Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity

Abstract
: Understanding the mechanisms that control stress is central to realize how cells respond to environmental and physiological insults. All the more important is to reveal how tumour cells withstand their harsher growth conditions and cope with drug-induced apoptosis, since resistance to chemotherapy is the foremost complication when curing cancer. Intensive research on tumour biology over the past number of years has provided significant insights into the molecular events that occur during oncogenesis, and resistance to anti-cancer drugs has been shown to often rely on stress response and expression of inducible heat shock proteins (HSPs). However, with respect to the mechanisms guarding cancer cells against proteotoxic stresses and the modulatory effects that allow their survival, much remains to be defined. Heat shock proteins are molecules responsible for folding newly synthesized polypeptides under physiological conditions and misfolded proteins under stress, but their role in maintaining the transformed phenotype often goes beyond their conventional chaperone activity. Expression of inducible HSPs is known to correlate with limited sensitivity to apoptosis induced by diverse cytotoxic agents and dismal prognosis of several tumour types, however whether cancer cells survive because of the constitutive expression of heat shock proteins or the ability to induce them when adapting to the hostile microenvironment remains to be elucidated. Clear is that tumours appear nowadays more “addicted” to heat shock proteins than previously envisaged, and targeting HSPs represents a powerful approach and a future challenge for sensitizing tumours to therapy. This review will focus on the anti-apoptotic role of heat shock 70kDa protein (Hsp70), and how regulatory factors that control inducible Hsp70 synthesis, expression and activity may be relevant for response to stress and survival of cancer cells.1. Introduction
Growth, proliferation and differentiation of prokaryotic and eukaryotic cells are processes continuously regulated by a strict control of the intracellular protein homeostasis, and deregulation of such processes can have severe consequences on cell survival. The problems of protein folding continuously challenge the cells, and these problems become more critical when cells are subjected to extended levels of stress. Under extreme growth conditions the cells miss the capacity to activate the apoptotic cascade and may undergo necrosis, whereas at lower levels of injury they activate the apoptotic program and undergo a relatively ordered form of death. Necrosis is an unprogrammed, disorderly form of cell death which occurs via rupture of cell walls and organelles, whereas apoptosis is a highly regulated, programmed form of cell death. However, if the level of stress is sufficiently low cells may attempt to survive by producing a set of highly conserved protecting proteins, namely heat shock proteins (HSPs), which assist folding of damaged proteins, preserve them from degradation, and impede toxic intracellular aggregates from threatening cell survival. Heat shock proteins are molecular chaperones that protect cells from extreme physiological, pathological and environmental insults, as well as under normal conditions they are involved in changing protein conformation by promoting multiprotein complex assembly and disassembly. Their rapid induction results in a coordinated series of biochemical and genetic events that are collectively referred as the “heat shock response” (HSR) [1] which involve the maintenance of the steady-state for the majority of the intracellular proteins by either providing stability or driving their proteasomal degradation, the proper folding of denatured or damaged polypeptides, and the inhibition of the apoptotic cascade at multiple levels [2-5]. This explains why proteins adapted to stress like heat shock proteins are so important for the growth and survival of cancer cells, but also why HSPs play critical roles during development and differentiation of untransformed cells [6-10]. A large body of evidence supports the role of the heat shock proteins in maintaining the cancer phenotype, and stressor conditions such those deriving from aneuploidy, copy number variation, and transcriptional alterations have been shown to be responsible for their high expression in tumours. Such a dependence on the normal cellular functions of genes that act in oncogenic pathways in a rate-limiting manner is known as “non-oncogene addiction” (NOA), and heat shock proteins, although not oncogenic per se, represent examples of NOA genes for their unusual high expression in cancer and the efficacy of selective therapeutic interventions resulting from their impairment [11-13].
2. Structure, Activity and Regulation of Heat Shock Proteins
Mammalian heat shock proteins are generally classified by their relative molecular weight and function into five major families, including the HSP90, HSP70, HSP60, HSP40 and small heat shock proteins family. Members of each family may be expressed both constitutively and induced during stress, and are characterized by chaperone activity controlled, or not, by reaction cycles of ATP binding, hydrolysis and release (Table I). Intracellular localization varies according to function, and heat shock proteins may localize both in the cytosol and nucleus (Hsp27, Hsc70, Hsp70 and Hsp90), in the lumen of the endoplasmic reticulum (Grp78 and Grp94), or reside in the mitochondrial space (Grp75, Hsp60 and Hsp90 homolog TRAP1) [14-16]. In general, HSPs share a molecular architecture consisting of an N-terminal adenine nucleotide binding domain (NBD), a C-terminal domain that includes a peptide/substrate binding domain (PBD/SBD), and a flexible interdomain linker that may both facilitate chaperone ATPase activity (Hsp70) and client recognition (Hsp90) [17-19]. Binding of substrates to the PBD domain occurs through a carboxyl-terminal chaperone motif (EEVD), and is regulated both intramolecularly by NBD nucleotide occupancy, hydrolysis and release, and intermolecularly by the activity of diverse classes of co-factors that interact with both the NBD and the PBD domain in a coordinated sequential manner [20,21]. Co-chaperones and nucleotide exchanging factors (NEFs) regulate substrate affinity and fulfil the ATPase driven cycle of major chaperone proteins, as shown for Hsp70 proteins which bind substrates delivered by co-chaperone J protein Hsp40 and release them upon the interaction with specific NEF co-factors [22,23]. Effective chaperoning activity of Hsp70 in the ‘open’ binding conformation of its PBD/SBD domain is rapid and transient when orchestrated by co-chaperones and NEFs, but iterative if the native state of the client protein is not attained on release [24,25]. J proteins are responsible for substrate binding and Hsp70 ATPase activity, whereas NEF factors catalyze the subsequent ADP release, dictating the duration over which a substrate remains associated to the chaperone. BAG proteins, for example, are nucleotide-exchanging factors that together with Hsp40 help regulate Hsp70 ATPase activity, but also work with Hsp70 in the targeting and selection of certain substrates. BAGs bind the NBD domain of Hsp70 once ATP hydrolysis has occurred and induce nucleotide exchange by stimulating ADP release. The regulatory domain at the C-terminus provides also the site of interaction for tetracopeptide repeat (TPR)-containing proteins, a class of different co-factors that bind and orchestrate nucleotide turnover and substrate binding/affinity using TPR, a 34-aminoacid motif forming an anti-parallel α-helical hairpin, as structural motif. TPR proteins exert multiple functions, and they interact with multiple chaperone proteins to either assist substrates transfer and folding (HOP) or to promote their degradation (CHIP) [26-29].
Co-chaperones generally rise to HSP levels when protein folding is required, and their exact exploitation depends on the type of protein conformational requirements. Raising co-chaperone and NEF levels without a concomitant increase in HSP expression, instead, reduces chaperone activity or may cause a premature release of captured client proteins in the non-native state. In addition, binding specificity of HSPs may be conditioned by structural elements of the non-native protein, such as an increased exposure of hydrophobic amino acids or specific peptide sequences. Hsp70, for example, binds a wide range of unfolded proteins with low sequence selectivity during the early stages of protein folding and misfolding, recognizing extended stretches of hydrophobic nonpolar amino acids in polypeptides undergoing synthesis on ribosomes or in proteins denatured by stress conditions [30,31]. Hsp90, instead, is a heat shock protein that possesses a higher degree of specificity for metastable signalling proteins, including protein kinases, cell cycle regulators and transcription factors, recognizing and binding specific conformational epitopes [32]. Such a diverse substrate specificity depends on the sequence selectivity of the SBD domain, which may recognize unstable polypeptide chains by either the aminoacid composition or the surface features [33-37]. In both cases, molecular chaperones prevent unwanted intermolecular and intramolecular interactions of unfolded proteins, and mask them to premature degradation.
3. HSF1 and Modulation of the Heat Shock Response
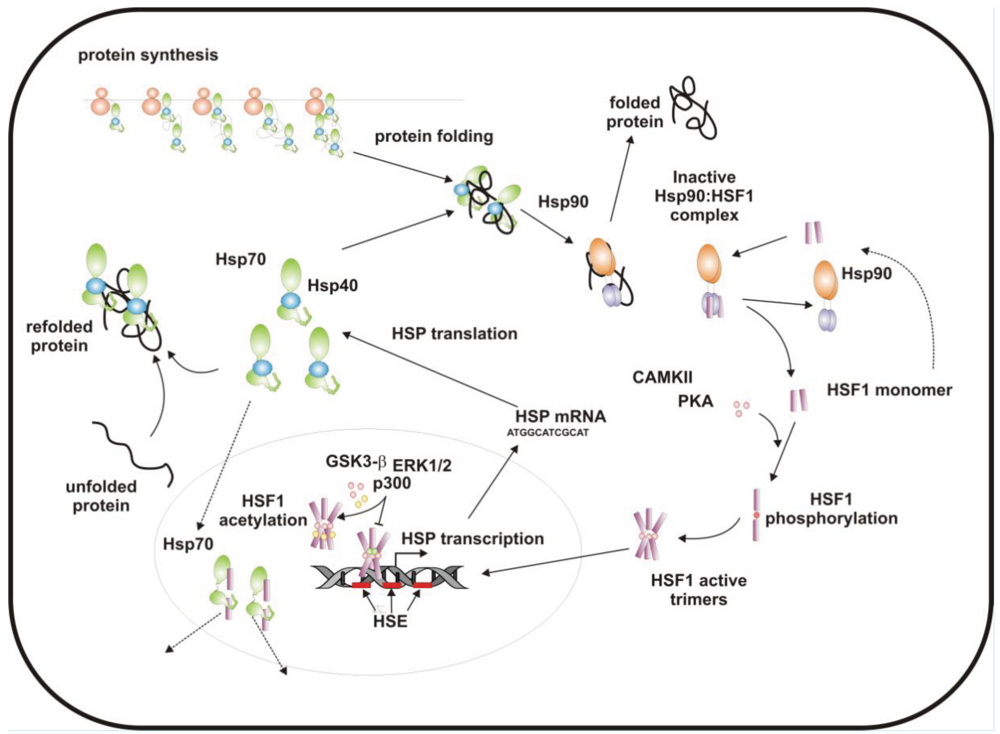
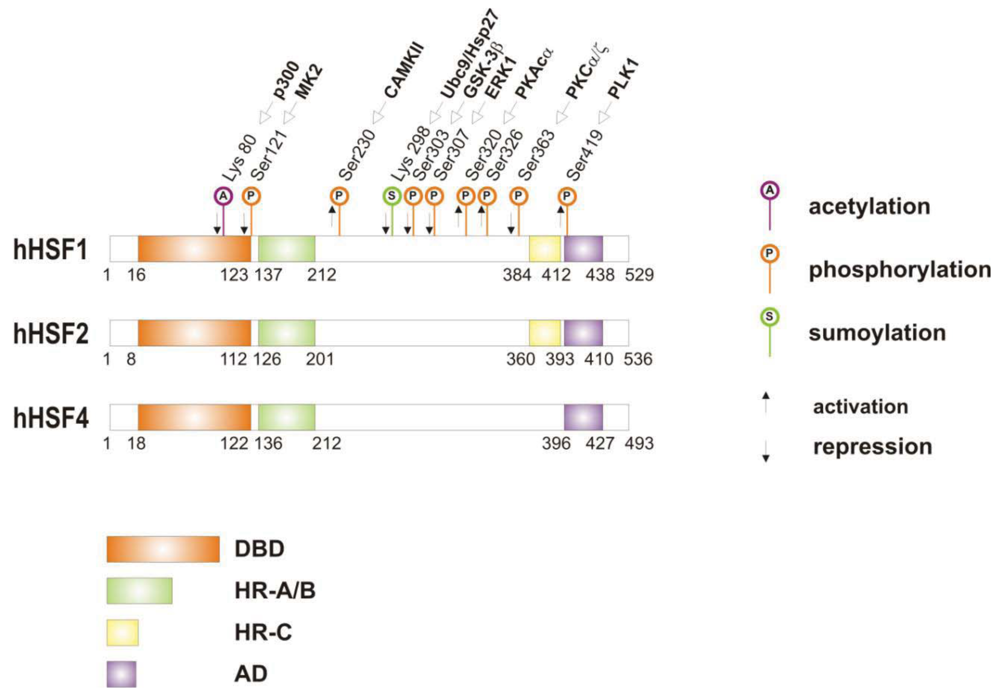
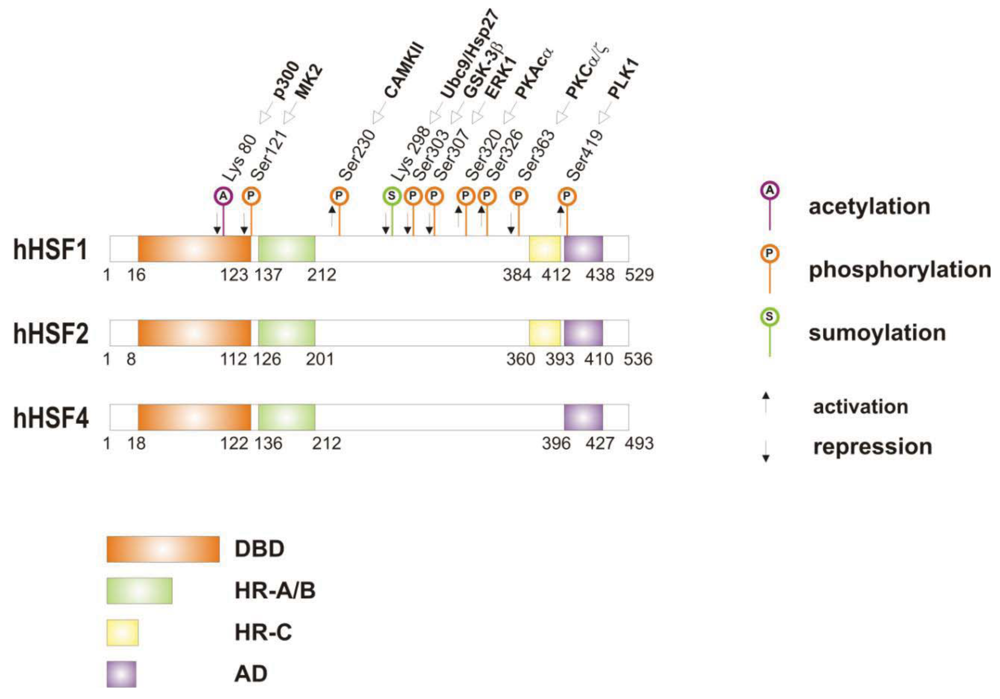
When cells are subjected to high temperatures (heat shock) or are exposed to stress, such as oxidative stress, heavy metals or proteotoxic drugs, they respond by increasing heat shock gene expression through the activation of stress-inducible transcription factors (heat shock factors, HSFs). HSFs are transcription factors that bind specific cis-acting sequences upstream of the heat shock gene promoters called heat shock elements (HSE), which consist of multiple adjacent inverted arrays of the 5′-nGAAn-3′ binding site [39-41]. Intermingled to HSE elements there are conventional AT-rich (TATA) and GC-rich (SP1) start sites, CCAAT boxes and AP1/AP2 control elements, suggesting that heat shock genes may be both induced in response to stress and expressed under non-stress conditions [42-44]. TBP (TATA-binding protein) and GAGA are transcription factors that bind HSP gene promoters under physiological conditions, while heat shock factors, like HSF1, regulate HSP expression in response to stressful insults [45-47]. However, binding sites for HSFs may be unmasked by TBP and GAGA proteins as well [48,49], which allow transcriptionally paused RNA polymerase II (RNA Pol II) to remain in place and prime heat shock genes for transcription immediately after stress induction [50-59] (Figure 1). Heat shock factors are transcription factor proteins composed of several functional domains, including a DNA-binding domain (DBD) and two oligomerization domains (HR-A/B) at the protein amino-terminus, a third oligomerization motif (HR-C) and an activation domain (AD) at the carboxyl-terminus, and one or more regulatory domains intermingled to these [58,60,61] (Figure 2). Four major heat shock factors (HSF1-4) have been identified in vertebrates, and three of these, HSF1, HSF2 and HSF4 have been characterized in humans on the base on their function, expression, post-translational modifications (PTMs) and partner protein interactions [38]. HSF1 has a critical role in the regulation of the heat shock stress response and in the expression of HSPs, HSF2 and HSF4 are involved in immunity and differentiation processes, while HSF3 is inactive and maintained in human cells as pseudogene [69,70]. Sequencing of the predicted amino acid regions of HSF1 has provided evidence that exons coding for DBD and HR-A/B domains have been highly conserved among orthologues during evolution, while differences have been accumulating in the HR-C, AD and regulatory domains [68]. The DBD domain consists of helix-turn-helix motifs which form compact globular structures that regulate DNA binding and target-gene recognition [61,63]. The hydrophobic heptad repeat (HR)-A/B domains, instead, mediate both spontaneous and inducible oligomerization of HSF1, by forming a coiled coil conformation typical of leucine zipper-containing proteins [64-67]. HSF1 contributes to many physiological functions during cell growth and differentiation processes, and regulates genes critical for energy production, signal transduction and cytoskeletal organization [71,72]. However, it is much more important for modulating HSP synthesis and stress response, as cells lacking this protein do not develop thermotolerance and are susceptible to stress-induced apoptosis [73,74].
Genetic knockout of HSF1 in transgenic mice, indeed, does not impair organ systems development and birth, but mice lacking HSF1 that are exposed to inflammatory insults or other types of stress have elevated levels of apoptosis [75-77]. Consistently, HSF1 protects cells experiencing constant intrinsic high levels of stress, such as during ageing or tumorigenesis. Both age-dependent collapse of cell proteostasis and tumour-associated increase of protein misfolding are events regulated by HSF1, strengthening the evidence of the unique role of this protein in preventing global instability of proteome in several pathophysiological conditions [78-81].
HSF1 activation occurs as a general response to stress, such as heat shock, oxidative stress and exposure to proteotoxic chemicals, through a series of regulatory events that include dissociation from inhibitor heat shock proteins Hsp70 and Hsp90, HSF1 transition from monomer to trimer, localization to the nucleus, inducible serine phosphorylation, and binding to HSE elements of target gene promoters. Under non-stress conditions HSF1 exists as an inactive monomer in the cytoplasm due to intramolecular bonds between HR-C and HR-A/B domains and its association with Hsp90 and Hsp70 [82-84], whereas in response to stress HSF1 is released from Hsp70 and Hsp90 proteins and forms homotrimers that bind to cis-acting DNA elements HSEs upon phosphorylation [85-91]. Under circumstances of prolonged stress, however, HSF1 is inactivated by negative feedback mechanisms which include the association with newly synthesized HSPs, and additional posttranslational modifications [92-94]. Posttranslational modifications have a critical impact on the HSF1 activation-attenuation cycle, and are finely modulated according to HSF1 subcellular localization and partner-protein interaction [71,95]. Sumoylation is a modification that occurs rapidly and transiently during stress, which is triggered by Hsp27 binding to nuclear HSF1 and HSF1 phosphorylation at Ser303 prior to SUMO (small ubiquitin-related modifier) conjugation at Lys298 [96-98]. Acetylation, by contrast, proceeds more slowly and requires stress-induced p300-CBP (CREB-binding protein) acetyltransferase activity [81,99,100] (Figure 2). While sumoylation impairs HSF1 transactivation activity without affecting DNA binding [101,102], acetylation of the DBD domain attenuates HSF1 binding to HSE elements of stress-responsive genes and inhibits heat shock gene transcription at comparable level. However, when recruited directly at the heat shock gene promoters p300-CBP acetylates histones and participates to HSF1 transcription activity. This occurs in conjunction with STRAP (stress response activator protein), a DNA damage-responsive protein that regulates chromatin-bound levels of both phosphorylated HSF1 and active p300-CBP. Cells depleted of STRAP, indeed, do not activate HSF1-dependent transcription of heat shock proteins, and have impaired survival under stressful conditions [103-107]. As regard to phosphorylation, 12 serine residues participate in the regulation of HSF1 transcriptional activity [108], and among these Ser230, Ser320, Ser326 and Ser419 activate HSF1, whereas Ser121, 303, 307 and 363 contribute to its repression [88,109,110]. Deletion mapping analysis has demonstrated that Ser307 and Ser303 are constitutively phosphorylated in a sequential manner by GSK3B and ERK1/2 MAP kinases, respectively, and inhibit HSF1 transcriptional activity during stress recovery, whereas serine 363 undergoes phosphorylation upon PKCα and ζ expression [109] and inhibits HSF1 transactivation activity rather than HSF1 binding to HSE elements [111].
By comparison, HSF1 is activated by phosphorylation at Ser230 and Ser326, which stimulates the transcriptional activity of DNA-bound HSF1, and by phosphorylation at Ser419 and Ser320 that modulates HSF1 oligomerization, nuclear translocation and DNA binding, respectively [88,108]. Protein kinases that regulate Ser230, Ser419, and Ser320 phosphorylation are calcium/calmodulin-dependent protein kinase II (CAMKII), PLK1 (polo-like kinase 1), and protein kinase A (PKAcα), respectively, whereas protein kinases responsible for phosphorylation of HSF1 at Ser326 are not known [88,112]. In this context, modulation of HSF1 phosphorylation may be of therapeutic benefit in the treatment of diseases that have an underlying abnormality in protein conformation and activity, as demonstrated in cancer cells where HSPs help to maintain the malignant phenotype, and their levels may further induced by chemotherapy as side effect of treatment. This has been demonstrated for several stress-related kinases, including p38MAP kinase, GSK3β and JNK [113-118]. Consistently, proteasome inhibitors, a well-known class of potent anti-cancer agents, up-regulate HSPs synthesis in cancer cells by stimulating hyperphosphorylation and DNA binding of HSF1, and this occurs through enhanced expression and activity of p38MAPK kinase [119-121]. Likewise, exposure to heat shock and oxidative stress activate p38MAPK and enhance HSF1 hyperphosphorylation, while p38MAPK inhibition reduces the rise of Hsp70 levels brought about by these harmful conditions and enhances stress-induced apoptosis (). Treatment with compounds that activate GSK3β, JNK and ERK1/2 MAP kinases may be equally effective at inhibiting Hsp70 expression and chaperone-mediated cytoprotection in transformed cells, as these protein kinases affect negatively HSF1 transcriptional activity and heat shock gene expression, as previously demonstrated [122-125].
4. Regulation and Expression of Inducible Hsp70 Chaperone Protein
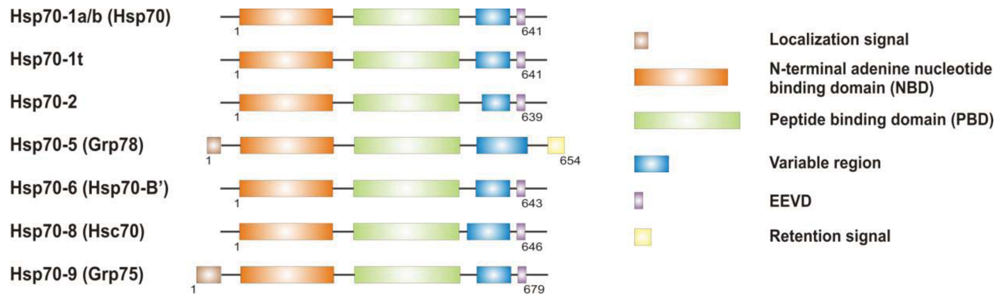
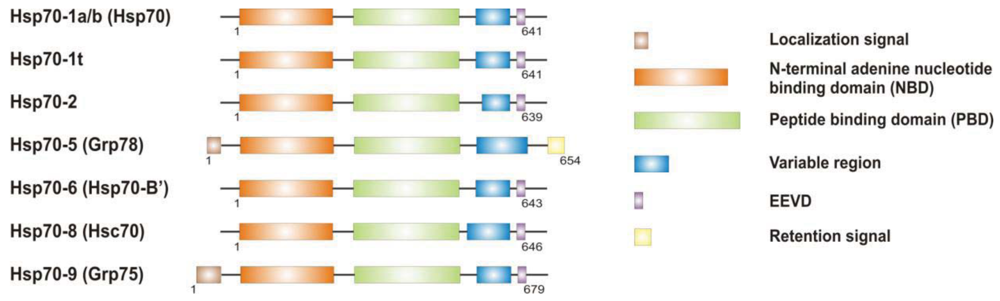
It is now clear that cancer arises through a multistep, mutagenic process whereby transformed cells acquire a common set of properties including unlimited proliferation potential, self-sufficiency on growth signals, and resistance to antiproliferative and apoptotic cues. Many of these phenotypic traits can be brought about by numerous genetic alterations, which alongside gain-of-function mutation and amplification of oncogenes, or loss of function of tumour suppressor proteins, include overexpression of key proteins with non-oncogenic functions. Besides, cancer cells have extensively rewired pathways for growth and survival that underline their malignant phenotype, and thus it appears important to identify either critical nodes or functional regulator proteins in the oncogenic network whose inhibition may result in system failure, that is apoptosis. As previously mentioned, tumours exhibit proteotoxic stress evidenced by their frequent constitutive activation of the heat shock response, and heat shock proteins are key players of many stress support pathways that help cancer cells to cope with the stress of their oncogenic state [126]. Among them, Hsp70 proteins are critical for growth and survival of transformed cells, because they serve both as chaperones, assisting folding of nascent polypeptides and the intracellular localization of native proteins, as they act to protect cells to repeat exposure of noxious stimuli which would otherwise cause lethal molecular damage and induce apoptosis. Eukaryotic cells possess several structurally similar Hsp70 family member proteins, but the fact that their simultaneous deletion is lethal for the cells suggests that Hsp70 proteins may serve several overlapping chaperone functions but also perform specific activities that cannot be compensated by others. In most of the cell types heat shock 70kDa proteins are detectable at significant levels in unstressed cells and further increased in abundance on stress [127,128]. Whereas inducible protein expression is regulated by HSF1 during stress, in unstressed conditions transcription factors different from HSF1 (i.e., STAT, CCAAT-box and SP1) modulate basal protein expression [129]. Eight Hsp70 family member proteins have been identified, and distinguished on the base of their amino acid composition, expression rate, subcellular localization and stress responsiveness. Among them, Hsp70 (Hsp70-1a; Hsp70-1b) and Hsp70-6 (Hsp70-B′) are stress-inducible family member proteins, whereas Hsp70-1t, Hsp70-2, Hsp70-5 (Bip, Grp78), Hsp70-8 (Hsc70) and Hsp70-9 (Grp75, mortalin) are chaperone protein expressed constitutively in a tissue specific-manner [126] (Figure 3).
Under normal growth conditions Hsp70s function as ATP-molecular chaperones and assist protein folding. On stress Hsp70 proteins cope with the increased concentration of unfolded and denatured proteins, and prevent toxic aggregates from threatening cell survival by inducing apoptosis [130,131]. Such an ability to disable apoptosis relies on the capacity of Hsp70s to interact with multiple unstable components of the programmed death machinery, including pro- and anti-apoptotic proteins, DNA-damaging factors, or stress protein kinases. Most of the studies published on these proteins deal with the two major members of the Hsp70 family, the inducible Hsp70 molecular chaperone (also known as Hsp72) and cognate protein Hsc70. Inducible Hsp70 is critical for transformation, proliferation, poor differentiation and invasion of cancer cells, whereas Hsc70 provides general assistance to normal cells [132-134]. Unlike Hsc70, which is constitutively expressed in non-tumor cells and tissues, Hsp70 is present at relatively low levels in untransformed cells and it's rapidly induced upon stress. Depletion of inducible Hsp70 however does not affect cell survival but results critical to withstand stressful insults induced by UV exposure, osmosis, and hyperthermia [135-138]. Conversely, constitutive high levels of Hsp70 are frequently observed in cancer cells, in which Hsp70 confers resistance to stress-induced apoptosis, serves in suppression of default senescence, and is associated with metastasis development and drug resistance [139-141]. In tumors Hsp70 may be also expressed regardless HSF1 transcriptional activity, consistent with the notion that different factors other than HSF1 may regulate Hsp70 status and activity [142]. STAT1, STAT3 (signal transducer and activator of transcription) and NF-IL6/CEBPB are the transcription factors responsible for Hsp70 synthesis in the absence of stress [143,144], as inhibition of both STAT and NF-IL6 activity reduces basal expression of Hsp70 chaperone protein in cancer cells lacking HSF1 or expressing a defective protein [142,145-147]. Whereas STAT3 binds sequences at the hsp70 promoter (5′-CTGGRA-3′) different from HSE elements and competes with HSF1 for Hsp70 expression on stress [148-150], STAT1 induces Hsp70 expression by recognizing different elements in the promoter region (GAS, interferon Gamma Activated Sequences), and does not compete with HSF1 for transcription [151]. By comparison, NF-IL6 (nuclear factors IL6) proteins (i.e., CCAT/enhancer-binding protein, C/EBP) recognize STAT-responsive elements and cooperate with HSF1 for inducing Hsp70 [152], but they can also bind Hsp70 proximal promoter regions together with STATs and produce stronger activation of Hsp70 transcription in the absence of stress [153,154].
5. Anti-Apoptotic Activity of Hsp70
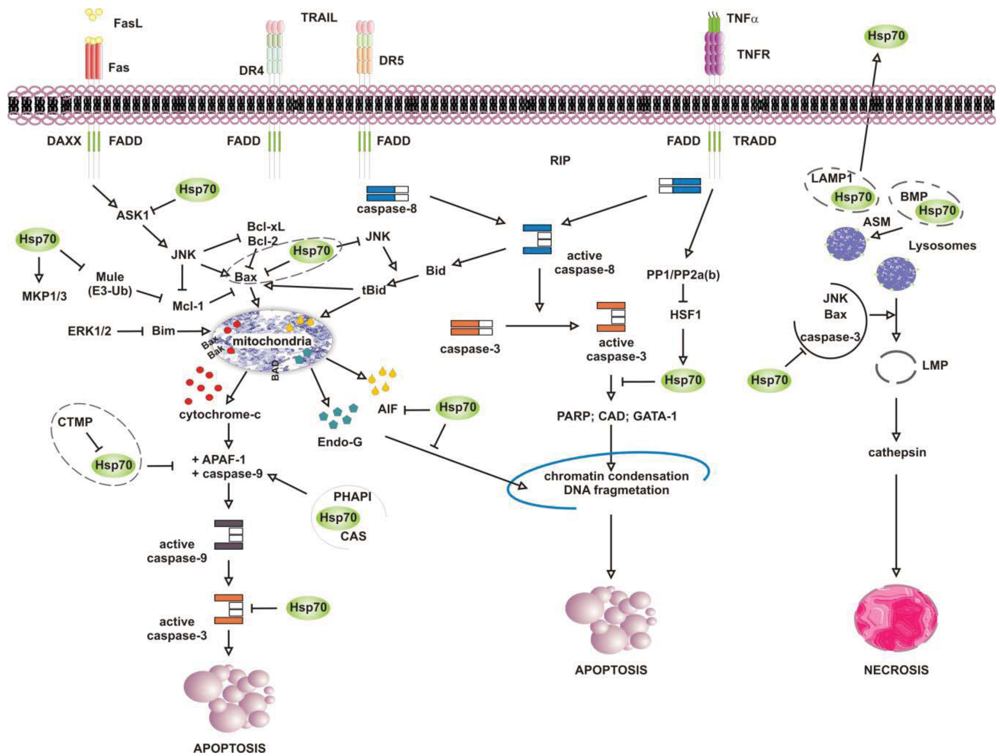
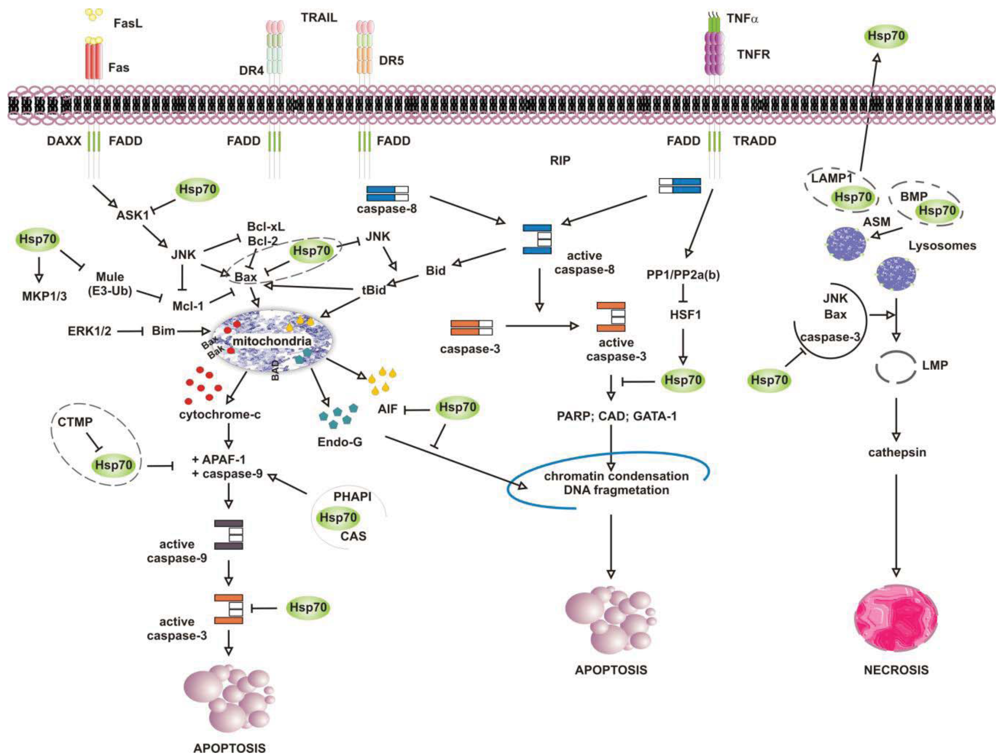
Numerous studies have attributed the survival function of Hsp70 to the ability to suppress the engagement of apoptosis, since cells that contain elevated levels of this chaperone have an increased ability to tolerate different types of stress at several point of the apoptotic cascade [155-157] (Figure 4). Stress-inducible Hsp70, in fact, acts as a natural inhibitor of several stress-kinases at the beginning of cell death, as well as it protects cells from apoptosis by binding and modulating the activity of various pro- and anti-apoptotic proteins at transcriptional and posttranslational level. By impairing Janus kinase (JNK) activity, in fact, Hsp70 prevents JNK-induced phosphorylation and inhibition of Bcl-2 and Bcl-×L anti-apoptotic proteins, and promotes cell survival through the maintenance of mitochondria stability. Similarly, Hsp70 when induced prevents Bid-mediated damage to mitochondria and inhibits the release of pro-apoptotic cytochrome c and SMAC from these organelles through the inhibition of SEK1-dependent phosphorylation of JNK and JNK-mediated activation of Bid [158-160]. Hsp70, thus, can prevent stress-induced activation of JNK maintaining the levels of inactive dephosphorylated JNK or, alternatively, inhibiting its activating phosphorylation by SEK1. Either scenario, however, leads to a block in JNK signaling and helps to prevent stress-induced JNK-mediated loss of survival. Conversely, JNK counteracts Hsp70 anti-apoptotic activity and facilitates cell death by phosphorylating HSF1 and inhibiting the transcription of Hsp70 [160-163]. In a similar fashion, Hsp70 modulates ERK1/2 MAP kinase activity during stress, through the activation of MKP1 and MKP3 phosphatases [164]. Tumor cells that overexpress Hsp70 are more resistant to stress because of MKP-dependent dephosphorylation and inactivation of pro-apoptotic ERK1/2, and do not undergo stress-induced cell death [165-168]. In contrast, the accumulation of phospho-ERK1/2 is typical of Hsp70 knock-out cells undergoing apoptosis that do not exhibit MKP1 and MKP3 activity. Likewise, Hsp70 competes with RAF for binding co-chaperone Bag-1, which supports RAF-induced activation of ERK1/2. Through the association with Bag-1, Hsp70 prevents phosphorylation of ERK1/2 by RAF and helps to maintain survival of cancer cells exposed to stress [169-170].
Release of pro-apoptotic factors from mitochondria is a second pivotal point within the intrinsic apoptotic pathway which is regulated by Hsp70, since Hsp70 binds and alter expression and activity of several pro- and anti-apoptotic molecules, including Bcl-2 family member proteins Bax and Bak [171-174]. Exposure to heat has been shown to alter multidomain pro-apoptotic Bax protein, which oligomerizes and inserts into mitochondria, creating transition pores at the outer membrane that serve for the release of apoptogenic factors cytochrome-c, SMAC/Diablo and AIF at the beginning of the apoptoic cascade [175-177]. Under these circumstances, JNK-dependent phosphorylation and inhibition of 14-3-3 proteins renders Bax free and competent for mitochondrial insertion, unless recognized and maintained inert in the cytoplasm by Hsp70 [178,179]. At the same time, JNK phosphorylates and primes anti-apoptotic Mcl-1 protein for degradation via the interaction with Mule ubiquitin ligase, preventing Mcl-1 from binding and inhibiting activated Bax. By binding JNK and Mule, Hsp70 prevents Mcl-1 degradation, which in turn binds and impedes Bax from forming transition pores at the mitochondrial membrane [180]. Indeed, chronic myeloid leukemia (CML) cells that express BCR-ABL oncogenic kinase exhibit increased levels of hyperphosphorylated HSF1, inducible Hsp70 and anti-apoptotic Mcl-1 proteins, while cells in which BCR-ABL is inhibited have less Hsp70, and the activation of pro-apoptotic Bax induced by imatinib leads to cell death [181]. Patients who relapse to imatinib have an even larger increase in Hsp70 and Mcl-1 levels, and drug resistance is largely ascribed to the anti-apoptotic activity of these two proteins [182-188].
Downstream to mitochondria Hsp70 possesses the ability to inhibit the formation of functionally competent apoptosome by directly associating to Apaf-1 (apoptotic protease activating factor 1) CARD domain (caspase recruitment domain) and masking it to initiator caspase 9 [189,190]. The ATPase activity of Hsp70 is crucial for binding Apaf-1, since complex formation does not occur in the absence of ATP or in the presence of an ATPase dead Hsp70 protein [191]. However, survival activity of Hsp70 may be counteracted by proteins located in the cytoplasm or released from mitochondria on stress, such as carboxyl-terminal modulator CTMP protein, CAS and PHAPI. CTMP localizes at the plasma membrane, where it acts as tumor suppressor protein of oncogenic PKB/Akt kinase, or resides in the intermembrane mitochondrial space, whereas CAS and PHAPI localize mainly in the cytoplasm. Upon phosphorylation CMTP is released from mitochndria and binds Hsp70 prior to its association with both Apaf-1 and cytochrome c [192]; cellular apoptosis susceptibility protein CAS and tumor suppressor PHAPI, instead, associate and prevent Hsp70 protein from binding Apaf-1 in conditions of low-expression Hsp70 levels, since on stress inducible Hsp70 binds Apaf-1 and cytochrome c despite CAS and PHAPI expression [193]. Hsp70 overexpression provides also resistance to Apaf-1-independent apoptosis, like that promoted by AIF (apoptosis inducible factor). AIF plays a role in survival of several tumour cell types, and like cytochrome c it effluxes from mitochondria in response to damages induced by noxious stimuli. Cytoplasmic AIF translocates into the nucleus where it induces caspase-independent chromatin condensation and DNA fragmentation [194-196] In the presence of Hsp70 nuclear import and death-inducing activity of AIF are prevented, whereas silencing of Hsp70 or inhibition of its ATPase activity enhance AIF pro-apoptotic functions and stimulate cells killing [191,197,198]. Notably, AIF mutants that lack the N-terminal portion of the protein are not inhibited by Hsp70, suggesting that this region is recognized by Hsp70 when AIF effluxes from mitochondria [199,200]. Mapping more in detail the AIF binding domain has permitted to design AIF-derived decoys for Hsp70, capable of sensitizing transformed cells to drug-induced apoptosis by competing with endogenous AIF for binding Hsp70 [201]. Similarly, Hsp70 associates and inhibits endonuclease G (EndoG), a DNAse responsible for caspase-independent disruption of nuclear integrity [202], although since EndoG forms a complex with AIF, the Hsp70/EndoG association might be an indirect consequence of Hsp70 binding to AIF [203].
At the plasma membrane the scenario is further complicated by what is known as the “heat shock paradox”, the seemingly ability of Hsp70 to induce both cytoprotection and cytotoxicity in response to different exogenous stimuli that activate TRAIL-R, TNFR1 and FasR death receptors. Death receptors are protein tyrosine kinases that oligomerize upon ligand binding and recruit a multitude of downstream proteins in what is known as “death-inducing signaling complex” (DISC). Proteins at DISC that bind death receptors directly or through specific adaptor proteins (i.e., FADD and TRADD) engage multiple intracellular signals that promote apoptosis through activation of either caspase 8 or the ASK-1/JNK pathway. With respect to this, Hsp70 has been reported to interact with DR4 and DR5 TRAIL receptors and affect ligand-induced assembly of DISC complex in both normal and tumor cells [204,205]. However, unlike normal cells that appear to be refractory to death receptor-induced apoptosis, tumor cells are sensitive to TRAIL receptors activity and undergo apoptosis if not protected by Hsp70 [206-208]. This occurs in cells that require amplification of the apoptotic signals through mitochondria injury (type II), whereas not in the cells that activate the apoptotic cascade without mitochondrial involvement (type II cells) [209]. This is consistent with the findings that Hsp70 does not bind and modulate initiator caspases at the plasma membrane (i.e., caspase-8) but it functions downstream to mitochondria where it prevents the activity of effector caspase 3. In a similar fashion, Hsp70 inhibits apoptosis induced by TNFα and Fas ligands, through the inhibition of receptor-induced JNK-mediated mitochondrial injury and caspase 9 activity [210-212]. Under these conditions Hsp70 prevents cell death induced by TNFR1 receptor through the inhibition of ASK-1 (apoptosis signal-regulating kinase 1) and JNK kinase activity [163,213]. This confers protection from JNK-induced phosphorylation of BH-3 pro-apoptotic protein Bid and Bid-mediated mitochondrial damage, giving long term survival advantage to cancer cells [214,215]. TNFR1, however, may counteract Hsp70 anti-apoptotic activity under the same circumstances, as the activation of PP1/PP2a and PP2b phosphatases by TNFR1 can modulate phosphorylation and activation of HSF1 and thereby HSF1-dependent Hsp70 transcription [216]. Under some circumstances, however, apoptosis may be enhanced by Hsp70 rather than being prevented, as shown when Hsp70 binds and stimulates mutant p53 to bind TRAIL-R1 and -R2 gene promoter regions, or when Hsp70 accumulates at the plasma membrane of tumour cells [208,217]. In the first case, pro-apoptotic activity of Hsp70 results in p53-mediated enhanced transcription and expression of TRAIL receptor proteins, whereas with respect to Hsp70 localization at the plasma membrane the underlying mechanism appears to involve an enhanced uptake of death-inducing granzyme B protein through its association with Hsp70. Similarly, Hsp70 may promote TNF-dependent FADD-mediated activation of caspase-8, by limiting anti-apoptotic proteins FLIP (FLICE inhibitory protein) and RIP1 (receptor-interacting protein 1) recruitment at the plasma membrane, or by inhibiting IKK-mediated phosphorylation and proteasomal degradation of NF-kB inhibitor IkB and, thus, NF-κB pro-survival signaling [218-221].
6. Role of Inducible Hsp70 Chaperone in Lysosome-Mediated Cell Death
It is well established that lysosomes play an active role in the execution of both apoptosis and necrosis, and a range of stress stimuli coming from outside and inside the cells lead to lysosomal membrane permeabilization (LMP) [222-225]. The primary function of lysosomes is degradation of proteins, and for such a purpose these organelles are filled with more than 50 different types of acid proteases [226,227]. Trafficking and stability of lysosomes is important for tumour growth and progression, since lysosomes rupture and intracellular release of their content is lethal, while the pericellular and extracellular release of it may facilitate matrix disruption, angiogenesis and tumour invasion [228,229]. In this context, Hsp70 has been shown to accumulate at the lysosomes of many tumour cell types, acting as safeguard of membrane integrity [230,231]. By interacting with LMP-inducers Bax, JNK and p53, as previously mentioned, Hsp70 may prevent lysosomal membrane permeabilization and proteolytic hydrolases release from damaged organelles, preserving cells from self-distruction [232,233]. Accordingly, Hsp70 downregulation in cancer cells causes both intracellular Ca2+ increase and loss of lysosomes integrity, resulting in massive cell death by LMP [234,235] (Figure 4). This has been shown in glioblastoma, breast and colon carcinoma cells, in which the inhibition of Hsp70 leads to both cathepsin-mediated lysosomes leakage and stress-induced cell death [236,237]. Neuronal apoptosis and necrosis following cerebral ischemia also occur via Hsp70-dependent regulation of lysosomes stability, like the upregulation of Hsp70 is effective at reducing cerebral infarction after oxidative ischemia through protection of lysosomal integrity [231].
At a molecular level Hsp70 is shuttled to late endosomal/lysosomal membranes by endo-lysosomal anionic phospholipid bis(monoacylglicerol)phosphate BMP protein, an essential co-factor for sphingomyelin metabolism [238-240]. Herein, Hsp70 sustains the activity of acid sphingomyelinase protein ASM, which controls phospholipids concentration and steric conformation of lysosomal membranes, as well as trafficking of these organelles across the cell [241]. Consistent with these findings, Hsp70, BMP and ASM are frequently overexpressed in tumour cells resistant against stress-induced lysosomal damages, whereas Hsp70 expression and activity is significantly reduced in patients affected by severe lysosomal storage disorders. Furthermore, whereas cognate Hsc70 chaperone protein localizes at lysosomes bound to lysosome-associated membrane protein 2a (LAMP2a) and permits misfolded proteins to be imported and degraded, inducible Hsp70 chaperone may also interact with LAMP1 protein, and be transported by this at the plasma membrane to be released outside during inflammation and immunity processes [222,242-244]. Accordingly, heat shocked cell lysates have enhanced vaccine activity compared to non-heat shocked controls, and forced expression of Hsp70 in tumours has been demonstrated to induce a powerful T cell mediated immune response [245,246]. When expressed at the surface of tumour-derived exosomes (TDE), indeed, Hsp70 may promote myeloid-derived suppressor cells (MDSCs) activity and restrain tumour immune surveillance of CD8+ cells [247,248]. TDE-associated Hsp70 binds Toll-like receptor 2 (TLR2) on MDSC plasma membrane and triggers MSDC immunosuppressive function through IL-6 production and STAT3 activation [249]. Tumour growth and drug resistance are both increased in TDE-presenting Hsp70-expressing cells, whereas exosomes depletion by efflux pump inhibitors reduces extracellular Hsp70 levels and enhances chemotherapy efficacy [250].
7. Targeting Hsp70
Considering the possible therapeutic potential of modulating Hsp70 activity in tumours, extensive work in recent years has focused on the identification of Hsp70 inhibitors to use alone or in combination with existing anti-neoplastic drugs. Agents which modulate Hsp70 expression and activity may be of particular benefit for the treatment of cancer, consistent with the notion that transformed cells are extremely dependent on heat shock proteins to maintain their constitutive state of stress, and several anti-cancer agents, including classical DNA intercalating molecules and more selective targeted therapeutics, induce HSP expression as a side effect of treatment. In this context, inhibitors of HSF1 and direct modulators of Hsp70 activity have been successfully tested as anti-cancer drugs, offering therapeutic benefits and selective effects on transformed versus untransformed cells [158,230,251-253]. With respect to this, the naturally occurring flavonoid quercetin (3,3′,4′,5,7-pentahydroxyflavone) is one of the first Hsp70 inhibitors that was shown to reduce the expression of Hsp70 by the inhibition of HSF1 phosphorylation and transcriptional activity, and to sensitize tumour cells to apoptosis induced by anti-cancer drugs by inhibiting Hsp70 and Mcl-1 expression, while activating pro-apoptic Bax [254-257]. However, quercetin effectiveness was found to be dependent on the concomitant expression of several functional proteins, including the multidrug resistance protein MRP [258-260]. HSF1, in fact, plays a role in P-glycoprotein (P-gp) expression and activity, and incubation with quercetin prior to administration of anti-cancer drugs appears to be more effective at inhibiting MRP-mediated drug efflux and to enhance drugs‧ effectiveness [261,262]. In addition, quercetin inhibits expression and activity of protein kinases involved in growth and survival of cancer cells, including few critical for HSF1 transcriptional activity and Hsp70 expression [263]. Casein kinase 2 (CK2) and calcium/calmodulin kinase II (CamKII) are among these proteins, as their inhibition by quercetin prevents activating phosphorylation of HSF1 at Thr142 and Ser230, respectively. Good inhibition of Hsp70 induction correlates with good inhibition of both CKII and CamKII kinases, whereas quercetin derivatives that show poor inhibition of either or both proteins have little or no ability to inhibit stress-induced Hsp70 expression [88,264]. Besides, quercetin may also act by inhibiting Hsp70 expression post-transcriptionally without direct effect on HSF1 activity. It has been shown that exposure of HeLa cells to quercetin favours AMP-activated protein kinase (AMPK) activity and reduces Hsp70 levels [265,266]. While in the absence of quercetin AMPK is inhibited by phosphatase 2A (PP2A) protein and Hsp70 is expressed, in the presence of quercetin, activated AMPK reduces Hsp70 mRNA stability and sensitizes cancer cells to stress-induced apoptosis [267]. Parallel to inhibition of Hsp70 expression, however, quercetin may cause phosphorylation of Hsp27 (Ser78) in the absence of heat shock, which counteracts the lethal effects achieved by Hsp70 depletion [266]. Quercetin derivatives that enhance phosphorylation of Hsp27 are indeed not as good stress inhibitors as derivatives capable of inhibiting both CK2 and CamKII without stimulating Hsp27 activity, since phosphorylation is a pre-requisite for Hsp27 anti-apoptotic activity [266].
Hsp70 can also be modulated more directly, as observed in cells treated with N-formyl-3,4-methylenedioxy-benzylidene-γ-butyrolaetam (KNK437) and diterpene triepoxide triptolide inhibitors. Interestingly, KNK437 alters stress-inducible Hsp70 binding to Hsp40, whereas triptolide inhibits the expression of Hsp70 by interfering with the activity of HSF1 C-terminal transactivation domain [268-270]. However, in combination with cytotoxic agents such as proteasome inhibitors and modulators of Hsp90 ATPase activity (i.e., 17-allylaminodemethoxygeldanamycin) both KNK437 and triptolide enhance drug-induced apoptosis and increase the loss of clonogenic survival of transformed cells, by downregulating Hsp70 expression and activity prior to the drug treatment [271-280]. This has been shown also by using two other inhibitors, NZ28 and emunin, which prevent Hsp70 expression and activity in both heat shocked cells and in cells treated with either proteasome or Hsp90 inhibitors [281-283]. These inhibitors are relatively specific compared to those described above, as both emunin and NZ28 inhibit Hsp70 translation rather than HSF1 activity, precluding Hsp70 expression only.
Blocking HSF1-dependent pTEFb recruitment at Hsp70 promoter may be an even more interesting approach to impede inducible Hsp70 expression, using decoys that interfere with the HSF1/cdk9 complex formation. Since HSF1 is required for p-TEFb protein (positive transcription elongation factor b) to be placed at the stress-responsive gene promoters primed with paused RNA polymerase II, molecules that associate with HSF1 and inhibit pTEFb recruitment may reduce Hsp70 transcription, thereby compromising cell survival [284,285]. Based on these findings, the possibility of modulating the Hsp70 ATPase activity may have even more promising therapeutic potential than the inhibition of chaperone expression. This has been shown with 15-deoxyspergualin (15-DSG) derivatives and different adenosine-derived inhibitors, which bind the C-terminal EEVD motif and the ATPase domain of Hsp70, respectively, and affect both the endogenous and Hsp40-stimulated ATPase cycle of Hsp70.
One interesting aspect of Hsp70 biology that remain to be more fully elucidated is the ability of Hsp70 to form multiprotein complexes with a wide range of molecules, including co-chaperones partner proteins and signal transducers. Targeting the interaction between Hsp70 and its regulatory partners would be expected to control specific chaperone activities, and have more limited toxicity because of reduced global impairment of protein homeostasis. In support of this idea, peptide aptamers that inhibit the anti-apoptotic function of Hsp70 by acting as substrate mimetic peptides have been recently developed, like ADD70, a construct encoding the minimum binding region of the Hsp70 substrate AIF. ADD70 when used alone is not cytotoxic per se, because it does not bind to Hsc70 or any other heat shock protein with housekeeping function. However when transduced in tumour cells ADD70 promotes stress-induced apoptosis by its unique capacity to interact with and sequester inducible Hsp70 chaperone protein from binding AIF [201,286]. Similar to this, a small-molecule Hsp70 inhibitor called 2-phenylethylenesulfonamide (PES) has been identified, and shown to alter the activity of Hsp70 in multiple cellular processes. PES interacts with the C-terminal SBD domain of Hsp70 and plays a critical role in determining Hsp70 partner selectivity. In the presence of PES, Hsp70 does not bind Hsp90, Hsp40, BAG and CHIP chaperones and co-chaperones anymore, and the normally cytoprotective role of Hsp70 is disabled. In this regard, cancer cells that overexpress Hsp70 and experience high levels of metabolic stresses are extremely sensitive to PES administration, and undergo readily cell death through caspase-independent mechanisms that involve increased protein aggregation, impairment of lysosomal functions, and inhibited autophagy.
8. Concluding Remarks
Nowadays, great clinical interest is focused on the design of novel therapeutic strategies that simultaneously inhibit multiple signaling pathways in cancer cells, since drug resistance frequently occurs in cells that are characterized by high genomic instability and the accumulation of somatic mutations. At any given time, a living cell is a multi-tasking system in which a plethora of external and internal stimuli continuously challenges its survival. Tumor cells are particularly threatened by harmful growth conditions, and a specific and rapid response to stress becomes necessary to maintain transmission of signals across different pathways that protect cells from death. Multiple perturbations of homeostasis can have severe consequences for the cell, and an excessive cell death may be very costly for the entire organism as well. In such a context, multiple checks need to be guaranteed by the cell to survive, and heat shock proteins, by the nature of their functions, provide such critical links between signaling networks and molecules. Contrary to their generic name HSPs do not just reverse stress-induced protein misfolding or help nascent polypeptides to fold properly, but have a more global role in cell metabolism, including protection from cell death. While stress-inducible heat shock proteins are barely detectable in normal cells, their expression is enhanced in many tumors, where they favor adaptation to adverse microenviromental conditions and contribute to the development of resistance to a wide range of pharmacological agents. Therefore, heat shock proteins may be considered potential drug targets, given their widespread roles in both nonstress and stressful conditions. Hsp70 has gathered significant attention as a potential drug target over the past few years, and the importance of its modulatory function has been demonstrated in a wide range of pathophysiological states, including cancer and neurodegenerative diseases. Hsp70, in fact, broadly shapes cell homeostasis, by affecting protein quality control and turnover, and when this occurs upon stress induction, as observed constantly in transformed cells, it provides survival advantages by binding and inhibiting multiple components of the caspase-dependent and –independent apoptotic pathways. Hsp70 plays roles also in lysosomes integrity, consistent with its known substrate promiscuity and pleiotrophic activity. Such a seemingly ubiquitous nature of the anti-apoptotic effects of Hsp70 may be interpreted as a lack of specificity and biological relevance, but it's now clear that in this way Hsp70 provides major support to tumorigenicity and resistance of cancer cells to noxious insults. The fact that the cytotoxic effects of Hsp70 down-modulation are so important for cancer cells survival is consistent with the notion that tumors are particularly dependent on HSP expression and activity, and that their constitutive stressed phenotype requires the continuous cytoprotective action of molecular chaperones like Hsp70. As a consequence, extensive efforts from many laboratories have provided insights of the complex regulatory mechanisms that control Hsp70 expression and activity, as well as novel cancer therapeutics that target Hsp70 activity have been developed. However, many questions remain before Hsp70 can be considered a fully validated drug target, and how to practically manipulate Hsp70 activity to have more limited toxicity remains a mandatory challenge that has to be addressed to make Hsp70 a good therapeutic target for clinical intervention.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Organism | Chaperones | Location | Function | |
|---|---|---|---|---|---|
| Hsp100 | E.coli | ClpA, B, C | cytosol |  | Role in stress tolerance; it helps the resolubilization of heat-inactivated proteins from insoluble aggregates |
| S.cervisiae | Hsp104 | cytosol | |||
| Hsp90 | E.coli | HtpG | cytosol |  | Role in signal transduction (e.g., interaction with steroid hormone receptors, tyrosine kinases, serine/threonine kinases); regulation of heat shock response; role in cell cycle and proliferation; maintenance of mitochondrial integrity |
| S.cervisiae | Hsp83 | cytosol | |||
| Humans | Hsp90 | cytosol, | |||
| GRP94 | nucleus | ||||
| TRAP1 | ER | ||||
| mitochondria | |||||
| Hsc/Hsp70 | E.coli | DnaK | cytosol |  | Roles in lambda phage replication; regulation of heat shock response; interaction with nascent chain polypeptides; functions in interorganellar transport; roles in signal transduction; refolding of denatured proteins in vitro; roles in cell cycle and proliferation; anti-apoptotic activity; potential antigen-presenting molecule in tumour cells |
| S.cervisiae | Ssa1-4, | cytosol | |||
| Ssb1,2 | ER, | ||||
| Kar2, Ssc1 | mitochondria | ||||
| Humans | |||||
| Hsc70, Hsp70 | cytosol, | ||||
| BIP, mHsp70 | nucleus | ||||
| ER, | |||||
| mitochondria | |||||
| Hsp60 | E.coli | GroEL/ES | cytosol |  | Roles in folding and stability of denatured protein in vitro; cofactor in diverse proteolytic systems; role in the assembly of bacteriophages and plant proteins (Rubisco) |
| S.cervisiae | Hsp60 | mitochondria | |||
| Plants | Cpn60 | chloroplast | |||
| Humans | Hsp60 | mitochondria | |||
| Hsp40 | E.coli | dnaJ | cytosol |  | Essential co-chaperone for Hsp70 ATPase activity, substrate binding and release |
| S.cervisiae | Ydj1 | cytosol | |||
| Humans | Hdj1, Hdj2 | nucleus | |||
| Small Hsps | E.coli | IbpA, IpbB | cytosol |  | Inhibition of in vitro protein aggregation and heat inactivation in S. cervisiae; Role in cell thermotolerance through stabilization of actin microfilaments; anti-apoptotic activity |
| Humans | Hsp27 | cytosol | |||
| crystallin | cytosol |
Acknowledgements
Our works were supported by Fondazione Città Della Speranza and MIUR (Ministero Istruzione Università e Ricerca).
References
- Nykanen, P.; Alastalo, T.P.; Ahlskog, J.; Horelli-Kuitunen, N.; Pirkkala, L.; Sistonen, L. Genomic organization and promoter analysis of the human heat shock factor 2 gene. Cell Stress Chaperones 2001, 6, 377–385. [Google Scholar]
- Bercovich, B.; Stancovski, I.; Mayer, A.; Blumenfeld, N.; Laszlo, A.; Schwartz, A.L.; Ciechanover, A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem. 1997, 272, 9002–9010. [Google Scholar]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar]
- Calderwood, S.K.; Murshid, A.; Prince, T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging—A mini-review. Gerontology 2009, 55, 550–558. [Google Scholar]
- Jaattela, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar]
- Mosser, D.D.; Morimoto, R.I. Molecular chaperones and the stress of oncogenesis. Oncogene 2004, 23, 2907–2918. [Google Scholar]
- Sun, Y.; MacRae, T.H. Small heat shock proteins: Molecular structure and chaperone function. Cell. Mol. Life Sci. 2005, 62, 2460–2476. [Google Scholar]
- Tsutsumi, S.; Neckers, L. Extracellular heat shock protein 90: A role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007, 98, 1536–1539. [Google Scholar]
- Sangster, T.A.; Salathia, N.; Undurraga, S.; Milo, R.; Schellenberg, K.; Lindquist, S.; Queitsch, C. HSP90 affects the expression of genetic variation and developmental stability in quantitative traits. Proc. Natl. Acad. Sci. USA 2008, 105, 2963–2968. [Google Scholar]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2010. Epub ahead of print. [Google Scholar]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar]
- Kampinga, H.H. Chaperones in preventing protein denaturation in living cells and protecting against cellular stress. Handb. Exp. Pharmacol. 2006, 172, 1–42. [Google Scholar]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar]
- Bhattacharya, A.; Kurochkin, A.V.; Yip, G.N.; Zhang, Y.; Bertelsen, E.B.; Zuiderweg, E.R. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J. Mol. Biol. 2009, 388, 475–490. [Google Scholar]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar]
- Galluzzi, L.; Giordanetto, F.; Kroemer, G. Targeting HSP70 for cancer therapy. Mol. Cell 2009, 36, 176–177. [Google Scholar]
- Vogel, M.; Mayer, M.P.; Bukau, B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J. Biol. Chem. 2006, 281, 38705–38711. [Google Scholar]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 2007, 26, 27–39. [Google Scholar]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar]
- Mitra, A.; Shevde, L.A.; Samant, R.S. Multi-faceted role of HSP40 in cancer. Clin. Exp. Metastasis 2009, 26, 559–567. [Google Scholar]
- Mayer, M.P.; Rudiger, S.; Bukau, B. Molecular basis for interactions of the DnaK chaperone with substrates. Biol. Chem. 2000, 381, 877–885. [Google Scholar]
- Erbse, A.; Mayer, M.P.; Bukau, B. Mechanism of substrate recognition by Hsp70 chaperones. Biochem. Soc. Trans. 2004, 32, 617–621. [Google Scholar]
- Blatch, G.L.; Lassle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. Bioessays 1999, 21, 932–939. [Google Scholar]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000, 101, 199–210. [Google Scholar]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP27 and alphaB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox Signal. 2005, 7, 404–413. [Google Scholar]
- Shashidharamurthy, R.; Koteiche, H.A.; Dong, J.; McHaourab, H.S. Mechanism of chaperone function in small heat shock proteins: Dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J. Biol. Chem. 2005, 280, 5281–5289. [Google Scholar]
- Craig, E.A.; Huang, P.; Aron, R.; Andrew, A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 1–21. [Google Scholar]
- Jiang, J.; Maes, E.G.; Taylor, A.B.; Wang, L.; Hinck, A.P.; Lafer, E.M.; Sousa, R. Structural basis of J cochaperone binding and regulation of Hsp70. Mol. Cell 2007, 28, 422–433. [Google Scholar]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar]
- Rudiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Biol. 1997, 4, 342–349. [Google Scholar]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar]
- Prodromou, C.; Pearl, L.H. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar]
- Pearl, L.H.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar]
- Akerfelt, M.; Trouillet, D.; Mezger, V.; Sistonen, L. Heat shock factors at a crossroad between stress and development. Ann. NY Acad. Sci. 2007, 1113, 15–27. [Google Scholar]
- Amin, J.; Ananthan, J.; Voellmy, R. Key features of heat shock regulatory elements. Mol. Cell Biol. 1988, 8, 3761–3769. [Google Scholar]
- Schiller, P.; Amin, J.; Ananthan, J.; Brown, M.E.; Scott, W.A.; Voellmy, R. Cis-acting elements involved in the regulated expression of a human HSP70 gene. J. Mol. Biol. 1988, 203, 97–105. [Google Scholar]
- Fernandes, M.; Xiao, H.; Lis, J.T. Binding of heat shock factor to and transcriptional activation of heat shock genes in Drosophila. Nucleic Acids Res. 1995, 23, 4799–4804. [Google Scholar]
- Latchman, D.S. Cell stress genes and neuronal protection. Neuropathol. Appl. Neurobiol. 1995, 21, 475–477. [Google Scholar]
- Lebedeva, L.A.; Nabirochkina, E.N.; Kurshakova, M.M.; Robert, F.; Krasnov, A.N.; Evgen'ev, M.B.; Kadonaga, J.T.; Georgieva, S.G.; Tora, L. Occupancy of the Drosophila hsp70 promoter by a subset of basal transcription factors diminishes upon transcriptional activation. Proc. Natl. Acad. Sci. USA 2005, 102, 18087–18092. [Google Scholar]
- Wilkerson, D.C.; Sarge, K.D. RNA polymerase II interacts with the Hspa1b promoter in mouse epididymal spermatozoa. Reproduction 2009, 137, 923–929. [Google Scholar]
- Kurshakova, M.M.; Nabirochkina, E.N.; Lebedeva, L.A.; Georgieva, S.G.; Evgen'ev, M.B.; Krasnov, A.N. Involvement of general transcriptional factors in the regulation of transcription of the hsp70 gene in vivo. Dokl. Biol. Sci. 2006, 411, 475–478. [Google Scholar]
- Lebedeva, L.A.; Nabirochkina, E.N.; Evgen'ev, M.B.; Georgieva, S.G.; Krasnov, A.N. Role of general transcription factors and the TFTC complex in transcription activation in vivo as revealed with a model of the hsp70 gene. Genetika 2007, 43, 32–37. [Google Scholar]
- Ni, Z.; Schwartz, B.E.; Werner, J.; Suarez, J.R.; Lis, J.T. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Mol. Cell 2004, 13, 55–65. [Google Scholar]
- Benjamin, L.R.; Gilmour, D.S. Nucleosomes are not necessary for promoter-proximal pausing in vitro on the Drosophila hsp70 promoter. Nucleic Acids Res. 1998, 26, 1051–1055. [Google Scholar]
- Petesch, S.J.; Lis, J.T. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 2008, 134, 74–84. [Google Scholar]
- Boehm, A.K.; Saunders, A.; Werner, J.; Lis, J.T. Transcription factor and polymerase recruitment, modification, and movement on dhsp70 in vivo in the minutes following heat shock. Mol. Cell. Biol. 2003, 23, 7628–7637. [Google Scholar]
- Sullivan, E.K.; Weirich, C.S.; Guyon, J.R.; Sif, S.; Kingston, R.E. Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol. Cell. Biol. 2001, 21, 5826–5837. [Google Scholar]
- Yao, J.; Ardehali, M.B.; Fecko, C.J.; Webb, W.W.; Lis, J.T. Intranuclear distribution and local dynamics of RNA polymerase II during transcription activation. Mol. Cell 2007, 28, 978–990. [Google Scholar]
- Mason, P.B., Jr.; Lis, J.T. Cooperative and competitive protein interactions at the hsp70 promoter. J. Biol. Chem. 1997, 272, 33227–33233. [Google Scholar]
- Yuan, C.X.; Gurley, W.B. Potential targets for HSF1 within the preinitiation complex. Cell Stress Chaperones 2000, 5, 229–242. [Google Scholar]
- Jolly, C.; Robert-Nicoud, M.; Vourc'h, C. Contribution of growing RNA molecules to the nuclear transcripts foci observed by FISH. Exp. Cell Res. 1998, 238, 299–304. [Google Scholar]
- Jolly, C.; Metz, A.; Govin, J.; Vigneron, M.; Turner, B.M.; Khochbin, S.; Vourc'h, C. Stress-induced transcription of satellite III repeats. J. Cell Biol. 2004, 164, 25–33. [Google Scholar]
- Sandqvist, A.; Sistonen, L. Nuclear stress granules: The awakening of a sleeping beauty? J. Cell Biol. 2004, 164, 15–17. [Google Scholar]
- Zuo, J.; Rungger, D.; Voellmy, R. Multiple layers of regulation of human heat shock transcription factor 1. Mol. Cell. Biol. 1995, 15, 4319–4330. [Google Scholar]
- Sarge, K.D.; Murphy, S.P.; Morimoto, R.I. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol. Cell. Biol. 1993, 13, 1392–1407. [Google Scholar]
- Damberger, F.F.; Pelton, J.G.; Harrison, C.J.; Nelson, H.C.; Wemmer, D.E. Solution structure of the DNA-binding domain of the heat shock transcription factor determined by multidimensional heteronuclear magnetic resonance spectroscopy. Protein Sci. 1994, 3, 1806–1821. [Google Scholar]
- Harrison, C.J.; Bohm, A.A.; Nelson, H.C. Crystal structure of the DNA binding domain of the heat shock transcription factor. Science 1994, 263, 224–227. [Google Scholar]
- Fujimoto, M.; Inouye, S.; Nakai, A. Physiological roles of heat shock transcription factors. Seikagaku 2004, 76, 419–428. [Google Scholar]
- Vuister, G.W.; Kim, S.J.; Orosz, A.; Marquardt, J.; Wu, C.; Bax, A. Solution structure of the DNA-binding domain of Drosophila heat shock transcription factor. Nat. Struct. Biol. 1994, 1, 605–614. [Google Scholar]
- Clos, J.; Rabindran, S.; Wisniewski, J.; Wu, C. Induction temperature of human heat shock factor is reprogrammed in a Drosophila cell environment. Nature 1993, 364, 252–255. [Google Scholar]
- Goodson, M.L.; Sarge, K.D. Heat-inducible DNA binding of purified heat shock transcription factor 1. J. Biol. Chem. 1995, 270, 2447–2450. [Google Scholar]
- Larson, J.S.; Schuetz, T.J.; Kingston, R.E. In vitro activation of purified human heat shock factor by heat. Biochemistry 1995, 34, 1902–1911. [Google Scholar]
- Zhong, M.; Orosz, A.; Wu, C. Direct sensing of heat and oxidation by Drosophila heat shock transcription factor. Mol. Cell 1998, 2, 101–108. [Google Scholar]
- Izu, H.; Inouye, S.; Fujimoto, M.; Shiraishi, K.; Naito, K.; Nakai, A. Heat shock transcription factor 1 is involved in quality-control mechanisms in male germ cells. Biol. Reprod. 2004, 70, 18–24. [Google Scholar]
- Pirkkala, L.; Alastalo, T.P.; Nykanen, P.; Seppa, L.; Sistonen, L. Differentiation lineage-specific expression of human heat shock transcription factor 2. FASEB J. 1999, 13, 1089–1098. [Google Scholar]
- Min, J.N.; Zhang, Y.; Moskophidis, D.; Mivechi, N.F. Unique contribution of heat shock transcription factor 4 in ocular lens development and fiber cell differentiation. Genesis 2004, 40, 205–217. [Google Scholar]
- Trinklein, N.D.; Chen, W.C.; Kingston, R.E.; Myers, R.M. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 2004, 9, 21–28. [Google Scholar]
- Hahn, J.S.; Hu, Z.; Thiele, D.J.; Iyer, V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004, 24, 5249–5256. [Google Scholar]
- McMillan, D.R.; Xiao, X.; Shao, L.; Graves, K.; Benjamin, I.J. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 1998, 273, 7523–7528. [Google Scholar]
- Christians, E.S.; Benjamin, I.J. Heat shock response: Lessons from mouse knockouts. Handb. Exp. Pharmacol. 2006, 172, 139–152. [Google Scholar]
- Christians, E.S.; Yan, L.J.; Benjamin, I.J. Heat shock factor 1 and heat shock proteins: Critical partners in protection against acute cell injury. Crit. Care Med. 2002, 30, S43–S50. [Google Scholar]
- Steele, A.D.; Hutter, G.; Jackson, W.S.; Heppner, F.L.; Borkowski, A.W.; King, O.D.; Raymond, G.J.; Aguzzi, A.; Lindquist, S. Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc. Natl. Acad. Sci. USA 2008, 105, 13626–13631. [Google Scholar]
- Jedlicka, P.; Mortin, M.A.; Wu, C. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J. 1997, 16, 2452–2462. [Google Scholar]
- de Billy, E.; Powers, M.V.; Smith, J.R.; Workman, P. Drugging the heat shock factor 1 pathway: Exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell Cycle 2009, 8, 3806–3808. [Google Scholar]
- Staib, J.L.; Quindry, J.C.; French, J.P.; Criswell, D.S.; Powers, S.K. Increased temperature, not cardiac load, activates heat shock transcription factor 1 and heat shock protein 72 expression in the heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R432–R439. [Google Scholar]
- Brunet Simioni, M.; De Thonel, A.; Hammann, A.; Joly, A.L.; Bossis, G.; Fourmaux, E.; Bouchot, A.; Landry, J.; Piechaczyk, M.; Garrido, C. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 2009, 28, 3332–3344. [Google Scholar]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar]
- Bharadwaj, S.; Ali, A.; Ovsenek, N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 In vivo. Mol. Cell. Biol. 1999, 19, 8033–8041. [Google Scholar]
- Guo, Y.; Guettouche, T.; Fenna, M.; Boellmann, F.; Pratt, W.B.; Toft, D.O.; Smith, D.F.; Voellmy, R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001, 276, 45791–45799. [Google Scholar]
- Xiao, H.; Perisic, O.; Lis, J.T. Cooperative binding of Drosophila heat shock factor to arrays of a conserved 5 bp unit. Cell 1991, 64, 585–593. [Google Scholar]
- Yao, J.; Munson, K.M.; Webb, W.W.; Lis, J.T. Dynamics of heat shock factor association with native gene loci in living cells. Nature 2006, 442, 1050–1053. [Google Scholar]
- Fernandes, M.; Xiao, H.; Lis, J.T. Fine structure analyses of the Drosophila and Saccharomyces heat shock factor--heat shock element interactions. Nucleic Acids Res. 1994, 22, 167–173. [Google Scholar]
- Holmberg, C.I.; Hietakangas, V.; Mikhailov, A.; Rantanen, J.O.; Kallio, M.; Meinander, A.; Hellman, J.; Morrice, N.; MacKintosh, C.; Morimoto, R.I.; et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001, 20, 3800–3810. [Google Scholar]
- Jolly, C.; Morimoto, R.; Robert-Nicoud, M.; Vourc'h, C. HSF1 transcription factor concentrates in nuclear foci during heat shock: Relationship with transcription sites. J. Cell Sci. 1997, 110, 2935–2941. [Google Scholar]
- Bjork, J.K.; Sistonen, L. Regulation of the members of the mammalian heat shock factor family. FEBS J. 2010, 277, 4126–4139. [Google Scholar]
- Morano, K.A.; Thiele, D.J. Heat shock factor function and regulation in response to cellular stress, growth, and differentiation signals. Gene Expr. 1999, 7, 271–282. [Google Scholar]
- Abravaya, K.; Myers, M.P.; Murphy, S.P.; Morimoto, R.I. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 1992, 6, 1153–1164. [Google Scholar]
- Baler, R.; Welch, W.J.; Voellmy, R. Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J. Cell Biol. 1992, 117, 1151–1159. [Google Scholar]
- Goodson, M.L.; Park-Sarge, O.K.; Sarge, K.D. Tissue-dependent expression of heat shock factor 2 isoforms with distinct transcriptional activities. Mol. Cell. Biol. 1995, 15, 5288–5293. [Google Scholar]
- Rizzi, N.; Denegri, M.; Chiodi, I.; Corioni, M.; Valgardsdottir, R.; Cobianchi, F.; Riva, S.; Biamonti, G. Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol. Biol. Cell. 2004, 15, 543–551. [Google Scholar]
- Anckar, J.; Hietakangas, V.; Denessiouk, K.; Thiele, D.J.; Johnson, M.S.; Sistonen, L. Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol. Cell. Biol. 2006, 26, 955–964. [Google Scholar]
- Hietakangas, V.; Ahlskog, J.K.; Jakobsson, A.M.; Hellesuo, M.; Sahlberg, N.M.; Holmberg, C.I.; Mikhailov, A.; Palvimo, J.J.; Pirkkala, L.; Sistonen, L. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell. Biol. 2003, 23, 2953–2968. [Google Scholar]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.P.; Vourc'h, C.; Matthias, P.; Khochbin, S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar]
- Sadoul, K.; Boyault, C.; Pabion, M.; Khochbin, S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 2008, 90, 306–312. [Google Scholar]
- Hilgarth, R.S.; Hong, Y.; Park-Sarge, O.K.; Sarge, K.D. Insights into the regulation of heat shock transcription factor 1 SUMO-1 modification. Biochem. Biophys. Res. Commun. 2003, 303, 196–200. [Google Scholar]
- Hong, Y.; Rogers, R.; Matunis, M.J.; Mayhew, C.N.; Goodson, M.L.; Park-Sarge, O.K.; Sarge, K.D. Regulation of heat shock transcription factor 1 by stress-induced SUMO-1 modification. J. Biol. Chem. 2001, 276, 40263–40267. [Google Scholar]
- Thomson, S.; Hollis, A.; Hazzalin, C.A.; Mahadevan, L.C. Distinct stimulus-specific histone modifications at hsp70 chromatin targeted by the transcription factor heat shock factor-1. Mol. Cell 2004, 15, 585–594. [Google Scholar]
- Li, Y.; Xu, W.; McBurney, M.W.; Longo, V.D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008, 8, 38–48. [Google Scholar]
- Xu, D.; La Thangue, N.B. Strap: A versatile transcription co-factor. Cell Cycle 2008, 7, 2456–2457. [Google Scholar]
- Xu, D.; Zalmas, L.P.; La Thangue, N.B. A transcription cofactor required for the heat-shock response. EMBO Rep. 2008, 9, 662–669. [Google Scholar]
- Fritah, S.; Col, E.; Boyault, C.; Govin, J.; Sadoul, K.; Chiocca, S.; Christians, E.; Khochbin, S.; Jolly, C.; Vourc'h, C. Heat-shock factor 1 controls genome-wide acetylation in heat-shocked cells. Mol. Biol. Cell 2009, 20, 4976–4984. [Google Scholar]
- Guettouche, T.; Boellmann, F.; Lane, W.S.; Voellmy, R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 6, 4. [Google Scholar]
- Chu, B.; Zhong, R.; Soncin, F.; Stevenson, M.A.; Calderwood, S.K. Transcriptional activity of heat shock factor 1 at 37 degrees C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Calpha and Czeta. J. Biol. Chem. 1998, 273, 18640–18646. [Google Scholar]
- Kline, M.P.; Morimoto, R.I. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol. Cell. Biol. 1997, 17, 2107–2115. [Google Scholar]
- Batista-Nascimento, L.; Neef, D.W.; Liu, P.C.; Rodrigues-Pousada, C.; Thiele, D.J. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. PLoS one 2011, 6, e15976. [Google Scholar]
- Murshid, A.; Chou, S.D.; Prince, T.; Zhang, Y.; Bharti, A.; Calderwood, S.K. Protein kinase A binds and activates heat shock factor 1. PLoS one 2010, 5, e13830. [Google Scholar]
- Rafiee, P.; Theriot, M.E.; Nelson, V.M.; Heidemann, J.; Kanaa, Y.; Horowitz, S.A.; Rogaczewski, A.; Johnson, C.P.; Ali, I.; Shaker, R.; et al. Human esophageal microvascular endothelial cells respond to acidic pH stress by PI3K/AKT and p38 MAPK-regulated induction of Hsp70 and Hsp27. Am. J. Physiol. Cell Physiol. 2006, 291, C931–C945. [Google Scholar]
- Taylor, D.M.; De Koninck, P.; Minotti, S.; Durham, H.D. Manipulation of protein kinases reveals different mechanisms for upregulation of heat shock proteins in motor neurons and non-neuronal cells. Mol. Cell. Neurosci. 2007, 34, 20–33. [Google Scholar]
- Venkatakrishnan, C.D.; Tewari, A.K.; Moldovan, L.; Cardounel, A.J.; Zweier, J.L.; Kuppusamy, P.; Ilangovan, G. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2680–H2691. [Google Scholar]
- Ferreiro, I.; Barragan, M.; Gubern, A.; Ballestar, E.; Joaquin, M.; Posas, F. The p38 SAPK is recruited to chromatin via its interaction with transcription factors. J. Biol. Chem. 2010, 285, 31819–31828. [Google Scholar]
- Ferreiro, I.; Joaquin, M.; Islam, A.; Gomez-Lopez, G.; Barragan, M.; Lombardia, L.; Dominguez, O.; Pisano, D.G.; Lopez-Bigas, N.; Nebreda, A.R.; et al. Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genomics 2010, 11, 144. [Google Scholar]
- Roth, M.; Zhong, J.; Tamm, M.; Szilard, J. Mesothelioma cells escape heat stress by upregulating Hsp40/Hsp70 expression via mitogen-activated protein kinases. J. Biomed. Biotechnol. 2009, 2009, 451084. [Google Scholar]
- Combaret, V.; Boyault, S.; Iacono, I.; Brejon, S.; Rousseau, R.; Puisieux, A. Effect of bortezomib on human neuroblastoma: Analysis of molecular mechanisms involved in cytotoxicity. Mol. Cancer 2008, 7, 50. [Google Scholar]
- Hideshima, T.; Podar, K.; Chauhan, D.; Ishitsuka, K.; Mitsiades, C.; Tai, Y.T.; Hamasaki, M.; Raje, N.; Hideshima, H.; Schreiner, G.; et al. p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene 2004, 23, 8766–8776. [Google Scholar]
- Kim, H.P.; Wang, X.; Zhang, J.; Suh, G.Y.; Benjamin, I.J.; Ryter, S.W.; Choi, A.M. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: Involvement of p38 beta MAPK and heat shock factor-1. J. Immunol. 2005, 175, 2622–2629. [Google Scholar]
- Chakraborty, P.K.; Mustafi, S.B.; Ganguly, S.; Chatterjee, M.; Raha, S. Resveratrol induces apoptosis in K562 (chronic myelogenous leukemia) cells by targeting a key survival protein, heat shock protein 70. Cancer Sci. 2008, 99, 1109–1116. [Google Scholar]
- Han, S.I.; Oh, S.Y.; Woo, S.H.; Kim, K.H.; Kim, J.H.; Kim, H.D.; Kang, H.S. Implication of a small GTPase Rac1 in the activation of c-Jun N-terminal kinase and heat shock factor in response to heat shock. J. Biol. Chem. 2001, 276, 1889–1895. [Google Scholar]
- Liu, J.; Zhang, D.; Mi, X.; Xia, Q.; Yu, Y.; Zuo, Z.; Guo, W.; Zhao, X.; Cao, J.; Yang, Q.; et al. p27 suppresses arsenite-induced Hsp27/Hsp70 expression through inhibiting JNK2/c-Jun- and HSF-1-dependent pathways. J. Biol. Chem. 2010, 285, 26058–26065. [Google Scholar]
- Salehi, A.H.; Morris, S.J.; Ho, W.C.; Dickson, K.M.; Doucet, G.; Milutinovic, S.; Durkin, J.; Gillard, J.W.; Barker, P.A. AEG3482 is an antiapoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem. Biol. 2006, 13, 213–223. [Google Scholar]
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar]
- Morimoto, R.I.; Kline, M.P.; Bimston, D.N.; Cotto, J.J. The heat-shock response: Regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997, 32, 17–29. [Google Scholar]
- Greene, J.M.; Larin, Z.; Taylor, I.C.; Prentice, H.; Gwinn, K.A.; Kingston, R.E. Multiple basal elements of a human hsp70 promoter function differently in human and rodent cell lines. Mol. Cell. Biol. 1987, 7, 3646–3655. [Google Scholar]
- Nylandsted, J.; Brand, K.; Jaattela, M. Heat shock protein 70 is required for the survival of cancer cells. Ann. NY Acad. Sci. 2000, 926, 122–125. [Google Scholar]
- Shi, W.; Zhou, Y.; Wild, J.; Adler, J.; Gross, C.A. DnaK, DnaJ, and GrpE are required for flagellum synthesis in Escherichia coli. J. Bacteriol. 1992, 174, 6256–6263. [Google Scholar]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar]
- Luo, X.B.; Liu, K.; Yi, Y.X.; Wang, H.; Liu, M.D.; Xiao, X.Z.; Deng, G.H. Effect of HSP72 on acute injury of neonatal cardiomyocytes induced by oxidative stress. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2006, 31, 228–231. [Google Scholar]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar]
- Yaglom, J.A.; Gabai, V.L.; Sherman, M.Y. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007, 67, 2373–2381. [Google Scholar]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Meriin, A.B.; Sherman, M.Y.; Morimoto, R.I.; Massie, B. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 2000, 20, 7146–7159. [Google Scholar]
- Nollen, E.A.; Brunsting, J.F.; Roelofsen, H.; Weber, L.A.; Kampinga, H.H. In vivo chaperone activity of heat shock protein 70 and thermotolerance. Mol. Cell. Biol. 1999, 19, 2069–2079. [Google Scholar]
- Shim, E.H.; Kim, J.I.; Bang, E.S.; Heo, J.S.; Lee, J.S.; Kim, E.Y.; Lee, J.E.; Park, W.Y.; Kim, S.H.; Kim, H.S.; et al. Targeted disruption of hsp70.1 sensitizes to osmotic stress. EMBO Rep. 2002, 3, 857–861. [Google Scholar]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar]
- Gurbuxani, S.; Schmitt, E.; Cande, C.; Parcellier, A.; Hammann, A.; Daugas, E.; Kouranti, I.; Spahr, C.; Pance, A.; Kroemer, G.; et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 2003, 22, 6669–6678. [Google Scholar]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jaattela, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar]
- Wigmore, S.J.; Sangster, K.; McNally, S.J.; Harrison, E.M.; Ross, J.A.; Fearon, K.C.; Garden, O.J. De-repression of heat shock transcription factor-1 in interleukin-6- treated hepatocytes is mediated by downregulation of glycogen synthase kinase 3beta and MAPK/ERK-1. Int. J. Mol. Med. 2007, 19, 413–420. [Google Scholar]
- Stephanou, A.; Latchman, D.S. Transcriptional modulation of heat-shock protein gene expression. Biochem. Res. Int. 2011. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. [Google Scholar]
- Xie, Y.; Chen, C.; Stevenson, M.A.; Auron, P.E.; Calderwood, S.K. Heat shock factor 1 represses transcription of the IL-1beta gene through physical interaction with the nuclear factor of interleukin 6. J. Biol. Chem. 2002, 277, 11802–11810. [Google Scholar]
- Xie, Y.; Chen, C.; Stevenson, M.A.; Hume, D.A.; Auron, P.E.; Calderwood, S.K. NF-IL6 and HSF1 have mutually antagonistic effects on transcription in monocytic cells. Biochem. Biophys. Res. Commun. 2002, 291, 1071–1080. [Google Scholar]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar]
- Yamagishi, N.; Fujii, H.; Saito, Y.; Hatayama, T. Hsp105beta upregulates hsp70 gene expression through signal transducer and activator of transcription-3. FEBS J. 2009, 276, 5870–5880. [Google Scholar]
- Stephanou, A.; Brar, B.K.; Knight, R.A.; Latchman, D.S. Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-× promoters. Cell Death Differ. 2000, 7, 329–330. [Google Scholar]
- Stephanou, A.; Isenberg, D.A.; Akira, S.; Kishimoto, T.; Latchman, D.S. The nuclear factor interleukin-6 (NF-IL6) and signal transducer and activator of transcription-3 (STAT-3) signalling pathways co-operate to mediate the activation of the hsp90beta gene by interleukin-6 but have opposite effects on its inducibility by heat shock. Biochem. J. 1998, 330, 189–195. [Google Scholar]
- Stephanou, A.; Isenberg, D.A.; Nakajima, K.; Latchman, D.S. Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90beta gene promoters. J. Biol. Chem. 1999, 274, 1723–1728. [Google Scholar]
- Stephanou, A.; Latchman, D.S. Opposing actions of STAT-1 and STAT-3. Growth Factors 2005, 23, 177–182. [Google Scholar]
- Akira, S.; Isshiki, H.; Nakajima, T.; Kinoshita, S.; Nishio, Y.; Natsuka, S.; Kishimoto, T. Regulation of expression of the interleukin 6 gene: Structure and function of the transcription factor NF-IL6. Ciba Found. Symp. 1992, 167, 47–62, discussion 62-47. [Google Scholar]
- Kinoshita, S.; Akira, S.; Kishimoto, T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc. Natl. Acad. Sci. USA 1992, 89, 1473–1476. [Google Scholar]
- Arya, R.; Mallik, M.; Lakhotia, S.C. Heat shock genes—Integrating cell survival and death. J. Biosci. 2007, 32, 595–610. [Google Scholar]
- Didelot, C.; Lanneau, D.; Brunet, M.; Joly, A.L.; De Thonel, A.; Chiosis, G.; Garrido, C. Anti-cancer therapeutic approaches based on intracellular and extracellular heat shock proteins. Curr. Med. Chem. 2007, 14, 2839–2847. [Google Scholar]
- Didelot, C.; Schmitt, E.; Brunet, M.; Maingret, L.; Parcellier, A.; Garrido, C. Heat shock proteins: Endogenous modulators of apoptotic cell death. Handb. Exp. Pharmacol. 2006, 172, 171–198. [Google Scholar]
- Buzzard, K.A.; Giaccia, A.J.; Killender, M.; Anderson, R.L. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem. 1998, 273, 17147–17153. [Google Scholar]
- Gabai, V.L.; Sherman, M.Y. Invited review: Interplay between molecular chaperones and signaling pathways in survival of heat shock. J. Appl. Physiol. 2002, 92, 1743–1748. [Google Scholar]
- Nishitai, G.; Matsuoka, M. Differential regulation of HSP70 expression by the JNK kinases SEK1 and MKK7 in mouse embryonic stem cells treated with cadmium. J. Cell Biochem. 2008, 104, 1771–1780. [Google Scholar]
- Bienemann, A.S.; Lee, Y.B.; Howarth, J.; Uney, J.B. Hsp70 suppresses apoptosis in sympathetic neurones by preventing the activation of c-Jun. J. Neurochem. 2008, 104, 271–278. [Google Scholar]
- Kumar, Y.; Tatu, U. Stress protein flux during recovery from simulated ischemia: Induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics 2003, 3, 513–526. [Google Scholar]
- Park, H.S.; Lee, J.S.; Huh, S.H.; Seo, J.S.; Choi, E.J. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001, 20, 446–456. [Google Scholar]
- Seo, H.R.; Chung, D.Y.; Lee, Y.J.; Lee, D.H.; Kim, J.I.; Bae, S.; Chung, H.Y.; Lee, S.J.; Jeoung, D.; Lee, Y.S. Heat shock protein 25 or inducible heat shock protein 70 activates heat shock factor 1: Dephosphorylation on serine 307 through inhibition of ERK1/2 phosphorylation. J. Biol. Chem. 2006, 281, 17220–17227. [Google Scholar]
- Gorostizaga, A.; Brion, L.; Maloberti, P.; Maciel, F.C.; Podesta, E.J.; Paz, C. Heat shock triggers MAPK activation and MKP-1 induction in Leydig testicular cells. Biochem. Biophys. Res. Commun. 2005, 327, 23–28. [Google Scholar]
- Sanlorenzo, L.; Zhao, B.; Spight, D.; Denenberg, A.G.; Page, K.; Wong, H.R.; Shanley, T.P. Heat shock inhibition of lipopolysaccharide-mediated tumor necrosis factor expression is associated with nuclear induction of MKP-1 and inhibition of mitogen-activated protein kinase activation. Crit. Care Med. 2004, 32, 2284–2292. [Google Scholar]
- Yaglom, J.; O'Callaghan-Sunol, C.; Gabai, V.; Sherman, M.Y. Inactivation of dual-specificity phosphatases is involved in the regulation of extracellular signal-regulated kinases by heat shock and hsp72. Mol. Cell. Biol. 2003, 23, 3813–3824. [Google Scholar]
- Parcellier, A.; Gurbuxani, S.; Schmitt, E.; Solary, E.; Garrido, C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem. Biophys. Res. Commun. 2003, 304, 505–512. [Google Scholar]
- Song, J.; Takeda, M.; Morimoto, R.I. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat. Cell Biol. 2001, 3, 276–282. [Google Scholar]
- O'Reilly, L.A.; Kruse, E.A.; Puthalakath, H.; Kelly, P.N.; Kaufmann, T.; Huang, D.C.; Strasser, A. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J. Immunol. 2009, 183, 261–269. [Google Scholar]
- Powers, M.V.; Jones, K.; Barillari, C.; Westwood, I.; van Montfort, R.L.; Workman, P. Targeting HSP70: The second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010, 9, 1542–1550. [Google Scholar]
- Powers, M.V.; Clarke, P.A.; Workman, P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle 2009, 8, 518–526. [Google Scholar]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar]
- Robertson, J.D.; Datta, K.; Kehrer, J.P. Bcl-×L overexpression restricts heat-induced apoptosis and influences hsp70, bcl-2, and Bax protein levels in FL5.12 cells. Biochem. Biophys. Res. Commun. 1997, 241, 164–168. [Google Scholar]
- Pagliari, L.J.; Kuwana, T.; Bonzon, C.; Newmeyer, D.D.; Tu, S.; Beere, H.M.; Green, D.R. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc. Natl. Acad. Sci. USA 2005, 102, 17975–17980. [Google Scholar]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.; Radicioni, S.M.; Mosser, D.D. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar]
- Rahmani, M.; Reese, E.; Dai, Y.; Bauer, C.; Kramer, L.B.; Huang, M.; Jove, R.; Dent, P.; Grant, S. Cotreatment with suberanoylanilide hydroxamic acid and 17-allylamino 17-demethoxygeldanamycin synergistically induces apoptosis in Bcr-Abl+ Cells sensitive and resistant to STI571 (imatinib mesylate) in association with down-regulation of Bcr-Abl, abrogation of signal transducer and activator of transcription 5 activity, and Bax conformational change. Mol. Pharmacol. 2005, 67, 1166–1176. [Google Scholar]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar]
- Stankiewicz, A.R.; Livingstone, A.M.; Mohseni, N.; Mosser, D.D. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ. 2009, 16, 638–647. [Google Scholar]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Dupouy, M.; Lapaillerie, D.; Claverol, S.; Vilain, S.; Bonneu, M.; Turcq, B.; Mahon, F.X.; Pasquet, J.M. Proteomic analysis of an imatinib-resistant K562 cell line highlights opposing roles of heat shock cognate 70 and heat shock 70 proteins in resistance. Proteomics 2008, 8, 2394–2406. [Google Scholar]
- Diakos, C.; Krapf, G.; Gerner, C.; Inthal, A.; Lemberger, C.; Ban, J.; Dohnal, A.M.; Panzer-Grumayer, E.R. RNAi-mediated silencing of TEL/AML1 reveals a heat-shock protein-and survivin-dependent mechanism for survival. Blood 2007, 109, 2607–2610. [Google Scholar]
- Marucci, A.; Miscio, G.; Padovano, L.; Boonyasrisawat, W.; Florez, J.C.; Doria, A.; Trischitta, V.; Di Paola, R. The role of HSP70 on ENPP1 expression and insulin-receptor activation. J. Mol. Med. 2009, 87, 139–144. [Google Scholar]
- Myers, S.M.; Mulligan, L.M. The RET receptor is linked to stress response pathways. Cancer Res. 2004, 64, 4453–4463. [Google Scholar]
- Yao, Q.; Nishiuchi, R.; Li, Q.; Kumar, A.R.; Hudson, W.A.; Kersey, J.H. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003, 9, 4483–4493. [Google Scholar]
- Yoo, J.C.; Hayman, M.J. HSP70 binds to SHP2 and has effects on the SHP2-related EGFR/GAB1 signaling pathway. Biochem. Biophys. Res. Commun. 2006, 351, 979–985. [Google Scholar]
- Steiner, K.; Graf, M.; Hecht, K.; Reif, S.; Rossbacher, L.; Pfister, K.; Kolb, H.J.; Schmetzer, H.M.; Multhoff, G. High HSP70-membrane expression on leukemic cells from patients with acute myeloid leukemia is associated with a worse prognosis. Leukemia 2006, 20, 2076–2079. [Google Scholar]
- Ray, S.; Lu, Y.; Kaufmann, S.H.; Gustafson, W.C.; Karp, J.E.; Boldogh, I.; Fields, A.P.; Brasier, A.R. Genomic mechanisms of p210BCR-ABL signaling: Induction of heat shock protein 70 through the GATA response element confers resistance to paclitaxel-induced apoptosis. J. Biol. Chem. 2004, 279, 35604–35615. [Google Scholar]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000, 2, 476–483. [Google Scholar]
- Cui, N.J.; Hu, L.; Lao, S.X. The virtuoueffect of heat shock protein 70 and evil effect of nuclear factor-kappa B in patients with chronic gastric disease of PI-Wie damp-heat syndrome type. Zhongguo Zhong Xi Yi Jie He Za Zhi 2009, 29, 1130–1132. [Google Scholar]
- Piao, L.; Li, Y.; Yang, K.J.; Park, K.A.; Byun, H.S.; Won, M.; Hong, J.; Kim, J.L.; Kweon, G.R.; Hur, G.M.; et al. Heat shock protein 70-mediated sensitization of cells to apoptosis by Carboxyl-Terminal Modulator Protein. BMC Cell Biol. 2009, 10, 53. [Google Scholar]
- Kim, H.E.; Jiang, X.; Du, F.; Wang, X. PHAPI, CAS, and Hsp70 promote apoptosome form194. ation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol. Cell 2008, 30, 239–247. [Google Scholar]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jaattela, M.; Penninger, J.M.; Garrido, C.; Kroemer, G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar]
- Corbiere, C.; Liagre, B.; Terro, F.; Beneytout, J.L. Induction of antiproliferative effect by diosgenin through activation of p53, release of apoptosis-inducing factor (AIF) and modulation of caspase-3 activity in different human cancer cells. Cell Res. 2004, 14, 188–196. [Google Scholar]
- Yuste, V.J.; Sanchez-Lopez, I.; Sole, C.; Moubarak, R.S.; Bayascas, J.R.; Dolcet, X.; Encinas, M.; Susin, S.A.; Comella, J.X. The contribution of apoptosis-inducing factor, caspase-activated DNase, and inhibitor of caspase-activated DNase to the nuclear phenotype and DNA degradation during apoptosis. J. Biol. Chem. 2005, 280, 35670–35683. [Google Scholar]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar]
- Arnoult, D.; Gaume, B.; Karbowski, M.; Sharpe, J.C.; Cecconi, F.; Youle, R.J. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003, 22, 4385–4399. [Google Scholar]
- Delettre, C.; Yuste, V.J.; Moubarak, R.S.; Bras, M.; Lesbordes-Brion, J.C.; Petres, S.; Bellalou, J.; Susin, S.A. AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J. Biol. Chem. 2006, 281, 6413–6427. [Google Scholar]
- Delettre, C.; Yuste, V.J.; Moubarak, R.S.; Bras, M.; Robert, N.; Susin, S.A. Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. J. Biol. Chem. 2006, 281, 18507–18518. [Google Scholar]
- Schmitt, E.; Parcellier, A.; Gurbuxani, S.; Cande, C.; Hammann, A.; Morales, M.C.; Hunt, C.R.; Dix, D.J.; Kroemer, R.T.; Giordanetto, F.; et al. Chemosensitization by a non-apoptogenic heat shock protein 70-binding apoptosis-inducing factor mutant. Cancer Res. 2003, 63, 8233–8240. [Google Scholar]
- Kalinowska, M.; Garncarz, W.; Pietrowska, M.; Garrard, W.T.; Widlak, P. Regulation of the human apoptotic DNase/RNase endonuclease G: Involvement of Hsp70 and ATP. Apoptosis 2005, 10, 821–830. [Google Scholar]
- Chinnathambi, S.; Tomanek-Chalkley, A.; Bickenbach, J.R. HSP70 and EndoG modulate cell death by heat in human skin keratinocytes in vitro. Cells Tissues Organs 2008, 187, 131–140. [Google Scholar]
- Goel, G.; Guo, M.; Ding, J.; Dornbos, D., 3rd.; Ali, A.; Shenaq, M.; Guthikonda, M.; Ding, Y. Combined effect of tumor necrosis factor (TNF)-alpha and heat shock protein (HSP)-70 in reducing apoptotic injury in hypoxia: A cell culture study. Neurosci. Lett. 2010, 483, 162–166. [Google Scholar]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar]
- Ashkenazi, A.; Dixit, V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 1999, 11, 255–260. [Google Scholar]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar]
- Ozoren, N.; El-Deiry, W. Heat shock protects HCT116 and H460 cells from TRAIL-induced apoptosis. Exp. Cell Res. 2002, 281, 175–181. [Google Scholar]
- Ozoren, N.; El-Deiry, W.S. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar]
- Clemons, N.J.; Buzzard, K.; Steel, R.; Anderson, R.L. Hsp72 inhibits Fas-mediated apoptosis upstream of the mitochondria in type II cells. J. Biol. Chem. 2005, 280, 9005–9012. [Google Scholar]
- Liossis, S.N.; Ding, X.Z.; Kiang, J.G.; Tsokos, G.C. Overexpression of the heat shock protein 70 enhances the TCR/CD3- and Fas/Apo-1/CD95-mediated apoptotic cell death in Jurkat T cells. J. Immunol. 1997, 158, 5668–5675. [Google Scholar]
- Musch, M.W.; Kaplan, B.; Chang, E.B. Role of increased basal expression of heat shock protein 72 in colonic epithelial c2BBE adenocarcinoma cells. Cell Growth Differ. 2001, 12, 419–426. [Google Scholar]
- Gao, Y.; Han, C.; Huang, H.; Xin, Y.; Xu, Y.; Luo, L.; Yin, Z. Heat shock protein 70 together with its co-chaperone CHIP inhibits TNF-alpha induced apoptosis by promoting proteasomal degradation of apoptosis signal-regulating kinase1. Apoptosis 2010, 15, 822–833. [Google Scholar]
- Jaattela, M.; Wissing, D.; Kokholm, K.; Kallunki, T.; Egeblad, M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998, 17, 6124–6134. [Google Scholar]
- Steel, R.; Doherty, J.P.; Buzzard, K.; Clemons, N.; Hawkins, C.J.; Anderson, R.L. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J. Biol. Chem. 2004, 279, 51490–51499. [Google Scholar]
- Schett, G.; Steiner, C.W.; Xu, Q.; Smolen, J.S.; Steiner, G. TNFalpha mediates susceptibility to heat-induced apoptosis by protein phosphatase-mediated inhibition of the HSF1/hsp70 stress response. Cell Death Differ. 2003, 10, 1126–1136. [Google Scholar]
- Clemons, N.J.; Anderson, R.L. TRAIL-induced apoptosis is enhanced by heat shock protein 70 expression. Cell Stress Chaperones 2006, 11, 343–355. [Google Scholar]
- Dai, C.; Whitesell, L.; Rogers, A.B. Lindquist, S, Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar]
- Tran, S.E.; Meinander, A.; Holmstrom, T.H.; Rivero-Muller, A.; Heiskanen, K.M.; Linnau, E.K.; Courtney, M.J.; Mosser, D.D.; Sistonen, L.; Eriksson, J.E. Heat stress downregulates FLIP and sensitizes cells to Fas receptor-mediated apoptosis. Cell Death Differ. 2003, 10, 1137–1147. [Google Scholar]
- Feng, X.; Bonni, S.; Riabowol, K. HSP70 induction by ING proteins sensitizes cells to tumor necrosis factor alpha receptor-mediated apoptosis. Mol. Cell. Biol. 2006, 26, 9244–9255. [Google Scholar]
- Imao, M.; Nagaki, M.; Moriwaki, H. Dual effects of heat stress on tumor necrosis factor-alpha-induced hepatocyte apoptosis in mice. Lab. Invest. 2006, 86, 959–967. [Google Scholar]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar]
- Luke, C.J.; Pak, S.C.; Askew, Y.S.; Naviglia, T.L.; Askew, D.J.; Nobar, S.M.; Vetica, A.C.; Long, O.S.; Watkins, S.C.; Stolz, D.B.; et al. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 2007, 130, 1108–1119. [Google Scholar]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cysteine cathepsins: Signaling pathways in apoptosis. Biol. Chem. 2007, 388, 555–560. [Google Scholar]
- Turk, B.; Stoka, V. Protease signalling in cell death: Caspases versus cysteine cathepsins. FEBS Lett. 2007, 581, 2761–2767. [Google Scholar]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar]
- Vasiljeva, O.; Turk, B. Dual contrasting roles of cysteine cathepsins in cancer progression, apoptosis versus tumour invasion. Biochimie 2008, 90, 380–386. [Google Scholar]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar]
- Doulias, P.T.; Kotoglou, P.; Tenopoulou, M.; Keramisanou, D.; Tzavaras, T.; Brunk, U.; Galaris, D.; Angelidis, C. Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: The role of lysosomes and iron. Free Radic. Biol. Med. 2007, 42, 567–577. [Google Scholar]
- Nylandsted, J.; Jaattela, M.; Hoffmann, E.K.; Pedersen, S.F. Heat shock protein 70 inhibits shrinkage-induced programmed cell death via mechanisms independent of effects on cell volume-regulatory membrane transport proteins. Pflugers Arch. 2004, 449, 175–185. [Google Scholar]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jaattela, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar]
- Dudeja, V.; Mujumdar, N.; Phillips, P.; Chugh, R.; Borja-Cacho, D.; Dawra, R.K.; Vickers, S.M.; Saluja, A.K. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology 2009, 136, 1772–1782. [Google Scholar]
- Nylandsted, J.; Wick, W.; Hirt, U.A.; Brand, K.; Rohde, M.; Leist, M.; Weller, M.; Jaattela, M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002, 62, 7139–7142. [Google Scholar]
- Bivik, C.; Rosdahl, I.; Ollinger, K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis 2007, 28, 537–544. [Google Scholar]
- Rammer, P.; Groth-Pedersen, L.; Kirkegaard, T.; Daugaard, M.; Rytter, A.; Szyniarowski, P.; Hoyer-Hansen, M.; Povlsen, L.K.; Nylandsted, J.; Larsen, J.E.; Jaattela, M. BAMLET activates a lysosomal cell death program in cancer cells. Mol. Cancer Ther. 2010, 9, 24–32. [Google Scholar]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar]
- Petersen, N.H.; Kirkegaard, T. HSP70 and lysosomal storage disorders: Novel therapeutic opportunities. Biochem. Soc. Trans 2010, 38, 1479–1483. [Google Scholar]
- Petersen, N.H.; Kirkegaard, T.; Olsen, O.D.; Jaattela, M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 2010, 9, 2305–2309. [Google Scholar]
- Utermohlen, O.; Herz, J.; Schramm, M.; Kronke, M. Fusogenicity of membranes: The impact of acid sphingomyelinase on innate immune responses. Immunobiology 2008, 213, 307–314. [Google Scholar]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta 2008, 1780, 1291–1303. [Google Scholar]
- Mambula, S.S.; Calderwood, S.K. Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int. J. Hyperth. 2006, 22, 575–585. [Google Scholar]
- Calderwood, S.K.; Theriault, J.R.; Gong, J. How is the immune response affected by hyperthermia and heat shock proteins? Int. J. Hyperth. 2005, 21, 713–716. [Google Scholar]
- Calderwood, S.K.; Theriault, J.R.; Gong, J. Message in a bottle: Role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur. J. Immunol. 2005, 35, 2518–2527. [Google Scholar]
- Gabrilovich, D.I. Molecular mechanisms and therapeutic reversal of immune suppression in cancer. Curr. Cancer Drug Targets 2007, 7, 1. [Google Scholar]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425–426. [Google Scholar]
- Chalmin, F.; Mignot, G.; Ghiringhelli, F. Myeloid-derived suppressor cells: A key player in cancer. Med. Sci. (Paris) 2010, 26, 576–579. [Google Scholar]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 2010, 120, 457–471. [Google Scholar]
- Gehrmann, M.; Radons, J.; Molls, M.; Multhoff, G. The therapeutic implications of clinically applied modifiers of heat shock protein 70 (Hsp70) expression by tumor cells. Cell Stress Chaperones 2008, 13, 1–10. [Google Scholar]
- Jaattela, M.; Wissing, D. Heat-shock proteins protect cells from monocyte cytotoxicity: Possible mechanism of self-protection. J. Exp. Med. 1993, 177, 231–236. [Google Scholar]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Denis-Larose, C.; Massie, B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol. 1997, 17, 5317–5327. [Google Scholar]
- Debes, A.; Oerding, M.; Willers, R.; Gobel, U.; Wessalowski, R. Sensitization of human Ewing's tumor cells to chemotherapy and heat treatment by the bioflavonoid quercetin. Anticancer Res. 2003, 23, 3359–3366. [Google Scholar]
- Granado-Serrano, A.B.; Martin, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar]
- Nguyen, T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar]
- van Erk, M.J.; Roepman, P.; van der Lende, T.R.; Stierum, R.H.; Aarts, J.M.; van Bladeren, P.J.; van Ommen, B. Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro. Eur. J. Nutr. 2005, 44, 143–156. [Google Scholar]
- Larocca, L.M.; Ranelletti, F.O.; Maggiano, N.; Rutella, S.; La Barbera, E.O.; Rumi, C.; Serra, F.; Voso, M.T.; Piantelli, M.; Teofili, L.; Leone, G. Differential sensitivity of leukemic and normal hematopoietic progenitors to the killing effect of hyperthermia and quercetin used in combination: Role of heat-shock protein-70. Int. J. Cancer 1997, 73, 75–83. [Google Scholar]
- Jakubowicz-Gil, J.; Paduch, R.; Piersiak, T.; Glowniak, K.; Gawron, A.; Kandefer-Szerszen, M. The effect of quercetin on pro-apoptotic activity of cisplatin in HeLa cells. Biochem. Pharmacol. 2005, 69, 1343–1350. [Google Scholar]
- Jakubowicz-Gil, J.; Pawlikowska-Pawlega, B.; Piersiak, T.; Pawelec, J.; Gawron, A. Quercetin suppresses heat shock-induced nuclear translocation of Hsp72. Folia Histochem. Cytobiol. 2005, 43, 123–128. [Google Scholar]
- Kim, S.H.; Yeo, G.S.; Lim, Y.S.; Kang, C.D.; Kim, C.M.; Chung, B.S. Suppression of multi262. drug resistance via inhibition of heat shock factor by quercetin in MDR cells. Exp. Mol. Med. 1998263, 30, 87–92. [Google Scholar]
- Vilaboa, N.E.; Galan, A.; Troyano, A.; de Blas, E.; Aller, P. Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1). J. Biol. Chem. 2000, 275, 24970–24976. [Google Scholar]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar]
- Soncin, F.; Zhang, X.; Chu, B.; Wang, X.; Asea, A.; Ann Stevenson, M.; Sacks, D.B.; Calderwood, S.K. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem. Biophys. Res. Commun. 2003, 303, 700–706. [Google Scholar]
- Jung, J.H.; Lee, J.O.; Kim, J.H.; Lee, S.K.; You, G.Y.; Park, S.H.; Park, J.M.; Kim, E.K.; Suh, P.G.; An, J.K.; et al. Quercetin suppresses HeLa cell viability via AMPK-induced HSP70 and EGFR down-regulation. J. Cell Physiol. 2010, 223, 408–414. [Google Scholar]
- Wang, R.E.; Kao, J.L.; Hilliard, C.A.; Pandita, R.K.; Roti Roti, J.L.; Hunt, C.R.; Taylor, J.S. Inhibition of heat shock induction of heat shock protein 70 and enhancement of heat shock protein 27 phosphorylation by quercetin derivatives. J. Med. Chem. 2009, 52, 1912–1921. [Google Scholar]
- Piantelli, M.; Tatone, D.; Castrilli, G.; Savini, F.; Maggiano, N.; Larocca, L.M.; Ranelletti, F.O.; Natali, P.G. Quercetin and tamoxifen sensitize human melanoma cells to hyperthermia. Melanoma Res. 2001, 11, 469–476. [Google Scholar]
- Westerheide, S.D.; Kawahara, T.L.; Orton, K.; Morimoto, R.I. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 2006, 281, 9616–9622. [Google Scholar]
- Koishi, M.; Yokota, S.; Mae, T.; Nishimura, Y.; Kanamori, S.; Horii, N.; Shibuya, K.; Sasai, K.; Hiraoka, M. The effects of KNK437, a novel inhibitor of heat shock protein synthesis, on the acquisition of thermotolerance in a murine transplantable tumor in vivo. Clin. Cancer Res. 2001, 7, 215–219. [Google Scholar]
- Yokota, S.; Kitahara, M.; Nagata, K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000, 60, 2942–2948. [Google Scholar]
- Davenport, E.L.; Zeisig, A.; Aronson, L.I.; Moore, H.E.; Hockley, S.; Gonzalez, D.; Smith, E.M.; Powers, M.V.; Sharp, S.Y.; Workman, P.; et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia 2010, 24, 1804–1807. [Google Scholar]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar]
- Nimmanapalli, R.; Gerbino, E.; Dalton, W.S.; Gandhi, V.; Alsina, M. HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br. J. Haematol. 2008, 142, 551–561. [Google Scholar]
- Wu, Y.C.; Yen, W.Y.; Lee, T.C.; Yih, L.H. Heat shock protein inhibitors, 17-DMAG and KNK437, enhance arsenic trioxide-induced mitotic apoptosis. Toxicol. Appl. Pharmacol. 2009, 236, 231–238. [Google Scholar]
- Ohnishi, K.; Takahashi, A.; Yokota, S.; Ohnishi, T. Effects of a heat shock protein inhibitor KNK437 on heat sensitivity and heat tolerance in human squamous cell carcinoma cell lines differing in p53 status. Int. J. Radiat. Biol. 2004, 80, 607–614. [Google Scholar]
- Sahin, E.; Sahin, M.; Sanlioglu, A.D.; Gumuslu, S. KNK437, a benzylidene lactam compound, sensitises prostate cancer cells to the apoptotic effect of hyperthermia. Int. J. Hyperth. 2011, 27, 63–73. [Google Scholar]
- Inoue, H.; Uyama, T.; Hayashi, J.; Watanabe, A.; Kobayashi, K.; Tadokoro, T.; Yamamoto, Y. N-Formyl-3,4-methylenedioxy-benzylidene-gamma-butyrolaetam, KNK437 induces caspase-3 activation through inhibition of mTORC1 activity in Cos-1 cells. Biochem. Biophys. Res. Commun. 2010, 395, 56–60. [Google Scholar]
- Antonoff, M.B.; Chugh, R.; Borja-Cacho, D.; Dudeja, V.; Clawson, K.A.; Skube, S.J.; Sorenson, B.S.; Saltzman, D.A.; Vickers, S.M.; Saluja, A.K. Triptolide therapy for neuroblastoma decreases cell viability in vitro and inhibits tumor growth in vivo. Surgery 2009, 146, 282–290. [Google Scholar]
- Clawson, K.A.; Borja-Cacho, D.; Antonoff, M.B.; Saluja, A.K.; Vickers, S.M. Triptolide and TRAIL combination enhances apoptosis in cholangiocarcinoma. J. Surg. Res. 2010, 163, 244–249. [Google Scholar]
- Phillips, P.A.; Dudeja, V.; McCarroll, J.A.; Borja-Cacho, D.; Dawra, R.K.; Grizzle, W.E.; Vickers, S.M.; Saluja, A.K. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007, 67, 9407–9416. [Google Scholar]
- Zaarur, N.; Gabai, V.L.; Porco, J.A., Jr.; Calderwood, S.; Sherman, M.Y. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006, 66, 1783–1791. [Google Scholar]
- Chauhan, D.; Li, G.; Shringarpure, R.; Podar, K.; Ohtake, Y.; Hideshima, T.; Anderson, K.C. Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res. 2003, 63, 6174–6177. [Google Scholar]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar]
- Lis, J.T.; Mason, P.; Peng, J.; Price, D.H.; Werner, J. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000, 14, 792–803. [Google Scholar]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.M.; Han, D.C. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar]
- Schmitt, E.; Maingret, L.; Puig, P.E.; Rerole, A.L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar]
Abbreviations
| AD | activation domain |
| AIF | apoptosis inducible factor |
| AMPK | AMP-activated protein kinase |
| Apaf-1 | apoptotic protease activating factor 1 |
| ASK-1 | apoptosis signal-regulating kinase 1 |
| BAG | Bcl2-associated athanogene |
| CAS | cellular apoptosis susceptibility protei |
| CBP | CREB binding protein |
| CEBPB | CCAAT/enhancer binding protein beta |
| CARD | caspase recruitment domain |
| CHIP | C-terminus of Hsp70-interacting protein |
| CKII | casein kinase II |
| CamKII | calcium/calmodulin kinase II |
| CTMP | carboxyl-terminal modulator protein |
| DBD | DNA binding domain |
| Diablo | IAP-binding mitochondrial protein |
| DSG | deoxyspergualin |
| ERK | extracellular regulated MAP kinase |
| FADD | Fas-associated death domain protein |
| FLIP | Flice-like inhibitor protein |
| GSK3β | glycogen synthase kinase 3 beta |
| HOP | Hsc70/Hsp90-organizing protein |
| HR | heptad repeat |
| HSE | heat shock elements |
| HSF | heat shock factor |
| HSP | heat shock protein |
| HSR | heat shock response |
| IkB | inhibitor of NF-kB |
| KNK437 | N-formyl-3,4-methylenedioxy-benzyldiene-γ-butyroletam |
| JNK | Janus kinase |
| LAMP | lysosome-associated membrane protein |
| LMP | lysosomal membrane permeabilization |
| MAPK | mitogen activated protein kinase |
| MDSC | myeloid-derived suppressor cells |
| Mcl-1 | myeloid leukaemia sequence 1 |
| MKP | mitogen-activated protein kinase phosphatase |
| MRP | multidrug resistance protein |
| NEF | nucleotide exchanging factor |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NF-IL6 | nuclear factor of IL6 gene |
| NOA | non-oncogene addiction |
| PBD | peptide binding domain |
| PES | 2-phenylethylenesulfonamide |
| PHAPI | putative HLA class II associated protein |
| PTM | posttranslational modification |
| SBD | substrate binding domain |
| RIP1 | receptor-interacting protein 1 |
| SEK1 | SAPK/ERK kinase 1 |
| SMAC | second mitochondrial-derived activator of caspases |
| STAT | signal-transducer and activator of transcription protein |
| STRAP | stress-responsive activator of p300 |
| p-TEFb | positive transcription elongation factor b |
| TLR2 | Toll-like receptor 2 |
| TNFR1 | tumour necrosis factor receptor 1 |
| TPR | tetracopeptide repeat |
| TRADD | TNFR1-associated death domain protein |
| TRAIL | TNF-related apoptosis-inducing ligand |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zorzi, E.; Bonvini, P. Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity. Cancers 2011, 3, 3921-3956. https://doi.org/10.3390/cancers3043921
Zorzi E, Bonvini P. Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity. Cancers. 2011; 3(4):3921-3956. https://doi.org/10.3390/cancers3043921
Chicago/Turabian StyleZorzi, Elisa, and Paolo Bonvini. 2011. "Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity" Cancers 3, no. 4: 3921-3956. https://doi.org/10.3390/cancers3043921
APA StyleZorzi, E., & Bonvini, P. (2011). Inducible Hsp70 in the Regulation of Cancer Cell Survival: Analysis of Chaperone Induction, Expression and Activity. Cancers, 3(4), 3921-3956. https://doi.org/10.3390/cancers3043921