
Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases: A Comprehensive Review


Simple Summary
Abstract
1. Introduction
1.1. Research Methodology for Article Selection
1.2. Historical Perspective of Spatial Transcriptomics
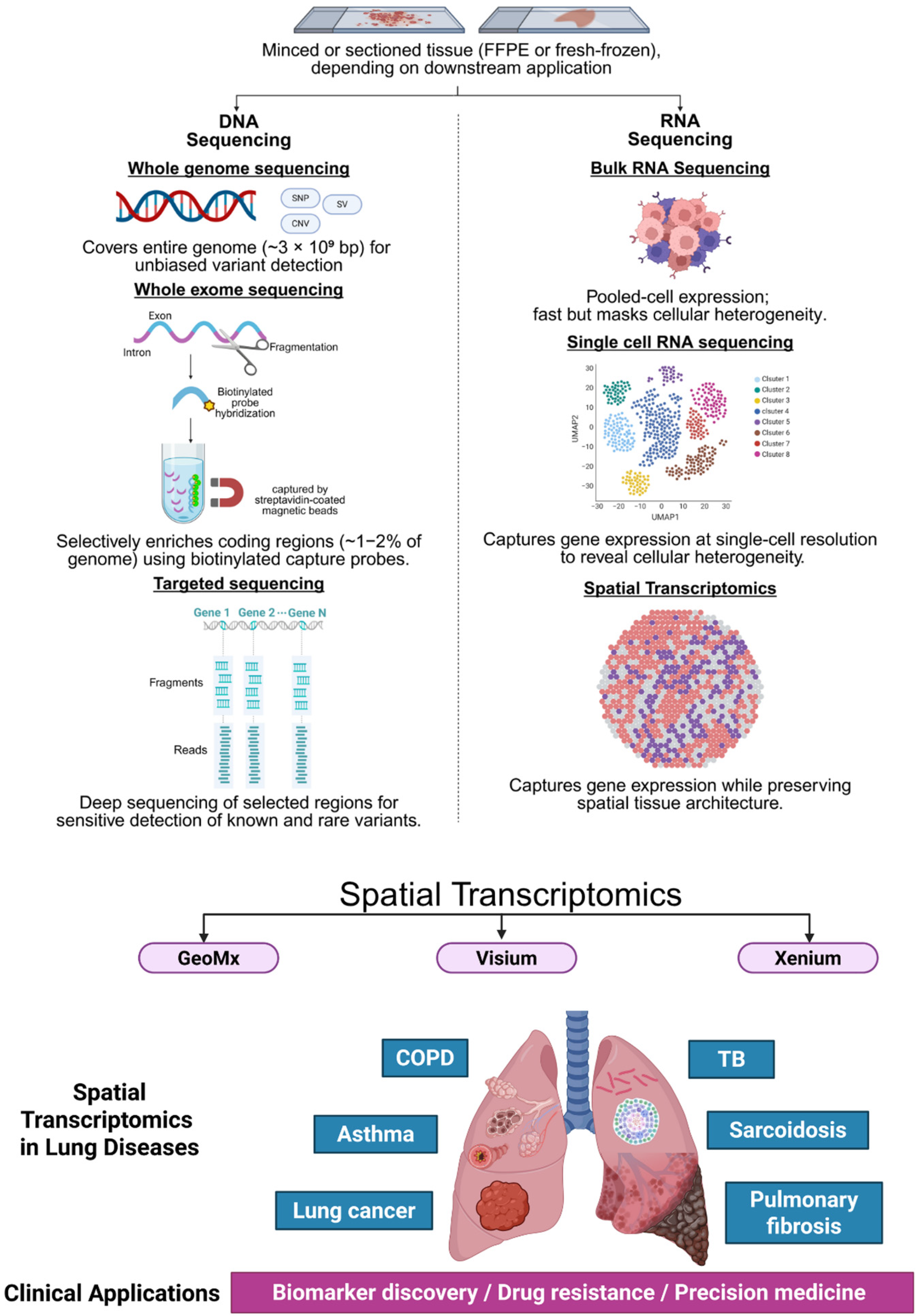
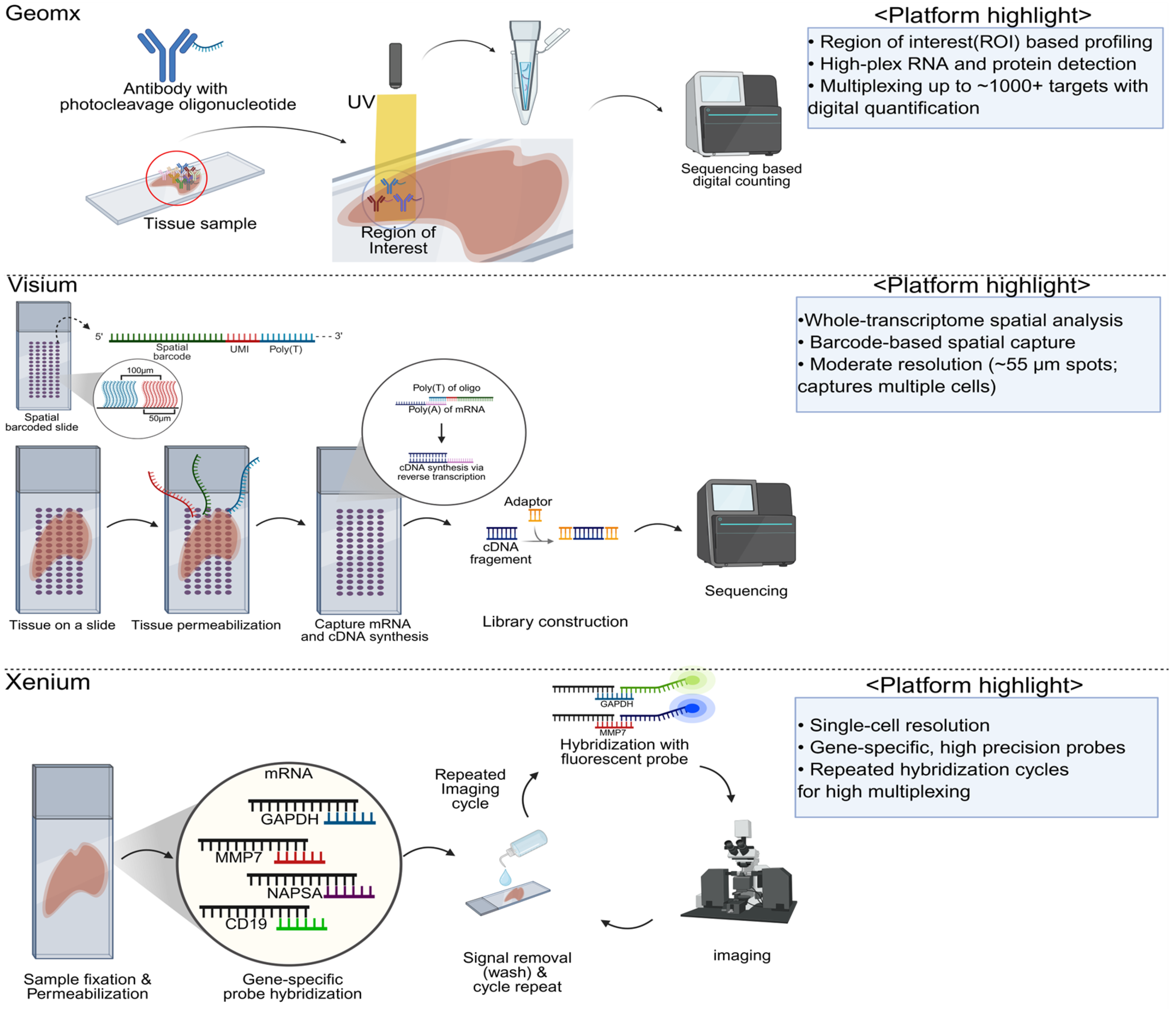
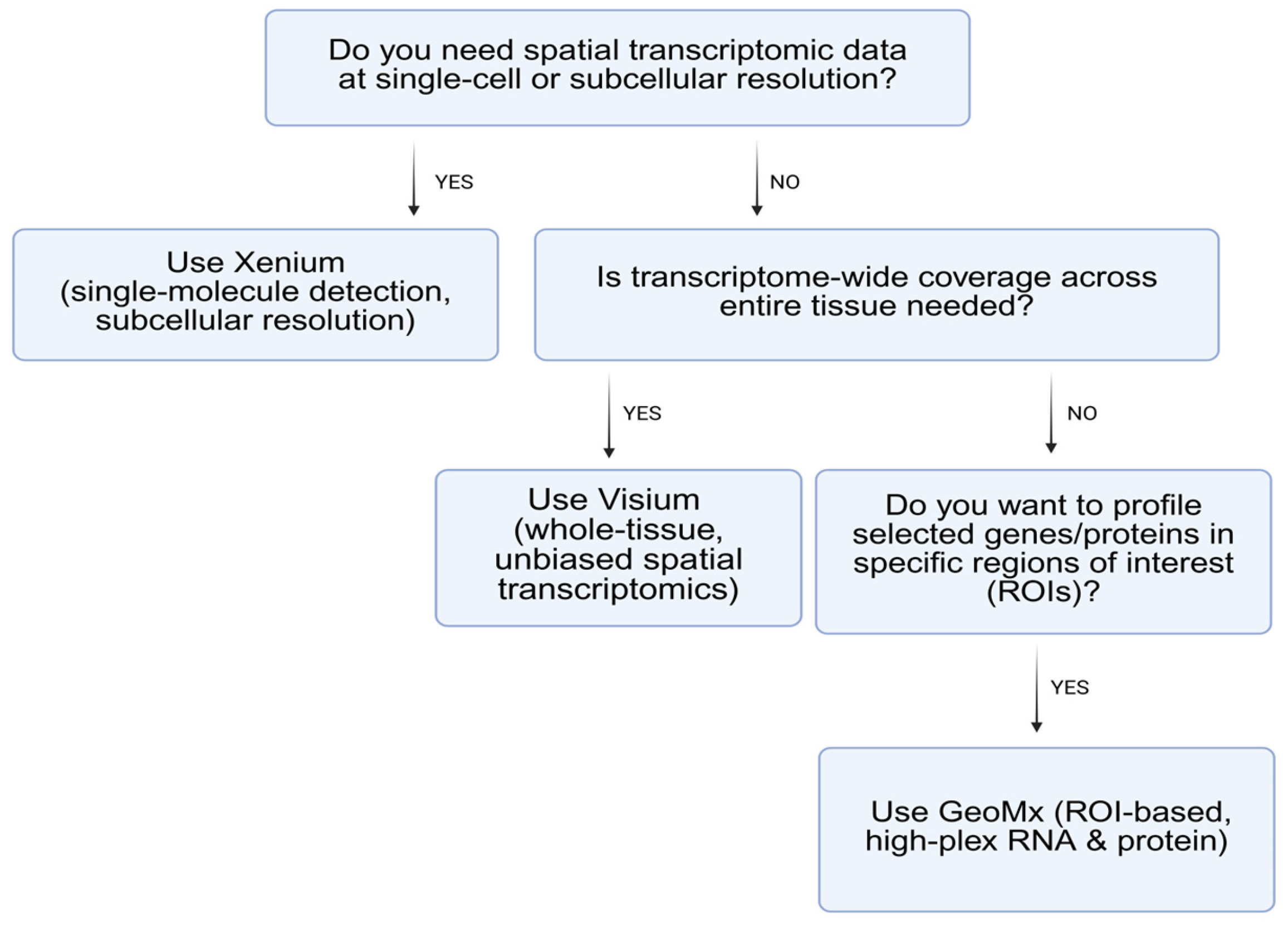
2. Major Platforms and Methodologies in Spatial Transcriptomics
Summary and Comparative Insights
3. Published Data on Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases
3.1. Spatial Transcriptomics in Lung Cancer: Tumor Heterogeneity, Microenvironment, and Therapeutic Implications
3.2. Spatial Transcriptomics in Non-Malignant Pulmonary Diseases: Inflammatory Niches and Fibrotic Remodeling
4. Ongoing Clinical Trials Utilizing Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases
5. Future Perspectives and Clinical Significance
5.1. Spatial Transcriptomics and Multomics Integration
5.2. Cutting-Edge Advances in Spatial Transcriptomics Methodologies
5.3. Spatial Transcriptomics and Artificial Intelligence
6. Challenges and Considerations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Piñeiro, A.J.; Houser, A.E.; Ji, A.L. Research Techniques Made Simple: Spatial Transcriptomics. J. Investig. Dermatol. 2022, 142, 993–1001.e1. [Google Scholar] [CrossRef]
- Robles-Remacho, A.; Sanchez-Martin, R.M.; Diaz-Mochon, J.J. Spatial Transcriptomics: Emerging Technologies in Tissue Gene Expression Profiling. Anal. Chem. 2023, 95, 15450–15460. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Xu, H.; Wang, S.; Fang, M.; Luo, S.; Chen, C.; Wan, S.; Wang, R.; Tang, M.; Xue, T.; Li, B.; et al. SPACEL: Deep learning-based characterization of spatial transcriptome architectures. Nat. Commun. 2023, 14, 7603. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Molla Desta, G.; Birhanu, A.G. Advancements in single-cell RNA sequencing and spatial transcriptomics: Transforming biomedical research. Acta Biochim. Pol. 2025, 72, 13922. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Luo, J.; Gao, Q.; Wang, M.; Liu, H.; Zhu, H. Single-cell and spatial transcriptome characterize coinhibitory cell-cell communications during histological progression of lung adenocarcinoma. Front. Immunol. 2024, 15, 1430163. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheng, M.; Jiang, Y.; Xu, J.; Mentis, A.A.; Wang, S.; Zheng, H.; Sahu, S.K.; Liu, L.; Xu, X. Spatially resolved transcriptomics: A comprehensive review of their technological advances, applications, and challenges. J. Genet. Genom. 2023, 50, 625–640. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Rong, D.; Zhang, H.; Shi, X.; Yang, W.; Zheng, W.; Sun, G.; Wu, F.; Cao, H.; et al. Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther. Oncolytics 2021, 21, 183–206. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gracia Villacampa, E.; Larsson, L.; Mirzazadeh, R.; Kvastad, L.; Andersson, A.; Mollbrink, A.; Kokaraki, G.; Monteil, V.; Schultz, N.; Appelberg, K.S.; et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genom. 2021, 1, 100065. [Google Scholar] [CrossRef]
- Cao, J.; Li, C.; Cui, Z.; Deng, S.; Lei, T.; Liu, W.; Yang, H.; Chen, P. Spatial Transcriptomics: A Powerful Tool in Disease Understanding and Drug Discovery. Theranostics 2024, 14, 2946–2968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Warr, A.; Robert, C.; Hume, D.; Archibald, A.; Deeb, N.; Watson, M. Exome Sequencing: Current and Future Perspectives. G3 2015, 5, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mahajan, M.C.; McLellan, A.S. Whole-Exome Sequencing (WES) for Illumina Short Read Sequencers Using Solution-Based Capture. Methods Mol. Biol. 2020, 2076, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ou, Y.N.; Wu, B.S.; Liu, W.S.; Deng, Y.T.; He, X.Y.; Chen, Y.L.; Kang, J.; Fei, C.J.; Zhu, Y.; et al. Large-scale whole-exome sequencing analyses identified protein-coding variants associated with immune-mediated diseases in 350,770 adults. Nat. Commun. 2024, 15, 5924. [Google Scholar] [CrossRef] [PubMed]
- Bagger, F.O.; Borgwardt, L.; Jespersen, A.S.; Hansen, A.R.; Bertelsen, B.; Kodama, M.; Nielsen, F.C. Whole genome sequencing in clinical practice. BMC Med. Genom. 2024, 17, 39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zahir, F.R.; Mwenifumbo, J.C.; Chun, H.E.; Lim, E.L.; Van Karnebeek, C.D.M.; Couse, M.; Mungall, K.L.; Lee, L.; Makela, N.; Armstrong, L.; et al. Comprehensive whole genome sequence analyses yields novel genetic and structural insights for Intellectual Disability. BMC Genom. 2017, 18, 403. [Google Scholar] [CrossRef]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yang, Y.C.; An, Z.J.; Zhang, M.H.; Fu, X.H.; Huang, Z.F.; Yuan, Y.; Hou, J. Advances in spatial transcriptomics and related data analysis strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Yue, L.; Liu, F.; Hu, J.; Yang, P.; Wang, Y.; Dong, J.; Shu, W.; Huang, X.; Wang, S. A guidebook of spatial transcriptomic technologies, data resources and analysis approaches. Comput. Struct. Biotechnol. J. 2023, 21, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Duggan, W.P.; Kisakol, B.; Woods, I.; Azimi, M.; Dussmann, H.; Fay, J.; O’Grady, T.; Maguire, B.; Reynolds, I.S.; Salvucci, M.; et al. Spatial transcriptomic analysis reveals local effects of intratumoral fusobacterial infection on DNA damage and immune signaling in rectal cancer. Gut Microbes 2024, 16, 2350149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Levy-Jurgenson, A.; Tekpli, X.; Kristensen, V.N.; Yakhini, Z. Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer. Sci. Rep. 2020, 10, 18802. [Google Scholar] [CrossRef] [PubMed]
- Rusch, M.; Nakitandwe, J.; Shurtleff, S.; Newman, S.; Zhang, Z.; Edmonson, M.N.; Parker, M.; Jiao, Y.; Ma, X.; Liu, Y. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018, 9, 3962. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, F.; Zhang, Y.; Zhang, L.; Li, Z.; Fang, Q.; Gao, R.; Zhang, Z. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019, 20, 242. [Google Scholar] [CrossRef]
- Larroquette, M.; Guegan, J.P.; Besse, B.; Cousin, S.; Brunet, M.; Le Moulec, S.; Le Loarer, F.; Rey, C.; Soria, J.C.; Barlesi, F.; et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J. Immunother. Cancer 2022, 10, e003890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, Y.; Zuo, Y.; Li, G.; Liu, W.; Pan, Y.; Fan, T.; Fu, X.; Yao, X.; Peng, Y. Advances in spatial transcriptomics and its applications in cancer research. Mol. Cancer 2024, 23, 129. [Google Scholar] [CrossRef]
- Van, T.M.; Blank, C.U. A user’s perspective on GeoMxTM digital spatial profiling. Immuno-Oncol. Technol. 2019, 1, 11–18. [Google Scholar] [CrossRef]
- Koldej, R.M.; Ritchie, D.S. High multiplex analysis of the immune microenvironment in bone marrow trephine samples using GeoMX™ digital spatial profiling. Immuno-Oncol. Technol. 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Wang, N.; Li, X.; Ding, Z. High-Plex Spatial Profiling of RNA and Protein Using Digital Spatial Profiler. Methods Mol. Biol. 2023, 2660, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, X.; Liu, Y.; Zhang, Y.; Yang, Z.; Dong, J.; Wang, N.; Ying, J.; Zhou, M.; Yang, L. Spatial Transcriptome-Wide Profiling of Small Cell Lung Cancer Reveals Intra-Tumoral Molecular and Subtype Heterogeneity. Adv. Sci. 2024, 11, e2402716. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, N.; Hong, W.; Wu, Y.; Chen, Z.S.; Bai, M.; Wang, W.; Zhu, J. Next-generation spatial transcriptomics: Unleashing the power to gear up translational oncology. MedComm 2024, 5, e765. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, L.; Chen, D.; Song, D.; Liu, X.; Zhang, Y.; Xu, X.; Wang, X. Clinical and translational values of spatial transcriptomics. Signal Transduct. Target. Ther. 2022, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Lundberg, E.; Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 2022, 23, 741–759. [Google Scholar] [CrossRef]
- Anderson, A.C.; Yanai, I.; Yates, L.R.; Wang, L.; Swarbrick, A.; Sorger, P.; Santagata, S.; Fridman, W.H.; Gao, Q.; Jerby, L.; et al. Spatial transcriptomics. Cancer Cell 2022, 40, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Zaman, T.; Chowdhury, F.; Mraiche, F.; Tariq, M.; Ahmad, I.S.; Hasan, A. Single-Cell RNA Sequencing with Spatial Transcriptomics of Cancer Tissues. Int. J. Mol. Sci. 2022, 23, 3042. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Franzén, L.; Olsson Lindvall, M.; Hühn, M.; Ptasinski, V.; Setyo, L.; Keith, B.P.; Collin, A.; Oag, S.; Volckaert, T.; Borde, A. Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis. Nat. Genet. 2024, 56, 1725–1736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lim, H.J.; Wang, Y.; Buzdin, A.; Li, X. A practical guide for choosing an optimal spatial transcriptomics technology from seven major commercially available options. BMC Genom. 2025, 26, 47. [Google Scholar] [CrossRef] [PubMed]
- Cilento, M.A.; Sweeney, C.J.; Butler, L.M. Spatial transcriptomics in cancer research and potential clinical impact: A narrative review. J. Cancer Res. Clin. Oncol. 2024, 150, 296. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vannan, A.; Lyu, R.; Williams, A.L.; Negretti, N.M.; Mee, E.D.; Hirsh, J.; Hirsh, S.; Hadad, N.; Nichols, D.S.; Calvi, C.L.; et al. Spatial transcriptomics identifies molecular niche dysregulation associated with distal lung remodeling in pulmonary fibrosis. Nat. Genet. 2025, 57, 647–658. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krausgruber, T.; Redl, A.; Barreca, D.; Doberer, K.; Romanovskaia, D.; Dobnikar, L.; Guarini, M.; Unterluggauer, L.; Kleissl, L.; Atzmüller, D.; et al. Single-cell and spatial transcriptomics reveal aberrant lymphoid developmental programs driving granuloma formation. Immunity 2023, 56, 289–306.e7. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhong, P.; Yue, L.; Li, C.; Yun, Z.; Si, G.; Li, M.; Chen, Z.; Tan, Y.; Bao, P. Spatial transcriptomic sequencing reveals immune microenvironment features of Mycobacterium tuberculosis granulomas in lung and omentum. Theranostics 2024, 14, 6185–6201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rojas-Quintero, J.; Ochsner, S.A.; New, F.; Divakar, P.; Yang, C.X.; Wu, T.D.; Robinson, J.; Shimoga Chandrashekar, D.; Banovich, N.E.; Rosas, I.O.; et al. Spatial Transcriptomics Resolve an Emphysema-specific Lymphoid Follicle B Cell Signature in COPD. Am. J. Respir. Crit. Care Med. 2023, 209, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Joulia, R.; Patti, S.; Traves, W.J.; Loewenthal, L.; Yates, L.; Walker, S.A.; Puttur, F.; Al-Sahaf, M.; Cahill, K.N.; Lai, J.; et al. A single-cell spatial chart of the airway wall reveals proinflammatory cellular ecosystems and their interactions in health and asthma. Nat. Immunol. 2025, 26, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, B.; Min, Q.; Yang, X.; Yan, S.; Ma, Y.; Li, S.; Fan, J.; Wang, Y.; Dong, B.; et al. Spatial transcriptomics delineates molecular features and cellular plasticity in lung adenocarcinoma progression. Cell Discov. 2023, 9, 96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Choi, H.; Na, K.J.; Jung, Y.; Lim, M.; Lee, D.; Lee, J.E.; Im, H.J.; Lee, D.; Koh, J.; Kim, Y.T. Spatial Transcriptomics Reveals Spatially Diverse Cancer-Associated Fibroblast in Lung Squamous Cell Carcinoma Linked to Tumor Progression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Xie, L.; Kong, H.; Yu, J.; Sun, M.; Lu, S.; Zhang, Y.; Hu, J.; Du, F.; Lian, Q.; Xin, H. Spatial transcriptomics reveals heterogeneity of histological subtypes between lepidic and acinar lung adenocarcinoma. Clin. Transl. Med. 2024, 14, e1573. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Q.; Abdo, R.; Iosef, C.; Kaneko, T.; Cecchini, M.; Han, V.K.; Li, S.S.C. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat. Commun. 2022, 13, 5983. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peyraud, F.; Guégan, J.P.; Rey, C.; Lara, O.; Odin, O.; Del Castillo, M.; Vanhersecke, L.; Coindre, J.M.; Clot, E.; Brunet, M.; et al. Spatially resolved transcriptomics reveal the determinants of primary resistance to immunotherapy in NSCLC with mature tertiary lymphoid structures. Cell Rep. Med. 2025, 6, 101934. [Google Scholar] [CrossRef] [PubMed]
- Lubo, I.; Hernandez, S.; Wistuba, I.I.; Solis Soto, L.M. Novel Spatial Approaches to Dissect the Lung Cancer Immune Microenvironment. Cancers 2024, 16, 4145. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wong-Rolle, A.; Dong, Q.; Zhu, Y.; Divakar, P.; Hor, J.L.; Kedei, N.; Wong, M.; Tillo, D.; Conner, E.A.; Rajan, A. Spatial meta-transcriptomics reveal associations of intratumor bacteria burden with lung cancer cells showing a distinct oncogenic signature. J. Immunother. Cancer 2022, 10, e004698. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, N.; Li, X.; Wang, R.; Ding, Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol. J. 2021, 16, e2100041. [Google Scholar] [CrossRef] [PubMed]
- De Zuani, M.; Xue, H.; Park, J.S.; Dentro, S.C.; Seferbekova, Z.; Tessier, J.; Curras-Alonso, S.; Hadjipanayis, A.; Athanasiadis, E.I.; Gerstung, M.; et al. Single-cell and spatial transcriptomics analysis of non-small cell lung cancer. Nat. Commun. 2024, 15, 4388. [Google Scholar] [CrossRef]
- Arora, R.; Cao, C.; Kumar, M.; Sinha, S.; Chanda, A.; McNeil, R.; Samuel, D.; Arora, R.K.; Matthews, T.W.; Chandarana, S.; et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat. Commun. 2023, 14, 5029. [Google Scholar] [CrossRef]
- Chen, C.; Guo, Q.; Liu, Y.; Hou, Q.; Liao, M.; Guo, Y.; Zhang, Y.; Wang, F.; Liu, H.; Luan, X.; et al. Single-cell and spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin. Transl. Med. 2023, 13, e1515. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, Y.; Li, S.; Zhao, Y.; Zhang, X.; Mao, X.; Xu, R. Integrative single-cell and spatial transcriptomics analysis reveals MDK-NCL pathway’s role in shaping the immunosuppressive environment of lung adenocarcinoma. Front. Immunol. 2025, 16, 1546382. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tagore, S.; Caprio, L.; Amin, A.D.; Bestak, K.; Luthria, K.; D’Souza, E.; Barrera, I.; Melms, J.C.; Wu, S.; Abuzaid, S.; et al. Single-cell and spatial genomic landscape of non-small cell lung cancer brain metastases. Nat. Med. 2025, 31, 1351–1363. [Google Scholar] [CrossRef]
- Carow, B.; Hauling, T.; Qian, X.; Kramnik, I.; Nilsson, M.; Rottenberg, M.E. Spatial and temporal localization of immune transcripts defines hallmarks and diversity in the tuberculosis granuloma. Nat. Commun. 2019, 10, 1823. [Google Scholar] [CrossRef] [PubMed]
- Joulia, R.; Puttur, F.; Stölting, H.; Traves, W.J.; Entwistle, L.J.; Voitovich, A.; Garcia Martín,, M.; Al-Sahaf, M.; Bonner, K.; Scotney, E.; et al. Mast cell activation disrupts interactions between endothelial cells and pericytes during early life allergic asthma. J. Clin. Investig. 2024, 134, e173676. [Google Scholar] [CrossRef]
- Berlanda, S.F.; Breitfeld, M.; Dietsche, C.L.; Dittrich, P.S. Recent Advances in Microfluidic Technology for Bioanalysis and Diagnostics. Anal. Chem. 2021, 93, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Jin, K.; Yao, Y.; Jin, L.; Shao, X.; Li, C.; Lu, X.; Fan, X. Spatial integration of multi-omics single-cell data with SIMO. Nat. Commun. 2025, 16, 1265. [Google Scholar] [CrossRef]
- Wess, M.; Andersen, M.K.; Midtbust, E.; Guillem, J.C.C.; Viset, T.; Størkersen, Ø.; Krossa, S.; Rye, M.B.; Tessem, M.B. Spatial integration of multi-omics data from serial sections using the novel Multi-Omics Imaging Integration Toolset. Gigascience 2025, 14, giaf035. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lv, T.; Zhang, Y.; Liu, J.; Kang, Q.; Liu, L. Multi-omics integration for both single-cell and spatially resolved data based on dual-path graph attention auto-encoder. Brief. Bioinform. 2024, 25, bbae450. [Google Scholar] [CrossRef]
- Nichterwitz, S.; Benitez, J.A.; Hoogstraaten, R.; Deng, Q.; Hedlund, E. LCM-Seq: A Method for Spatial Transcriptomic Profiling Using Laser Capture Microdissection Coupled with PolyA-Based RNA Sequencing. Methods Mol. Biol. 2018, 1649, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Androvic, P.; Schifferer, M.; Perez Anderson, K.; Cantuti-Castelvetri, L.; Jiang, H.; Ji, H.; Liu, L.; Gouna, G.; Berghoff, S.A.; Besson-Girard, S.; et al. Spatial Transcriptomics-correlated Electron Microscopy maps transcriptional and ultrastructural responses to brain injury. Nat. Commun. 2023, 14, 4115. [Google Scholar] [CrossRef]
- Sansone, A. Spatial transcriptomics levels up. Nat. Methods 2019, 16, 458. [Google Scholar] [CrossRef]
- Gimondi, S.; Ferreira, H.; Reis, R.L.; Neves, N.M. Microfluidic Devices: A Tool for Nanoparticle Synthesis and Performance Evaluation. ACS Nano 2023, 17, 14205–14228. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rajdeo, P.; Aronow, B.; Surya Prasath, V.B. Deep learning-based multimodal spatial transcriptomics analysis for cancer. Adv. Cancer Res. 2024, 163, 1–38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bi, W.L.; Hosny, A.; Schabath, M.B.; Giger, M.L.; Birkbak, N.J.; Mehrtash, A.; Allison, T.; Arnaout, O.; Abbosh, C.; Dunn, I.F.; et al. Artificial intelligence in cancer imaging: Clinical challenges and applications. CA Cancer J. Clin. 2019, 69, 127–157. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.; Wang, C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | WES | WGS | Bulk RNA-seq | scRNA-seq | Spatial Transcriptomics |
|---|---|---|---|---|---|
| Data Type | DNA (exons only) | DNA (whole genome) | RNA (pooled sample) | RNA (individual cells) | RNA in intact tissue (spots or single cells) |
| Coverage | Protein-coding regions (~1–2% of genome) | Entire genome | Transcriptome (averaged) | Transcriptome (per cell) | Whole/partial transcriptome; spatial resolution varies by platform (spot to subcellular) |
| Resolution | Detects exon variants only | Detects all variants (SNVs, SVs, etc.) | Average expression across mixed cells | Expression profiles at single-cell level | Multi-/single-cell gene expression + tissue context |
| Applications | Disease-causing coding variants, driver mutations | Comprehensive variant analysis, structural variation | Differential expression, biomarker discovery | Heterogeneity, rare cell types, lineage tracing | Cell–cell interaction, tissue microenvironment, histology integration |
| Complexity | Moderate; targeted capture + NGS | High; large data, complex variant calling | Moderate; standard RNA-seq pipeline | High; single-cell isolation, large datasets | High; specialized platforms, data integration (image + transcriptome) |
| Typical Cost | Medium (less than WGS) | Highest among options | Lower (per sample) | Higher (per sample), specialized library prep | High (instrumentation, reagents, large data) |
| Key Limitations | Misses noncoding variants, no expression data | Expensive, high storage/computing needs | Loses single-cell detail, masks rare populations | No spatial info, can alter cell states upon dissociation | Technology still evolving, fewer high-throughput options, costlier |
| Platform | GeoMx (NanoString) | Xenium (10x Genomics) | Visium (10x Genomics) |
|---|---|---|---|
| Technical Principle | Photocleavable oligo tags bound to probes or antibodies; UV-based ROI illumination | In situ hybridization of barcoded probes for single-molecule detection | Spatially barcoded capture spots on slides (oligo-dT or targeted probes) |
| Resolution | Multi-cell to near single-cell (depends on ROI size) | Single-molecule or single-cell resolution | Multi-cell per spot (55–100 µm diameter) |
| Molecular Targets | RNA (up to 18,000-plex) or proteins (100+ markers) | RNA transcripts (single-molecule sensitivity) | Poly(A)+ RNA; a targeted probe approach available for FFPE |
| Sample Compatibility | FFPE or fresh/frozen | Primarily fresh/frozen; must be compatible with in situ protocols | Fresh/frozen standard; FFPE version uses targeted capture |
| Key Strengths |
|
|
|
| Key Limitations |
|
|
|
| Use Cases |
|
|
|
| Diseases | Key Findings | ST Platforms | Sample Size | Reference |
|---|---|---|---|---|
| Pulmonary Fibrosis (PF) | Mapped alveolar epithelial dysregulation (SFTPC↓, KRT17↑) and macrophage polarization (SPP1/CHI3L1) across remodeling gradients in idiopathic PF. | Xenium, Visium | 35 unique lungs | [43] |
| Pulmonary Fibrosis, | IPF lungs show arrested alveolar cell regeneration, unlike the active repair seen in the BLM model, due to differences in signaling molecules (TGF-β, APOE, YAP1, TEAD) and immune cell profiles. | Visium | 4 Human IPF, 6 bleomycin-induced mouse lungs | [40] |
| Sarcoidosis | Metabolically reprogrammed macrophages, cytokine-producing Th17.1 cells, and fibroblasts with inflammatory and tissue-remodeling phenotypes are key players in granuloma formation | Visium | 12 patients | [44] |
| Tuberculosis | Mtb infection can activate TGF-β signaling by inducing the expression of THBS1/2 and CD36 | Visium | 2 patients, 2 controls | [45] |
| COPD, emphysema | The extent of centrilobular emphysema was significantly associated with genes involved in B cell maturation and antibody production. | GeoMx DSP | 40 patients | [46] |
| Asthma | The asthma airway mucosa exhibited a distinct remodeling program within these cellular ecosystems, marked by increased proximity between key cell types | Xenium, GeoMx DSP | 20 patients, 8 controls | [47] |
| Lung Adenocarcinoma (LUAD) | Identified co-inhibitory ligand-receptor interactions (NECTIN2/TIGIT, PVR/TIGIT) in solid histological patterns, correlating with poor immunotherapy response. | Visium | 2 tumor samples | [7] |
| LUAD Progression | Linked dedifferentiation trajectories (lepidic → micropapillary) to KRT17 overexpression and macrophage spatial heterogeneity. | Visium | 10 patients | [48] |
| Lung Squamous Cell Carcinoma (LUSC) | Revealed spatially distinct CAF subtypes (POSTN+/COL11A1+) driving tumor invasion and metabolic reprogramming (HK2/LDHA). | MERFISH, Visium | 33 patients | [49] |
| LUAD Histologic Subtypes | Tumor endothelial cells that express PD-L1 in stage IA LUAD suppress immune-responsive CD8+ T cells. | Visium | 11 postoperative LUAD patients | [50] |
| NSCLC Brain Metastasis | Characterized transcriptomic divergence between primary tumor cores (PanCK+) and brain TIME/TBMEs. | GeoMx DSP | 44 patients | [51] |
| Trial ID | Phase | Focus | Disease |
|---|---|---|---|
| NCT06893354 | 4 | Explore the Mechanisms Underlying Disease Resistance and Potential Primary Resistance Mechanism | ALK (+) NSCLC |
| NCT04789252 | Observational | Heterogeneity of Dendritic Cells in NSCLC | NSCLC |
| NCT06987734 | 2 | ST explores the changes in the iTME before and after suglizumab administration | NSCLC |
| NCT05055947 | Observational | ST related biomarkers to predict the efficacy of Atezolizumab plus etoposide and platinium | SCLC [ES] |
| NCT06396910 | NA | Immunological micro-environments of granulomas | Tuberculosis and sarcoidosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.H.; Kim, Y.; Lee, J.H.; Kang, H.S.; Chung, C. Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases: A Comprehensive Review. Cancers 2025, 17, 1912. https://doi.org/10.3390/cancers17121912
Kang DH, Kim Y, Lee JH, Kang HS, Chung C. Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases: A Comprehensive Review. Cancers. 2025; 17(12):1912. https://doi.org/10.3390/cancers17121912
Chicago/Turabian StyleKang, Da Hyun, Yoonjoo Kim, Ji Hyeon Lee, Hyeong Seok Kang, and Chaeuk Chung. 2025. "Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases: A Comprehensive Review" Cancers 17, no. 12: 1912. https://doi.org/10.3390/cancers17121912
APA StyleKang, D. H., Kim, Y., Lee, J. H., Kang, H. S., & Chung, C. (2025). Spatial Transcriptomics in Lung Cancer and Pulmonary Diseases: A Comprehensive Review. Cancers, 17(12), 1912. https://doi.org/10.3390/cancers17121912