Nanocatalysts Containing Direct Electron Transfer-Capable Oxidoreductases: Recent Advances and Applications


Abstract
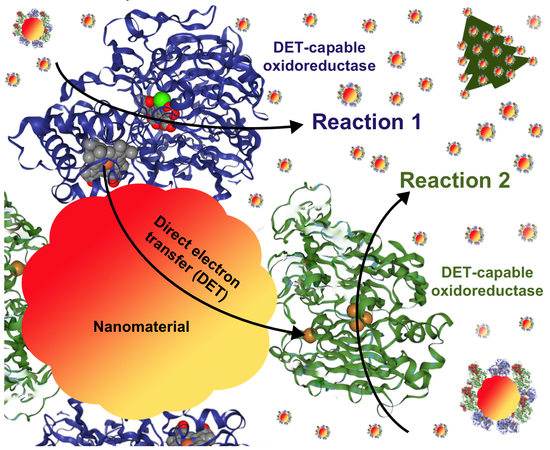
1. General Overview of the Enzyme-Based Heterogeneous Catalysis
2. Application of Direct Electron Transfer (DET)-Capable Oxidoreductases
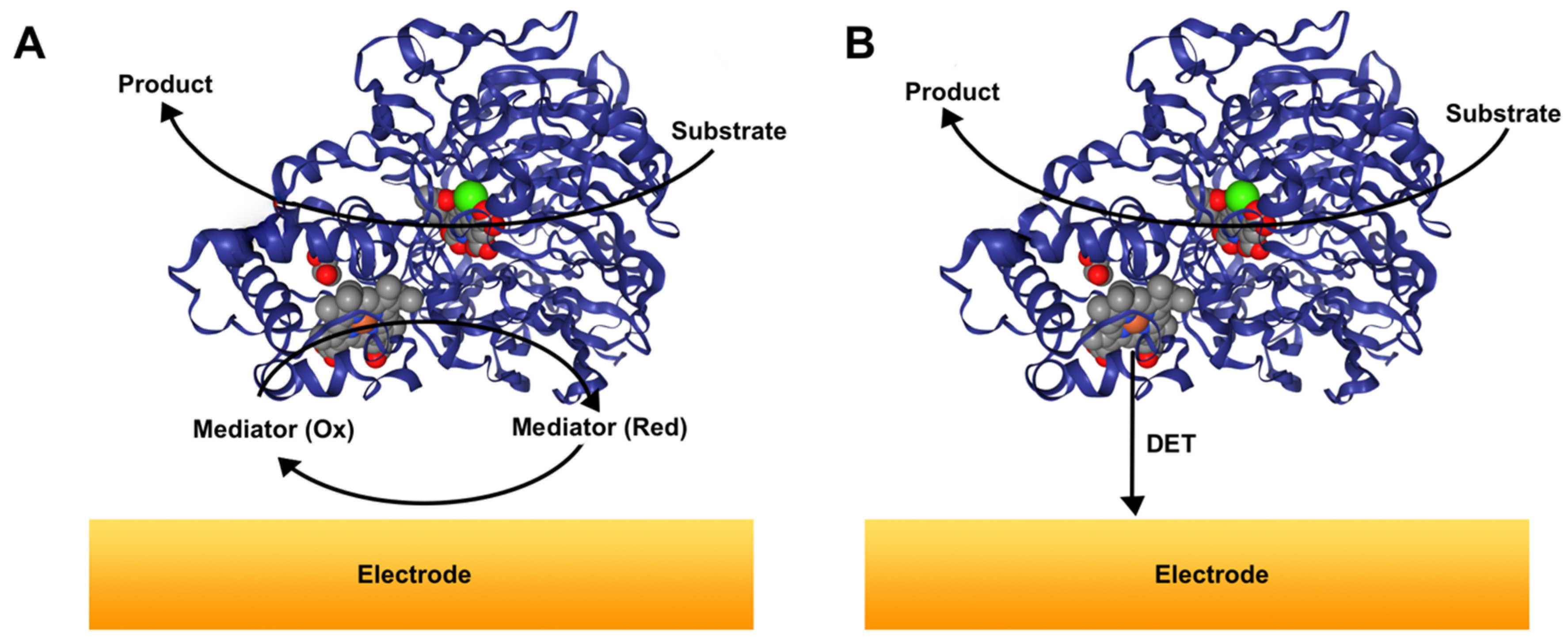
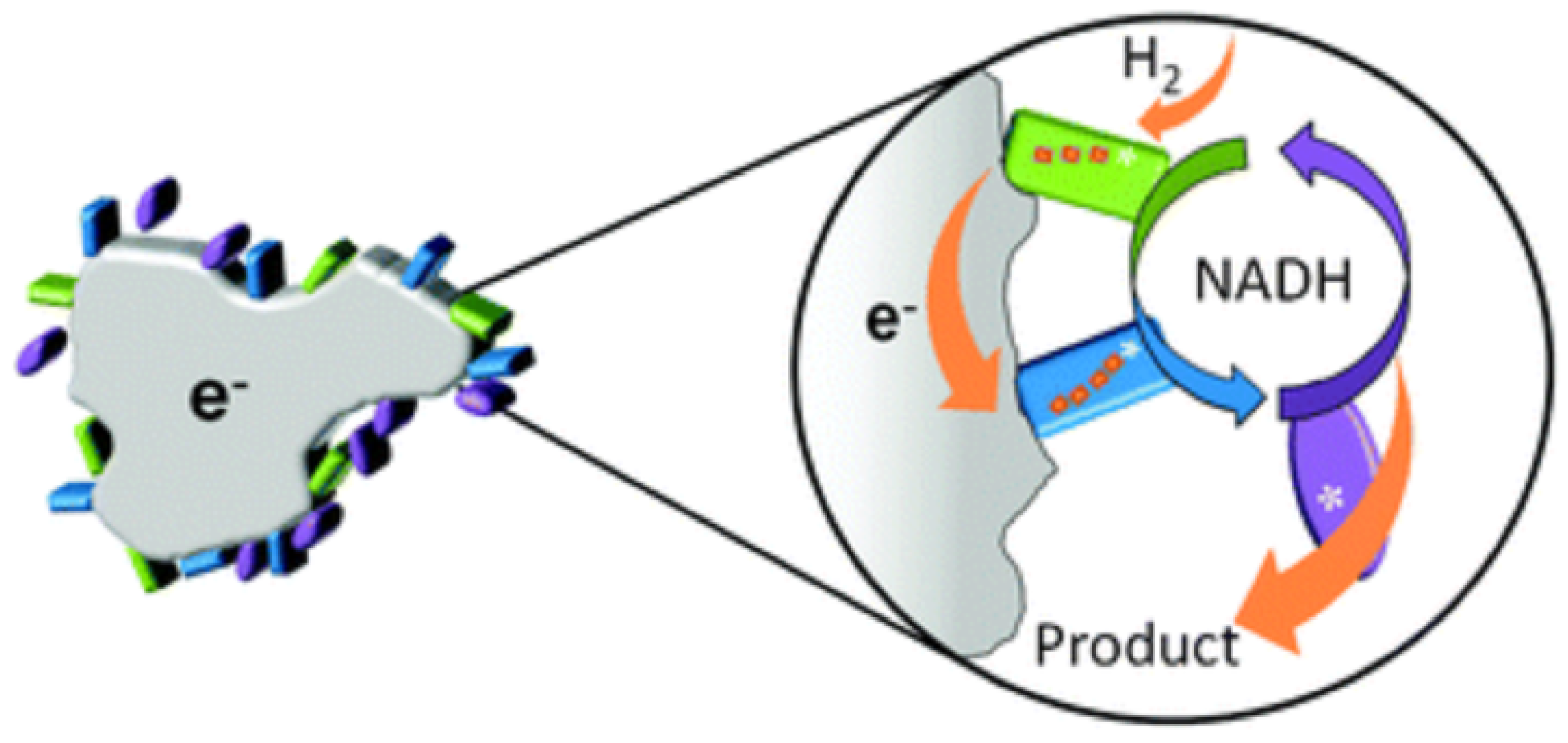
2.1. Principles of the DET Mechanism
2.2. Development of Biosensors and Biofuel Cells
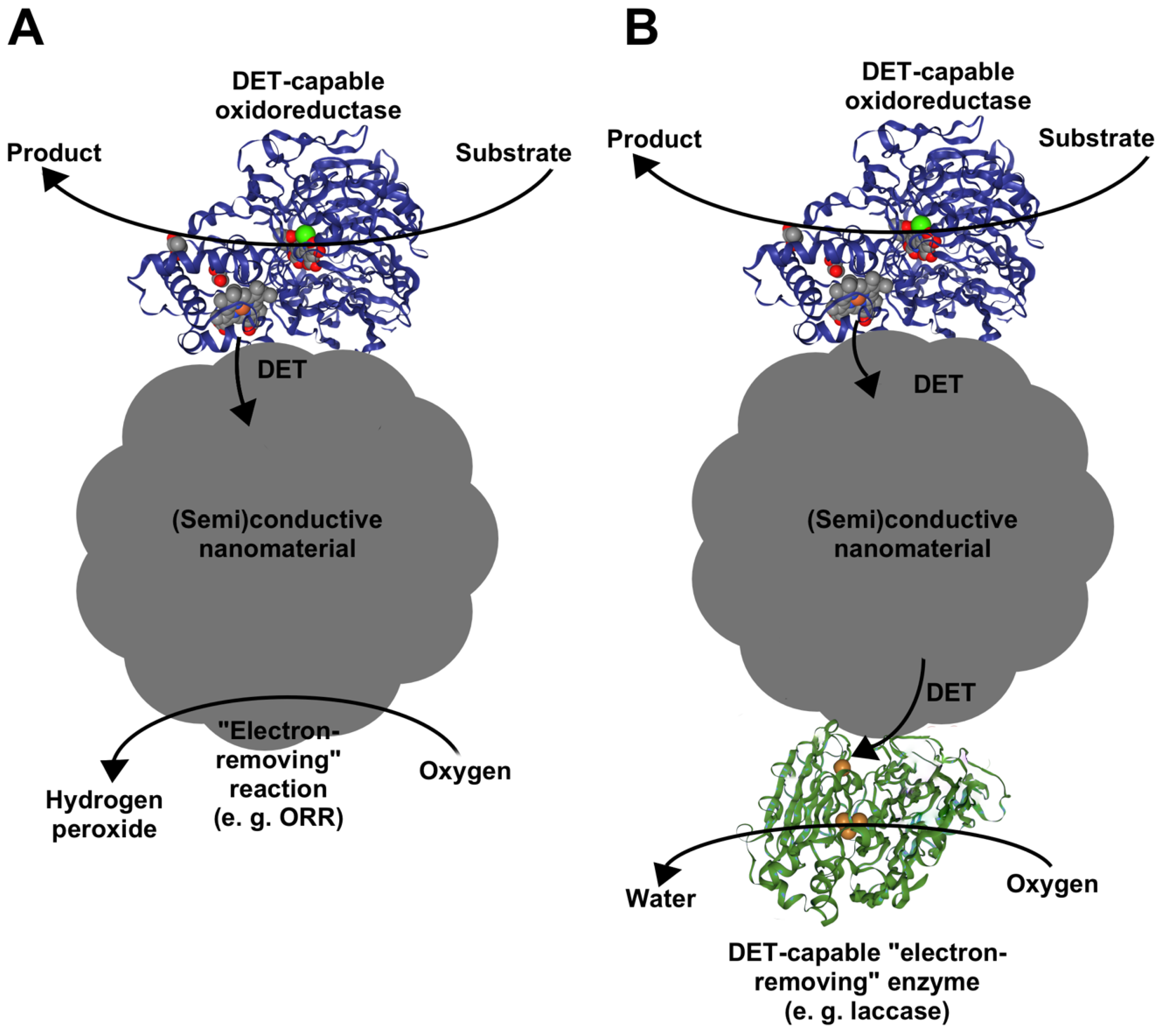
3. Performance of DET-Capable Oxidoreductases at Nanomaterial Interface
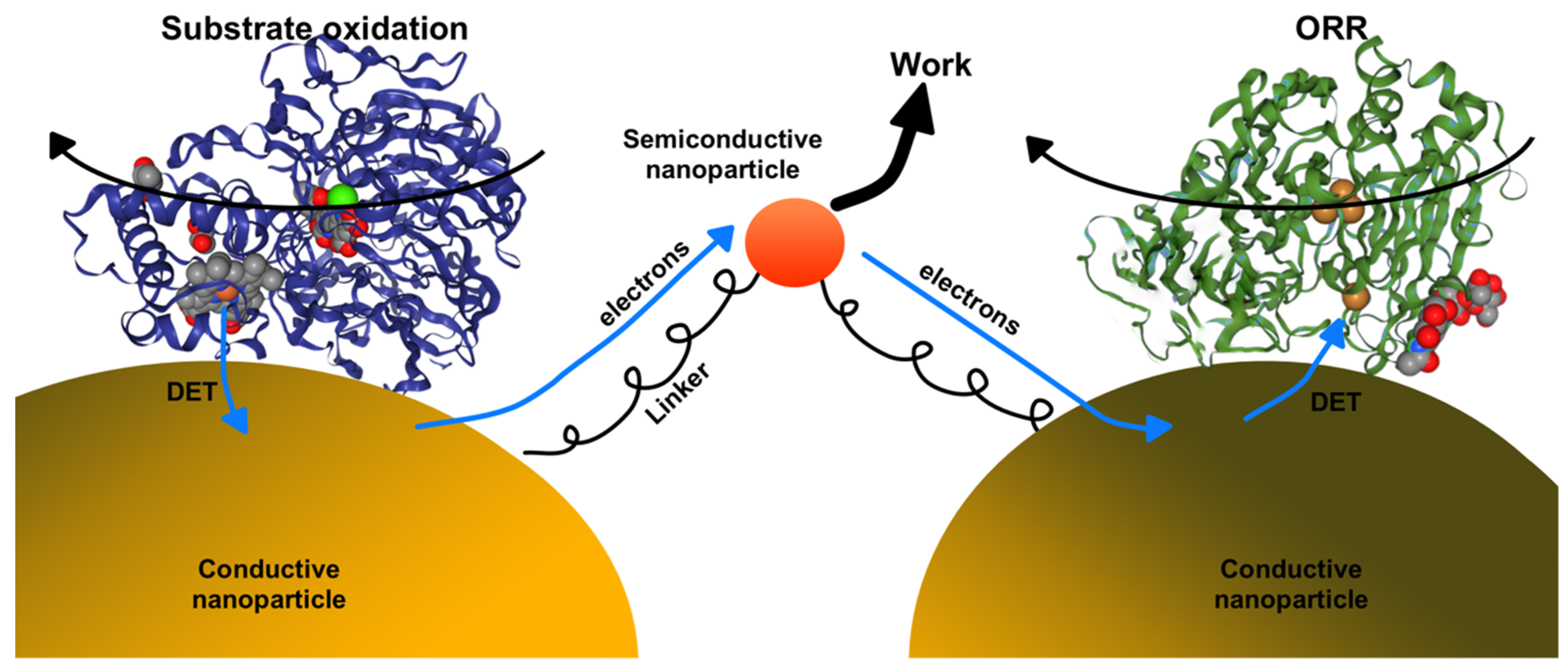
3.1. Design Challenges of Enzyme-Nanoparticle Catalysts Containing DET-Capable Oxidoreductases
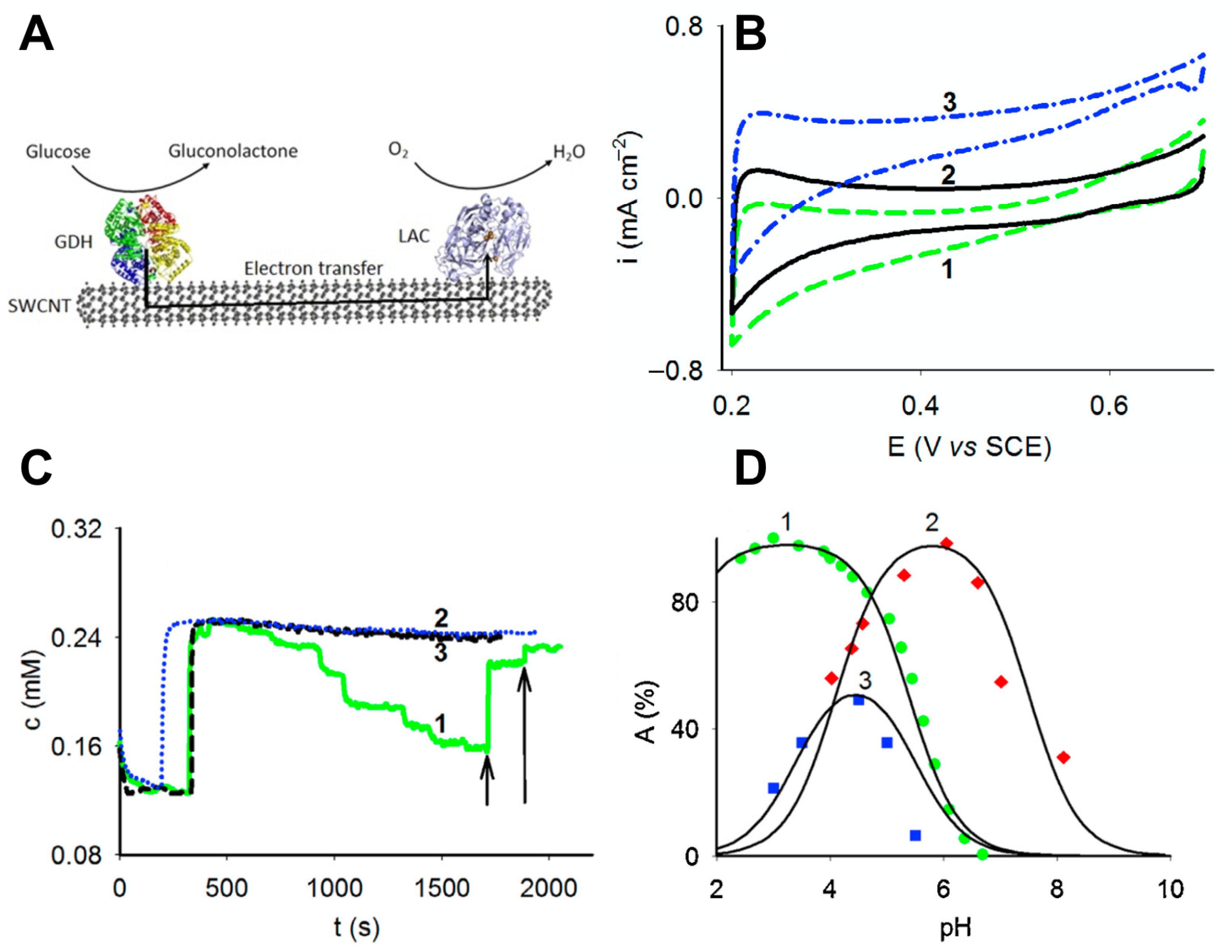
3.1.1. Designing Enzyme–Nanoparticle Catalysts with Enzymes Immobilized on Carbon Nanomaterials
3.1.2. Designing Enzyme–Nanoparticle Catalysts with Enzymes Immobilized on Metallic Nanoparticles
3.2. Challenges for Enzyme–Nanoparticle Catalysts Based on DET-Capable Enzymes
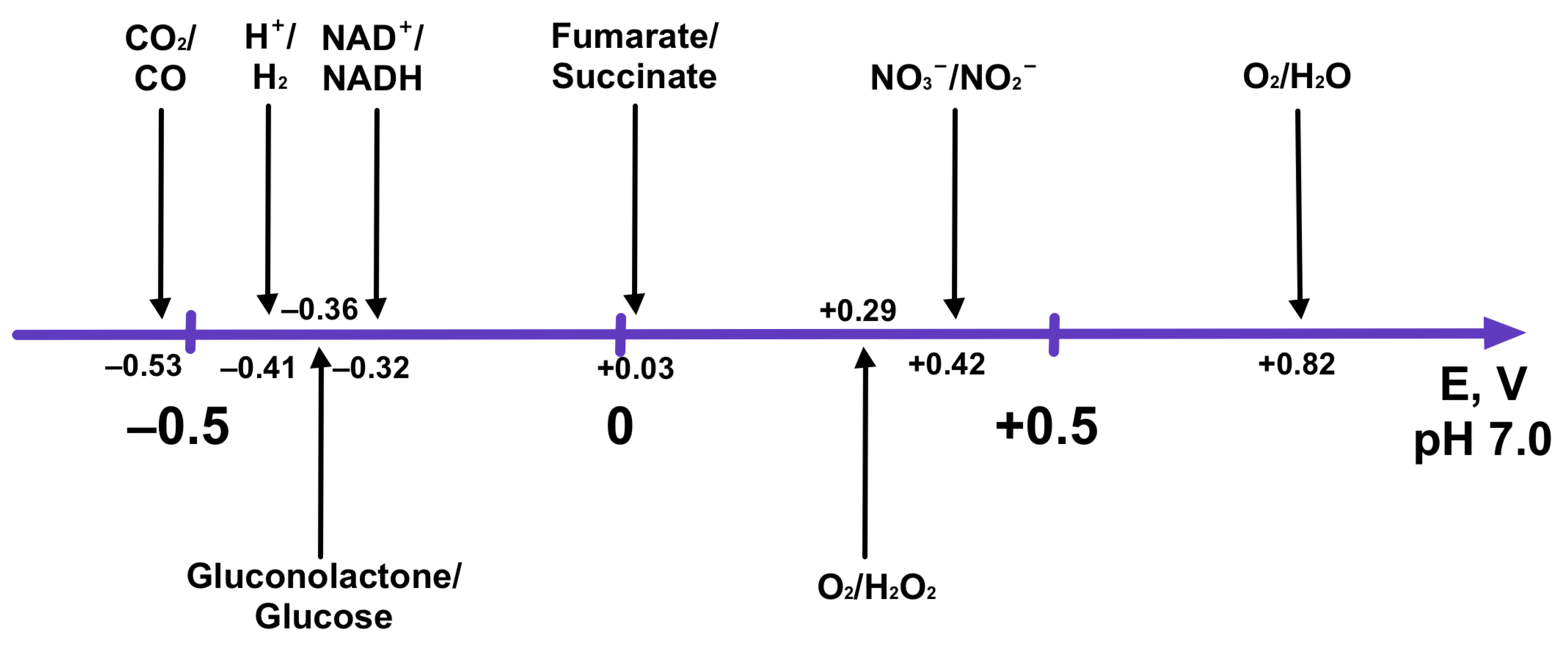
3.2.1. Compatibility of the Oxidoreductase Activity Dependence on pH
3.2.2. Compatibility of the Operational Electrochemical Potential of Enzymes
4. Concluding Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADH | Alcohol dehydrogenase |
| AuNP(s) | Gold nanoparticle(s) |
| CODH | CO2-reducing enzyme |
| DET | Direct electron transfer |
| FAD-GDH | FAD-depended glucose dehydrogenase |
| GDH | Glucose dehydrogenase |
| GOx | Glucose oxidase |
| HER | Hydrogen evolution reaction |
| LAC | Laccase |
| MET | Mediated electron transfer |
| ORR | Oxygen reduction reaction |
| SCE | Saturated calomel electrode (0.244 V vs. SHE) |
| SHE | Standard hydrogen electrode |
| SWCNT | Single-walled carbon nanotubes |
References
- Albery, W.J.; Knowles, J.R. Efficiency and Evolution of Enzyme Catalysis. Angew. Chem. Int. Ed. Engl. 1977, 16, 285–293. [Google Scholar] [CrossRef]
- Northrup, S.H.; Reynolds, J.C.L.; Miller, C.M.; Forrest, K.J.; Boles, J.O. Diffusion-controlled association rate of cytochrome c and cytochrome c peroxidase in a simple electrostatic model. J. Am. Chem. Soc. 1986, 108, 8162–8170. [Google Scholar] [CrossRef]
- Reetz, M.T.; Wiesenhöfer, W.; Franciò, G.; Leitner, W. Continuous Flow Enzymatic Kinetic Resolution and Enantiomer Separation using Ionic Liquid/Supercritical Carbon Dioxide Media. Adv. Synth. Catal. 2003, 345, 1221–1228. [Google Scholar] [CrossRef]
- Bodnár, J.; Gubicza, L.; Szabó, L.P. Enantiomeric separation of 2-chloropropionic acid by enzymatic esterification in organic solvents. J. Mol. Catal. 1990, 61, 353–361. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; Elsevier Science: 2014; Wiley-Blackwell: Weinheim, Germany, 2012; ISBN 978-1-322-33429-5. [Google Scholar]
- Iyer, P.V.; Ananthanarayan, L. Enzyme stability and stabilization—Aqueous and non-aqueous environment. Process Biochem. 2008, 43, 1019–1032. [Google Scholar] [CrossRef]
- Larsson, K.M.; Adlercreutz, P.; Mattiasson, B.; Olsson, U. Enzymatic catalysis in microemulsions: Enzyme reuse and product recovery. Biotechnol. Bioeng. 1990, 36, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, H.; Campbell, C.T. Nanoparticles for Catalysis. Acc. Chem. Res. 2013, 46, 1671–1672. [Google Scholar] [CrossRef]
- Kreyling, W.G.; Semmler-Behnke, M.; Chaudhry, Q. A complementary definition of nanomaterial. Nano Today 2010, 5, 165–168. [Google Scholar] [CrossRef]
- Uddin, J. Macro to Nano Spectroscopy; Intech: Rijeka, Croatia, 2012; ISBN 978-953-51-0664-7. [Google Scholar]
- Gruskiene, R.; Krivorotova, T.; Staneviciene, R.; Ratautas, D.; Serviene, E.; Sereikaite, J. Preparation and characterization of iron oxide magnetic nanoparticles functionalized by nisin. Colloids Surf. B Biointerfaces 2018, 169, 126–134. [Google Scholar] [CrossRef]
- Sharma, N.; Ojha, H.; Bharadwaj, A.; Pathak, D.P.; Sharma, R.K. Preparation and catalytic applications of nanomaterials: A review. RSC Adv. 2015, 5, 53381–53403. [Google Scholar] [CrossRef]
- Lin, C.; Compton, R.G. Size Effects in Nanoparticle Catalysis at Nanoparticle Modified Electrodes: The Interplay of Diffusion and Chemical Reactions. J. Phys. Chem. C 2017, 121, 2521–2528. [Google Scholar] [CrossRef]
- Luo, W.; Zhu, C.; Su, S.; Li, D.; He, Y.; Huang, Q.; Fan, C. Self-Catalyzed, Self-Limiting Growth of Glucose Oxidase-Mimicking Gold Nanoparticles. ACS Nano 2010, 4, 7451–7458. [Google Scholar] [CrossRef] [PubMed]
- Lee, H. Utilization of shape-controlled nanoparticles as catalysts with enhanced activity and selectivity. RSC Adv. 2014, 4, 41017–41027. [Google Scholar] [CrossRef]
- Tominaga, M.; Shimazoe, T.; Nagashima, M.; Taniguchi, I. Electrocatalytic oxidation of glucose at gold nanoparticle-modified carbon electrodes in alkaline and neutral solutions. Electrochem. Commun. 2005, 7, 189–193. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, N.; Yu, H.; Niu, Y.; Sun, C. Covalent attachment of glucose oxidase to an Au electrode modified with gold nanoparticles for use as glucose biosensor. Bioelectrochemistry 2005, 67, 15–22. [Google Scholar] [CrossRef]
- Lynch, I.; Dawson, K.A. Protein-nanoparticle interactions. Nano Today 2008, 3, 40–47. [Google Scholar] [CrossRef]
- Cagliani, R.; Gatto, F.; Bardi, G. Protein Adsorption: A Feasible Method for Nanoparticle Functionalization? Materials 2019, 12, 1991. [Google Scholar] [CrossRef]
- Barbero, F.; Russo, L.; Vitali, M.; Piella, J.; Salvo, I.; Borrajo, M.L.; Busquets-Fité, M.; Grandori, R.; Bastús, N.G.; Casals, E.; et al. Formation of the Protein Corona: The Interface between Nanoparticles and the Immune System. Semin. Immunol. 2017, 34, 52–60. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Vilanova, O.; Mittag, J.J.; Kelly, P.M.; Milani, S.; Dawson, K.A.; Rädler, J.O.; Franzese, G. Understanding the Kinetics of Protein-Nanoparticle Corona Formation. ACS Nano 2016, 10, 10842–10850. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zeng, G.; Xu, P.; Lai, C.; Tang, L. How Do Enzymes ‘Meet’ Nanoparticles and Nanomaterials? Trends Biochem. Sci. 2017, 42, 914–930. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.J.; Russ Algar, W.; Malanoski, A.P.; Ancona, M.G.; Medintz, I.L. Understanding enzymatic acceleration at nanoparticle interfaces: Approaches and challenges. Nano Today 2014, 9, 102–131. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, Y.; Ran, X.; Ren, J.; Qu, X. A semipermeable enzymatic nanoreactor as an efficient modulator for reversible pH regulation. Nanoscale 2014, 6, 11328–11335. [Google Scholar] [CrossRef]
- Delaittre, G.; Reynhout, I.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Cascade Reactions in an All-Enzyme Nanoreactor. Chem. A Eur. J. 2009, 15, 12600–12603. [Google Scholar] [CrossRef]
- Li, H.; Ma, L.; Zhou, L.; Gao, J.; Huang, Z.; He, Y.; Jiang, Y. An integrated nanocatalyst combining enzymatic and metal-organic framework catalysts for cascade degradation of organophosphate nerve agents. Chem. Commun. 2018, 54, 10754–10757. [Google Scholar] [CrossRef]
- Aquino Neto, S.; Almeida, T.S.; Palma, L.M.; Minteer, S.D.; de Andrade, A.R. Hybrid nanocatalysts containing enzymes and metallic nanoparticles for ethanol/O2 biofuel cell. J. Power Sources 2014, 259, 25–32. [Google Scholar] [CrossRef]
- Parolo, C.; de la Escosura-Muñiz, A.; Merkoçi, A. Enhanced lateral flow immunoassay using gold nanoparticles loaded with enzymes. Biosens. Bioelectron. 2013, 40, 412–416. [Google Scholar] [CrossRef]
- Phadtare, S.; Kumar, A.; Vinod, V.P.; Dash, C.; Palaskar, D.V.; Rao, M.; Shukla, P.G.; Sivaram, S.; Sastry, M. Direct Assembly of Gold Nanoparticle “Shells” on Polyurethane Microsphere “Cores” and Their Application as Enzyme Immobilization Templates. Chem. Mater. 2003, 15, 1944–1949. [Google Scholar] [CrossRef]
- May, S.W. Applications of oxidoreductases. Curr. Opin. Biotechnol. 1999, 10, 370–375. [Google Scholar] [CrossRef]
- Razumiene, J.; Barkauskas, J.; Kubilius, V.; Meskys, R.; Laurinavicius, V. Modified graphitized carbon black as transducing material for reagentless H2O2 and enzyme sensors. Talanta 2005, 67, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Razumiene, J.; Vilkanauskyte, A.; Gureviciene, V.; Barkauskas, J.; Meskys, R.; Laurinavicius, V. Direct electron transfer between PQQ dependent glucose dehydrogenases and carbon electrodes: An approach for electrochemical biosensors. Electrochim. Acta 2006, 51, 5150–5156. [Google Scholar] [CrossRef]
- Yaropolov, A.I.; Varfolomeev, S.D.; Berezin, I.V. Bioelectrocatalysis. Activation of a cathode oxygen reduction in the peroxidase-mediator carbon electrode system. FEBS Lett. 1976, 71, 306–308. [Google Scholar] [CrossRef][Green Version]
- Eddowes, M.J.; Hill, H.A.O. Novel method for the investigation of the electrochemistry of metalloproteins: Cytochrome c. J. Chem. Soc. Chem. Commun. 1977, 771b. [Google Scholar] [CrossRef]
- Kulys, J.J.; Švirmickas, G.J.S. Reagentless lactate sensor based on cytochrome b2. Anal. Chim. Acta 1980, 117, 115–120. [Google Scholar] [CrossRef]
- Ferapontova, E.E.; Gorton, L. Direct electrochemistry of heme multicofactor-containing enzymes on alkanethiol-modified gold electrodes. Bioelectrochemistry 2005, 66, 55–63. [Google Scholar] [CrossRef]
- Sakinyte, I.; Barkauskas, J.; Gaidukevic, J.; Razumiene, J. Thermally reduced graphene oxide: The study and use for reagentless amperometric d-fructose biosensors. Talanta 2015, 144, 1096–1103. [Google Scholar] [CrossRef]
- Christenson, A.; Dimcheva, N.; Ferapontova, E.E.; Gorton, L.; Ruzgas, T.; Stoica, L.; Shleev, S.; Yaropolov, A.I.; Haltrich, D.; Thorneley, R.N.F.; et al. Direct Electron Transfer Between Ligninolytic Redox Enzymes and Electrodes. Electroanalysis 2004, 16, 1074–1092. [Google Scholar] [CrossRef]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.D.; Ruzgas, T.; Gazaryan, I. Direct electron transfer between heme-containing enzymes and electrodes as basis for third generation biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar] [CrossRef]
- Razumiene, J.; Niculescu, M.; Ramanavicius, A.; Laurinavicius, V.; Csöregi, E. Direct bioelectrocatalysis at carbon electrodes modified with quinohemoprotein alcohol dehydrogenase from Gluconobacter sp. 33. Electroanalysis 2002, 14, 43–49. [Google Scholar] [CrossRef]
- Sakurai, T.; Kataoka, K. Basic and applied features of multicopper oxidases, CueO, bilirubin oxidase, and laccase. Chem. Rec. 2007, 7, 220–229. [Google Scholar] [CrossRef]
- Lojou, É.; Luo, X.; Brugna, M.; Candoni, N.; Dementin, S.; Giudici-Orticoni, M.T. Biocatalysts for fuel cells: Efficient hydrogenase orientation for H2 oxidation at electrodes modified with carbon nanotubes. JBIC J. Biol. Inorg. Chem. 2008, 13, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Shleev, S.; Andoralov, V.; Pankratov, D.; Falk, M.; Aleksejeva, O.; Blum, Z. Oxygen Electroreduction versus Bioelectroreduction: Direct Electron Transfer Approach. Electroanalysis 2016, 28, 2270–2287. [Google Scholar] [CrossRef]
- Ruzgas, T.; Larpant, N.; Shafaat, A.; Sotres, J. Wireless, Battery-Less Biosensors Based on Direct Electron Transfer Reactions. ChemElectroChem 2019, 6, 5167–5171. [Google Scholar] [CrossRef]
- Okuda-Shimazaki, J.; Yoshida, H.; Sode, K. FAD dependent glucose dehydrogenases—Discovery and engineering of representative glucose sensing enzymes-. Bioelectrochemistry 2019, 107414. [Google Scholar] [CrossRef]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef]
- Wilson, G.S. Native glucose oxidase does not undergo direct electron transfer. Biosens. Bioelectron. 2016, 82, vii–viii. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Al-Lolage, F.A. There is no evidence to support literature claims of direct electron transfer (DET) for native glucose oxidase (GOx) at carbon nanotubes or graphene. J. Electroanal. Chem. 2018, 819, 26–37. [Google Scholar] [CrossRef]
- Courjean, O.; Gao, F.; Mano, N. Deglycosylation of Glucose Oxidase for Direct and Efficient Glucose Electrooxidation on a Glassy Carbon Electrode. Angew. Chem. Int. Ed. 2009, 48, 5897–5899. [Google Scholar] [CrossRef]
- Ma, S.; Ludwig, R. Direct Electron Transfer of Enzymes Facilitated by Cytochromes. ChemElectroChem 2019, 6, 958–975. [Google Scholar] [CrossRef]
- Rottenberg, H. The Thermodynamic Description of Enzyme-Catalyzed Reactions. Biophys. J. 1973, 13, 503–511. [Google Scholar] [CrossRef]
- Gallop, P.M.; Paz, M.A.; Flückiger, R.; Kagan, H.M. PQQ, the elusive coenzyme. Trends Biochem. Sci. 1989, 14, 343–346. [Google Scholar] [CrossRef]
- Walsh, C. Flavin coenzymes: At the crossroads of biological redox chemistry. Acc. Chem. Res. 1980, 13, 148–155. [Google Scholar] [CrossRef]
- Saleh, F.S.; Rahman, M.R.; Okajima, T.; Mao, L.; Ohsaka, T. Determination of formal potential of NADH/NAD+ redox couple and catalytic oxidation of NADH using poly(phenosafranin)-modified carbon electrodes. Bioelectrochemistry 2011, 80, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ansell, R.J.; Lowe, C.R. Artificial redox coenzymes: Biomimetic analogues of NAD+. Appl. Microbiol. Biotechnol. 1999, 51, 703–710. [Google Scholar] [CrossRef]
- Vaitkutė, G.; Bratkovskaja, I.; Časaitė, V.; Stankevičiūtė, J.; Meškys, R.; Tetianec, L. Electron transfer mediators for PQQ dependent soluble glucose dehydrogenase catalyzed lactose oxidation reaction. Chemija 2019, 30, 194–200. [Google Scholar] [CrossRef]
- Tetianec, L.; Chaleckaja, A.; Kulys, J.; Janciene, R.; Marcinkeviciene, L.; Meskiene, R.; Stankeviciute, J.; Meskys, R. Characterization of methylated azopyridine as a potential electron transfer mediator for electroenzymatic systems. Process Biochem. 2017, 54, 41–48. [Google Scholar] [CrossRef]
- Ramonas, E.; Ratautas, D.; Dagys, M.; Meškys, R.; Kulys, J. Highly sensitive amperometric biosensor based on alcohol dehydrogenase for determination of glycerol in human urine. Talanta 2019, 200, 333–339. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Bradford, V.Q.; Whitaker, R.G. Enzyme electrode studies of glucose oxidase modified with a redox mediator. Talanta 1991, 38, 57–63. [Google Scholar] [CrossRef]
- Besharati, M.; Hamedi, J.; Hosseinkhani, S.; Saber, R. A novel electrochemical biosensor based on TetX2 monooxygenase immobilized on a nano-porous glassy carbon electrode for tetracycline residue detection. Bioelectrochemistry 2019, 128, 66–73. [Google Scholar] [CrossRef]
- Tsujimura, S.; Murata, K.; Akatsuka, W. Exceptionally High Glucose Current on a Hierarchically Structured Porous Carbon Electrode with “Wired” Flavin Adenine Dinucleotide-Dependent Glucose Dehydrogenase. J. Am. Chem. Soc. 2014, 136, 14432–14437. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.T.; Kao, D.Y.; Hickey, D.; Schmidtke, D.W.; Glatzhofer, D.T. High Current Density Ferrocene-Modified Linear Poly(ethylenimine) Bioanodes and Their Use in Biofuel Cells. J. Electrochem. Soc. 2011, 158, B166–B174. [Google Scholar] [CrossRef]
- Shao, M.; Zafar, M.N.; Falk, M.; Ludwig, R.; Sygmund, C.; Peterbauer, C.K.; Guschin, D.A.; MacAodha, D.; Ó Conghaile, P.; Leech, D.; et al. Optimization of a Membraneless Glucose/Oxygen Enzymatic Fuel Cell Based on a Bioanode with High Coulombic Efficiency and Current Density. ChemPhysChem 2013, 14, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, W. Functionalized polypyrrole. A new material for the construction of biosensors. Synth. Met. 1991, 41, 429–432. [Google Scholar] [CrossRef]
- Schuhmann, W. Non-leaking amperometric biosensors based on high-molecular ferrocene derivatives. Biosens. Bioelectron. 1993, 8, 191–196. [Google Scholar] [CrossRef]
- Freire, R.S.; Pessoa, C.A.; Mello, L.D.; Kubota, L.T. Direct electron transfer: An approach for electrochemical biosensors with higher selectivity and sensitivity. J. Braz. Chem. Soc. 2003, 14, 230–243. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L.; Antiochia, R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors 2018, 18, 1319. [Google Scholar] [CrossRef]
- Hush, N.S. Distance Dependence of Electron Transfer Rates. Coord. Chem. Rev. 1985, 64, 135–157. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Electron Transfer in Proteins. Annu. Rev. Biochem. 1996, 65, 537–561. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Long-range electron transfer. Proc. Natl. Acad. Sci. USA 2005, 102, 3534–3539. [Google Scholar] [CrossRef]
- Zhang, W.; Li, G. Third-Generation Biosensors Based on the Direct Electron Transfer of Proteins. Anal. Sci. 2004, 20, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Blum, Z.; Shleev, S. Direct electron transfer based enzymatic fuel cells. Electrochim. Acta 2012, 82, 191–202. [Google Scholar] [CrossRef]
- Koopal, C.G.J.; Nolte, R.J.M. Kinetic study of the performance of third-generation biosensors. Bioelectrochem. Bioenerg. 1994, 33, 45–53. [Google Scholar] [CrossRef]
- Stoica, L.; Ludwig, R.; Haltrich, D.; Gorton, L. Third-Generation Biosensor for Lactose Based on Newly Discovered Cellobiose Dehydrogenase. Anal. Chem. 2006, 78, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Loew, N.; Tsugawa, W.; Ikebukuro, K.; Sode, K. Development of a third-generation glucose sensor based on the open circuit potential for continuous glucose monitoring. Biosens. Bioelectron. 2019, 124–125, 216–223. [Google Scholar] [PubMed]
- Ito, Y.; Okuda-Shimazaki, J.; Tsugawa, W.; Loew, N.; Shitanda, I.; Lin, C.E.; La Belle, J.; Sode, K. Third generation impedimetric sensor employing direct electron transfer type glucose dehydrogenase. Biosens. Bioelectron. 2019, 129, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Liu, Y.; Xu, Y.; Shi, L.; Peng, J.; Jin, W. In-situ fabrication of well-distributed gold nanocubes on thiol graphene as a third-generation biosensor for ultrasensitive glucose detection. Electrochim. Acta 2015, 176, 162–171. [Google Scholar] [CrossRef]
- Treu, B.L.; Minteer, S.D. Isolation and purification of PQQ-dependent lactate dehydrogenase from Gluconobacter and use for direct electron transfer at carbon and gold electrodes. Bioelectrochemistry 2008, 74, 73–77. [Google Scholar] [CrossRef]
- Larpant, N.; Pham, A.D.; Shafaat, A.; Gonzalez-Martinez, J.F.; Sotres, J.; Sjöholm, J.; Laiwattanapaisal, W.; Faridbod, F.; Ganjali, M.R.; Arnebrant, T.; et al. Sensing by wireless reading Ag/AgCl redox conversion on RFID tag: Universal, battery-less biosensor design. Sci. Rep. 2019, 9, 12948. [Google Scholar] [CrossRef]
- Musameh, M.M.; Dunn, C.J.; Uddin, M.H.; Sutherland, T.D.; Rapson, T.D. Silk provides a new avenue for third generation biosensors: Sensitive, selective and stable electrochemical detection of nitric oxide. Biosens. Bioelectron. 2018, 103, 26–31. [Google Scholar] [CrossRef]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent advances on developing 3rd generation enzyme electrode for biosensor applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef]
- Díaz Nieto, C.H.; Granero, A.M.; Lopez, J.C.; Pierini, G.D.; Levin, G.J.; Fernández, H.; Zon, M.A. Development of a third generation biosensor to determine hydrogen peroxide based on a composite of soybean peroxidase/chemically reduced graphene oxide deposited on glassy carbon electrodes. Sens. Actuators B Chem. 2018, 263, 377–386. [Google Scholar] [CrossRef]
- Ghosh, T.; Sarkar, P.; Turner, A.P.F. A novel third generation uric acid biosensor using uricase electro-activated with ferrocene on a Nafion coated glassy carbon electrode. Bioelectrochemistry 2015, 102, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanchez, C.; Shleev, S.; De Lacey, A.L.; Pita, M. Third-generation oxygen amperometric biosensor based on Trametes hirsuta laccase covalently bound to graphite electrode. Chem. Pap. 2015, 69, 237–240. [Google Scholar] [CrossRef][Green Version]
- Dagys, M.; Haberska, K.; Shleev, S.; Arnebrant, T.; Kulys, J.; Ruzgas, T. Laccase-gold nanoparticle assisted bioelectrocatalytic reduction of oxygen. Electrochem. Commun. 2010, 12, 933–935. [Google Scholar] [CrossRef]
- Gutiérrez-Sánchez, C.; Pita, M.; Vaz-Domínguez, C.; Shleev, S.; De Lacey, A.L. Gold Nanoparticles as Electronic Bridges for Laccase-Based Biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220. [Google Scholar] [CrossRef]
- Shleev, S. Quo Vadis, Implanted Fuel Cell? ChemPlusChem 2017, 82, 522–539. [Google Scholar] [CrossRef]
- Pita, M.; Mate, D.M.; Gonzalez-Perez, D.; Shleev, S.; Fernandez, V.M.; Alcalde, M.; De Lacey, A.L. Bioelectrochemical Oxidation of Water. J. Am. Chem. Soc. 2014, 136, 5892–5895. [Google Scholar] [CrossRef]
- Dagys, M.; Laurynėnas, A.; Ratautas, D.; Kulys, J.; Vidžiūnaitė, R.; Talaikis, M.; Niaura, G.; Marcinkevičienė, L.; Meškys, R.; Shleev, S. Oxygen electroreduction catalysed by laccase wired to gold nanoparticles via the trinuclear copper cluster. Energy Environ. Sci. 2017, 10, 498–502. [Google Scholar] [CrossRef]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Nöll, G. Direct Electron Transfer at Cellobiose Dehydrogenase Modified Anodes for Biofuel Cells. J. Phys. Chem. C 2008, 112, 9956–9961. [Google Scholar] [CrossRef]
- Ludwig, R.; Harreither, W.; Tasca, F.; Gorton, L. Cellobiose Dehydrogenase: A Versatile Catalyst for Electrochemical Applications. ChemPhysChem 2010, 11, 2674–2697. [Google Scholar] [CrossRef]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Enhanced Direct Electron Transfer of Fructose Dehydrogenase Rationally Immobilized on a 2-Aminoanthracene Diazonium Cation Grafted Single-Walled Carbon Nanotube Based Electrode. ACS Catal. 2018, 8, 10279–10289. [Google Scholar] [CrossRef]
- Flexer, V.; Durand, F.; Tsujimura, S.; Mano, N. Efficient Direct Electron Transfer of PQQ-glucose Dehydrogenase on Carbon Cryogel Electrodes at Neutral pH. Anal. Chem. 2011, 83, 5721–5727. [Google Scholar] [CrossRef] [PubMed]
- Ratautas, D.; Laurynėnas, A.; Dagys, M.; Marcinkevičienė, L.; Meškys, R.; Kulys, J. High current, low redox potential mediatorless bioanode based on gold nanoparticles and glucose dehydrogenase from Ewingella Americana. Electrochim. Acta 2016, 199, 254–260. [Google Scholar] [CrossRef]
- Muguruma, H.; Iwasa, H.; Hidaka, H.; Hiratsuka, A.; Uzawa, H. Mediatorless Direct Electron Transfer between Flavin Adenine Dinucleotide-Dependent Glucose Dehydrogenase and Single-Walled Carbon Nanotubes. ACS Catal. 2017, 7, 725–734. [Google Scholar] [CrossRef]
- Aquino Neto, S.; Suda, E.L.; Xu, S.; Meredith, M.T.; De Andrade, A.R.; Minteer, S.D. Direct electron transfer-based bioanodes for ethanol biofuel cells using PQQ-dependent alcohol and aldehyde dehydrogenases. Electrochim. Acta 2013, 87, 323–329. [Google Scholar] [CrossRef]
- Ratautas, D.; Tetianec, L.; Marcinkevičienė, L.; Meškys, R.; Kulys, J. Bioanode with alcohol dehydrogenase undergoing a direct electron transfer on functionalized gold nanoparticles for an application in biofuel cells for glycerol conversion. Biosens. Bioelectron. 2017, 98, 215–221. [Google Scholar] [CrossRef]
- Gineitytė, J.; Meškys, R.; Dagys, M.; Ratautas, D. Highly efficient direct electron transfer bioanode containing glucose dehydrogenase operating in human blood. J. Power Sources 2019, 441, 227163. [Google Scholar] [CrossRef]
- Vaitheeswaran, S.; Garcia, A.E. Protein stability at a carbon nanotube interface. J. Chem. Phys. 2011, 134, 125101. [Google Scholar] [CrossRef]
- Razumiene, J.; Sakinyte, I.; Barkauskas, J.; Baronas, R. Nano-structured carbon materials for improved biosensing applications. Appl. Surf. Sci. 2015, 334, 185–191. [Google Scholar] [CrossRef]
- Setkus, A.; Galdikas, A.; Laurinavicius, V.; Meskys, R.; Mironas, A.; Razumiene, J. Electric charge transport in the symmetric metal-enzyme junctions affected by biochemical interaction. Colloids Surf. A Physicochem. Eng. Asp. 2004, 249, 141–143. [Google Scholar]
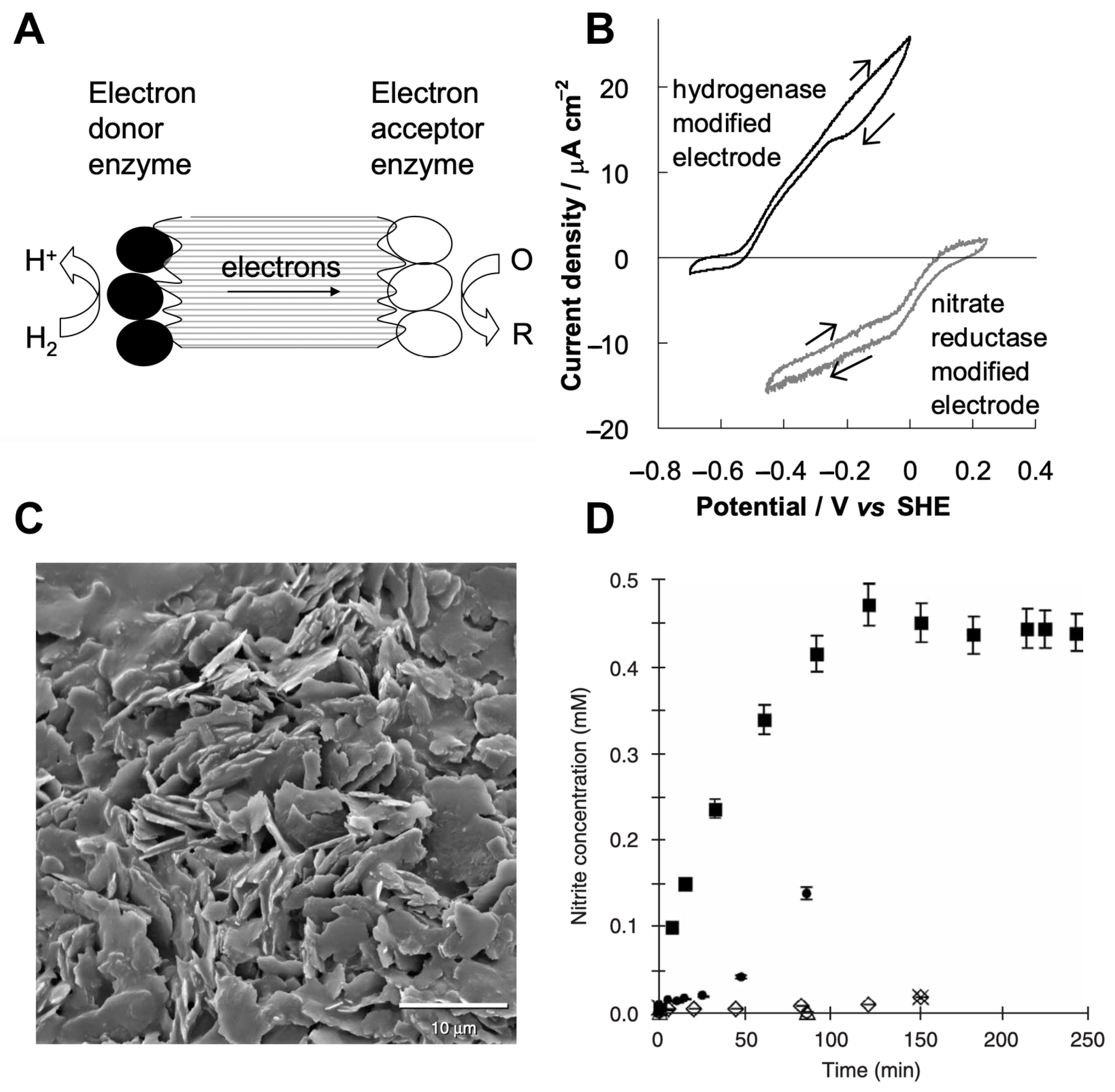
- Vincent, K.A.; Li, X.; Blanford, C.F.; Belsey, N.A.; Weiner, J.H.; Armstrong, F.A. Enzymatic catalysis on conducting graphite particles. Nat. Chem. Biol. 2007, 3, 761–762. [Google Scholar] [CrossRef]
- Reeve, H.A.; Lauterbach, L.; Ash, P.A.; Lenz, O.; Vincent, K.A. A modular system for regeneration of NADcofactors using graphite particles modified with hydrogenase and diaphorase moieties. Chem. Commun. 2012, 48, 1589–1591. [Google Scholar] [CrossRef] [PubMed]
- Kragl, U.; Kruse, W.; Hummel, W.; Wandrey, C. Enzyme engineering aspects of biocatalysis: Cofactor regeneration as example. Biotechnol. Bioeng. 2000, 52, 309–319. [Google Scholar] [CrossRef]
- Baronas, R.; Kulys, J.; Petrauskas, K.; Razumiene, J. Modelling carbon nanotubes-based mediatorless biosensor. Sensors (Switzerland) 2012, 12, 9146–9160. [Google Scholar] [CrossRef] [PubMed]
- Razumiene, J.; Gureviciene, V.; Sakinyte, I.; Barkauskas, J.; Petrauskas, K.; Baronas, R. Modified SWCNTs for Reagentless Glucose Biosensor: Electrochemical and Mathematical Characterization. Electroanalysis 2013, 25, 166–173. [Google Scholar] [CrossRef]
- Ratautas, D.; Marcinkevičienė, L.; Meškys, R.; Kulys, J. Mediatorless electron transfer in glucose dehydrogenase/laccase system adsorbed on carbon nanotubes. Electrochim. Acta 2015, 174, 940–944. [Google Scholar] [CrossRef]
- Casals, E.; Pfaller, T.; Duschl, A.; Oostingh, G.J.; Puntes, V. Time Evolution of the Nanoparticle Protein Corona. ACS Nano 2010, 4, 3623–3632. [Google Scholar] [CrossRef]
- Galdikas, A.; Bukauskas, V.; Kaciulis, S.; Laurinavicius, V.; Meskys, R.; Mezzi, A.; Mironas, A.; Razumiene, J.; Setkus, A. Properties of the planar ADH-dry-layer structures based on electrically controlled coupling between enzyme molecules and metal surfaces. Sens. Actuators B Chem. 2006, 118, 60–66. [Google Scholar] [CrossRef]
- Agarwal, J.; Fujita, E.; Schaefer, H.F.; Muckerman, J.T. Mechanisms for CO Production from CO2 Using Reduced Rhenium Tricarbonyl Catalysts. J. Am. Chem. Soc. 2012, 134, 5180–5186. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer-Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Yoneda, N.; Kusano, S.; Yasui, M.; Pujado, P.; Wilcher, S. Recent advances in processes and catalysts for the production of acetic acid. Appl. Catal. A Gen. 2001, 221, 253–265. [Google Scholar] [CrossRef]
- Jeong, H.Y.; Balamurugan, M.; Choutipalli, V.S.K.; Jeong, E.; Subramanian, V.; Sim, U.; Nam, K.T. Achieving highly efficient CO2 to CO electroreduction exceeding 300 mA cm−2 with single-atom nickel electrocatalysts. J. Mater. Chem. A 2019, 7, 10651–10661. [Google Scholar] [CrossRef]
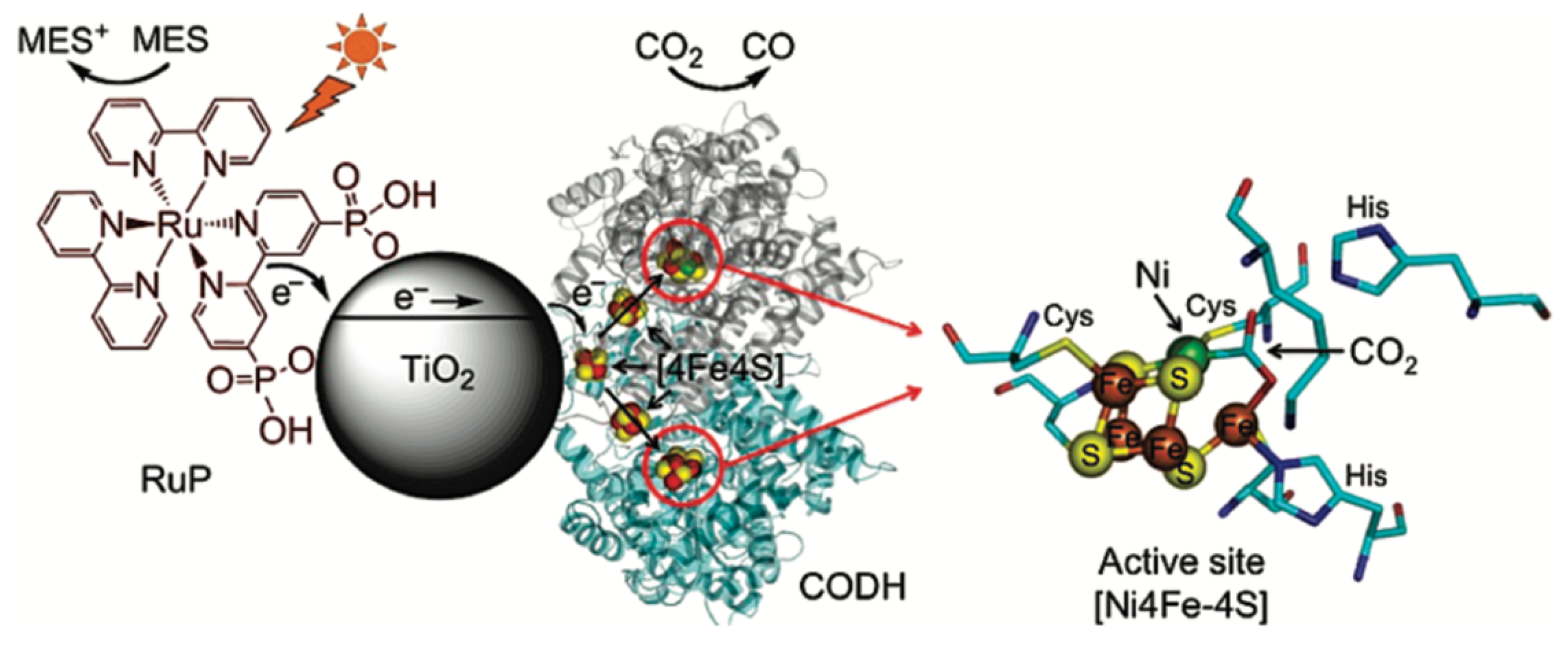
- Woolerton, T.W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S.W.; Armstrong, F.A. Efficient and Clean Photoreduction of CO2 to CO by Enzyme-Modified TiO2 Nanoparticles Using Visible Light. J. Am. Chem. Soc. 2010, 132, 2132–2133. [Google Scholar] [CrossRef]
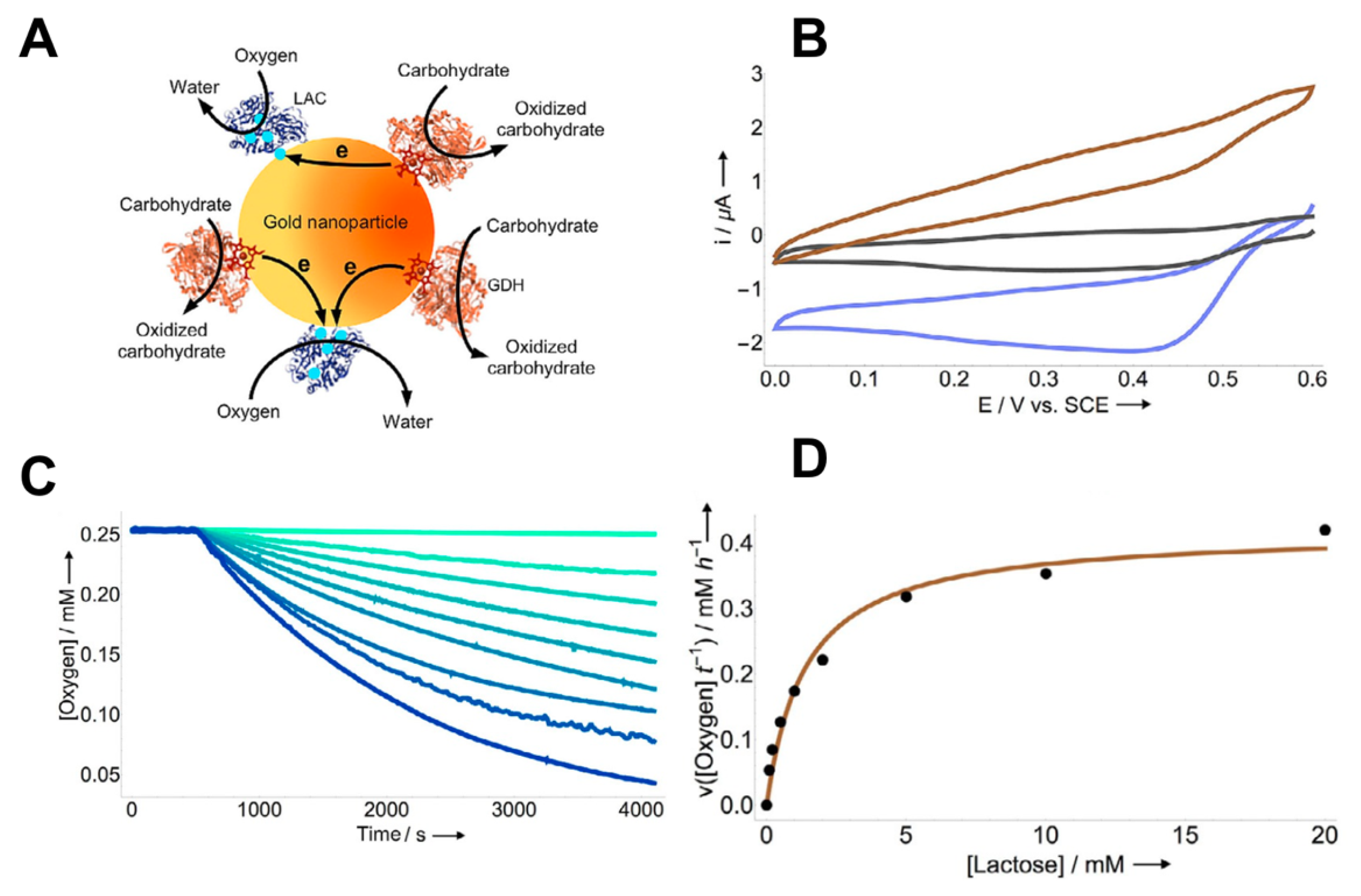
- Ratautas, D.; Ramonas, E.; Marcinkevičienė, L.; Meškys, R.; Kulys, J. Wiring Gold Nanoparticles and Redox Enzymes: A Self-Sufficient Nanocatalyst for the Direct Oxidation of Carbohydrates with Molecular Oxygen. ChemCatChem 2018, 10, 971–974. [Google Scholar] [CrossRef]
- Gutiérrez, L.F.; Hamoudi, S.; Belkacemi, K. Lactobionic acid: A high value-added lactose derivative for food and pharmaceutical applications. Int. Dairy J. 2012, 26, 103–111. [Google Scholar] [CrossRef]
- Gružauskaitė, J.; Jasinskaitė, J.; Meškys, R.; Gaidamavičienė, G.; Žalga, A.; Laurynėnas, A.; Tetianec, L.; Dagys, M. Gold-coated magnetic nanocatalyst containing wired oxidoreductases for mediatorless catalysis of carbohydrate oxidation by oxygen. Catal. Commun. 2020, 135, 105848. [Google Scholar] [CrossRef]
- Thomas, P.G.; Russell, A.J.; Fersht, A.R. Tailoring the pH dependence of enzyme catalysis using protein engineering. Nature 1985, 318, 375–376. [Google Scholar] [CrossRef]
- Marangoni, A.G. Enzyme Kinetics: A Modern Approach; Wiley-Interscience: Hoboken, NJ, USA, 2003; ISBN 978-0-471-15985-8. [Google Scholar]
- McDermid, A.S.; McKee, A.S.; Marsh, P.D. Effect of environmental pH on enzyme activity and growth of Bacteroides gingivalis W50. Infect. Immun. 1988, 56, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.E.; Chow, M.T.C. Immobilized enzyme catalysis with reaction-generated pH change. Biotechnol. Bioeng. 1974, 16, 1345–1357. [Google Scholar] [CrossRef]
- Russell, A.J.; Fersht, A.R. Rational modification of enzyme catalysis by engineering surface charge. Nature 1987, 328, 496–500. [Google Scholar] [CrossRef]
- Reeve, H.A.; Ash, P.A.; Park, H.; Huang, A.; Posidias, M.; Tomlinson, C.; Lenz, O.; Vincent, K.A. Enzymes as modular catalysts for redox half-reactions in H2-powered chemical synthesis: From biology to technology. Biochem. J. 2017, 474, 215–230. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers; Zhang, J., Ed.; Springer: London, UK, 2008; pp. 89–134. ISBN 978-1-84800-935-6. [Google Scholar]
- Armstrong, F.A.; Hirst, J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 14049–14054. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Patolsky, F.; Katz, E.; Hainfeld, J.F.; Willner, I. “Plugging into Enzymes”: Nanowiring of redox enzymes by a gold nanoparticle. Science 2003, 299, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, O.; Pasi, M.; Dandela, R.; Meijler, M.M.; Alfonta, L. Electron transfer rate analysis of a site-specifically wired copper oxidase. Phys. Chem. Chem. Phys. 2018, 20, 6159–6166. [Google Scholar] [CrossRef] [PubMed]
- Zeradjanin, A.R.; Grote, J.P.; Polymeros, G.; Mayrhofer, K.J.J. A Critical Review on Hydrogen Evolution Electrocatalysis: Re-exploring the Volcano-relationship. Electroanalysis 2016, 28, 2256–2269. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, W.Q.; Guo, X.C.; Ren, H.Y.; Wu, J.T.; Cao, G.L.; Wang, A.J.; Ren, N.Q. Continuous hydrogen production from glucose/xylose by an anaerobic sequential batch reactor to maximize the energy recovery efficiency. RSC Adv. 2018, 8, 20712–20718. [Google Scholar] [CrossRef]
- Kaida, Y.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Ultimate downsizing of D-fructose dehydrogenase for improving the performance of direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2019, 98, 101–105. [Google Scholar] [CrossRef]
- Wang, G.X.; Wang, M.; Wu, Z.Q.; Bao, W.J.; Zhou, Y.; Xia, X.H. Dependence of the direct electron transfer activity and adsorption kinetics of cytochrome c on interfacial charge properties. Analyst 2013, 138, 5777. [Google Scholar] [CrossRef]
- Lattuada, M.; Hatton, T.A. Synthesis, properties and applications of Janus nanoparticles. Nano Today 2011, 6, 286–308. [Google Scholar] [CrossRef]
- Staff, R.H.; Willersinn, J.; Musyanovych, A.; Landfester, K.; Crespy, D. Janus nanoparticles with both faces selectively functionalized for click chemistry. Polym. Chem. 2014, 5, 4097. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanomaterial | Anodic Half-Reaction | Cathodic Half-Reaction | Total Catalytic Process, E0 (pH 7.0) | Reference |
|---|---|---|---|---|
| Conductive graphite particles | Hydrogen oxidation catalyzed by hydrogenase | Nitrate reduction catalyzed by nitrate reductase | [105] | |
| Conductive graphite particles | Hydrogen oxidation catalyzed by hydrogenase moiety | NAD+ reduction to NADH catalyzed by diaphorase moiety | [106] | |
| Single-walled carbon nanotubes | Glucose oxidation catalyzed by GDH | Oxygen reduction catalyzed by LAC | [110] | |
| TiO2 nanoparticles | MES oxidation by RuP complex enhanced by visible light | CO2 reduction to CO catalyzed by CODH | E = 0.1–0.4 1 | [117] |
| AuNPs | Lactose oxidation catalyzed by GDH | Oxygen reduction catalyzed by LAC | [118] | |
| Magnetic Fe3O4 particles covered with gold casing | Lactose oxidation catalyzed by GDH | Oxygen reduction catalyzed by LAC | [120] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratautas, D.; Dagys, M. Nanocatalysts Containing Direct Electron Transfer-Capable Oxidoreductases: Recent Advances and Applications. Catalysts 2020, 10, 9. https://doi.org/10.3390/catal10010009
Ratautas D, Dagys M. Nanocatalysts Containing Direct Electron Transfer-Capable Oxidoreductases: Recent Advances and Applications. Catalysts. 2020; 10(1):9. https://doi.org/10.3390/catal10010009
Chicago/Turabian StyleRatautas, Dalius, and Marius Dagys. 2020. "Nanocatalysts Containing Direct Electron Transfer-Capable Oxidoreductases: Recent Advances and Applications" Catalysts 10, no. 1: 9. https://doi.org/10.3390/catal10010009
APA StyleRatautas, D., & Dagys, M. (2020). Nanocatalysts Containing Direct Electron Transfer-Capable Oxidoreductases: Recent Advances and Applications. Catalysts, 10(1), 9. https://doi.org/10.3390/catal10010009