1. Introduction
Lithium-ion batteries (LIBs) are among the most predominant energy storage systems for portable to stationary electronic devices. LIBs are indispensable to laptops, mobile phones, and electric vehicles due to their high energy/power density and long cycle life [
1]. Accordingly, there is a continuing increase in the technical demand for developing higher capacity/power LIBs. In particular, silicon (Si) has been intensively pursued as one of the most promising anode materials because of its high specific capacity (4200 mAh/g for Li
22Si
5), in comparison with the conventional graphite (372 mAh/g for LiC
6), and its abundance [
2]. Despite its high capacity, Si suffers from fast capacity loss caused by its large volume change (>300%), unstable solid electrolyte interphase (SEI) and the physical disintegration (cracking and crumbling) of the electrode structure during lithiation/delithiation processes [
3,
4]. Therefore, there are various research activities to control the electrochemical performance of Si anode materials.
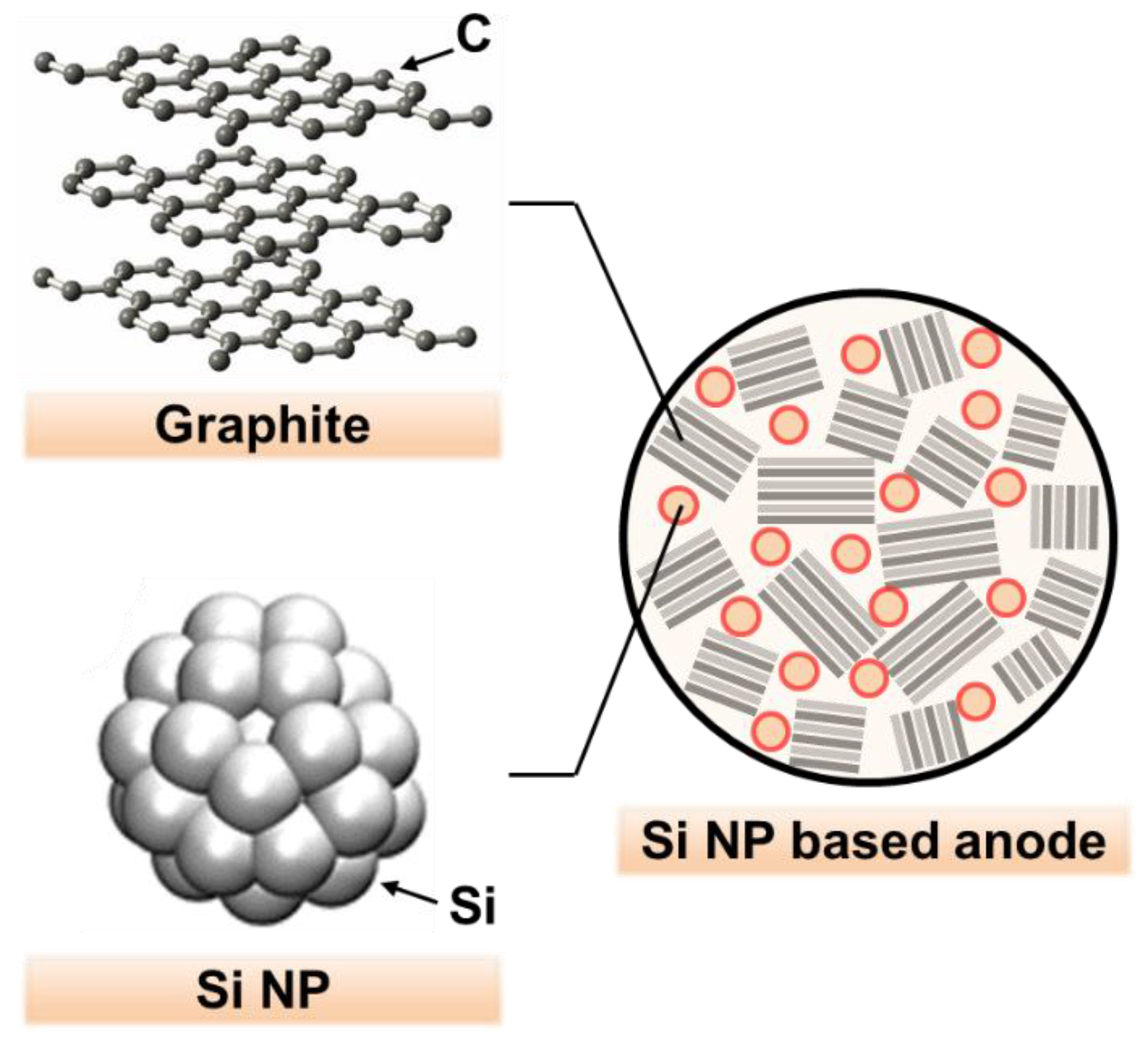
The engineering of Si nanostructures proved to be an effective method for improving capacity and cycling stability, since nano-sized Si can alleviate mechanical fractures during volume changes. It also allows fast charge transfer for a high rate capacity, having a large surface area in contact with the electrolyte [
5,
6]. There were various types of nanostructured Si materials, including nanoparticles, nano-wires, nano-tubes, and hollow nano-spheres, etc. These nanostructures have been designed and synthesized, and, subsequently, encouraging results have been achieved. Among them, nanoparticles have attracted particular interest, since the synthesis of silicon nanoparticles (Si NPs) can feasibly become commercially available, unlike other nano-shaped materials [
7]. Moreover, an in-situ transmission electron microscopy (TEM) analysis of Si NPs revealed a strong size dependence of fracture during the first lithiation process, demonstrating that nanoparticles are indeed better in terms of averting the adverse mechanical consequence accompanying electrochemical reactions [
8,
9]. In this regard, as illustrated in
Figure 1, Si NPs can be added into the void region in the polycrystalline graphite matrix, resulting in an effective increase in the overall energy density of the anode.
There has been abundant theoretical research focusing on the detailed lithiation mechanism for Si anode materials using various atomistic scale simulation methods, such as first-principle quantum mechanics (QM) and molecular dynamics (MD) approaches. Furthermore, this research is usually based on the bulk-phase crystal or amorphous silicon materials [
10,
11]. Several QM studies based on Si NPs have been reported [
12,
13]. However, due to the high computational burden of the QM method, there is still a lack of systematic address for a theoretical understanding of the lithiation process of various Si NPs. In this work, we designed reasonable structural models of Si NPs in terms of particle size and lithiation ratio through Monte Carlo (MC) simulations, and we conducted a series of first-principle simulations to understand their intrinsic electrochemical properties.
2. Computational Details
To capture the reasonable atomistic structure of pure and lithiated Silicon nanoparticles using the MC method, we employed the reactive force field (ReaxFF) [
14] with the parameters reported by Fan et al., which provided accurate predictions of a set of fundamental properties for Li
xSi alloys [
15]. We considered various atomic structures of pure and lithiated Si NPs in terms of particle size and lithiation ratio, using the in-house MC code, coupled with the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) [
16] software, to evaluate the potential energy based on the ReaxFF parameters. The acceptance of a new trial structure was determined by the Metropolis algorithm. The initial structures of pure and lithiated Si NPs were prepared from the bulk crystalline Si structure, and the lithium atoms at the surface of the optimized final structure of the Si NPs were manually located. To overcome the trapping problem in local minima while searching for a global optimization state, we carried out a simulated annealing process by adjusting the temperature in accordance with the Boltzmann factor of Metropolis criteria from 400 K to 1600 K at least five times for each case. The trial move for one temperature annealing cycle was 100,000 times, and the most stable structures of pure and lithiated Si NPs were selected for further analysis.
The first-principle density functional theory (DFT) calculations were conducted for evaluating statistical atomic distributions and theoretical lithiation potentials of the sampled lithiated Si NP models from the MC step. We used the Vienna Ab Initio Simulation Package (VASP) [
17] with Projector Augmented Wave (PAW) type pseudo-potentials for core electrons, and the generalized gradient approximation (GGA) of Perdew–Becke–Ernzerhof (PBE) [
18] was used for the exchange-correlation functional. Note that DFT calculations based on the VASP have often been used for the electrochemical characterization of battery electrodes [
19,
20,
21,
22]. An energy cutoff of 400 eV and gamma-point only k-point sampling schemes were applied for the cluster system of Si NPs. The radial distribution function analysis was conducted using the 5 ps ab initio molecular dynamics (AIMD) trajectory obtained at 300 K with the Nosé–Hoover thermostat. The radial distribution function (RDF) between Si–Si and Si–Li pairs was calculated using the Visual Molecular Dynamics (VMD) [
23] software with the following formula:
where
is the number of atoms between
and
from a given atom;
is the total number density of the system. We calculated the RDF under a periodic boundary condition with 0.1
spacing.
The most stable structures during the AIMD procedure were then fully optimized to evaluate the ground state electronic energy for each lithiated Si NP model. During the geometry optimization, all atoms were fully relaxed using the conjugate gradient method until residual forces on constituent atoms became smaller than 5 × 10
−2 eV/Å. From ground state electronic energies for pure and lithiated Si NPs and bulk crystal structures, we derived a theoretical lithiation potential using the following formula:
where
n is 18, 36, or 54,
E(Li
xSi
n) is the energy of the molecular cluster of pure and lithiated Si NP models, and
E(Li) is the energy of the body-centered cubic (BCC) phase of lithium metal.
3. Results and Discussion
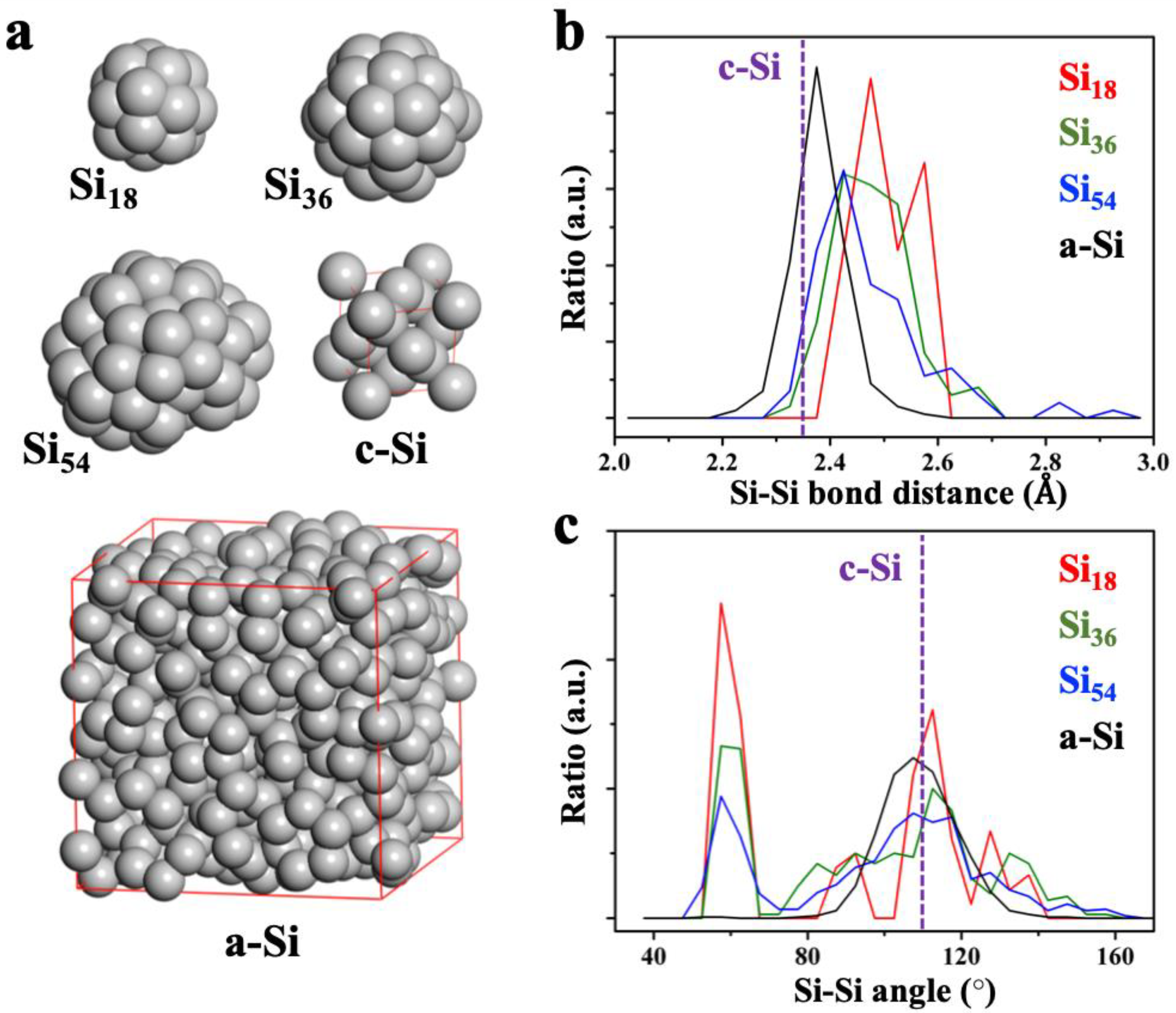
We produced three optimized pure Si nanoparticle (NP) structures with 18 (Si
18), 36 (Si
36), and 54 (Si
54) Si atoms using the MC sampling method based on the ReaxFF potential (
Figure 2a). The sizes of the Si NPs were approximately 0.7, 1.0, and 1.3 nm for Si
18, Si
36, and Si
54, respectively, including the Shannon radii for Si atoms (0.04 nm) [
24]. We found that the structures of Si NPs are highly symmetrized and that these Si–Si bonding networks are closer to the bulk amorphous Si structure (a-Si) than to the bulk crystalline Si structure (c-Si). As shown in
Figure 2b,c, the bond and angle distributions of the nearest neighbor Si atoms clearly show the structural similarity between Si NPs and a-Si (the structure of a-Si in
Figure 2a was obtained from other literature [
25]). As the size of the Si NP increases from Si
18 to Si
54, the overall distances of Si–Si bonds are shortened, getting close to the distribution of the a-Si (the average Si–Si bond distances are 2.50, 2.48, 2.47, 2.38, and 2.35 Å for Si
18, Si
36, Si
54, a-Si, and c-Si, respectively), by the decreased contribution of the surface Si atoms that have low coordination numbers (the Connolly surface [
26] to atomic volume ratios are 0.69, 0.58, and 0.52 Å
−1 for Si
18, Si
36, and Si
54, respectively). For the same reason, the angle distributions around 60° (contributions from the surface Si atoms) are decreased as the size of the NPs increases. The rest of the angle distribution shows an almost similar shape to that of a-Si (average angles are 90.83, 95.99, 97.59, 109.02, and 109.47° for Si
18, Si
36, Si
54, a-Si, and c-Si, respectively). Due to the nature of this Si bonding network, the lithiation behavior of Si NPs could be closer to a-Si than c-Si so that the faster Li diffusion is expected inside the Si NPs, as reported in previous theoretical studies [
27,
28,
29] that are based on bulk a-Si structures. In this regard, we theoretically investigated the lithiation behavior in Si NP models with high surface ratios.
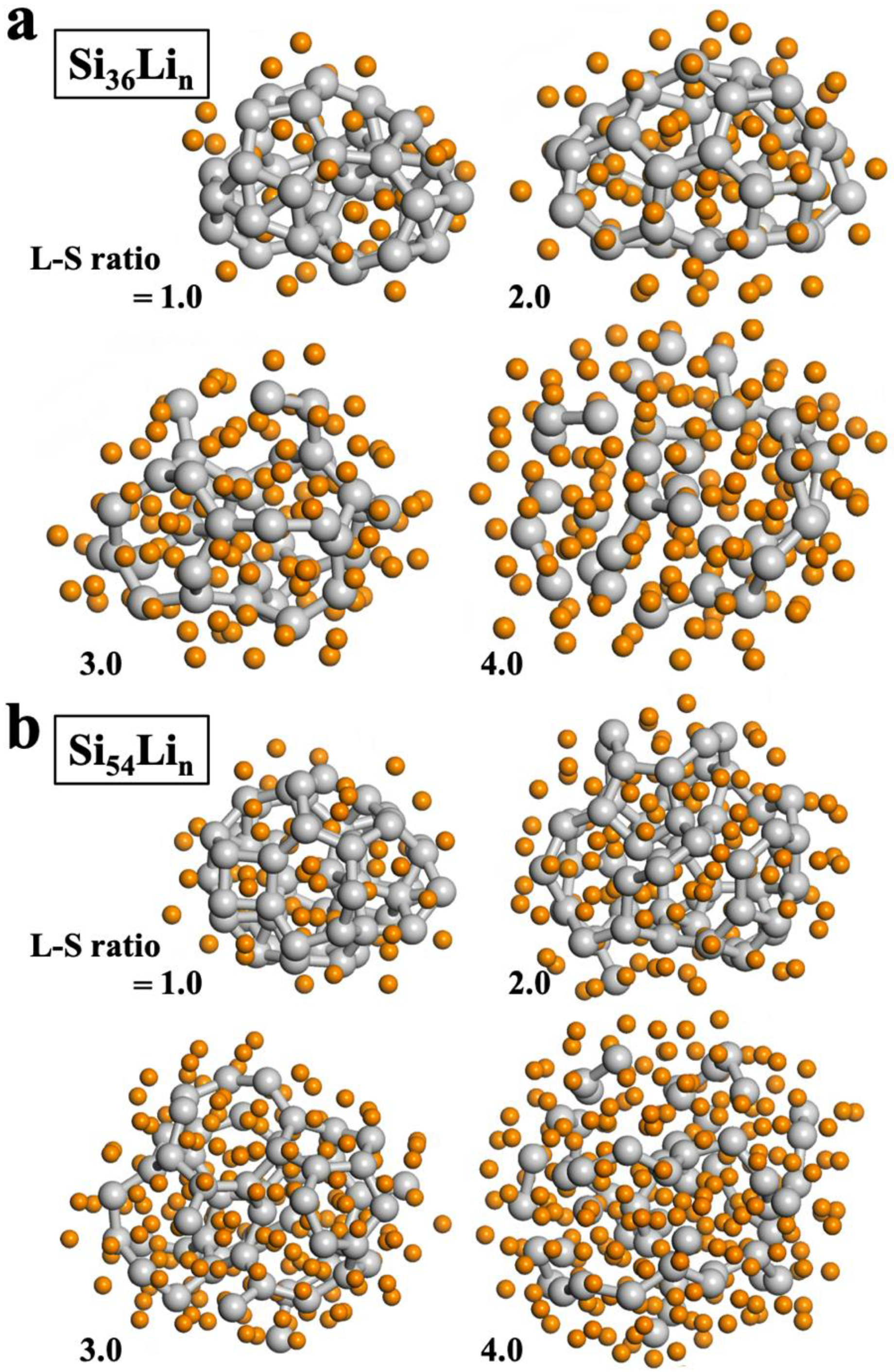
Based on the three Si NP structures, we sampled stable lithiated structures in terms of a lithium to silicon (L–S) ratio, using the MC sampling method based on the ReaxFF potential. We produced lithiated structures of Si NPs with an L–S ratio of 1.0 to 4.0. In particular, for the smallest Si
18 structure, we finely generated the lithiated Si NPs with an L–S ratio of 0.1 to 0.8 to clearly observe the characteristics of the initial lithiation process of the Si NP. The most stable lithiated Si
18 structures from an L–S ratio of 0.1 to 4.0 are shown in
Figure 3. At the initial stage of the lithiation process (0.1~0.3 of L–S ratio) of Si
18, Si NPs retain the main bonding network, almost similar to that of the original one, and most of the lithium atoms tend to form a cluster at the outer surface. At the L–S ratio of 0.2 and 0.3, Si NP structures with one lithium atom penetrated into the center were sampled to be stable. In the real lithiation process, however, the penetration of a single lithium atom into the center site would require a considerable equilibrium temperature and time because the lithium diffusion through a compact triangular or rectangular Si bonding network has a large thermodynamic barrier.
For the L–S ratio of 0.5 and above, lithium insertion results in significant change in the electronic structure of a Si NP, accompanied by change in the Si bonding network. For instance, the original Si bonding networks of the triangles and squares of Si
18 expand to form pentagonal, hexagonal, and more than heptagonal (yellow, red, and blue colored bonds in
Figure 3, respectively) Si rings as the number of lithium atoms penetrated into the particle center increases. Facile lithium diffusion could occur through the systematically expanded Si bonding networks, with two expanded Si rings (pentagons or hexagons) in an L–S ratio between 0.5 and 0.8, three hexagons in 1.0, and four hexagons and three pentagons in 2.0. In the L–S ratio of 3.0 and 4.0, heptagonal or greater Si rings are formed due to the greatly weakened Si–Si bonds by lithiation and start to generate a dangling Si bond (cyan colored bond in
Figure 3) in the final stage of lithiation.
For the Si NPs larger than Si
18, bonds between Si atoms are more easily broken during lithiation. The most stable sampled structures for Si
36 and Si
54 during the lithiation are shown in
Figure 4. For the Si
36 case, dangling Si bonds start to form at an L–S ratio of 3.0, and subsequent Si–Si bond breaking occurs at 4.0, thereby resulting in a large number of Si dumbbells and isolated Si atoms surrounded by Li atoms. In the case of Si
54, Si dangling bonds form from a lower L–S ratio of 2.0, and a large number of Si dumbbells and isolated Si atoms form at 4.0. As shown in
Figure 5, this tendency to break the Si bonding network is even more prominent in the fully equilibrated bulk-phase lithiated silicon crystal structures, where the number of Si atoms constituting the Si clusters in the crystal gradually decreases as the L–S ratio increases from 1.7 to 3.8. Therefore, the Si bonding network in a Si NP structure is relatively hard to break compared to the bulk structure, because the unstable Si atoms formed after the breakage of the Si network cannot be sufficiently stabilized by the lithium atoms on the particle surface. From these structural characteristics of the lithiation process of extremely fine Si NPs, it would seem that a certain level of content in an anode could be one of the reasons for failing to achieve the theoretical capacity. Furthermore, it could drive undesired metal plating on the surface of Si NPs, possibly causing lithium dendrite formation [
30].
For each sampled lithiated Si NP from the MC simulations, we also performed DFT calculations to analyze more precise structural and thermodynamic characteristics. First, we analyzed the radial distribution function (RDF) between atomic pairs inside Si
18, Si
36, and Si
54 NPs, using the L–S ratio 1.0 to 4.0, by obtaining ab initio molecular dynamics (AIMD) trajectories at 300 K (
Figure 6). From the position of the first peak of RDF, the bond distances and the coordination numbers between the nearest neighbor atoms of Si–Si and Si–Li pairs are almost unchanged during the lithiation of all Si NPs. The second nearest neighbor distribution patterns of Si–Si RDF show a tendency to gradually increase the Si–Si distance and decrease the broadness of the Si–Si distance distribution depending on the degree of lithiation. Considering that this tendency is obvious in large size Si NPs, the extent to which the Si bonding network is broken to form isolated silicon dumbbells or atoms occurs dominantly in the large Si NPs, as observed in the above MC simulation results.
Next, we estimated the theoretical lithiation potential profile of Si NPs by calculating the ground-state DFT energies based on the most stable structures captured during the AIMD simulations and compared them with the corresponding values from the homogeneous bulk lithiated silicon crystal structures (Si
7Li
12, Si
3Li
7, Si
4Li
13, and Si
4Li
15 phases in
Figure 5 and the additional Si
5Li
21 phase). As shown in
Figure 7, the lithiation (charging process) potential profiles of Si NPs are considerably lower than those of ideal bulk crystals because of the overall nanosized effect including the insufficient breakage of Si bonding networks. Specifically, it is expected that the lithiation capacity of Si NPs will not be able to obtain more than 3/4 of the bulk-phase capacity because the lithiation potentials become less than 0 V before the L–S ratio of 3. As discussed earlier, if the lithiation potential is lowered below 0 V versus the Li reduction potential, lithium metal deposition on the surface of Si NPs could be feasible and therefore the lithium dendrite could be easily formed on the surface of Si NPs, which may adversely affect LIB operations. This tendency is expected to be greater as the size of the Si NP is further reduced. There is little difference between Si
54 and Si
36 NPs, but for the all L–S ratios, their lithiation potentials increase by about 0.1 V in comparison with the Si
18 case. Compared to the larger Si NPs, this clearly reflects a characteristic of the hard-to-break Si bonding network of Si
18 observed through lithiated Si NP structures from MC simulations. Since the actual lithiation potential of a Si NP is expected to be lower due to the various overpotentials in actual battery operation, the same result can be expected in Si NPs larger than the size covered in this study. Therefore, more in-depth experimental and theoretical studies on the side effects of the nanonization of silicon materials on the performance of LIBs are necessary.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






