyl–CoA Pathway Is the Most Ancient and Energy–Efficient CO2 Fixation Pathway
2.4. The 3-Hydroxypropionate Bi-Cycle Producing Pyruvate from CO2
2.5. The HP/HB Cycle and DC/HB Cycle Are the Most Recently Discovered CO2 Fixation Pathway
3. Enzymes Catalyzing CO2 to Organic Compounds
3.1. Rubisco Is the Main Carboxylase for CO2 Fixation
3.1.1. Screening and Recombinant Expression of Efficient Rubiscos to Enhance CO2 Fixation
3.1.2. Enhancing CO2 Fixation through Constructing Synthetic Phosphoglycolate Salvage Pathway
3.1.3. Concentrating CO2 for Fixation by Enzyme Engineering
3.2. Biotin-Dependent Carboxylase
3.3. Carbon Monoxide Dehydrogenase/acetyl-CoA Synthase and the Application for Biosynthesis
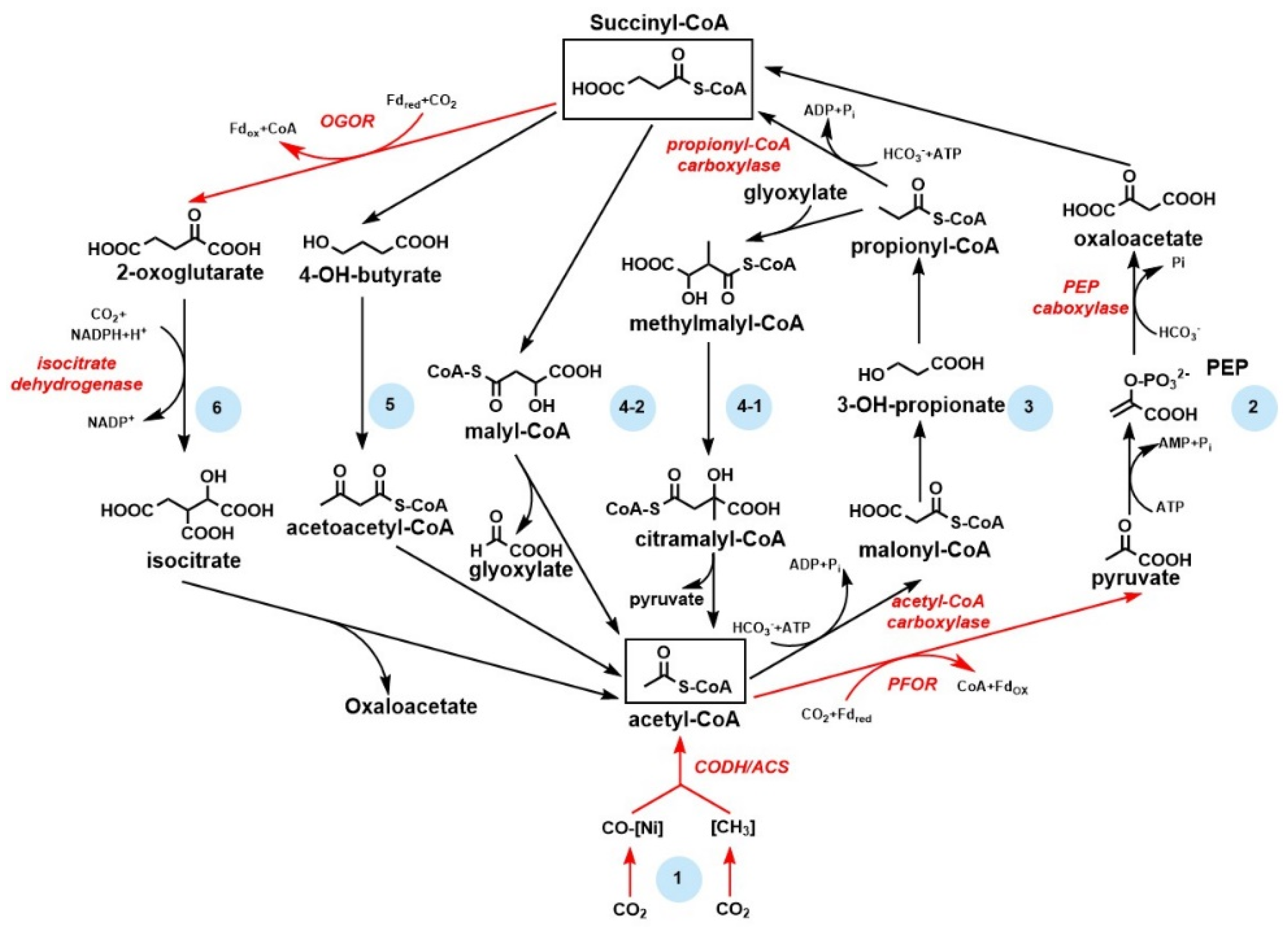
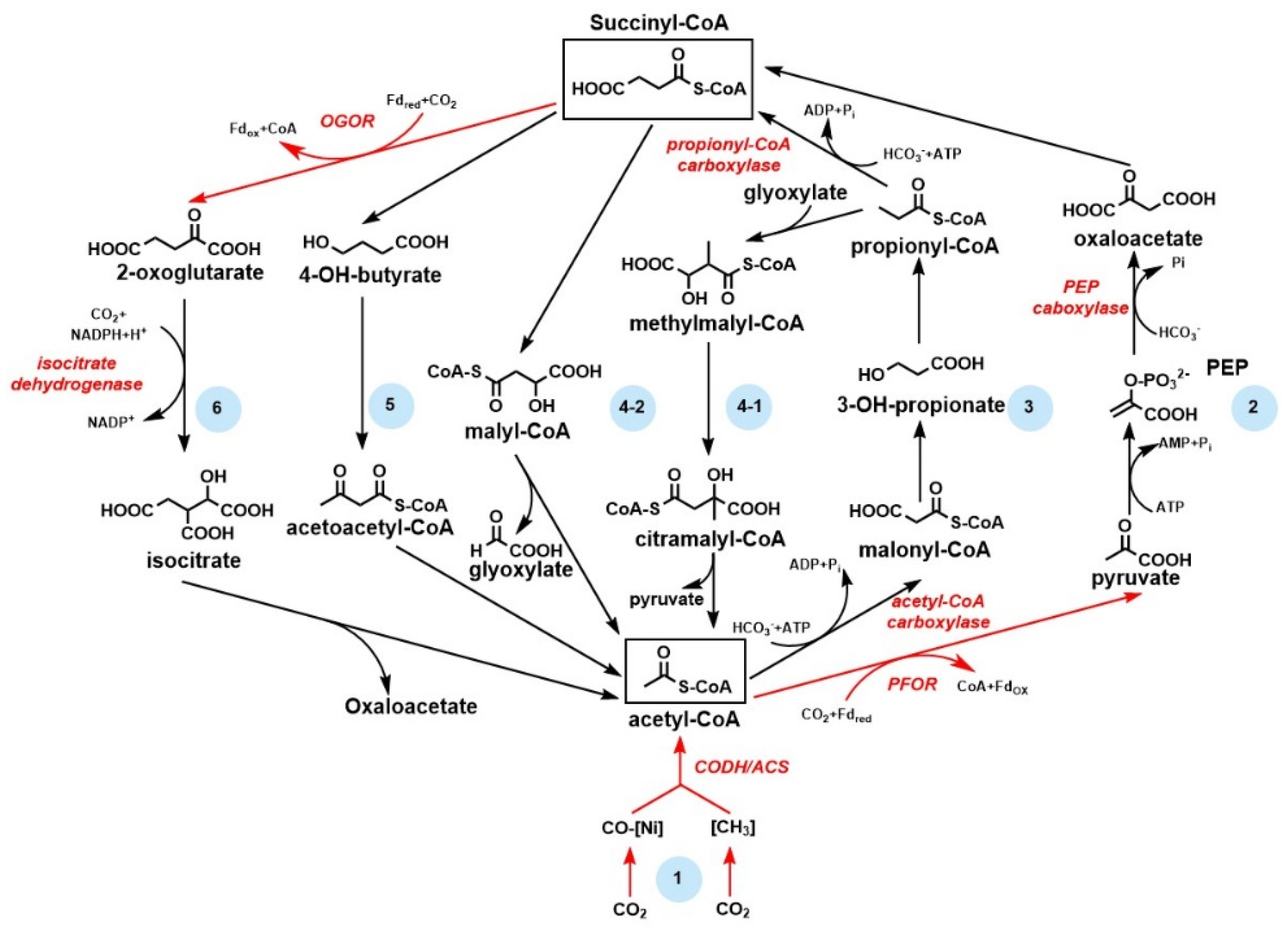
3.4. 2-Oxoacid:Ferredoxin Oxidoreductase and the Application in CO2 Fixation
3.5. Artificial Pathways for CO2 Fixation and Biosynthesis
4. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, J.; Jiang, Y.; Jiang, Z.; Wang, X.; Wang, X.; Zhang, S.; Han, P.; Yang, C. Enzymatic conversion of carbon dioxide. Chem. Soc. Rev. 2015, 44, 5981–6000. [Google Scholar] [CrossRef] [PubMed]
- Haszeldine, R.S. Carbon capture and storage: How green can black be? Science 2009, 325, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.H.; Long, J.R. Carbon dioxide capture in metal–organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chong, Z.R.; Qureshi, M.F.; Linga, P. Carbon Dioxide Sequestration via Gas Hydrates: A Potential Pathway toward Decarbonization. Energy Fuels 2020, 34, 10529–10546. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, S.; Row, K.H.; Ahn, W.-S. Amine–silica composites for CO2 capture: A short review. J. Energy Chem. 2017, 26, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.; Santos, S.; Bordado, J. Choosing amine–based absorbents for CO2 capture. Environ. Technol. 2015, 36, 19–25. [Google Scholar] [CrossRef]
- Yeung, C.S. Photoredox Catalysis as a Strategy for CO2 Incorporation: Direct Access to Carboxylic Acids from a Renewable Feedstock. Angew. Chem. Int. Ed. 2019, 58, 5492–5502. [Google Scholar] [CrossRef]
- Xu, S.; Carter, E.A. Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction. Chem Rev. 2019, 119, 6631–6669. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.G.C.N.; Andrade, L.H.; Toma, H.E. Carbon dioxide/methanol conversion cycle based on cascade enzymatic reactions supported on superparamagnetic nanoparticles. An. Acad. Bras. Cienc. 2018, 90, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Kelly, R.M.; Han, Y. Sequential processing with fermentative Caldicellulosiruptor kronotskyensis and chemolithoautotrophic Cupriavidus necator for converting rice straw and CO2 to polyhydroxybutyrate. Biotechnol. Bioeng. 2018, 115, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.; Milo, R. A feeling for the numbers in biology. Proc. Natl. Acad. Sci. USA 2009, 106, 21465–21471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb, T.J.; Zarzycki, J. A short history of RubisCO: The rise and fall (?) of Nature’s predominant CO2 fixing enzyme. Curr. Opin. Biotechnol. 2018, 49, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Kopke, M.; Mihalcea, C.; Bromley, J.C.; Simpson, S.D. Fermentative production of ethanol from carbon monoxide. Curr. Opin. Biotechnol. 2011, 22, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Angermayr, S.A.; Gorchs Rovira, A.; Hellingwerf, K.J. Metabolic engineering of cyanobacteria for the synthesis of commodity products. Trends Biotechnol. 2015, 33, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Nybo, S.E.; Khan, N.E.; Woolston, B.M.; Curtis, W.R. Metabolic engineering in chemolithoautotrophic hosts for the production of fuels and chemicals. Metab. Eng. 2015, 30, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangle, S.N.; Ziesack, M.; Buckley, S.; Trivedi, D.; Loh, D.M.; Nocera, D.G.; Silver, P.A. Valorization of CO2 through lithoautotrophic production of sustainable chemicals in Cupriavidus necator. Metab. Eng. 2020, 62, 207–220. [Google Scholar] [CrossRef]
- Tang, R.H.; Peng, X.W.; Weng, C.H.; Han, Y.J. The overexpression of phasin and regulator genes promoting the synthesis of polyhydroxybutyrate in Cupriavidus necator H16 under nonstress conditions. Appl. Environ. Microbiol. 2022, 88. [Google Scholar] [CrossRef]
- Tang, R.; Weng, C.; Peng, X.; Han, Y. Metabolic engineering of Cupriavidus necator H16 for improved chemoautotrophic growth and PHB production under oxygen–limiting conditions. Metab. Eng. 2020, 61, 11–23. [Google Scholar] [CrossRef]
- Lee, K.; Evans, J.P.; Satagopan, S.; Sun, Y.; Parquette, J.R.; Sundaresan, V.B.; Tabita, F.R.; Bakshi, B.R. Carbon Footprint of Biomimetic Carbon Fixation by Immobilizing Nature’s CO2–sequestering Enzyme and Regenerating Its Energy Carrier. ACS Sustain. Chem. Eng. 2020, 8, 16833–16841. [Google Scholar] [CrossRef]
- Berg, I.A. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 2011, 77, 1925–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvin, M.; Benson, A.A. The Path of Carbon in Photosynthesis. Science 1948, 107, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, H.G.; Gottschalk, G.; Bartha, R.V. Formation and Utilization of Poly–β–Hydroxybutyric Acid by Knallgas Bacteria (Hydrogenomonas). Nature 1961, 191, 463–465. [Google Scholar] [CrossRef]
- Claassens, N.J.; Scarinci, G.; Fischer, A.; Flamholz, A.I.; Newell, W.; Frielingsdorf, S.; Lenz, O.; Bar-Even, A. Phosphoglycolate salvage in a chemolithoautotroph using the Calvin cycle. Proc. Natl. Acad. Sci. USA 2020, 117, 22452–22461. [Google Scholar] [CrossRef] [PubMed]
- Tcherkez, G. The mechanism of Rubisco–catalysed oxygenation. Plant. Cell Environ. 2016, 39, 983–997. [Google Scholar] [CrossRef]
- Sharkey, T.D. Estimating the rate of photorespiration in leaves. Physiol. Plant. 1988, 73, 147–152. [Google Scholar] [CrossRef]
- Walker, B.J.; VanLoocke, A.; Bernacchi, C.J.; Ort, D.R. The Costs of Photorespiration to Food Production Now and in the Future. Annu. Rev. Plant. Biol. 2016, 67, 107–129. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.C.; Buchanan, B.B.; Arnon, D.I. A new ferredoxin–dependent carbon reduction cycle in a photosynthetic bacterium. Proc. Natl. Acad. Sci. USA 1966, 55, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, G. Alternative Pathways of Carbon Dioxide Fixation: Insights into the Early Evolution of Life? Annu. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar] [CrossRef]
- Erb, T.J. Carboxylases in Natural and Synthetic Microbial Pathways. Appl. Environ. Microbiol. 2011, 77, 8466–8477. [Google Scholar] [CrossRef] [Green Version]
- Moulis, J.M.; Davasse, V.; Meyer, J.; Gaillard, J. Molecular mechanism of pyruvate–ferredoxin oxidoreductases based on data obtained with the Clostridium pasteurianum enzyme. Febs. Lett. 1996, 380, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Bock, A.K.; Kunow, J.; Glasemacher, J.; Schonheit, P. Catalytic properties, molecular composition and sequence alignments of pyruvate:ferredoxin oxidoreductase from the methanogenic archaeon Methanosarcina barkeri (strain Fusaro). Eur. J. Biochem. 1996, 237, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.V.; Malinina, N.V.; Ivanovsky, R.N. A comparative study of the isocitrate dehydrogenases of Chlorobium limicola forma thiosulfatophilum and Rhodopseudomonas palustris. Microbiology 2002, 71, 657–661. [Google Scholar] [CrossRef]
- Aoshima, M.; Igarashi, Y. Nondecarboxylating and decarboxylating isocitrate dehydrogenases: Oxalosuccinate reductase as an ancestral form of isocitrate dehydrogenase. J. Bacteriol. 2008, 190, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoshima, M.; Igarashi, Y. A novel oxalosuccinate–forming enzyme involved in the reductive carboxylation of 2–oxoglutarate in Hydrogenobacter thermophilus TK–6. Mol. Microbiol. 2006, 62, 748–759. [Google Scholar] [CrossRef]
- Aoshima, M.; Ishii, M.; Igarashi, Y. A novel biotin protein required for reductive carboxylation of 2–oxoglutarate by isocitrate dehydrogenase in Hydrogenobacter thermophilus TK–6. Mol. Microbiol. 2004, 51, 791–798. [Google Scholar] [CrossRef]
- Nunoura, T.; Chikaraishi, Y.; Izaki, R.; Suwa, T.; Sato, T.; Harada, T.; Mori, K.; Kato, Y.; Miyazaki, M.; Shimamura, S.; et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 2018, 359, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Schulman, M.; Parker, D.; Ljungdahl, L.G.; Wood, H.G. Total Synthesis of Acetate from CO2 V. Determination by Mass Analysis of the Different Types of Acetate Formed from 13CO2 by Heterotrophic Bacteria. J. Bacteriol. 1972, 109, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Martin, W.F. Hydrogen, metals, bifurcating electrons, and proton gradients: The early evolution of biological energy conservation. FEBS Lett. 2012, 586, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta–Proteins Proteom. 2008, 1784, 1873–1898. [Google Scholar] [CrossRef] [Green Version]
- Ragsdale, S.W. Enzymology of the Wood–Ljungdahl pathway of acetogenesis. Ann. N. Y. Acad. Sci. 2008, 1125, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spormann, A.M.; Thauer, R.K. Anaerobic acetate oxidation to CO2 by Desulfotomaculum acetoxidans. Arch. Microbiol. 1988, 150, 374–380. [Google Scholar] [CrossRef]
- Schauder, R.; Preuß, A.; Jetten, M.; Fuchs, G. Oxidative and reductive acetyl CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. Arch. Microbiol. 1988, 151, 84–89. [Google Scholar] [CrossRef]
- Strauss, G.; Fuchs, G. Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3–hydroxypropionate cycle. Eur. J. Biochem. 1993, 215, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Alber, B.E.; Fuchs, G. Propionyl–coenzyme A synthase from Chloroflexus aurantiacus, a key enzyme of the 3–hydroxypropionate cycle for autotrophic CO2 fixation. J. Biol. Chem. 2002, 277, 12137–12143. [Google Scholar] [CrossRef] [Green Version]
- Zarzycki, J.; Brecht, V.; Mueller, M.; Fuchs, G. Identifying the missing steps of the autotrophic 3–hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc. Natl. Acad. Sci. USA 2009, 106, 21317–21322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, H.; Gallenberger, M.; Jahn, U.; Eylert, E.; Berg, I.A.; Kockelkorn, D.; Eisenreich, W.; Fuchs, G. A dicarboxylate/4–hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc. Natl. Acad. Sci. USA 2008, 105, 7851–7856. [Google Scholar] [CrossRef] [Green Version]
- Berg, I.A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. A 3–Hydroxypropionate/4–Hydroxybutyrate Autotrophic Carbon Dioxide Assimilation Pathway in Archaea. Science 2007, 318, 1782–1786. [Google Scholar] [CrossRef] [Green Version]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef]
- Flamholz, A.I.; Prywes, N.; Moran, U.; Davidi, D.; Bar-On, Y.M.; Oltrogge, L.M.; Alves, R.; Savage, D.; Milo, R. Revisiting Trade–offs between Rubisco Kinetic Parameters. Biochemistry 2019, 58, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Tabita, F.R.; Satagopan, S.; Hanson, T.E.; Kreel, N.E.; Scott, S.S. Distinct form I, II, III, and IV Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships. J. Exp. Bot. 2008, 59, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Badger, M.R.; Bek, E.J. Multiple Rubisco forms in proteobacteria: Their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 2008, 59, 1525–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spreitzer, R.J. Role of the small subunit in ribulose–1,5–bisphosphate carboxylase/oxygenase. Arch. Biochem. Biophys. 2003, 414, 141–149. [Google Scholar] [CrossRef]
- Banda, D.M.; Pereira, J.H.; Liu, A.K.; Orr, D.J.; Hammel, M.; He, C.; Parry, M.A.J.; Carmo-Silva, E.; Adams, P.D.; Banfield, J.F.; et al. Novel bacterial clade reveals origin of form I Rubisco. Nat. Plants 2020, 6, 1158–1166. [Google Scholar] [CrossRef]
- Davidi, D.; Shamshoum, M.; Guo, Z.; Bar-On, Y.M.; Prywes, N.; Oz, A.; Jablonska, J.; Flamholz, A.; Wernick, D.G.; Antonovsky, N.; et al. Highly active rubiscos discovered by systematic interrogation of natural sequence diversity. EMBO J. 2020, 39, e104081. [Google Scholar] [CrossRef]
- Aono, R.; Sato, T.; Imanaka, T.; Atomi, H. A pentose bisphosphate pathway for nucleoside degradation in Archaea. Nat. Chem. Biol. 2015, 11, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Hanson, T.E.; Tabita, F.R. A ribulose–1,5–bisphosphate carboxylase/oxygenase (RubisCO)–like protein from Chlorobium tepidum that is involved with sulfur metabolism and the response to oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 4397–4402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, G.; Carvajal, A.I.; Wilson, R.H.; Cai, Z.; Li, Y. Discovery of a readily heterologously expressed Rubisco from the deep sea with potential for CO2 capture. Bioresour. Bioprocess. 2021, 8, 86. [Google Scholar] [CrossRef]
- Cotton, C.A.R.; Edlich-Muth, C.; Bar-Even, A. Reinforcing carbon fixation: CO2 reduction replacing and supporting carboxylation. Curr. Opin. Biotechnol. 2018, 49, 49–56. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef]
- Furdui, C.; Ragsdale, S.W. The role of pyruvate ferredoxin oxidoreductase in pyruvate synthesis during autotrophic growth by the Wood–Ljungdahl pathway. J. Biol. Chem. 2000, 275, 28494–28499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamby, H.; Li, B.; Shinopoulos, K.E.; Keller, H.R.; Elliott, S.J.; Dukovic, G. Light–driven carbon–carbon bond formation via CO2 reduction catalyzed by complexes of CdS nanorods and a 2–oxoacid oxidoreductase. Proc. Natl. Acad. Sci. USA 2020, 117, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ikeda, T.; Arai, H.; Ishii, M.; Igarashi, Y. Carboxylation reaction catalyzed by 2–oxoglutarate:ferredoxin oxidoreductases from Hydrogenobacter thermophilus. Extremophiles 2010, 14, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Tcherkez, G.G.B.; Farquhar, G.D.; Andrews, T.J. Despite slow catalysis and confused substrate specificity, all ribulose bisphosphate carboxylases may be nearly perfectly optimized. Proc. Natl. Acad. Sci. USA 2006, 103, 7246–7251. [Google Scholar] [CrossRef] [Green Version]
- Bracher, A.; Whitney, S.M.; Hartl, F.U.; Hayer-Hartl, M. Biogenesis and Metabolic Maintenance of Rubisco. Annu. Rev. Plant Biol. 2017, 68, 29–60. [Google Scholar] [CrossRef]
- Li, Z.; Xin, X.; Xiong, B.; Zhao, D.; Zhang, X.; Bi, C. Engineering the Calvin–Benson–Bassham cycle and hydrogen utilization pathway of Ralstonia eutropha for improved autotrophic growth and polyhydroxybutyrate production. Microb. Cell Fact. 2020, 19, 228. [Google Scholar] [CrossRef]
- Yu, H.; Li, X.; Duchoud, F.; Chuang, D.S.; Liao, J.C. Augmenting the Calvin–Benson–Bassham cycle by a synthetic malyl–CoA–glycerate carbon fixation pathway. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Scheffen, M.; Marchal, D.G.; Beneyton, T.; Schuller, S.K.; Klose, M.; Diehl, C.; Lehmann, J.; Pfister, P.; Carrillo, M.; He, H.; et al. A new–to–nature carboxylation module to improve natural and synthetic CO2 fixation. Nat. Catal. 2021, 4, 105–115. [Google Scholar] [CrossRef]
- Flamholz, A.; Shih, P.M. Cell biology of photosynthesis over geologic time. Curr. Biol. 2020, 30, R490–R494. [Google Scholar] [CrossRef]
- Ignatova, L.; Rudenko, N.; Zhurikova, E.; Borisova-Mubarakshina, M.; Ivanov, B. Carbonic Anhydrases in Photosynthesizing Cells of C3 Higher Plants. Metabolites 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Brigham, C.J.; Li, S.; Sinskey, A.J. Ralstonia eutropha H16 as a Platform for the Production of Biofuels, Biodegradable Plastics, and Fine Chemicals from Diverse Carbon Resources. In Biotechnology for Biofuel Production and Optimization; Elsevier: Amsterdam, The Netherlands, 2016; pp. 325–351. [Google Scholar] [CrossRef]
- Yasumoto, K.; Sakata, T.; Yasumoto, J.; Yasumoto-Hirose, M.; Sato, S.-I.; Mori-Yasumoto, K.; Jimbo, M.; Kusumi, T.; Watabe, S. Atmospheric CO2 captured by biogenic polyamines is transferred as a possible substrate to Rubisco for the carboxylation reaction. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Borden, J.S.; Savage, D.F. New discoveries expand possibilities for carboxysome engineering. Curr. Opin. Microbiol. 2021, 61, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Sage, R.F.; Sage, T.L.; Kocacinar, F. Photorespiration and the evolution of C4 photosynthesis. Annu. Rev. Plant. Biol. 2012, 63, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, M.; Arrivault, S.; Giuliani, R.; Danila, F.; Alonso-Cantabrana, H.; Vlad, D.; Ishihara, H.; Feil, R.; Guenther, M.; Borghi, G.L.; et al. Installation of C4 photosynthetic pathway enzymes in rice using a single construct. Plant. Biotechnol. J. 2021, 19, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Arrivault, S.; Coe, R.A.; Karki, S.; Covshoff, S.; Bagunu, E.; Lunn, J.E.; Stitt, M.; Furbank, R.T.; Hibberd, J.M.; et al. A Partial C–4 Photosynthetic Biochemical Pathway in Rice. Front. Plant. Sci. 2020, 11, 564463. [Google Scholar] [CrossRef] [PubMed]
- Flamholz, A.I.; Dugan, E.; Blikstad, C.; Gleizer, S.; Ben-Nissan, R.; Amram, S.; Antonovsky, N.; Ravishankar, S.; Noor, E.; Bar-Even, A.; et al. Functional reconstitution of a bacterial CO2 concentrating mechanism in Escherichia coli. eLife 2020, 9, e59882. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.B.; Dou, Z.; Heinhorst, S.; Shively, J.M.; Cannon, G.C. Halothiobacillus neapolitanus carboxysomes sequester heterologous and chimeric RubisCO species. PLoS ONE 2008, 3, e3570. [Google Scholar] [CrossRef] [Green Version]
- Oltrogge, L.M.; Chaijarasphong, T.; Chen, A.W.; Bolin, E.R.; Marqusee, S.; Savage, D.F. Multivalent interactions between CsoS2 and Rubisco mediate alpha–carboxysome formation. Nat. Struct. Mol. Biol. 2020, 27, 281–287. [Google Scholar] [CrossRef]
- Iniguez, C.; Aguilo-Nicolau, P.; Galmes, J. Improving photosynthesis through the enhancement of Rubisco carboxylation capacity. Biochem. Soc. Trans. 2021, 49, 2007–2019. [Google Scholar] [CrossRef]
- Lombard, J.; Moreira, D. Early evolution of the biotin–dependent carboxylase family. BMC Evol. Biol. 2011, 11, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Jitrapakdee, S.; Wallace, J.C. The biotin enzyme family: Conserved structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 2003, 4, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, X.; Ding, Y.; Gao, W.; Xian, M.; Wang, J.; Zhao, G. Characterization and directed evolution of propionyl–CoA carboxylase and its application in succinate biosynthetic pathway with two CO2 fixation reactions. Metab. Eng. 2020, 62, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, H. Directed Evolution of Propionyl–CoA Carboxylase for Succinate Biosynthesis. Trends Biotechnol. 2021, 39, 330–331. [Google Scholar] [CrossRef]
- Xu, G.; Wu, M.; Jiang, L. Site–saturation engineering of proline 474 in pyruvate carboxylase from Rhizopus oryzae to elevate fumaric acid production in engineered Saccharomyces cerevisiae cells. Biochem. Eng. J. 2017, 117, 36–42. [Google Scholar] [CrossRef]
- Xu, G.; Shi, X.; Gao, Y.; Wang, J.; Cheng, H.; Liu, Y.; Chen, Y.; Li, J.; Xu, X.; Zha, J.; et al. Semi–rational evolution of pyruvate carboxylase from Rhizopus oryzae for elevated fumaric acid synthesis in Saccharomyces cerevisiae. Biochem. Eng. J. 2022, 177, 108238. [Google Scholar] [CrossRef]
- Xia, S.; Veony, E. Gas Phase Carbon Dioxide as an Optimum Substrate for Isocitrate Dehydrogenase Reaction. In Proceedings of the International Conference on Green Energy and Environment Engineering (CGEEE), Seisa Dohto University, Kitahiroshima, Japan, 27–29 August 2019. [Google Scholar]
- Xia, S.; Zhao, X.; Frigo-Vaz, B.; Zheng, W.; Kim, J.; Wang, P. Cascade enzymatic reactions for efficient carbon sequestration. Bioresour. Technol. 2015, 182, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.E.; Can, M.; Wittenborn, E.C.; Hendrickson, R.A.; Ragsdale, S.W.; Drennan, C.L. Crystallographic Characterization of the Carbonylated A–Cluster in Carbon Monoxide Dehydrogenase/Acetyl–CoA Synthase. ACS Catal. 2020, 10, 9741–9746. [Google Scholar] [CrossRef]
- Cohen, S.E.; Brignole, E.J.; Wittenborn, E.C.; Can, M.; Thompson, S.; Ragsdale, S.W.; Drennan, C.L. Negative–Stain Electron Microscopy Reveals Dramatic Structural Rearrangements in Ni–Fe–S–Dependent Carbon Monoxide Dehydrogenase/Acetyl–CoA Synthase. Structure 2021, 29, 43–49.e3. [Google Scholar] [CrossRef]
- Lemaire, O.N.; Wagner, T. Gas channel rerouting in a primordial enzyme: Structural insights of the carbon–monoxide dehydrogenase/acetyl–CoA synthase complex from the acetogen Clostridium autoethanogenum. Biochim. Biophys. Acta–Bioenerg. 2021, 1862, 148330. [Google Scholar] [CrossRef]
- Jain, S.; Katsyv, A.; Basen, M.; Mueller, V. The monofunctional CO dehydrogenase CooS is essential for growth of Thermoanaerobacter kivui on carbon monoxide. Extremophiles 2022, 26, 1–12. [Google Scholar] [CrossRef]
- Mock, J.; Zheng, Y.; Mueller, A.P.; Ly, S.; Tran, L.; Segovia, S.; Nagaraju, S.; Kopke, M.; Durre, P.; Thauer, R.K. Energy Conservation Associated with Ethanol Formation from H2 and CO2 in Clostridium autoethanogenum Involving Electron Bifurcation. J. Bacteriol. 2015, 197, 2965–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopke, M.; Mihalcea, C.; Liew, F.; Tizard, J.H.; Ali, M.S.; Conolly, J.J.; Al-Sinawi, B.; Simpson, S.D. 2,3–butanediol production by acetogenic bacteria, an alternative route to chemical synthesis, using industrial waste gas. Appl. Environ. Microbiol. 2011, 77, 5467–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopke, M.; Gerth, M.L.; Maddock, D.J.; Mueller, A.P.; Liew, F.; Simpson, S.D.; Patrick, W.M. Reconstruction of an acetogenic 2,3–butanediol pathway involving a novel NADPH–dependent primary–secondary alcohol dehydrogenase. Appl. Environ. Microbiol. 2014, 80, 3394–3403. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Song, Y.; Jin, S.; Shin, J.; Bae, J.; Kim, D.R.; Lee, J.-K.; Kim, S.C.; Cho, S.; Cho, B.-K. Adaptive Laboratory Evolution of Eubacterium limosum ATCC 8486 on Carbon Monoxide. Front. Microbiol. 2020, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, B.; Oh, S.; Pathiraja, D.; Kim, J.-Y.; Jung, S.; Jeong, J.; Cha, M.; Park, Z.-Y.; Choi, I.-G.; et al. Metabolism perturbation Caused by the overexpression of carbon monoxide dehydrogenase/Acetyl–CoA synthase gene complex accelerated gas to acetate conversion rate of Eubacterium limosum KIST612. Bioresour. Technol. 2021, 341, 125879. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.E.; Jang, Y.-S. Metabolic engineering of microorganisms for the production of ethanol and butanol from oxides of carbon. Appl. Microbiol. Biotechnol. 2019, 103, 8283–8292. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Miller, A.-F.; Jones, A.K.; King, P.W.; Adams, M.W.W. Electron bifurcation. Curr. Opin. Chem. Biol. 2016, 31, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Liu, G.; Gong, F.; Cai, Z.; Li, Y. The reductive carboxylation activity of heterotetrameric pyruvate synthases from hyperthermophilic archaea. Biochem. Biophys. Res. Commun. 2021, 572, 151–156. [Google Scholar] [CrossRef]
- Li, B.; Elliott, S.J. The Catalytic Bias of 2–Oxoacid:ferredoxin Oxidoreductase in CO2: Evolution and reduction through a ferredoxin–mediated electrocatalytic assay. Electrochim. Acta 2016, 199, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Maruyama, A.; Arakawa, T.; Fushinobu, S.; Wakagi, T. Crystal structures of archaeal 2–oxoacid: Ferredoxin oxidoreductases from Sulfolobus tokodaii. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.-T.; Li, B.; Drennan, C.L.; Elliott, S.J. A Reverse TCA Cycle 2–Oxoacid: Ferredoxin Oxidoreductase that Makes C–C Bonds from CO2. Joule 2019, 3, 595–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, A.; Pozzi, R.; Diesch, S.; Haedicke, O.; Grammel, H. New light on ancient enzymes—In vitro CO2 Fixation by Pyruvate Synthase of Desulfovibrio africanus and Sulfolobus acidocaldarius. Febs. J. 2019, 286, 4494–4508. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A.; Noor, E.; Lewis, N.E.; Milo, R. Design and analysis of synthetic carbon fixation pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 8889–8894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwander, T.; Borzyskowski, L.S.v.; Burgener, S.; Cortina, N.S.; Erb, T.J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, S.; Diehl, C.; Cortina, N.S.; Bamberger, J.; Paczia, N.; Erb, T.J. A Modular In Vitro Platform for the Production of Terpenes and Polyketides from CO2. Angew. Chem. Int. Ed. Engl. 2021, 60, 16420–16425. [Google Scholar] [CrossRef]
- Li, Y.; Wen, L.; Tan, T.; Lv, Y. Sequential Co–immobilization of Enzymes in Metal–Organic Frameworks for Efficient Biocatalytic Conversion of Adsorbed CO2 to Formate. Front. Bioeng. Biotechnol. 2019, 7, 394. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Ao, S.; Deng, H.; Wang, M.; Qin, C.; Zhang, J.; Jia, Y.; Ye, P.; Ni, H. Ordered Coimmobilization of a Multienzyme Cascade System with a Metal Organic Framework in a Membrane: Reduction of CO2 to Methanol. ACS Appl. Mater. Interfaces 2019, 11, 33581–33588. [Google Scholar] [CrossRef]
- Satagopan, S.; Sun, Y.; Parquette, J.R.; Tabita, F.R. Synthetic CO2–fixation enzyme cascades immobilized on self–assembled nanostructures that enhance CO2/O2 selectivity of RubisCO. Biotechnol. Biofuels 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Bathellier, C.; Tcherkez, G.; Lorimer, G.H.; Farquhar, G.D. Rubisco is not really so bad. Plant Cell Environ. 2018, 41, 705–716. [Google Scholar] [CrossRef]
- Gassler, T.; Sauer, M.; Gasser, B.; Egermeier, M.; Troyer, C.; Causon, T.; Hann, S.; Mattanovich, D.; Steiger, M.G. The industrial yeast Pichia pastoris is converted from a heterotroph into an autotroph capable of growth on CO2. Nat. Biotechnol. 2020, 38, 210–216. [Google Scholar] [CrossRef]
- Gleizer, S.; Ben-Nissan, R.; Bar-On, Y.M.; Antonovsky, N.; Noor, E.; Zohar, Y.; Jona, G.; Krieger, E.; Shamshoum, M.; Bar-Even, A.; et al. Conversion of Escherichia coli to Generate All Biomass Carbon from CO2. Cell 2019, 179, 1255–1263.e1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leakey, A.D.B.; Ainsworth, E.A.; Bernacchi, C.J.; Rogers, A.; Long, S.P.; Ort, D.R. Elevated CO2 effects on plant carbon, nitrogen, and water relations: Six important lessons from FACE. J. Exp. Bot. 2009, 60, 2859–2876. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Losada, L.; Adam, D.; Paczia, N.; Buesen, D.; Steffler, F.; Sieber, V.; Erb, T.J.; Richter, M.; Plumere, N. Bioelectrocatalytic Cofactor Regeneration Coupled to CO2 Fixation in a Redox–Active Hydrogel for Stereoselective C–C Bond Formation. Angew. Chem. Int. Ed. Engl. 2021, 60, 21056–21061. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Beneyton, T.; Schwander, T.; Diehl, C.; Girault, M.; McLean, R.; Chotel, T.; Claus, P.; Cortina, N.S.; Baret, J.-C.; et al. Light–powered CO2 fixation in a chloroplast mimic with natural and synthetic parts. Science 2020, 368, 649–654. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Enzymes (EC Number) | Carbon Species | Oxygen Sensitivity | Km,C for Carbon Species (mM) a | kcat,c for Carbon Fixation (s−1) a | kcat,c/Km,C (×105 M−1 s−1) a | Specific Activity (μmol/min/mg) a | Co-Factor Requirements | Referrence |
|---|---|---|---|---|---|---|---|---|
| Rubisco (4.1.1.39) | CO2 | No but with side reaction | 0.05 (0.002–2.35) | 4.15 (0.2–22.2) | 1.22 (0.12–4.11) | 2.23 (0.023–9.9) | / | [55,58,59,60] |
| PFOR (1.2.7.1) | CO2 | Yes | 2 b | 3.2 b | 0.016 b | 1.6 b | Ferredoxin(red) | [61] |
| OGOR (1.2.7.3) | CO2 | Yes | 3 b | 3 (1.7–4.3) | / | 1.5 (0.45–2.5) | Ferredoxin(red) | [62,63] |
| CO dehydrogenase/acetyl–CoA synthase (2.3.1.169) | CO, CO2 | Yes | / | / | / | / | Ferredoxin(red) | |
| isocitrate dehydrogenase (1.1.1.41–42) | CO2 | No | 4.3 (0.02–13.82) | 33.4 (1.5–96.6) | 0.38 (0.003–0.69) | 18.98 (2–38) | NAD(P)H | [59,60] |
| 2–oxoglutarate carboxylase (6.4.1.7) | HCO3– | No | / | 30.6b | / | 14.6 b | ATP | [36] |
| PEP carboxylase (4.1.1.31) | HCO3– | No | 0.99 (0.02–7.6) | 43.8 (6.1–150) | 10.7 (1.7–15) | 32.7 (1.5–150) | / | [59,60] |
| pyruvate carboxylase (6.4.1.1) | HCO3– | No | 3.96 (0.22–29.9) | 55.7 (11.6–89.8) | 0.68 (0.25–1.8) | 22.0 (1.03–47.8) | ATP | [59,60] |
| acetyl–CoA/propionyl–CoA carboxylase (6.4.1.2/6.4.1.3) | HCO3– | No | 2.69 (0.3–12.8) | 18.6 (2.9–42.7) | 0.17 (0.059–0.37) | 12.5 (0.3–52.4) | ATP | [59,60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, X.; Han, Y. Promotion of Carbon Dioxide Biofixation through Metabolic and Enzyme Engineering. Catalysts 2022, 12, 399. https://doi.org/10.3390/catal12040399
Pu X, Han Y. Promotion of Carbon Dioxide Biofixation through Metabolic and Enzyme Engineering. Catalysts. 2022; 12(4):399. https://doi.org/10.3390/catal12040399
Chicago/Turabian StylePu, Xin, and Yejun Han. 2022. "Promotion of Carbon Dioxide Biofixation through Metabolic and Enzyme Engineering" Catalysts 12, no. 4: 399. https://doi.org/10.3390/catal12040399
APA StylePu, X., & Han, Y. (2022). Promotion of Carbon Dioxide Biofixation through Metabolic and Enzyme Engineering. Catalysts, 12(4), 399. https://doi.org/10.3390/catal12040399