
Genetic and Biochemical Characterizations of aLhr1 Helicase in the Thermophilic Crenarchaeon Sulfolobus acidocaldarius

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Prediction of Saci_0814 as a Putative Helicase
2.2. Construction of the alhr1-Deleted Strain of S. acidocaldarius
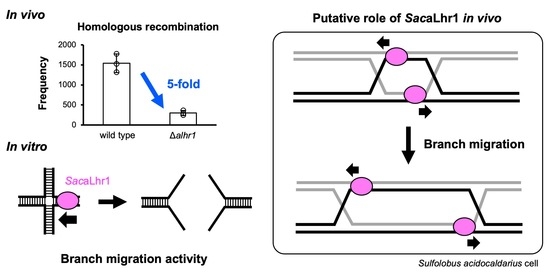
2.3. Estimation of HR Frequencies
2.4. Purification of the Recombinant Protein Encoded by alhr1
2.5. ss/dsDNA-Dependent ATPase Activity of SacaLhr1
2.6. SacaLhr1 Is a 3′-to-5′ DNA Helicase
2.7. ATP- and Mg2+-Dependent Helicase Activity of SacaLhr1
2.8. Unwinding Activity SacaLhr1 on Branched DNA
2.9. Binding Properties of SacaLhr1 to DNA with or without a Branch Point
2.10. Branch Migration Activity of SacaLhr1 against a Synthetic HJ
3. Discussion
4. Materials and Methods
4.1. Strains and Growth Conditions
4.2. Construction of Gene-Deleted Strain
4.3. Estimation of HR Frequencies
4.4. Cloning of Gene Encoding SacaLhr1 from S. acidocaldarius
4.5. Preparation of the Recombinant SacaLhr1 Protein
4.6. ATPase Assay
4.7. Preparation of DNA Substrates
4.8. DNA Unwinding Assay
4.9. Electrophoretic Mobility Shift Assay (EMSA)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Stetter, O.K. Hyperthermophiles in the history of life. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1837–1843. [Google Scholar] [CrossRef]
- Grogan, D.W. The question of DNA repair in hyperthermophilic archaea. Trends Microbiol. 2000, 8, 180–185. [Google Scholar] [CrossRef]
- Ishino, Y.; Nishino, T.; Morikawa, K. Mechanisms of Maintaining Genetic Stability by Homologous Recombination. Chem. Rev. 2006, 106, 324–339. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Homologous recombination in the archaea: The means justify the ends. Biochem. Soc. Trans. 2011, 39, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Grasso, S.; Tell, G. Base excision repair in Archaea: Back to the future in DNA repair. DNA Repair 2014, 21, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Grogan, D.W. Understanding DNA repair in hyperthermophilic archaea: Persistent gaps and other reactions to focus on the fork. Archaea 2015, 2015, 942605. [Google Scholar] [CrossRef]
- Ishino, Y.; Narumi, I. DNA repair in hyperthermophilic and hyperradioresistant microorganisms. Curr. Opin. Microbiol. 2015, 25, 103–112. [Google Scholar] [CrossRef]
- White, M.F.; Allers, T. DNA repair in the archaea—an emerging picture. FEMS Microbiol. Rev. 2018, 42, 514–526. [Google Scholar] [CrossRef]
- Craig, J.; Marshall, C.J.; Santangelo, T.J. Archaeal DNA Repair Mechanisms. Biomolecules 2020, 10, 1472. [Google Scholar]
- Fujikane, R.; Komori, K.; Shinagawa, H.; Ishino, Y. Identification of a Novel Helicase Activity Unwinding Branched DNAs from the Hyperthermophilic Archaeon, Pyrococcus furiosus. J. Biol. Chem. 2005, 280, 12351–12358. [Google Scholar] [CrossRef]
- Guy, C.P.; Bolt, E.L. Archaeal Hel308 helicase targets replication forks in vivo and in vitro and unwinds lagging strands. Nucleic Acids Res. 2005, 33, 3678–3690. [Google Scholar] [CrossRef]
- Fujikane, R.; Shinagawa, H.; Ishino, Y. The archaeal Hjm helicase has recQ-like functions, and may be involved in repair of stalled replication fork. Genes Cells 2006, 11, 99–110. [Google Scholar] [CrossRef]
- Tsuboi, K.; Sakai, H.D.; Nur, N.; Stedman, K.M.; Kurosawa, N.; Suwanto, A. Sulfurisphaera javensis sp. nov., a hyperther-mophilic and acidophilic archaeon isolated from Indonesian hot spring, and reclassification of Sulfolobus tokodaii Suzuki et al. 2002 as Sulfurisphaera tokodaii comb. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 1907–1913. [Google Scholar] [CrossRef]
- Li, Z.; Lu, S.; Hou, G.; Ma, X.; Sheng, D.; Ni, J.; Shen, Y. Hjm/Hel308A DNA helicase from Sulfolobus tokodaii promotes repli-cation fork regression and interacts with Hjc endonuclease in vitro. J. Bacteriol. 2008, 190, 3006–3017. [Google Scholar] [CrossRef]
- Zhai, B.; Duprez, K.; Doukov, T.I.; Li, H.; Huang, M.; Shang, G.; Ni, J.; Gu, L.; Shen, Y.; Fan, L. Structure and function of a novel ATPase that interacts with holloday junction resolvase Hjc and promotes branch migration. J. Mol. Biol. 2017, 429, 1009–1029. [Google Scholar] [CrossRef][Green Version]
- Yamada, K.; Miyata, T.; Tsuchiya, D.; Oyama, T.; Fujiwara, Y.; Ohnishi, T.; Iwasaki, H.; Shinagawa, H.; Ariyoshi, M.; Ma-yanagi, K.; et al. Crystal structure of the RuvA-RuvB complex: A structural basis for the Holliday junction migrating motor machinery. Mol. Cell 2002, 10, 671–681. [Google Scholar] [CrossRef]
- Zhai, B.; DuPrez, K.; Han, X.; Yuan, Z.; Ahmad, S.; Xu, C.; Gu, L.; Ni, J.; Fan, L.; Shen, Y. The archaeal ATPase PINA interacts with the helicase Hjm via its carboxyl terminal KH domain remodeling and processing replication fork and Holliday junction. Nucleic Acids Res. 2018, 46, 6627–6641. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Koonin, E.V.; Rudd, E.K.; Deutscher, M.P. The gene for the longest known Escherichia coli protein is a member of helicase superfamily II. J. Bacteriol. 1995, 177, 5393–5400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ordonez, H.; Shuman, S. Mycobacterium smegmatis Lhr is a DNA-dependent ATPase and a 3′-to-5′ DNA Translocase and Helicase That Prefers to Unwind 3′-Tailed RNA:DNA Hybrids. J. Biol. Chem. 2013, 288, 14125–14134. [Google Scholar] [CrossRef]
- De Felice, M.; Aria, V.; Esposito, L.; De Falco, M.; Pucci, B.; Rossi, M.; Pisani, F.M. A novel DNA helicase with strand-annealing activity from the crenarchaeon Sulfolobus solfataricus. Biochem. J. 2007, 408, 87–95. [Google Scholar] [CrossRef] [PubMed]
- van Wolferen, M.; Ma, X.; Albers, S.-V. DNA Processing Proteins Involved in the UV-Induced Stress Response of Sulfolobales. J. Bacteriol. 2015, 197, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.J.; Kramm, K.; Cooper, C.D.O.; Grohmann, D.; Bolt, E.L. Mechanistic insights into Lhr helicase function in DNA repair. Biochem. J. 2020, 477, 2935–2947. [Google Scholar] [CrossRef]
- Chamieh, H.; Ibrahim, H.; Kozah, J. Genome-wide identification of SF1 and SF2 helicases from archaea. Gene 2016, 576, 214–228. [Google Scholar] [CrossRef]
- Hajj, M.; Langendijk-Genevaux, P.; Batista, M.; Quentin, Y.; Laurent, S.; Abdel Razzak, Z.; Flament, D.; Chamieh, H.; Fichant, G.; Clouet-d’Orval, B.; et al. Phylogenetic Diversity of Lhr Proteins and Biochemical Activities of the Thermococcales aLhr2 DNA/RNA Helicase. Biomolecules 2021, 11, 950. [Google Scholar] [CrossRef]
- Murzin, A.G. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef]
- Chédin, F.; Seitz, E.M.; Kowalczykowski, S.C. Novel homologs of replication protein A in Archaea: Implications of the evolution of ssDNA-binding proteins. Trends Biochem. Sci. 1998, 23, 273–277. [Google Scholar] [CrossRef]
- Kerr, I.D.; Wadsworth, R.I.; Cubeddu, L.; Blankenfeldt, W.; Naismith, J.H.; White, M. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J. 2003, 22, 2561–2570. [Google Scholar] [CrossRef]
- Paytubi, S.; McMahon, S.A.; Graham, S.; Liu, H.; Botting, C.H.; Makarova, K.S.; Koonin, E.V.; Naismith, J.H.; White, M.F. Displacement of the canonical single-stranded DNA-binding protein in the Thermoproteales. Proc. Natl. Acad. Sci. USA 2012, 109, E398–E405. [Google Scholar] [CrossRef]
- Meyer, R.R.; Glassberg, J.; Kornberg, A. An Escherichia coli mutant defective in single-strand binding protein is defective in DNA replication. Proc. Natl. Acad. Sci. USA 1979, 76, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, J.; Meyer, R.R.; Kornberg, A. Mutant single-strand binding protein of Escherichia coli: Genetic and physiological characterization. J. Bacteriol. 1979, 140, 14–19. [Google Scholar] [CrossRef]
- Longhese, M.P.; Plevani, P.; Lucchini, G. Replication factor A is required in vivo for DNA replication, repair, and recombination. Mol. Cell. Biol. 1994, 14, 7884–7890. [Google Scholar]
- Muniyappa, K.; Shaner, S.L.; Tsang, S.S.; Radding, C.M. Mechanism of the concerted action of recA protein and he-lix-destabilizing proteins in homologous recombination. Proc. Natl. Acad. Sci. USA 1984, 81, 2757–2761. [Google Scholar] [CrossRef]
- Sugiyama, T.; Zaitseva, E.M.; Kowalczykowski, S.C. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J. Biol. Chem. 1997, 272, 7940–7945. [Google Scholar] [CrossRef]
- Komori, K.; Ishino, Y. Replication Protein A in Pyrococcus furiosus Is Involved in Homologous DNA Recombination. J. Biol. Chem. 2001, 276, 25654–25660. [Google Scholar] [CrossRef]
- Haseltine, C.A.; Kowalczykowski, S.C. A distinctive single-stranded DNA-binding protein from the Archaeon Sulfolobus solfataricus. Mol. Microbiol. 2002, 43, 1505–1515. [Google Scholar] [CrossRef]
- Rolfsmeier, M.L.; Haseltine, C.A. The Single-Stranded DNA Binding Protein of Sulfolobus solfataricus Acts in the Presynaptic Step of Homologous Recombination. J. Mol. Biol. 2010, 397, 31–45. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, S.; Zhu, S.; Sheng, D.; Ni, J.; Shen, Y. Physical and functional interaction between archaeal single-stranded DNA-binding protein and the 5′–3′ nuclease NurA. Biochem. Biophys. Res. Commun. 2008, 367, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Cubeddu, L.; White, M.F. DNA Damage Detection by an Archaeal Single-stranded DNA-binding Protein. J. Mol. Biol. 2005, 353, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kurosawa, N. Disruption of the gene encoding restriction endonuclease SuaI and development of a host–vector system for the thermoacidophilic archaeon Sulfolobus acidocaldarius. Extremophiles 2016, 20, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kurosawa, N. Development of the multiple gene knockout system with one-step PCR in thermophilic crenarchaeon Sulfolobus acidocaldarius. Archaea 2017, 2017, 7459310. [Google Scholar] [CrossRef]
- Ogino, H.; Ishino, S.; Haugland, G.T.; Birkeland, N.K.; Kohda, D.; Ishino, Y. Activation of the MCM helicase from the thermophilic archaeon, Thermoplasma acidophilum by interactions with GINS and Cdc6-2. Extremophiles 2014, 18, 915–924. [Google Scholar] [CrossRef]
- Muris, D.F.R.; Vreeken, K.; Schmidt, H.; Ostermann, K.; Clever, B.; Lohman, P.H.M.; Pastink, A. Homologous recombination in the fission yeast Schizosaccharomyces pombe: Different requirements for the rhp51 +, rhp54 + and rad22 + genes. Curr. Genet. 1997, 31, 248–254. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Morishita, T.; Iwasaki, H.; Toh, H.; Shinagawa, H. A Recombination Repair Gene of Schizosaccharomyces pombe, rhp57, Is a Functional Homolog of the Saccharomyces cerevisiae RAD57 Gene and Is Phylogenetically Related to the Human XRCC3 Gene. Genetics 2000, 154, 1451–1461. [Google Scholar] [CrossRef]
- Woods, W.G.; Dyall-Smith, M.L. Construction and analysis of a recombination-deficient (radA) mutant of Haloferax volcan-ii. Mol. Microbiol. 1997, 23, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Özsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- De Falco, M.; Catalano, F.; Rossi, M.; Ciaramella, M.; De Felice, M. NurA Is Endowed with Endo- and Exonuclease Activities that Are Modulated by HerA: New Insight into Their Role in DNA-End Processing. PLoS ONE 2015, 10, e0142345. [Google Scholar] [CrossRef] [PubMed]
- De Falco, M.; Massa, F.; Rossi, M.; De Felice, M. The Sulfolobus solfataricus RecQ-like DNA helicase Hel112 inhibits the Nu-rA/HerA complex exonuclease activity. Extremophiles 2018, 22, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Han, Y.-W.; Shibata, T.; Kubota, Y.; Ishino, Y.; Iwasaki, H.; Shinagawa, H. Role of the Escherichia coli RecQ DNA helicase in SOS signaling and genome stabilization at stalled replication forks. Genes Dev. 2004, 18, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, A.; Shuman, S. Characterization of Lhr-Core DNA helicase and manganese- dependent DNA nuclease components of a bacterial gene cluster encoding nucleic acid repair enzymes. J. Biol. Chem. 2018, 293, 17491–17504. [Google Scholar] [CrossRef]
- Warren, G.M.; Wang, J.; Patel, D.J.; Shuman, S. Oligomeric quaternary structure of Escherichia coli and Mycobacterium smegmatis Lhr helicases is nucleated by a novel C-terminal domain composed of five winged-helix modules. Nucleic Acids Res. 2021, 49, 3876–3887. [Google Scholar] [CrossRef]
- Song, X.; Huang, Q.; Ni, J.; Yu, Y.; Shen, Y. Knockout and functional analysis of two DExD/H-box family helicase genes in Sulfolobus islandicus REY15A. Extremophiles 2016, 20, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, A.; Ordonez, H.; Jacewicz, A.; Ferrao, R.; Shuman, S. Structure of mycobacterial 3′-to-5′ RNA:DNA helicase Lhr bound to a ssDNA tracking strand highlights distinctive features of a novel family of bacterial helicases. Nucleic Acids Res. 2018, 46, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Büttner, K.; Nehring, S.; Hopfner, K.-P. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007, 14, 647–652. [Google Scholar] [CrossRef]
- Schlegel, S.; Genevaux, P.; de Gier, J.-W. De-convoluting the Genetic Adaptations of E. coli C41(DE3) in Real Time Reveals How Alleviating Protein Production Stress Improves Yields. Cell Rep. 2015, 10, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Dumon-Seignovert, L.; Cariot, G.; Vuillard, L. The toxicity of recombinant proteins in Escherichia coli: A comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr. Purif. 2004, 37, 203–206. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains or DNAs | Relevant Characteristic (s) | Source or Reference |
|---|---|---|
| Strains | ||
| S. acidocaldarius | ||
| DP-1 | SK-1 [40] with Δphr (ΔpyrE ΔsuaI Δphr) | [41] |
| DP-17 | DP-1 with Δalhr1 (ΔpyrE ΔsuaI Δphr Δalhr1) | This study |
| Plasmids | ||
| placSpyrE | Plasmid DNA carrying 0.8 kb of 5′ and 3′ flanking regions of suaI locus at both ends of pyrE-lacS dual marker | [41] |
| pSAV2 | Sulfolobus-E. coli shuttle vector, based on pBluescript II KS (-) and pRN1, with the SsopyrEF maker | [40] |
| PCR products | ||
| MONSTER-alhr1 | Linear DNA carrying a 39 bp region of alhr1 as the Tg-arm, and the 39 bp 3′ and 30 bp 5′ flanking regions of alhr1 at both ends of pyrE-lacS dual marker | This study |
| pyrElacS800 | Linear DNA carrying 0.8 kb of 5′ and 3′ flanking regions of suaI locus at both ends of pyrE-lacS dual marker | [41] |
| Name | Sequence (5′-3′) or Combination for Annealing |
|---|---|
| Labeled oligonucleotides a | |
| Cy5-HJ4-59 | Cy5- GACCTAGGAACCACCAGAAACACGCCACAGCCAGGAAGCCGATTGCGAGGCCGTCCTAC |
| Cy5-HJ3-54 | Cy5-TCACTCCGCATCTGCCGATTCTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC |
| HJ3-54mer-Cy5 | TCACTCCGCATCTGCCGATTCTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC-Cy5 |
| DyLight 5-HJ2S | DyLight 5-CGTTGACATCTCGCGTGCTCGGTCAATCGGCAGATGCGGAGTGAAGTTCC |
| Oligonucleotides | |
| HJ4-34 | GACCTAGGAACCACCAGAAACACGCCACAGCCAG |
| HJ4-34-R | CTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC |
| HJ4 | GACCTAGGAACCACCAGAAACACGCCACAGCCAGGAAGCCGATTGCGAGGCCGTCCTACCATCCTGCAGG |
| HJ1S | GGTAGGACGGCCTCGCAATCGGCTTCGACCGAGCACGCGAGATGTCAACG |
| HJ3S | GGAACTTCACTCCGCATCTGCCGATTCTGGCTGTGGCGTGTTTCTGGTGG |
| HJ4S | CCACCAGAAACACGCCACAGCCAGGAAGCCGATTGCGAGGCCGTCCTACC |
| HJ2S-RC | GGAACTTCACTCCGCATCTGCCGATTGACCGAGCACGCGAGATGTCAACG |
| HJS1-2-trap | GGAACTTCACTCCGCATCTGCCGATTGAAGCCGATTGCGAGGCCGTCCTACC |
| HJS2-3-trap | CCACCAGAAACACGCCACAGCCAGGACCGAGCACGCGAGATGTCAACG |
| DNA substrates b | |
| 3′-overhang | Cy5-HJ4-59/HJ4-34-R |
| 5′-overhang | HJ3-54mer-Cy5/HJ4-34 |
| Splayed DNA1 | Cy5-HJ3-54/HJ4 |
| ssDNA | DyLight 5-HJ2S |
| dsDNA | DyLight 5-HJ2S/HJ2S-RC |
| Splayed DNA2 | DyLight 5-HJ2S/HJ3S |
| Y-junction1 | HJ1S/DyLight 5-HJ2S/HJS1-2-trap |
| Y-junction2 | DyLight 5-HJ2S/HJ3S/HJS2-3-trap |
| Three-strand DNA1 | DyLight 5-HJ2S/HJ3S/HJ4S |
| Three-strand DNA2 | HJ1S/DyLight 5-HJ2S/HJ4S |
| Three-strand DNA3 | HJ1S/DyLight 5-HJ2S/HJ3S |
| HJS | HJ1S/DyLight 5-HJ2S/HJ3S/HJ4S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, S.; Kurosawa, N.; Yamagami, T.; Matsumoto, S.; Numata, T.; Ishino, S.; Ishino, Y. Genetic and Biochemical Characterizations of aLhr1 Helicase in the Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Catalysts 2022, 12, 34. https://doi.org/10.3390/catal12010034
Suzuki S, Kurosawa N, Yamagami T, Matsumoto S, Numata T, Ishino S, Ishino Y. Genetic and Biochemical Characterizations of aLhr1 Helicase in the Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Catalysts. 2022; 12(1):34. https://doi.org/10.3390/catal12010034
Chicago/Turabian StyleSuzuki, Shoji, Norio Kurosawa, Takeshi Yamagami, Shunsuke Matsumoto, Tomoyuki Numata, Sonoko Ishino, and Yoshizumi Ishino. 2022. "Genetic and Biochemical Characterizations of aLhr1 Helicase in the Thermophilic Crenarchaeon Sulfolobus acidocaldarius" Catalysts 12, no. 1: 34. https://doi.org/10.3390/catal12010034
APA StyleSuzuki, S., Kurosawa, N., Yamagami, T., Matsumoto, S., Numata, T., Ishino, S., & Ishino, Y. (2022). Genetic and Biochemical Characterizations of aLhr1 Helicase in the Thermophilic Crenarchaeon Sulfolobus acidocaldarius. Catalysts, 12(1), 34. https://doi.org/10.3390/catal12010034