
SREBP1-Dependent Metabolism as a Potential Target for Breast Cancer Risk Reduction



Simple Summary
Abstract
1. Introduction
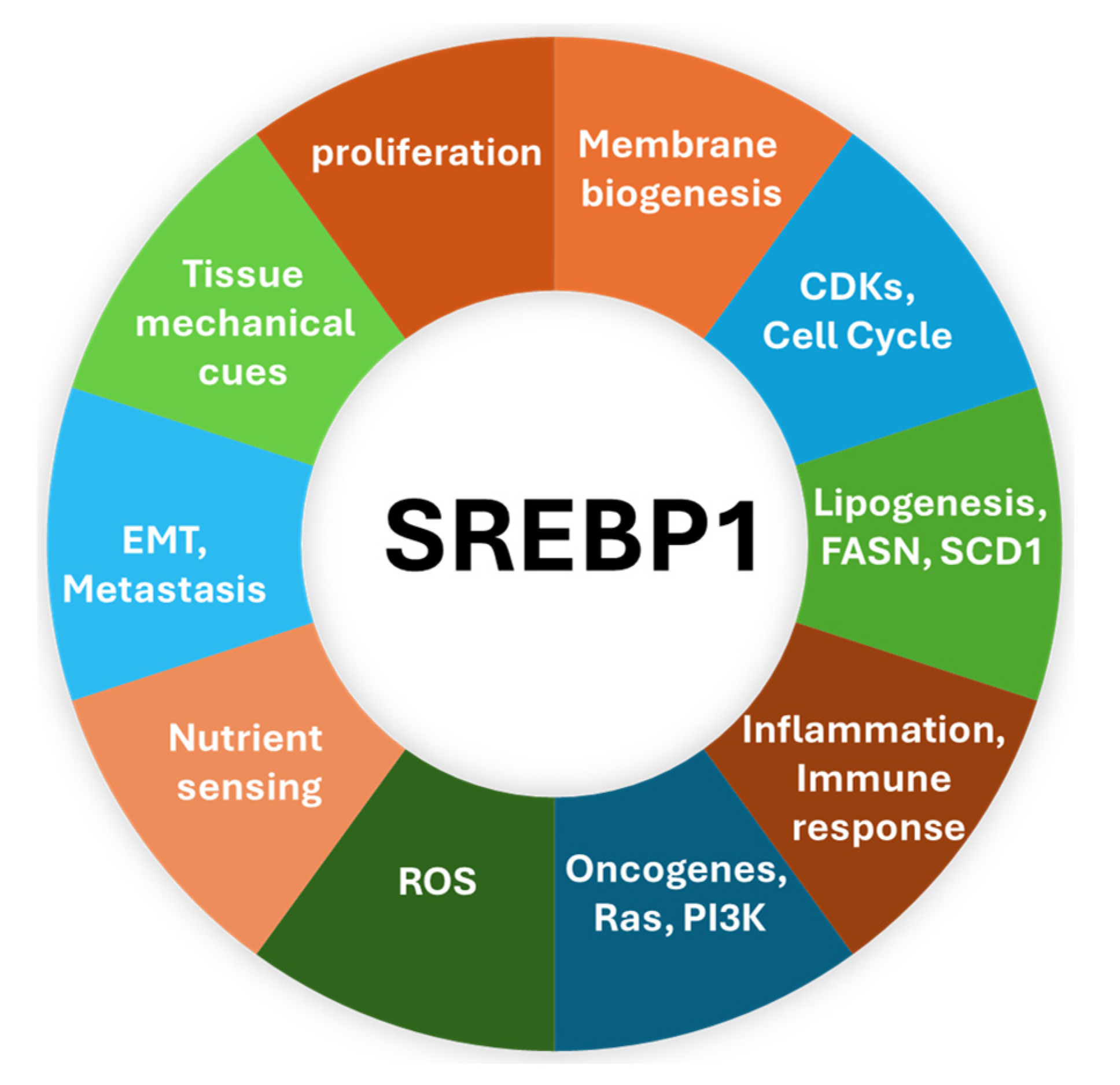
2. SREBP1—The Master Regulator of Lipogenic Metabolism
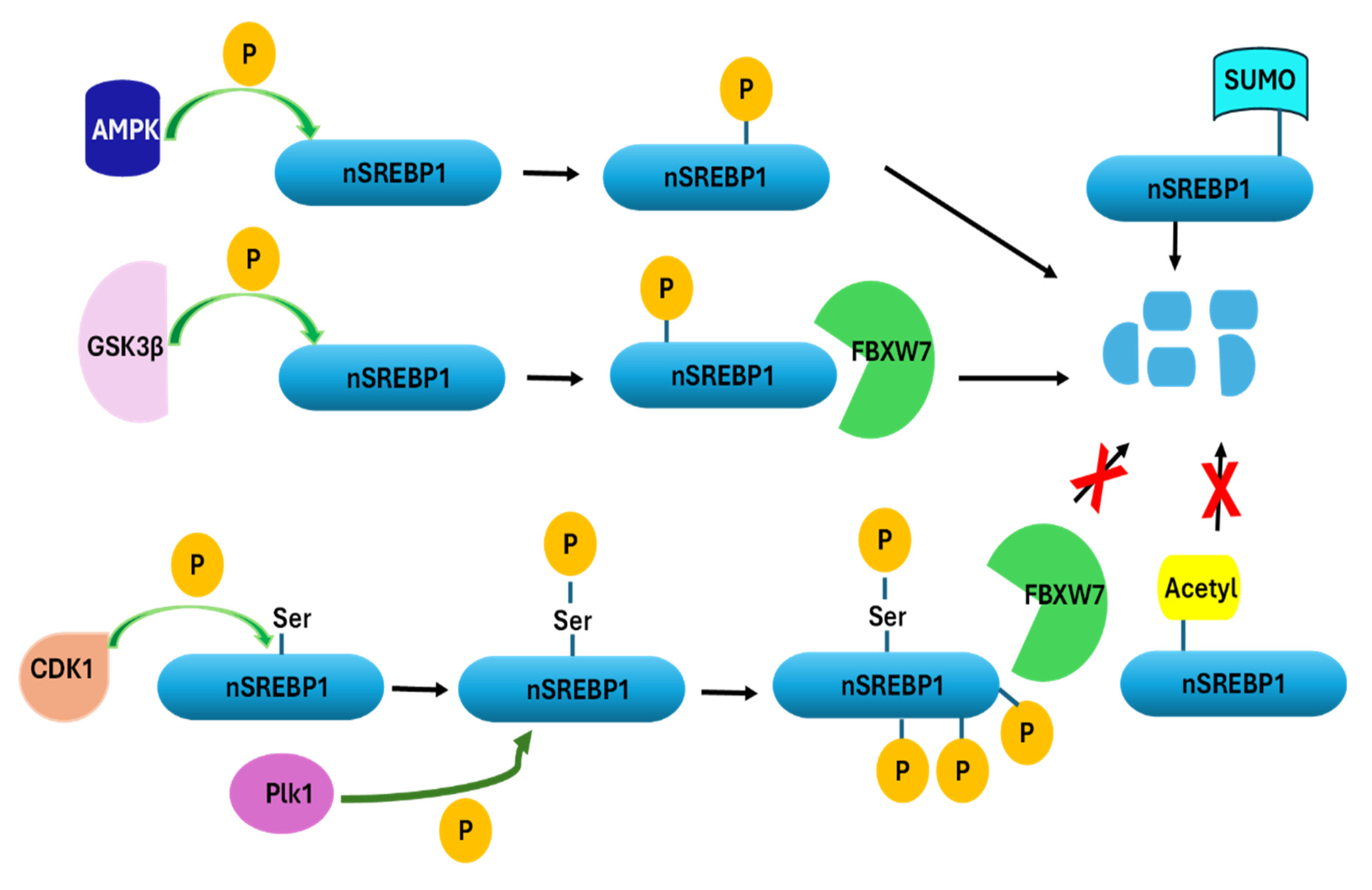
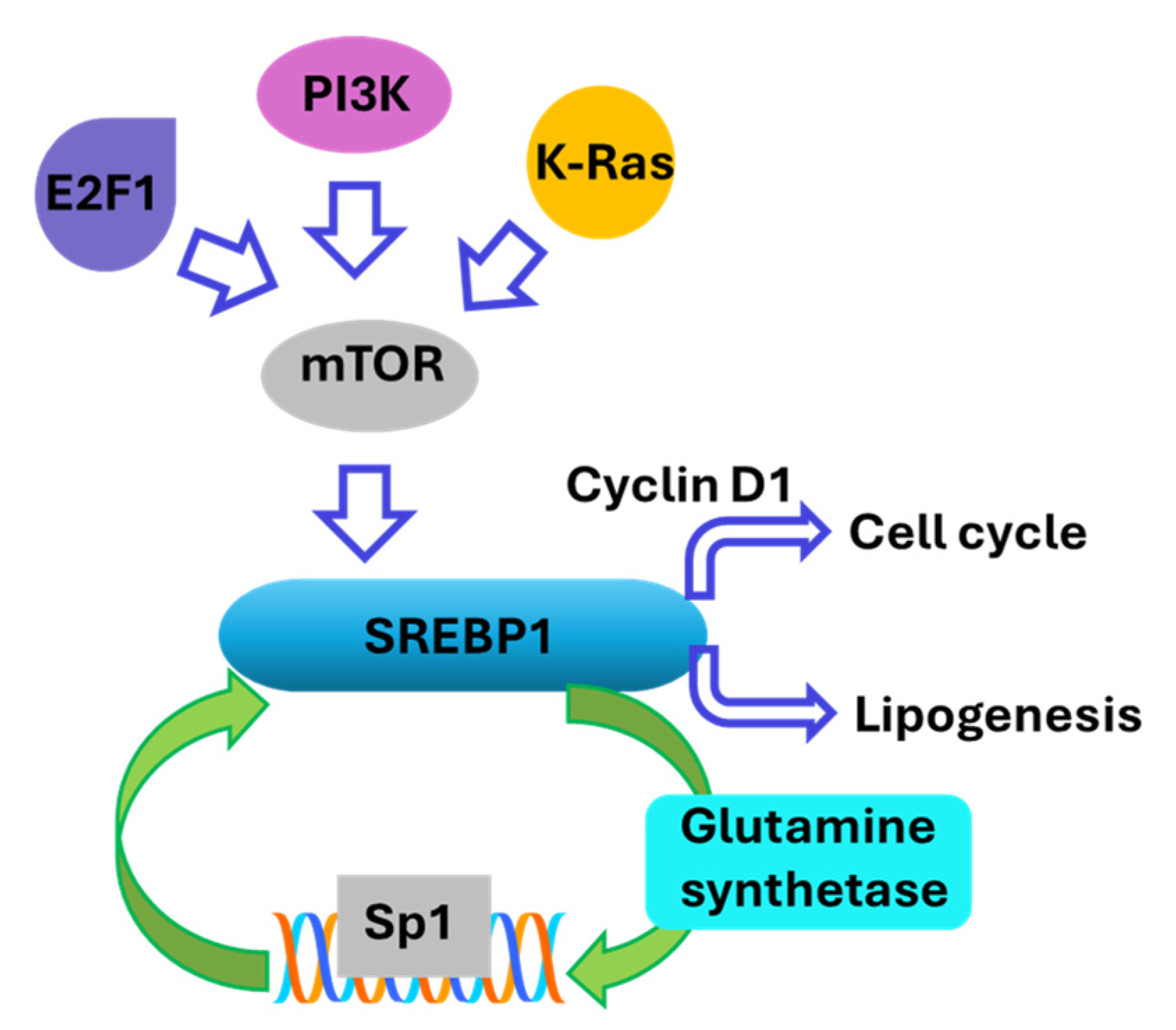
3. Regulation of SREBP1 Activity
4. The Implication of SREBP1 in Various Cancers
5. SREBP1 and Breast Cancer
5.1. SREBP1 and Ductal Carcinoma In Situ of the Breast
5.2. SREBP1 and Invasive Progression of Breast Cancer
5.3. SREBP1 and Oncogene-Driven Breast Cancer
5.4. SREBP1 and Obesity-Driven Breast Cancer
5.5. SREBP1 and Metastasis of Breast Cancer
6. SREBP1 and Lobular Breast Cancer
6.1. Differential Role of SREBP1 in Breast Cancer Subtypes
6.2. SREBP1 Inhibitors for Breast Cancer Prevention and Interception
6.2.1. Small Molecule Interventions
6.2.2. RNA-Based Interventions
6.2.3. Protein-Based Interventions
7. Deficiencies, Challenges, and Future Directions
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Form |
| ACI | August–Copenhagen–Irish (rat model) |
| ACC | Acetyl-CoA Carboxylase |
| ACLY | ATP Citrate Lyase |
| AH | Atypical Hyperplasia |
| AIs | Aromatase Inhibitors |
| AMPK | AMP-Activated Protein Kinase |
| BC | Breast Cancer |
| CBP | CREB-Binding Protein |
| CSCs | Cancer Stem-like Cells |
| DCIS | Ductal Carcinoma In Situ |
| DHA | Docosahexaenoic Acid |
| DMFS | Distant Metastasis-Free Survival |
| ECM | Extracellular Matrix |
| EMT | Epithelial–Mesenchymal Transition |
| ER | Estrogen Receptor |
| ER+ | Estrogen Receptor Positive |
| ER- | Estrogen Receptor Negative |
| FAD | Flavin Adenine Dinucleotide |
| FABP4 | Fatty Acid Binding Protein 4 |
| FASN | Fatty Acid Synthase |
| FBXW7 | F-box and WD repeat domain-containing 7 |
| FLAD1 | Flavin Adenine Dinucleotide Synthetase 1 |
| FOXM1 | Forkhead Box M1 |
| GCN2 | General Control Nonderepressible 2 |
| GPER | G Protein-Coupled Estrogen Receptor |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| H&E | Hematoxylin and Eosin |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HR+ | Hormone Receptor Positive |
| ILC | Invasive Lobular Carcinoma |
| LDLR | Low-Density Lipoprotein Receptor |
| LPS | Lipopolysaccharide |
| LSD1 | Lysine-Specific Histone Demethylase 1A |
| LXR | Liver X Receptor |
| MAGI2 | Membrane Associated Guanylate Kinase, WW And PDZ Domain Containing 2 |
| mTOR | Mechanistic Target of Rapamycin |
| mTORC1 | mTOR Complex 1 |
| mTORC2 | mTOR Complex 2 |
| MUFA | Monounsaturated Fatty Acids |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NEDD8 | Neural Precursor Cell Expressed Developmentally Downregulated Protein 8 |
| NCOR | Nuclear Receptor Corepressor |
| nSREBP | Nuclear SREBP |
| NSCLC | Non-Small Cell Lung Cancer |
| OA | Oleic Acid |
| OGT | O-GlcNAc Transferase |
| OS | Overall Survival |
| PAK | p21-Activated Kinase |
| PERK | Protein Kinase RNA-like Endoplasmic Reticulum Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| PRMT5 | Protein Arginine Methyltransferase 5 |
| PTEN | Phosphatase and Tensin Homolog |
| RARRES2 | Retinoic Acid Receptor Responder 2 |
| RBP7 | Retinol-Binding Protein 7 |
| RCT | Randomized Clinical Trial |
| RFS | Recurrence-Free Survival |
| ROCK | Rho-Associated Coiled-Coil Containing Protein Kinase |
| ROS | Reactive Oxygen Species |
| RXR | Retinoid X Receptor |
| SCAP | SREBP Cleavage-Activating Protein |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| SREBPs | Sterol Regulatory Element-Binding Proteins |
| SERMs | Selective Estrogen Receptor Modulators |
| SIM | SUMO-Interacting Motif |
| Sp1 | Specificity Protein 1 |
| SUMO | Small Ubiquitin-like Modifier |
| TAM | Tamoxifen |
| TG | Triglyceride |
| TNBC | Triple Negative Breast Cancer |
| TNM | Tumor Node Metastasis staging |
| UBC12 | Ubiquitin-Conjugating Enzyme E2M |
| VD3 | Vitamin D3 |
| VDR | Vitamin D Receptor |
References
- Lawal, K.O.; Nilan, L.; Amenta, J.; McGuinness, J.E.; Kukafka, R.; Crew, K.D. Comparing Breast Cancer and Cardiovascular Disease Risk and Use of Chemoprevention and Statins among Women with High-risk Breast Lesions. Cancer Prev. Res. 2023, 16, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A. Breast Cancer Risk Reduction: Current Status and Emerging Trends to Increase Efficacy and Reduce Toxicity of Preventive Medication. Surg. Oncol. Clin. N. Am. 2023, 32, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Britt, K.L.; Cuzick, J.; Phillips, K.A. Key steps for effective breast cancer prevention. Nat. Rev. Cancer 2020, 20, 417–436. [Google Scholar] [CrossRef]
- Trivedi, M.S.; Coe, A.M.; Vanegas, A.; Kukafka, R.; Crew, K.D. Chemoprevention Uptake among Women with Atypical Hyperplasia and Lobular and Ductal Carcinoma In Situ. Cancer Prev. Res. 2017, 10, 434–441. [Google Scholar] [CrossRef]
- Hum, S.; Wu, M.; Pruthi, S.; Heisey, R. Physician and Patient Barriers to Breast Cancer Preventive Therapy. Curr. Breast Cancer Rep. 2016, 8, 158–164. [Google Scholar] [CrossRef]
- Maresso, K.C.; Tsai, K.Y.; Brown, P.H.; Szabo, E.; Lippman, S.; Hawk, E.T. Molecular cancer prevention: Current status and future directions. CA Cancer J. Clin. 2015, 65, 345–383. [Google Scholar] [CrossRef]
- Martinez, K.A.; Fagerlin, A.; Witteman, H.O.; Holmberg, C.; Hawley, S.T. What Matters to Women When Making Decisions About Breast Cancer Chemoprevention? Patient 2016, 9, 149–159. [Google Scholar] [CrossRef]
- Jahan, N.; Jones, C.; Rahman, R.L. Endocrine prevention of breast cancer. Mol. Cell Endocrinol. 2021, 530, 111284. [Google Scholar] [CrossRef]
- Bychkovsky, B.; Laws, A.; Katlin, F.; Hans, M.; Knust Graichen, M.; Pace, L.E.; Scheib, R.; Garber, J.E.; King, T.A. Initiation and tolerance of chemoprevention among women with high-risk breast lesions: The potential of low-dose tamoxifen. Breast Cancer Res. Treat. 2022, 193, 417–427. [Google Scholar] [CrossRef]
- Jayasekera, J.; Zhao, A.; Schechter, C.; Lowry, K.; Yeh, J.M.; Schwartz, M.D.; O’Neill, S.; Wernli, K.J.; Stout, N.; Mandelblatt, J.; et al. Reassessing the Benefits and Harms of Risk-Reducing Medication Considering the Persistent Risk of Breast Cancer Mortality in Estrogen Receptor-Positive Breast Cancer. J. Clin. Oncol. 2023, 41, 859–870. [Google Scholar] [CrossRef]
- DeCensi, A.; Puntoni, M.; Guerrieri-Gonzaga, A.; Caviglia, S.; Avino, F.; Cortesi, L.; Taverniti, C.; Pacquola, M.G.; Falcini, F.; Gulisano, M.; et al. Randomized Placebo Controlled Trial of Low-Dose Tamoxifen to Prevent Local and Contralateral Recurrence in Breast Intraepithelial Neoplasia. J. Clin. Oncol. 2019, 37, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Padamsee, T.J.; Hils, M.; Muraveva, A. Understanding low chemoprevention uptake by women at high risk of breast cancer: Findings from a qualitative inductive study of women’s risk-reduction experiences. BMC Womens Health 2021, 21, 157. [Google Scholar] [CrossRef]
- Llombart-Cussac, A.; Perez-Garcia, J.M.; Ruiz Borrego, M.; Tolosa, P.; Blanch, S.; Fernandez-Ortega, A.; Urruticoechea, A.; Blancas, I.; Saura, C.; Rojas, B.; et al. Preventing alpelisib-related hyperglycaemia in HR+/HER2−/PIK3CA−mutated advanced breast cancer using metformin (METALLICA): A multicentre, open-label, single-arm, phase 2 trial. EClinicalMedicine 2024, 71, 102520. [Google Scholar] [CrossRef]
- Bitter, A.; Nussler, A.K.; Thasler, W.E.; Klein, K.; Zanger, U.M.; Schwab, M.; Burk, O. Human sterol regulatory element-binding protein 1a contributes significantly to hepatic lipogenic gene expression. Cell Physiol. Biochem. 2015, 35, 803–815. [Google Scholar] [CrossRef]
- Toth, J.I.; Datta, S.; Athanikar, J.N.; Freedman, L.P.; Osborne, T.F. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol. Cell Biol. 2004, 24, 8288–8300. [Google Scholar] [CrossRef]
- Osborne, T.F. Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- He, Y.; Qi, S.; Chen, L.; Zhu, J.; Liang, L.; Chen, X.; Zhang, H.; Zhuo, L.; Zhao, S.; Liu, S.; et al. The roles and mechanisms of SREBP1 in cancer development and drug response. Genes Dis. 2024, 11, 100987. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 484–493. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J. Biol. Chem. 2014, 289, 7547–7557. [Google Scholar] [CrossRef]
- Geng, F.; Guo, D. SREBF1/SREBP-1 concurrently regulates lipid synthesis and lipophagy to maintain lipid homeostasis and tumor growth. Autophagy 2024, 20, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Oh, Y.T.; Yue, P.; Khuri, F.R.; Sun, S.Y. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016, 35, 642–650. [Google Scholar] [CrossRef]
- Pinweha, P.; Rattanapornsompong, K.; Charoensawan, V.; Jitrapakdee, S. MicroRNAs and oncogenic transcriptional regulatory networks controlling metabolic reprogramming in cancers. Comput. Struct. Biotechnol. J. 2016, 14, 223–233. [Google Scholar] [CrossRef]
- Bengoechea-Alonso, M.T.; Ericsson, J. The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth. Cell Cycle 2016, 15, 2753–2765. [Google Scholar] [CrossRef]
- Naar, A.M.; Beaurang, P.A.; Robinson, K.M.; Oliner, J.D.; Avizonis, D.; Scheek, S.; Zwicker, J.; Kadonaga, J.T.; Tjian, R. Chromatin, TAFs, and a novel multiprotein coactivator are required for synergistic activation by Sp1 and SREBP-1a in vitro. Genes. Dev. 1998, 12, 3020–3031. [Google Scholar] [CrossRef]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin–RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef]
- Meng, H.; Shen, M.; Li, J.; Zhang, R.; Li, X.; Zhao, L.; Huang, G.; Liu, J. Novel SREBP1 inhibitor cinobufotalin suppresses proliferation of hepatocellular carcinoma by targeting lipogenesis. Eur. J. Pharmacol. 2021, 906, 174280. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Z.; Ding, W.; Xiao, C.; Zhang, Y.; Gao, S.; Gao, Y.; Cai, W. SREBP1 siRNA enhance the docetaxel effect based on a bone-cancer dual-targeting biomimetic nanosystem against bone metastatic castration-resistant prostate cancer. Theranostics 2020, 10, 1619–1632. [Google Scholar] [CrossRef]
- Ma, D.B.; Liu, X.Y.; Jia, H.; Zhang, Y.; Jiang, Q.; Sun, H.; Li, X.; Sun, F.; Chai, Y.; Feng, F.; et al. A Novel Small-Molecule Inhibitor of SREBP-1 Based on Natural Product Monomers Upregulates the Sensitivity of Lung Squamous Cell Carcinoma Cells to Antitumor Drugs. Front. Pharmacol. 2022, 13, 895744. [Google Scholar] [CrossRef]
- Tiong, T.Y.; Weng, P.W.; Wang, C.H.; Setiawan, S.A.; Yadav, V.K.; Pikatan, N.W.; Fong, I.H.; Yeh, C.T.; Hsu, C.H.; Kuo, K.T. Targeting the SREBP-1/Hsa-Mir-497/SCAP/FASN Oncometabolic Axis Inhibits the Cancer Stem-like and Chemoresistant Phenotype of Non-Small Cell Lung Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 7283. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, H.; Liu, Y.; Su, P.; Zhang, J.; Wang, X.; Sun, M.; Chen, B.; Zhao, W.; Wang, L.; et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death Differ. 2019, 26, 843–859. [Google Scholar] [CrossRef]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol. 2019, 21, 338–347. [Google Scholar] [CrossRef]
- Hu, Q.; Mao, Y.; Liu, M.; Luo, R.; Jiang, R.; Guo, F. The active nuclear form of SREBP1 amplifies ER stress and autophagy via regulation of PERK. FEBS J. 2020, 287, 2348–2366. [Google Scholar] [CrossRef]
- Jhu, J.W.; Yan, J.B.; Lin, Z.H.; Lin, S.C.; Peng, I.C. SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9814. [Google Scholar] [CrossRef]
- Aldaalis, A.; Bengoechea-Alonso, M.T.; Ericsson, J. The SREBP-dependent regulation of cyclin D1 coordinates cell proliferation and lipid synthesis. Front. Oncol. 2022, 12, 942386. [Google Scholar] [CrossRef]
- Liao, S.; Gollowitzer, A.; Bormel, L.; Maier, C.; Gottschalk, L.; Werz, O.; Wallert, M.; Koeberle, A.; Lorkowski, S. alpha-Tocopherol-13’-Carboxychromanol Induces Cell Cycle Arrest and Cell Death by Inhibiting the SREBP1-SCD1 Axis and Causing Imbalance in Lipid Desaturation. Int. J. Mol. Sci. 2023, 24, 9229. [Google Scholar] [CrossRef]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef]
- Crewe, C.; Zhu, Y.; Paschoal, V.A.; Joffin, N.; Ghaben, A.L.; Gordillo, R.; Oh, D.Y.; Liang, G.; Horton, J.D.; Scherer, P.E. SREBP-regulated adipocyte lipogenesis is dependent on substrate availability and redox modulation of mTORC1. JCI Insight 2019, 5, e129397. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Gao, Y.; Huang, Q.; Xuan, Y.; Yao, Y.; Gu, L.; Huang, Y.; Zhang, Y.; Li, P.; Fan, Y.; et al. E2F1 promotes proliferation and metastasis of clear cell renal cell carcinoma via activation of SREBP1-dependent fatty acid biosynthesis. Cancer Lett. 2021, 514, 48–62. [Google Scholar] [CrossRef]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef]
- Fei, X.; Huang, J.; Li, F.; Wang, Y.; Shao, Z.; Dong, L.; Wu, Y.; Li, B.; Zhang, X.; Lv, B.; et al. The Scap-SREBP1-S1P/S2P lipogenesis signal orchestrates the homeostasis and spatiotemporal activation of NF-kappaB. Cell Rep. 2023, 42, 112586. [Google Scholar] [CrossRef]
- Oishi, Y.; Koike, H.; Kumagami, N.; Nakagawa, Y.; Araki, M.; Taketomi, Y.; Miki, Y.; Matsuda, S.; Kim, H.; Matsuzaka, T.; et al. Macrophage SREBP1 regulates skeletal muscle regeneration. Front. Immunol. 2023, 14, 1251784. [Google Scholar] [CrossRef]
- Xu, X.; Jin, W.; Chang, R.; Ding, X. Research progress of SREBP and its role in the pathogenesis of autoimmune rheumatic diseases. Front. Immunol. 2024, 15, 1398921. [Google Scholar] [CrossRef]
- Cheng, Y.; Manabe, I.; Hayakawa, S.; Endo, Y.; Oishi, Y. Caspase-11 contributes to site-1 protease cleavage and SREBP1 activation in the inflammatory response of macrophages. Front. Immunol. 2023, 14, 1009973. [Google Scholar] [CrossRef]
- Gerlic, M.; Croker, B.A.; Cengia, L.H.; Moayeri, M.; Kile, B.T.; Masters, S.L. NLRP1a expression in Srebp-1a-deficient mice. Cell Metab. 2014, 19, 345–346. [Google Scholar] [CrossRef]
- Li, X.; Huang, W.; Gu, J.; Du, X.; Lei, L.; Yuan, X.; Sun, G.; Wang, Z.; Li, X.; Liu, G. SREBP-1c overactivates ROS-mediated hepatic NF-kappaB inflammatory pathway in dairy cows with fatty liver. Cell Signal 2015, 27, 2099–2109. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Puig, J.; Burcelin, R.; Dumas, M.E.; Barton, R.H.; Hoyles, L.; Federici, M.; Fernandez-Real, J.M. The APOA1bp-SREBF-NOTCH axis is associated with reduced atherosclerosis risk in morbidly obese patients. Clin. Nutr. 2020, 39, 3408–3418. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, X.; Ye, Q. Metabolism and immunity in breast cancer. Front. Med. 2021, 15, 178–207. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef]
- Li, J.; Yan, H.; Zhao, L.; Jia, W.; Yang, H.; Liu, L.; Zhou, X.; Miao, P.; Sun, X.; Song, S.; et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget 2016, 7, 52392–52403. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, L.; Wang, D.; Jiang, S.; Cao, D.; Zhao, Z.; Huang, M.; Jin, J. Lipidomics reveals that sustained SREBP-1-dependent lipogenesis is a key mediator of gefitinib-acquired resistance in EGFR-mutant lung cancer. Cell Death Discov. 2021, 7, 353. [Google Scholar] [CrossRef]
- Tan, M.; Lin, X.; Chen, H.; Ye, W.; Yi, J.; Li, C.; Liu, J.; Su, J. Sterol regulatory element binding transcription factor 1 promotes proliferation and migration in head and neck squamous cell carcinoma. PeerJ 2023, 11, e15203. [Google Scholar] [CrossRef]
- Wang, J.; Ling, R.; Zhou, Y.; Gao, X.; Yang, Y.; Mao, C.; Chen, D. SREBP1 silencing inhibits the proliferation and motility of human esophageal squamous carcinoma cells via the Wnt/beta-catenin signaling pathway. Oncol. Lett. 2020, 20, 2855–2869. [Google Scholar] [CrossRef]
- Zou, X.Z.; Hao, J.F.; Zhou, X.H. Inhibition of SREBP-1 Activation by a Novel Small-Molecule Inhibitor Enhances the Sensitivity of Hepatocellular Carcinoma Tissue to Radiofrequency Ablation. Front. Oncol. 2021, 11, 796152. [Google Scholar] [CrossRef]
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019, 10, 672. [Google Scholar] [CrossRef]
- Wang, T.B.; Geng, M.; Jin, H.; Tang, A.G.; Sun, H.; Zhou, L.Z.; Chen, B.H.; Shen, G.; Sun, Q. SREBP1 site 1 protease inhibitor PF-429242 suppresses renal cell carcinoma cell growth. Cell Death Dis. 2021, 12, 717. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, T.; Yan, H.; Guo, K.; Liu, Z.; Wei, L.; Lu, W.; Qiu, C.; Jiang, J. Fatostatin reverses progesterone resistance by inhibiting the SREBP1-NF-kappaB pathway in endometrial carcinoma. Cell Death Dis. 2021, 12, 544. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, R.; Wang, Y.; Xiao, H.; Lin, W.; Diao, M.; He, S.; Mei, P.; Liao, Y. G protein-coupled estrogen receptor activates PI3K/AKT/mTOR signaling to suppress ferroptosis via SREBP1/SCD1-mediated lipogenesis. Mol. Med. 2024, 30, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, D.; Zhen, Z.; Ao, J.; Yuan, X.; Gao, X. Annexin A2 positively regulates milk synthesis and proliferation of bovine mammary epithelial cells through the mTOR signaling pathway. J. Cell Physiol. 2018, 233, 2464–2475. [Google Scholar] [CrossRef]
- Xu, H.F.; Luo, J.; Zhao, W.S.; Yang, Y.C.; Tian, H.B.; Shi, H.B.; Bionaz, M. Overexpression of SREBP1 (sterol regulatory element binding protein 1) promotes de novo fatty acid synthesis and triacylglycerol accumulation in goat mammary epithelial cells. J. Dairy Sci. 2016, 99, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Harvatine, K.J.; Bauman, D.E. SREBP1 and thyroid hormone responsive spot 14 (S14) are involved in the regulation of bovine mammary lipid synthesis during diet-induced milk fat depression and treatment with CLA. J. Nutr. 2006, 136, 2468–2474. [Google Scholar] [CrossRef]
- Belkaid, A.; Duguay, S.R.; Ouellette, R.J.; Surette, M.E. 17β-estradiol induces stearoyl-CoA desaturase-1 expression in estrogen receptor-positive breast cancer cells. BMC Cancer 2015, 15, 440. [Google Scholar] [CrossRef]
- Li, Y.; Wu, S.; Zhao, X.; Hao, S.; Li, F.; Wang, Y.; Liu, B.; Zhang, D.; Wang, Y.; Zhou, H. Key events in cancer: Dysregulation of SREBPs. Front. Pharmacol. 2023, 14, 1130747. [Google Scholar] [CrossRef]
- Angelucci, C.; Maulucci, G.; Colabianchi, A.; Iacopino, F.; D’Alessio, A.; Maiorana, A.; Palmieri, V.; Papi, M.; De Spirito, M.; Di Leone, A.; et al. Stearoyl-CoA desaturase 1 and paracrine diffusible signals have a major role in the promotion of breast cancer cell migration induced by cancer-associated fibroblasts. Br. J. Cancer 2015, 112, 1675–1686. [Google Scholar] [CrossRef]
- Jung, J.H.; Yang, Y.; Kim, Y. Hypoxia-induced SREBP1-mediated lipogenesis and autophagy promote cell survival via fatty acid oxidation in breast cancer cells. Oncol. Lett. 2025, 29, 175. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef]
- Zhang, L.; La, X.; Tian, J.; Li, H.; Li, A.; Liu, Y.; Wu, C.; Li, Z. The phytochemical vitexin and syringic acid derived from foxtail fillet bran inhibit breast cancer cells proliferation via GRP78/SREBP-1/SCD1 signaling axis. J. Func. Foods 2021, 85, 104620. [Google Scholar] [CrossRef]
- Berthier, A.; Vinod, M.; Porez, G.; Steenackers, A.; Alexandre, J.; Yamakawa, N.; Gheeraert, C.; Ploton, M.; Marechal, X.; Dubois-Chevalier, J.; et al. Combinatorial regulation of hepatic cytoplasmic signaling and nuclear transcriptional events by the OGT/REV-ERBalpha complex. Proc. Natl. Acad. Sci. USA 2018, 115, E11033–E11042. [Google Scholar] [CrossRef]
- Sodi, V.L.; Bacigalupa, Z.A.; Ferrer, C.M.; Lee, J.V.; Gocal, W.A.; Mukhopadhyay, D.; Wellen, K.E.; Ivan, M.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene 2018, 37, 924–934. [Google Scholar] [CrossRef]
- Pandey, P.R.; Xing, F.; Sharma, S.; Watabe, M.; Pai, S.K.; Iiizumi-Gairani, M.; Fukuda, K.; Hirota, S.; Mo, Y.Y.; Watabe, K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene 2013, 32, 5111–5122. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Choi, S.K.; Moon, W.K.; Kim, S.J. Different Biological Action of Oleic Acid in ALDHhigh and ALDHlow Subpopulations Separated from Ductal Carcinoma In Situ of Breast Cancer. PLoS ONE 2016, 11, e0160835. [Google Scholar] [CrossRef]
- Wu, M.; Wang, J.; Zhou, W.; Wang, M.; Hu, C.; Zhou, M.; Jiao, K.; Li, Z. Vitamin D inhibits tamoxifen-induced non-alcoholic fatty liver disease through a nonclassical estrogen receptor/liver X receptor pathway. Chem. Biol. Interact. 2024, 389, 110865. [Google Scholar] [CrossRef]
- Ismail, A.; Doghish, A.S.; Elsadek, B.E.M.; Salama, S.A.; Mariee, A.D. Hydroxycitric acid potentiates the cytotoxic effect of tamoxifen in MCF-7 breast cancer cells through inhibition of ATP citrate lyase. Steroids 2020, 160, 108656. [Google Scholar] [CrossRef]
- Chen, Y.; Li, K.; Gong, D.; Zhang, J.; Li, Q.; Zhao, G.; Lin, P. ACLY: A biomarker of recurrence in breast cancer. Pathol. Res. Pract. 2020, 216, 153076. [Google Scholar] [CrossRef]
- Bandyopadhayaya, S.; Akimov, M.G.; Verma, R.; Sharma, A.; Sharma, D.; Kundu, G.C.; Gretskaya, N.M.; Bezuglov, V.V.; Mandal, C.C. N-arachidonoyl dopamine inhibits epithelial-mesenchymal transition of breast cancer cells through ERK signaling and decreasing the cellular cholesterol. J. Biochem. Mol. Toxicol. 2021, 35, e22693. [Google Scholar] [CrossRef]
- Jiang, W.; Xing, X.L.; Zhang, C.; Yi, L.; Xu, W.; Ou, J.; Zhu, N. MET and FASN as Prognostic Biomarkers of Triple Negative Breast Cancer: A Systematic Evidence Landscape of Clinical Study. Front. Oncol. 2021, 11, 604801. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, C.; Yin, H.; Yu, J.; Chen, S.; Fang, J.; Guo, F. Leucine deprivation inhibits proliferation and induces apoptosis of human breast cancer cells via fatty acid synthase. Oncotarget 2016, 7, 63679–63689. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-Q.; Yu, T.-J.; Ou-Yang, Y.; Ding, J.-H.; Jiang, Y.-Z.; Shao, Z.-M.; Xiao, Y. Copy number amplification of FLAD1 promotes the progression of triple-negative breast cancer through lipid metabolism. Nat. Commun. 2025, 16, 1241. [Google Scholar] [CrossRef] [PubMed]
- Perone, Y.; Farrugia, A.J.; Rodriguez-Meira, A.; Gyorffy, B.; Ion, C.; Uggetti, A.; Chronopoulos, A.; Marrazzo, P.; Faronato, M.; Shousha, S.; et al. SREBP1 drives Keratin-80-dependent cytoskeletal changes and invasive behavior in endocrine-resistant ERalpha breast cancer. Nat. Commun. 2019, 10, 2115. [Google Scholar] [CrossRef]
- Heo, M.J.; Kang, S.H.; Kim, Y.S.; Lee, J.M.; Yu, J.; Kim, H.R.; Lim, H.; Kim, K.M.; Jung, J.; Jeong, L.S.; et al. UBC12-mediated SREBP-1 neddylation worsens metastatic tumor prognosis. Int. J. Cancer 2020, 147, 2550–2563. [Google Scholar] [CrossRef]
- Mahmud, I.; Tian, G.; Wang, J.; Hutchinson, T.E.; Kim, B.J.; Awasthee, N.; Hale, S.; Meng, C.; Moore, A.; Zhao, L.; et al. DAXX drives de novo lipogenesis and contributes to tumorigenesis. Nat. Commun. 2023, 14, 1927. [Google Scholar] [CrossRef]
- Deng, S.; Chen, B.; Huo, J.; Liu, X. Therapeutic potential of NR4A1 in cancer: Focus on metabolism. Front. Oncol. 2022, 12, 972984. [Google Scholar] [CrossRef]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-gamma. Immunity 2019, 51, 381–397.e6. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef]
- Jin, Q.; Qi, D.; Zhang, M.; Qu, H.; Dong, Y.; Sun, M.; Quan, C. CLDN6 inhibits breast cancer growth and metastasis through SREBP1-mediated RAS palmitoylation. Cell Mol. Biol. Lett. 2024, 29, 112. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, Z.; Zhou, H.; Xu, R.; Xu, J.; Liu, W.; Wu, Y.; Qiu, Y.; Zhang, G.; Huang, X.; et al. RBP7 functions as a tumor suppressor in HR + breast cancer by inhibiting the AKT/SREBP1 pathway and reducing fatty acid. Cancer Cell Int. 2024, 24, 118. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhao, X.; Zhao, L.; Yang, H.; Liu, L.; Li, J.; Wu, J.; Yang, F.; Huang, G.; Liu, J. p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP-1A. Oncogene 2016, 35, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Zhang, Y.; Li, L.; Fang, R.; Li, Y.; Liu, Q.; Zhang, W.; Qiu, L.; Liu, F.; et al. Oncoprotein HBXIP Modulates Abnormal Lipid Metabolism and Growth of Breast Cancer Cells by Activating the LXRs/SREBP-1c/FAS Signaling Cascade. Cancer Res. 2016, 76, 4696–4707. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Y.; Wang, Y.; Li, Q.; Zeng, K.; Li, X.; Feng, X. Myc derived circRNA promotes triple-negative breast cancer progression via reprogramming fatty acid metabolism. Discov. Oncol. 2023, 14, 67. [Google Scholar] [CrossRef]
- Yang, Y.; Liao, J.; Pan, Z.; Meng, J.; Zhang, L.; Shi, W.; Wang, X.; Zhang, X.; Zhou, Z.; Luo, J.; et al. Dual Inhibition of CDK4/6 and CDK7 Suppresses Triple-Negative Breast Cancer Progression via Epigenetic Modulation of SREBP1-Regulated Cholesterol Metabolism. Adv. Sci. 2025, 12, e2413103. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Lee, K.H.; Cho, J.Y. ABCA9, an ER cholesterol transporter, inhibits breast cancer cell proliferation via SREBP-2 signaling. Cancer Sci. 2023, 114, 1451–1463. [Google Scholar] [CrossRef]
- Pham, D.V.; Park, P.H. Adiponectin triggers breast cancer cell death via fatty acid metabolic reprogramming. J. Exp. Clin. Cancer Res. 2022, 41, 9. [Google Scholar] [CrossRef]
- Pham, D.V.; Tilija Pun, N.; Park, P.H. Autophagy activation and SREBP-1 induction contribute to fatty acid metabolic reprogramming by leptin in breast cancer cells. Mol. Oncol. 2021, 15, 657–678. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, X.; Meng, L.; Dong, K.; Shang, S.; Jiang, T.; Liu, Z.; Gao, H. Single-cell RNA-sequencing reveals a unique landscape of the tumor microenvironment in obesity-associated breast cancer. Oncogene 2024, 43, 3277–3290. [Google Scholar] [CrossRef]
- Rajput, P.K.; Varghese, J.F.; Srivastava, A.K.; Kumar, U.; Yadav, U.C.S. Visfatin-induced upregulation of lipogenesis via EGFR/AKT/GSK3beta pathway promotes breast cancer cell growth. Cell Signal 2023, 107, 110686. [Google Scholar] [CrossRef]
- Ma, X.; Wang, A.; Wang, Y.; Ma, J.; Liu, Y.; Mei, Y. Zinc Finger SWIM-Type Containing 3 Reprograms Lipid Metabolism and Drives Breast Cancer Progression. Discov. Med. 2025, 37, 152–165. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Marce-Grau, A.; Muixi, L.; Nieva, C.; Marro, M.; Sebastian, D.; Munoz, J.P.; Zorzano, A.; Sierra, A. GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells. Int. J. Mol. Sci. 2019, 20, 3883. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Sun, F.-Z.; Li, C.-X.; Mo, H.-N.; Zhou, Y.-T.; Lv, D.; Zhai, J.-T.; Qian, H.-L.; Ma, F. RARRES2 regulates lipid metabolic reprogramming to mediate the development of brain metastasis in triple negative breast cancer. Mil. Med. Res. 2023, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- King, T.A.; Pilewskie, M.; Muhsen, S.; Patil, S.; Mautner, S.K.; Park, A.; Oskar, S.; Guerini-Rocco, E.; Boafo, C.; Gooch, J.C.; et al. Lobular Carcinoma in Situ: A 29-Year Longitudinal Experience Evaluating Clinicopathologic Features and Breast Cancer Risk. J. Clin. Oncol. 2015, 33, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.Y.; Brogi, E. Lobular Carcinoma In Situ. Surg. Pathol. Clin. 2018, 11, 123–145. [Google Scholar] [CrossRef]
- Du, T.; Sikora, M.J.; Levine, K.M.; Tasdemir, N.; Riggins, R.B.; Wendell, S.G.; Van Houten, B.; Oesterreich, S. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 2018, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Zipinotti Dos Santos, D.; de Souza, J.C.; Pimenta, T.M.; da Silva Martins, B.; Junior, R.S.R.; Butzene, S.M.S.; Tessarolo, N.G.; Cilas, P.M.L., Jr.; Silva, I.V.; Rangel, L.B.A. The impact of lipid metabolism on breast cancer: A review about its role in tumorigenesis and immune escape. Cell Commun. Signal 2023, 21, 161. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase: A druggable driver of breast cancer brain metastasis. Expert. Opin. Ther. Targets 2022, 26, 427–444. [Google Scholar] [CrossRef]
- Peluffo, G.; Subedee, A.; Harper, N.W.; Kingston, N.; Jovanovic, B.; Flores, F.; Stevens, L.E.; Beca, F.; Trinh, A.; Chilamakuri, C.S.R.; et al. EN1 Is a Transcriptional Dependency in Triple-Negative Breast Cancer Associated with Brain Metastasis. Cancer Res. 2019, 79, 4173–4183. [Google Scholar] [CrossRef]
- Carmona, A.; Mitri, S.; James, T.A.; Ubellacker, J.M. Lipidomics and metabolomics as potential biomarkers for breast cancer progression. Npj Metab. Health Dis. 2024, 2, 24. [Google Scholar] [CrossRef]
- Mosley, J.D.; Poirier, J.T.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/ErbB2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; dos Sarbassov, D.; Muller, W.J.; et al. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Vieira, R.; Xu, Z.; Colp, P.; Marignani, P.A. Loss of LKB1 expression reduces the latency of ErbB2-mediated mammary gland tumorigenesis, promoting changes in metabolic pathways. PLoS ONE 2013, 8, e56567. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Guerrero-Zotano, A.; Won, H.; Gonzalez Ericsson, P.; Servetto, A.; Huerta-Rosario, M.; Ye, D.; Lee, K.M.; Formisano, L.; Guo, Y.; et al. Hyperactivation of TORC1 Drives Resistance to the Pan-HER Tyrosine Kinase Inhibitor Neratinib in HER2-Mutant Cancers. Cancer Cell 2020, 37, 183–199.e5. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.V.; Matthews, S.B.; Fettig, L.M.; Riley, D.; Finlay-Schultz, J.; Paul, K.V.; Jackman, M.; Kabos, P.; MacLean, P.S.; Sartorius, C.A. Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism. Cancers 2022, 14, 1776. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Arca, A.; Madak-Erdogan, Z. Crosstalk between Estrogen Signaling and Breast Cancer Metabolism. Trends Endocrinol. Metab. 2019, 30, 25–38. [Google Scholar] [CrossRef]
- Jia, M.; Andreassen, T.; Jensen, L.; Bathen, T.F.; Sinha, I.; Gao, H.; Zhao, C.; Haldosen, L.A.; Cao, Y.; Girnita, L.; et al. Estrogen Receptor alpha Promotes Breast Cancer by Reprogramming Choline Metabolism. Cancer Res. 2016, 76, 5634–5646. [Google Scholar] [CrossRef]
- Santolla, M.F.; Lappano, R.; De Marco, P.; Pupo, M.; Vivacqua, A.; Sisci, D.; Abonante, S.; Iacopetta, D.; Cappello, A.R.; Dolce, V.; et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17β-estradiol in cancer cells and cancer-associated fibroblasts. J. Biol. Chem. 2012, 287, 43234–43245. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz Casiano, A.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef]
- Golden, E.; Rashwan, R.; Woodward, E.A.; Sgro, A.; Wang, E.; Sorolla, A.; Waryah, C.; Tie, W.J.; Cuyas, E.; Ratajska, M.; et al. The oncogene AAMDC links PI3K-AKT-mTOR signaling with metabolic reprograming in estrogen receptor-positive breast cancer. Nat. Commun. 2021, 12, 1920. [Google Scholar] [CrossRef]
- Wang, C.; Uray, I.P.; Mazumdar, A.; Mayer, J.A.; Brown, P.H. SLC22A5/OCTN2 expression in breast cancer is induced by estrogen via a novel intronic estrogen-response element (ERE). Breast Cancer Res. Treat. 2012, 134, 101–115. [Google Scholar] [CrossRef]
- Tang, J.-J.; Li, J.-G.; Qi, W.; Qiu, W.-W.; Li, P.-S.; Li, B.-L.; Song, B.-L. Inhibition of SREBP by a Small Molecule, Betulin, Improves Hyperlipidemia and Insulin Resistance and Reduces Atherosclerotic Plaques. Cell Metab. 2011, 13, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Engelking, L.J.; Kuriyama, H.; Hammer, R.E.; Horton, J.D.; Brown, M.S.; Goldstein, J.L.; Liang, G. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J. Clin. Investig. 2004, 113, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Xu, L.L.; Peng, J.; Zhang, D.H. Al-MPS Obstructs EMT in Breast Cancer by Inhibiting Lipid Metabolism via miR-215-5p/SREBP1. Endocrinology 2022, 163, bqac040. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Rong, S.; Xia, M.; Vale, G.; Wang, S.; Kim, C.W.; Li, S.; McDonald, J.G.; Radhakrishnan, A.; Horton, J.D. DGAT2 inhibition blocks SREBP-1 cleavage and improves hepatic steatosis by increasing phosphatidylethanolamine in the ER. Cell Metab. 2024, 36, 617–629.e7. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, W.; O-Sullivan, I.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143. [Google Scholar] [CrossRef]
- Tiwari, U.; Akhtar, S.; Mir, S.S.; Khan, M.K.A. Evaluation of selected indigenous spices- and herbs-derived small molecules as potential inhibitors of SREBP and its implications for breast cancer using MD simulations and MMPBSA calculations. Mol. Divers. 2025. ahead of print. [Google Scholar] [CrossRef]
- Palliyaguru, D.L.; Yang, L.; Chartoumpekis, D.V.; Wendell, S.G.; Fazzari, M.; Skoko, J.J.; Liao, Y.; Oesterreich, S.; Michalopoulos, G.K.; Kensler, T.W. Sulforaphane Diminishes the Formation of Mammary Tumors in Rats Exposed to 17β-Estradiol. Nutrients 2020, 12, 2282. [Google Scholar] [CrossRef]
- Huang, L.-H.; Chung, H.-Y.; Su, H.-M. Docosahexaenoic acid reduces sterol regulatory element binding protein-1 and fatty acid synthase expression and inhibits cell proliferation by inhibiting pAkt signaling in a human breast cancer MCF-7 cell line. BMC Cancer 2017, 17, 890. [Google Scholar] [CrossRef]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.B.; Chung, L.W.; Huang, W.C. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6, 41018–41032. [Google Scholar] [CrossRef] [PubMed]
- Siqingaowa; Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, D.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Induction of SREBP1 degradation coupled with suppression of SREBP1-mediated lipogenesis impacts the response of EGFR mutant NSCLC cells to osimertinib. Oncogene 2021, 40, 6653–6665. [Google Scholar] [CrossRef]
- Bhushan, B.; Banerjee, S.; Paranjpe, S.; Koral, K.; Mars, W.M.; Stoops, J.W.; Orr, A.; Bowen, W.C.; Locker, J.; Michalopoulos, G.K. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model. Hepatology 2019, 70, 1546–1563. [Google Scholar] [CrossRef]
- Xie, D.; Jiang, Y.; Wang, H.; Zhu, L.; Huang, S.; Liu, S.; Zhang, W.; Li, T. Formononetin triggers ferroptosis in triple-negative breast cancer cells by regulating the mTORC1/SREBP1/SCD1 pathway. Front. Pharmacol. 2024, 15, 1441105. [Google Scholar] [CrossRef]
- Singh, K.B.; Hahm, E.R.; Kim, S.H.; Singh, S.V. Withaferin A Inhibits Fatty Acid Synthesis in Rat Mammary Tumors. Cancer Prev. Res. 2023, 16, 5–16. [Google Scholar] [CrossRef]
- Tang, J.Y.; Chuang, Y.T.; Shiau, J.P.; Yang, K.H.; Chang, F.R.; Hou, M.F.; Farooqi, A.A.; Chang, H.W. Long Noncoding RNAs and Circular RNAs Regulate AKT and Its Effectors to Control Cell Functions of Cancer Cells. Cells 2022, 11, 2940. [Google Scholar] [CrossRef]
- Hu, P.; Zhou, P.; Sun, T.; Liu, D.; Yin, J.; Liu, L. Therapeutic protein PAK restrains the progression of triple negative breast cancer through degrading SREBP-1 mRNA. Breast Cancer Res. 2023, 25, 151. [Google Scholar] [CrossRef]
- Bu, W.; Li, Y. Advances in Immunocompetent Mouse and Rat Models. Cold Spring Harb. Perspect. Med. 2024, 14, a041328. [Google Scholar] [CrossRef]
- Hajirahimkhan, A.; Bartom, E.T.; Chung, C.H.; Guo, X.; Berkley, K.; Lee, O.; Chen, R.; Cho, W.; Chandrasekaran, S.; Clare, S.E.; et al. Reprogramming SREBP1-dependent lipogenesis and inflammation in high-risk breast with licochalcone A: A novel path to cancer prevention. bioRxiv 2024. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitory Effect on SREBP1 |
|---|---|
| SCAP–SREBP1 interaction [121] | translocation from endoplasmic reticulum to Golgi |
| SCAP–Insig interaction [122] | interaction with SCAP |
| miRNA overexpression [123] | expression |
| NAE [84] | stabilization |
| SOAT1 inhibition [124] | expression |
| DGAT2 inhibition [125] | cleavage in the endoplasmic reticulum |
| FoxO1 activation [126] | transcription |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajirahimkhan, A.; Brown, K.A.; Clare, S.E.; Khan, S.A. SREBP1-Dependent Metabolism as a Potential Target for Breast Cancer Risk Reduction. Cancers 2025, 17, 1664. https://doi.org/10.3390/cancers17101664
Hajirahimkhan A, Brown KA, Clare SE, Khan SA. SREBP1-Dependent Metabolism as a Potential Target for Breast Cancer Risk Reduction. Cancers. 2025; 17(10):1664. https://doi.org/10.3390/cancers17101664
Chicago/Turabian StyleHajirahimkhan, Atieh, Kristy A. Brown, Susan E. Clare, and Seema Ahsan Khan. 2025. "SREBP1-Dependent Metabolism as a Potential Target for Breast Cancer Risk Reduction" Cancers 17, no. 10: 1664. https://doi.org/10.3390/cancers17101664
APA StyleHajirahimkhan, A., Brown, K. A., Clare, S. E., & Khan, S. A. (2025). SREBP1-Dependent Metabolism as a Potential Target for Breast Cancer Risk Reduction. Cancers, 17(10), 1664. https://doi.org/10.3390/cancers17101664