
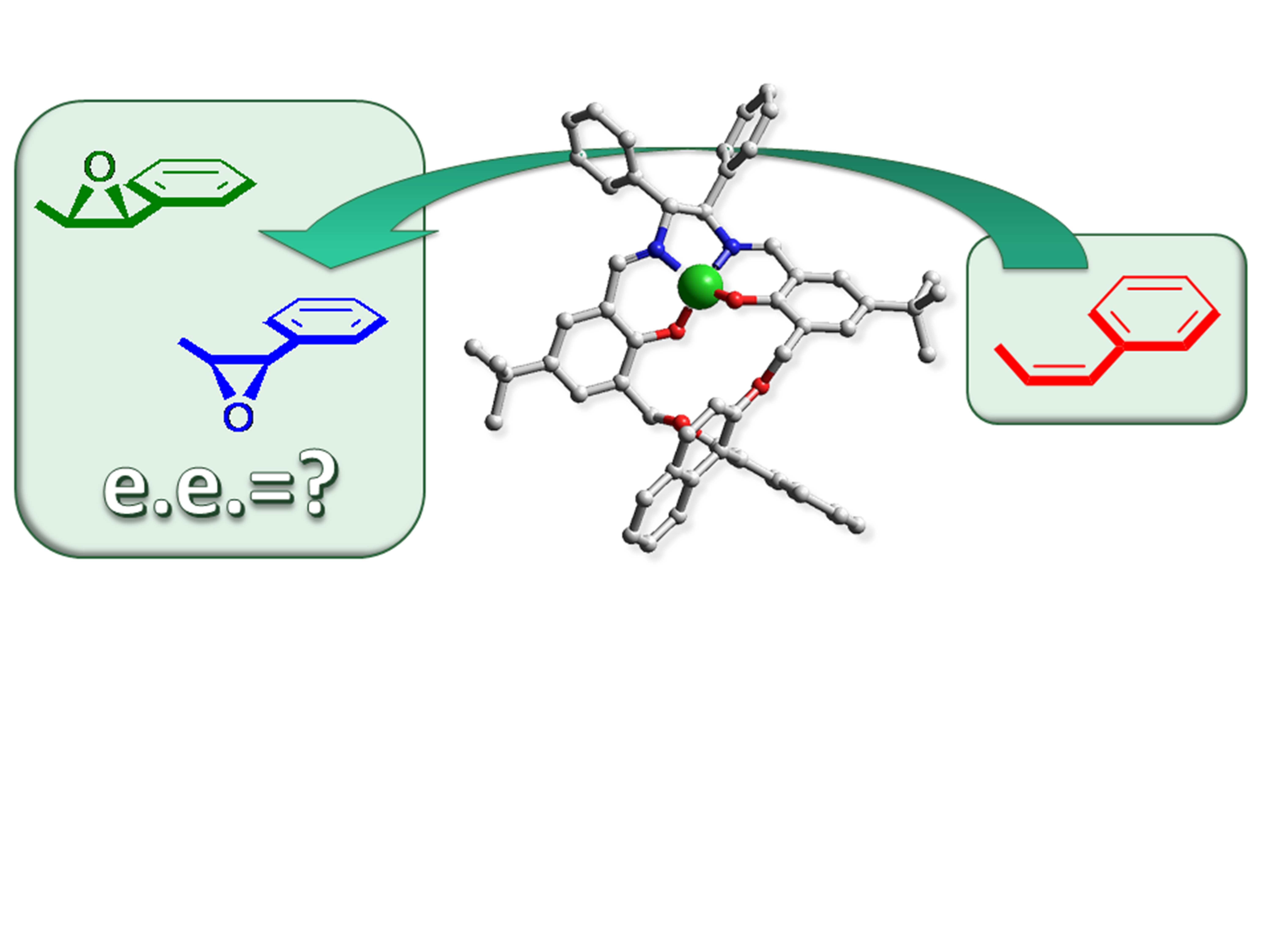
Alkene Epoxidations Mediated by Mn-Salen Macrocyclic Catalysts



 ,
, 

Abstract

1. Introduction
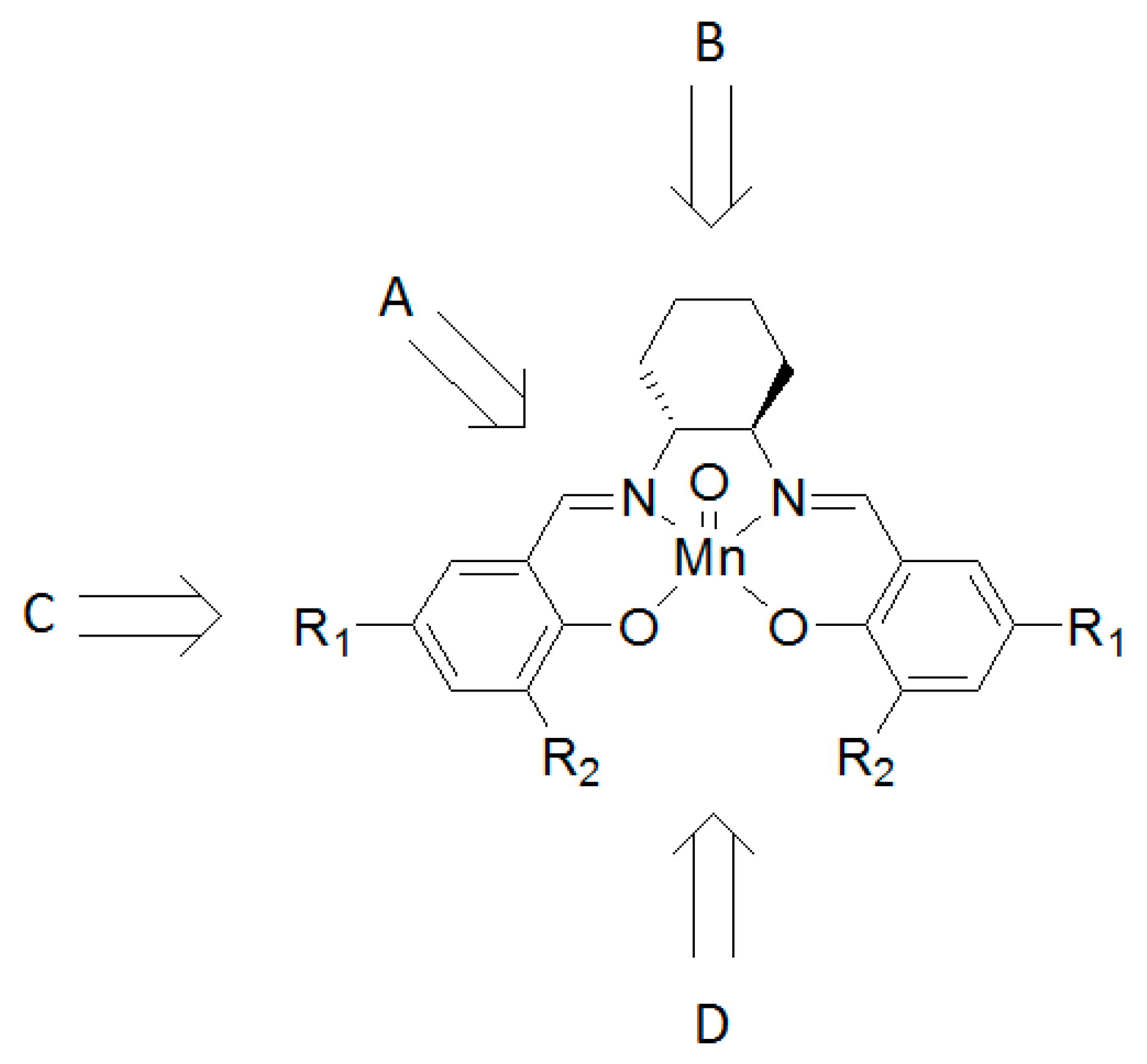
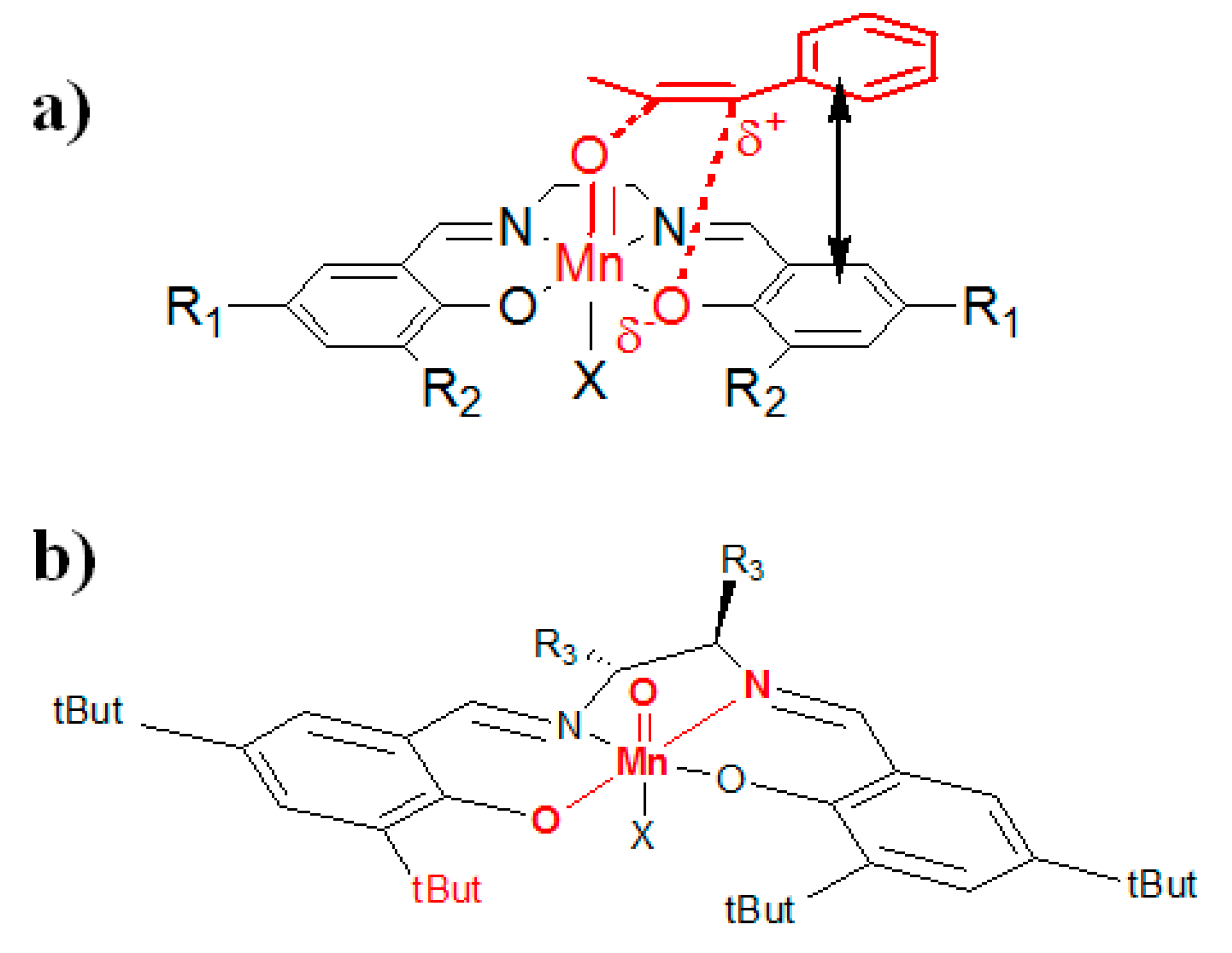
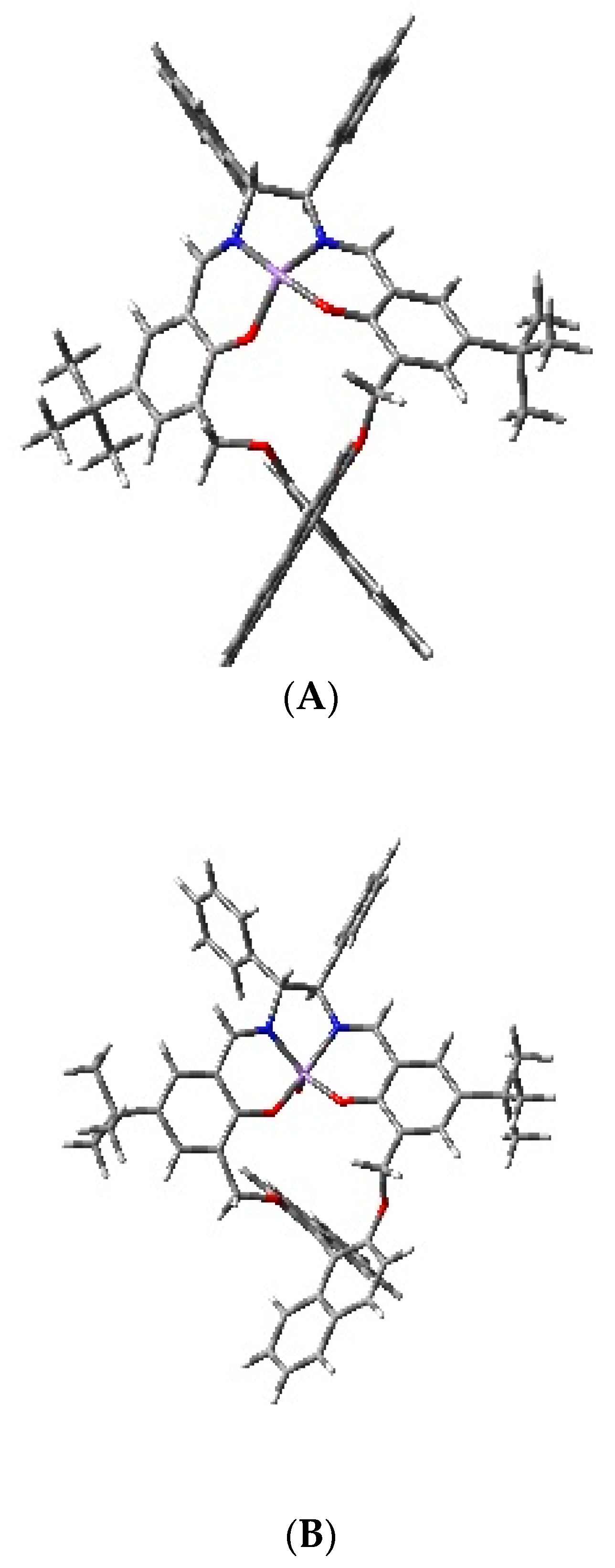
2. Results and Discussion
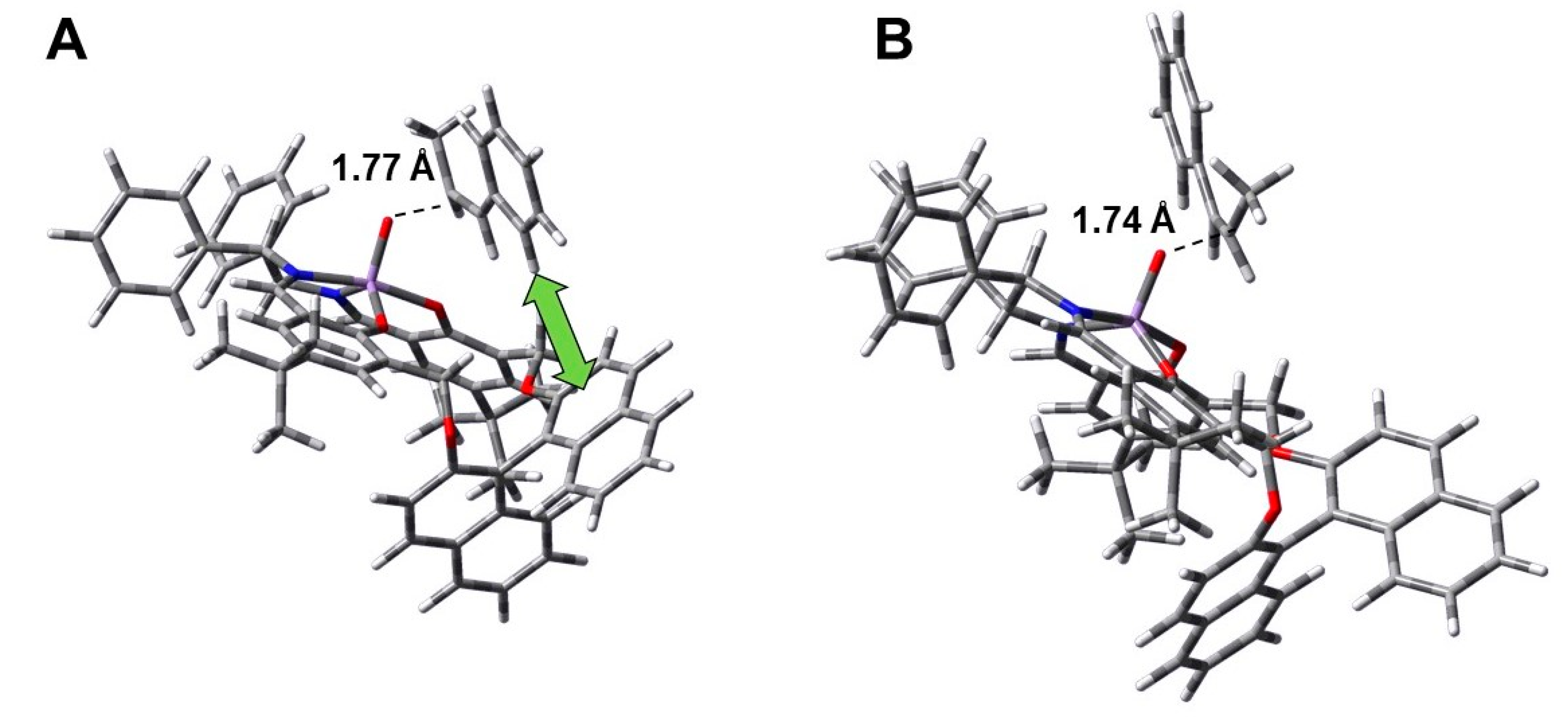
Theoretical Calculations
3. Materials and Methods
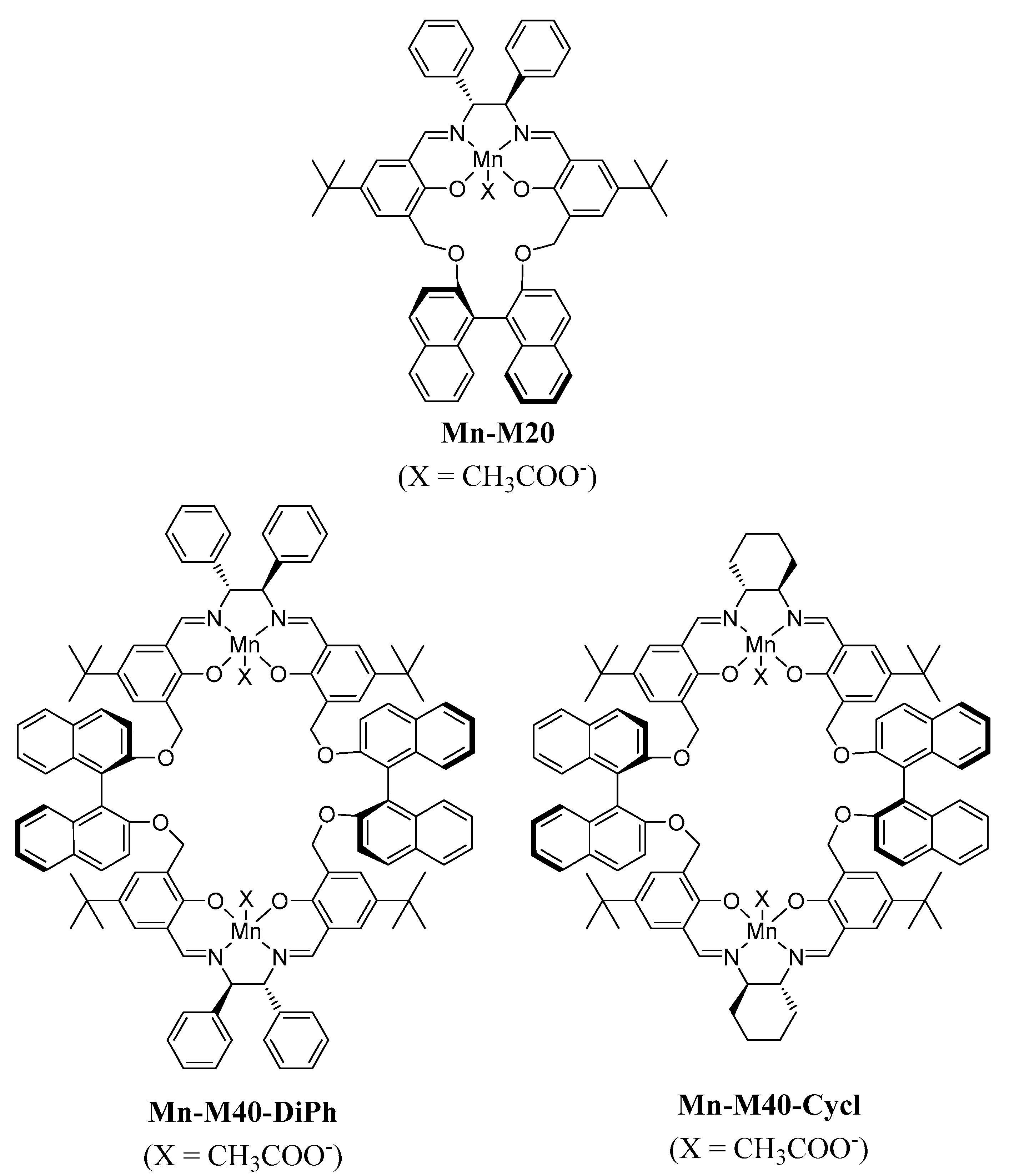
3.1. Synthesis of Mn Macrocycle Catalysts
3.2. General Procedure for Epoxidation Reactions
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mc Garrigle, E.M.; Gilheany, D.G. Chromium- and Manganese-salen Promoted Epoxidation of Alkenes. Chem. Rev. 2005, 105, 1563–1602. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.N.; Zhang, W.; Guler, M.L. Electronic tuning of asymmetric catalysts. J. Am. Chem. Soc. 1991, 113, 6703–6704. [Google Scholar] [CrossRef]
- Katsuki, T. Catalytic asymmetric oxidations using optically active (salen) manganese(III) complexes as catalysts. Coord. Chem. Rev. 1995, 140, 189–214. [Google Scholar] [CrossRef]
- Houk, K.N.; De Mello, N.C.; Condroski, K.; Fennen, J.; Kasuga, T. Origins of enantioselection in Jacobsen epoxidations. In Proceedings of the Electronic Conference on Heterocyclic Chemistry (ECHET96), London, UK, 24 June–22 July 1996. [Google Scholar]
- La Paglia Fragola, V.; Lupo, F.; Pappalardo, A.; Trusso Sfrazzetto, G.; Toscano, R.M.; Ballistreri, F.P.; Tomaselli, G.A.; Gulino, A. A surface-confined O=MnV(salen) oxene catalyst and high turnover values in asymmetric epoxidation of unfunctionalized olefins. J. Mat. Chem. 2012, 22, 20561. [Google Scholar] [CrossRef]
- Hosoya, N.; Hatayama, A.; Yanai, K.; Fujii, H.; Irie, R.; Katsuki, T. Asymmetric epoxidation with optically active (salen) manganese(III) complexes: From which direction does the olefin approach the metal- oxo bond? Synlett 1993, 9, 641–645. [Google Scholar] [CrossRef]
- Hamada, T.; Irie, R.; Katsuki, T. How does chiral oxo(salen) manganese(V) complex discriminate the enantioface of simple olefins? Scope and limitation of salen- catalyzed epoxidation. Synlett 1994, 7, 479–481. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1, 2- diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Pospisil, P.J.; Carsten, D.H.; Jacobsen, E.N. X- ray structural studies of highly enantioselective Mn(salen) epoxidation catalysts. Chem. A Eur. J. 1996, 2, 974–980. [Google Scholar] [CrossRef]
- Hashihayata, T.; Punniyamurthy, T.; Irie, R.; Katsuki, T.; Akita, M.; Moro-oka, Y. Conformational analysis of cationic (R, S)- and (R, R)-(salen) manganese complexes possessing axial chirality as a chiral element based on x-ray crystallography: An explanation of the effect of apical ligand on enantioselection by (salen) manganese catalyst. Tetrahedron 1999, 55, 14599–14610. [Google Scholar]
- Katsuki, T. Chiral metallosalen complexes: Structures and catalyst tuning for asymmetric epoxidation and cyclopropanation. Adv. Synth. Catal. 2002, 344, 131–147. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Jacobsen, H.; Cavallo, L. A possible mechanism for enantioselectivity in the chiral epoxidation of olefins with [Mn(salen)] catalysts. Chem. A Eur. J. 2001, 7, 800–807. [Google Scholar] [CrossRef]
- Kurti, L.; Blewett, M.M.; Corey, E.J. Origin of Enantioselectivity in the Jacobsen Epoxidation of Olefins. Org. Lett. 2009, 11, 4592–4595. [Google Scholar] [CrossRef]
- Brown, M.K.; Blewett, M.M.; Colombe, J.R.; Corey, E.J. Mechanism of the Enantioselective Oxidation of Racemic Secondary Alcohols Catalyzed by Chiral Mn(III)-Salen Complexes. J. Am. Chem. Soc. 2010, 132, 11165–11170. [Google Scholar] [CrossRef]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Sciotto, D.; Tomaselli, G.A.; Toscano, R.M. Synthesis and conformational aspects of 20- and 40-membered macrocyclic mono and dinuclear uranyl complexes incorporating salen and (R)-BINOL units. Tetrahedron 2007, 63, 9751–9757. [Google Scholar] [CrossRef]
- Amato, M.E.; Ballistreri, F.P.; Gentile, S.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M. Recognition of Achiral and Chiral Ammonium Salts by Neutral Ditopic Receptors Based on Chiral Salen-UO2 Macrocycles. J. Org. Chem. 2010, 75, 1437–1443. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S.; Ballistreri, F.P.; Trusso Sfrazzetto, G. Synthesis and Characterization of Some Chiral Metal-Salen Complexes Bearing a Ferrocenophane Substituent. Molecules 2009, 14, 4312–4325. [Google Scholar] [CrossRef] [PubMed]
- Trusso Sfrazzetto, G.; Millesi, S.; Pappalardo, A.; Toscano, R.M.; Ballistreri, F.P.; Tomaselli, G.A.; Gulino, A. Olefin epoxidation by a (salen)Mn(III) catalyst covalently grafted on glass beads. Catal. Sci. Technol. 2015, 5, 673–679. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Toscano, R.M.; Amato, M.E.; Pappalardo, A.; Gangemi, C.M.A.; Spidalieri, S.; Puglisi, R.; Trusso Sfrazzetto, G. A New Mn–Salen Micellar Nanoreactor for Enantioselective Epoxidation of Alkenes in Water. Catalysts 2018, 8, 129. [Google Scholar] [CrossRef]
- Zammataro, A.; Gangemi, C.M.A.; Pappalardo, A.; Toscano, R.M.; Puglisi, R.; Nicotra, G.; Fragalà, M.E.; Tuccitto, N.; Trusso Sfrazzetto, G. Covalently functionalized carbon nanoparticles with a chiral Mn-Salen: A new nanocatalyst for enantioselective epoxidation of alkenes. Chem. Commun. 2019, 55, 5255–5258. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Gangemi, M.A.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Trusso Sfrazzetto, G. (Salen)Mn(III) Catalyzed Asymmetric Epoxidation Reactions by Hydrogen Peroxide in Water: A Green Protocol. Int. J. Mol. Sci. 2016, 17, 1112. [Google Scholar] [CrossRef]
- Legnani, L.; Iannazzo, D.; Pistone, A.; Celesti, C.; Giofrè, S.; Romeo, R.; Di Pietro, A.; Visalli, G.; Fresta, M.; Bottino, P.; et al. Functionalized polyhedral oligosilsesquioxane (POSS) based composites for bone tissue engineering: Synthesis, computational and biological studies. RSC Adv. 2020, 10, 11325–11334. [Google Scholar] [CrossRef]
- Legnani, L.; Puglisi, R.; Pappalardo, A.; Chiacchio, M.A.; Trusso Sfrazzetto, G. Supramolecular recognition of phosphocholine by an enzyme-like cavitand receptor. Chem. Commun. 2020, 56, 539–542. [Google Scholar]
- Chiacchio, M.A.; Legnani, L.; Caramella, P.; Tejero, T.; Merino, P. Revealing carbocations in highly asynchronous concerted reactions: The ene-type reaction between dithiocarboxylic acids and alkenes. Tetrahedron 2018, 74, 5627–5634. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L.; Caramella, P.; Tejero, T.; Merino, P. Pivotal Neighboring-Group Participation in Substitution versus Elimination Reactions—Computational Evidence for Ion Pairs in the Thionation of Alcohols with Lawesson’s Reagent. Eur. J. Org. Chem. 2017, 14, 1952–1960. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3042. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Sasaki, H.; Irie, R.; Hamada, T.; Suzuki, K.; Katsuki, T. Rational design of Mn-salen catalyst. 2: Highly enantioselective epoxidation of conjugated cis-olefins. Tetrahedron 1994, 50, 11827–11838. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins using chiral (salen) manganese(III) complexes. Tetrahedron Asymmetry 1991, 2, 481–494. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkene | Entry | Catalyst | Time (min) | Conv. b (%) | Yield b (%) | EE b (%) |
|---|---|---|---|---|---|---|
| cis-β-methyl-styrene | 1 c | Mn-M20 | 300 | 80 | 88 | 16 |
| 2 | Mn-M20 | 240 | 90 | 88 | 27 | |
| 3 c | Mn-M40-DiPh | 420 | 78 | 84 | 17 | |
| 4 | Mn-M40-DiPh | 300 | 87 | 82 | 31 | |
| 5 c | Mn-M40-Cycl | 420 | 77 | 85 | 12 | |
| 6 | Mn-M40-Cycl | 300 | 89 | 84 | 30 | |
| 1,2-dihydro-naphthalene | 7 c | Mn-M20 | 60 | 92 | 93 | 23 |
| 8 | Mn-M20 | 30 | 95 | 94 | 38 | |
| 9 c | Mn-M40-DiPh | 90 | 90 | 91 | 25 | |
| 10 | Mn-M40-DiPh | 30 | 88 | 89 | 35 | |
| 11 c | Mn-M40-Cycl | 90 | 80 | 93 | 29 | |
| 12 | Mn-M40-Cycl | 30 | 90 | 99 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pappalardo, A.; Ballistreri, F.P.; Toscano, R.M.; Chiacchio, M.A.; Legnani, L.; Grazioso, G.; Veltri, L.; Trusso Sfrazzetto, G. Alkene Epoxidations Mediated by Mn-Salen Macrocyclic Catalysts. Catalysts 2021, 11, 465. https://doi.org/10.3390/catal11040465
Pappalardo A, Ballistreri FP, Toscano RM, Chiacchio MA, Legnani L, Grazioso G, Veltri L, Trusso Sfrazzetto G. Alkene Epoxidations Mediated by Mn-Salen Macrocyclic Catalysts. Catalysts. 2021; 11(4):465. https://doi.org/10.3390/catal11040465
Chicago/Turabian StylePappalardo, Andrea, Francesco P. Ballistreri, Rosa Maria Toscano, Maria Assunta Chiacchio, Laura Legnani, Giovanni Grazioso, Lucia Veltri, and Giuseppe Trusso Sfrazzetto. 2021. "Alkene Epoxidations Mediated by Mn-Salen Macrocyclic Catalysts" Catalysts 11, no. 4: 465. https://doi.org/10.3390/catal11040465
APA StylePappalardo, A., Ballistreri, F. P., Toscano, R. M., Chiacchio, M. A., Legnani, L., Grazioso, G., Veltri, L., & Trusso Sfrazzetto, G. (2021). Alkene Epoxidations Mediated by Mn-Salen Macrocyclic Catalysts. Catalysts, 11(4), 465. https://doi.org/10.3390/catal11040465