

EWSR1::ATF1 Orchestrates the Clear Cell Sarcoma Transcriptome in Human Tumors and a Mouse Genetic Model


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Genetic Model of CCS (mCCS)
2.2. Immunochemistry of Mouse Model Tumors
2.3. Human Models of CCS
2.4. Knockdown of EA1 by siRNA Transfection
2.5. RNA-Sequencing Acquisition and Data Analysis
2.6. ChIP-Sequencing of Mouse Model Tumors
2.7. ChIP-Sequencing of Human CCS Cell Lines
2.8. ChIP-Sequencing Analysis
2.9. Tumor Processing for Histology
3. Results
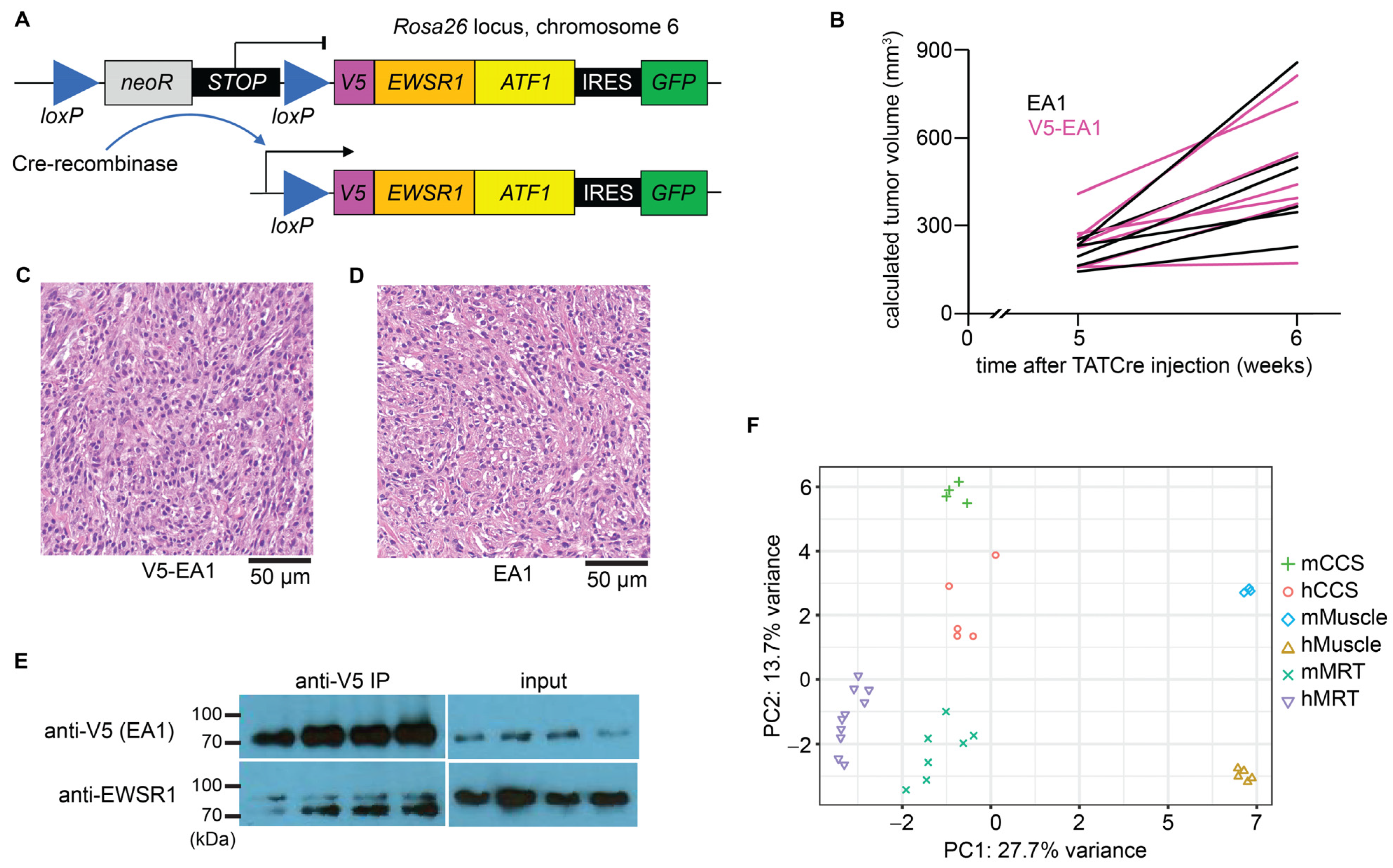
3.1. Conditional Expression of V5-Tagged EWSR1::ATF1 (EA1) Drives Tumorigenesis, Which Recapitulates the Human CCS Transcriptome
3.2. Transcriptomic Analysis of Mouse and Human CCS Identified Shared Differentially Expressed Genes Involved in Proliferation
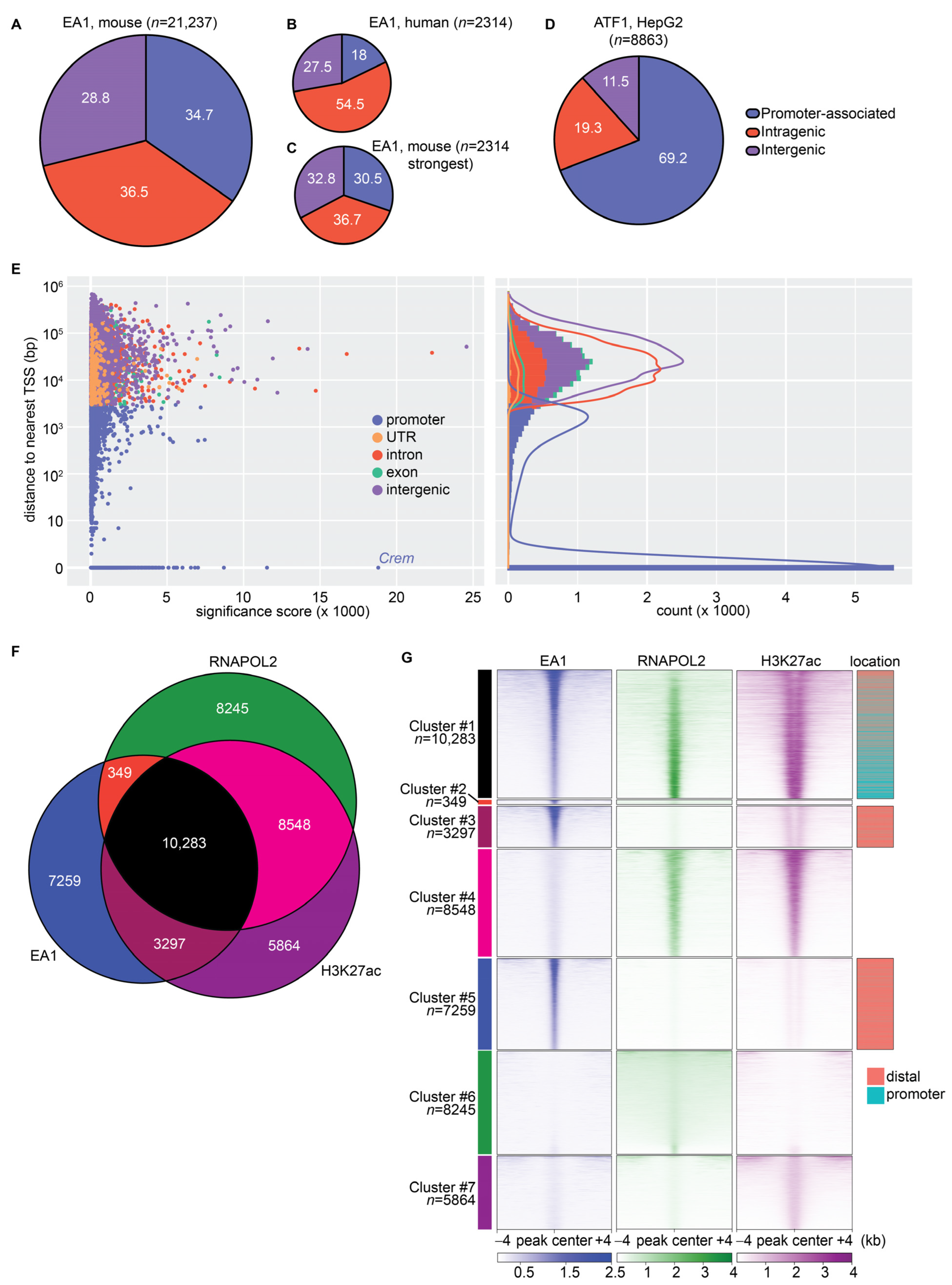
3.3. Genomic Binding of EWSR1::ATF1 Shows Predilection of Distal Regions and Correlation with RNAPOL2 and H3K27ac Signatures
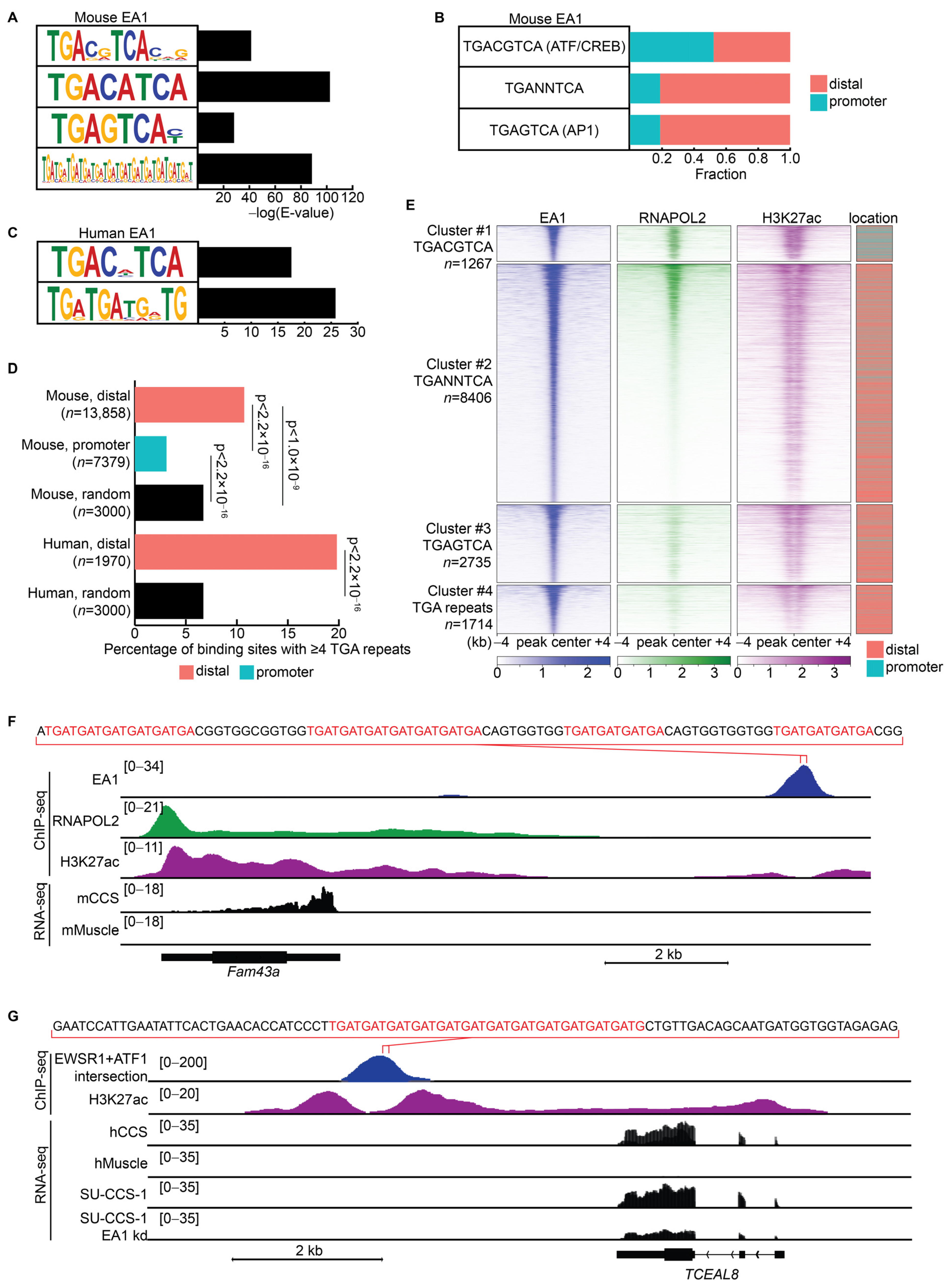
3.4. EWSR1::ATF1 Binds the Canonical ATF/CREB Motif at Promoters and Novel Variant Motifs at Distal Regions
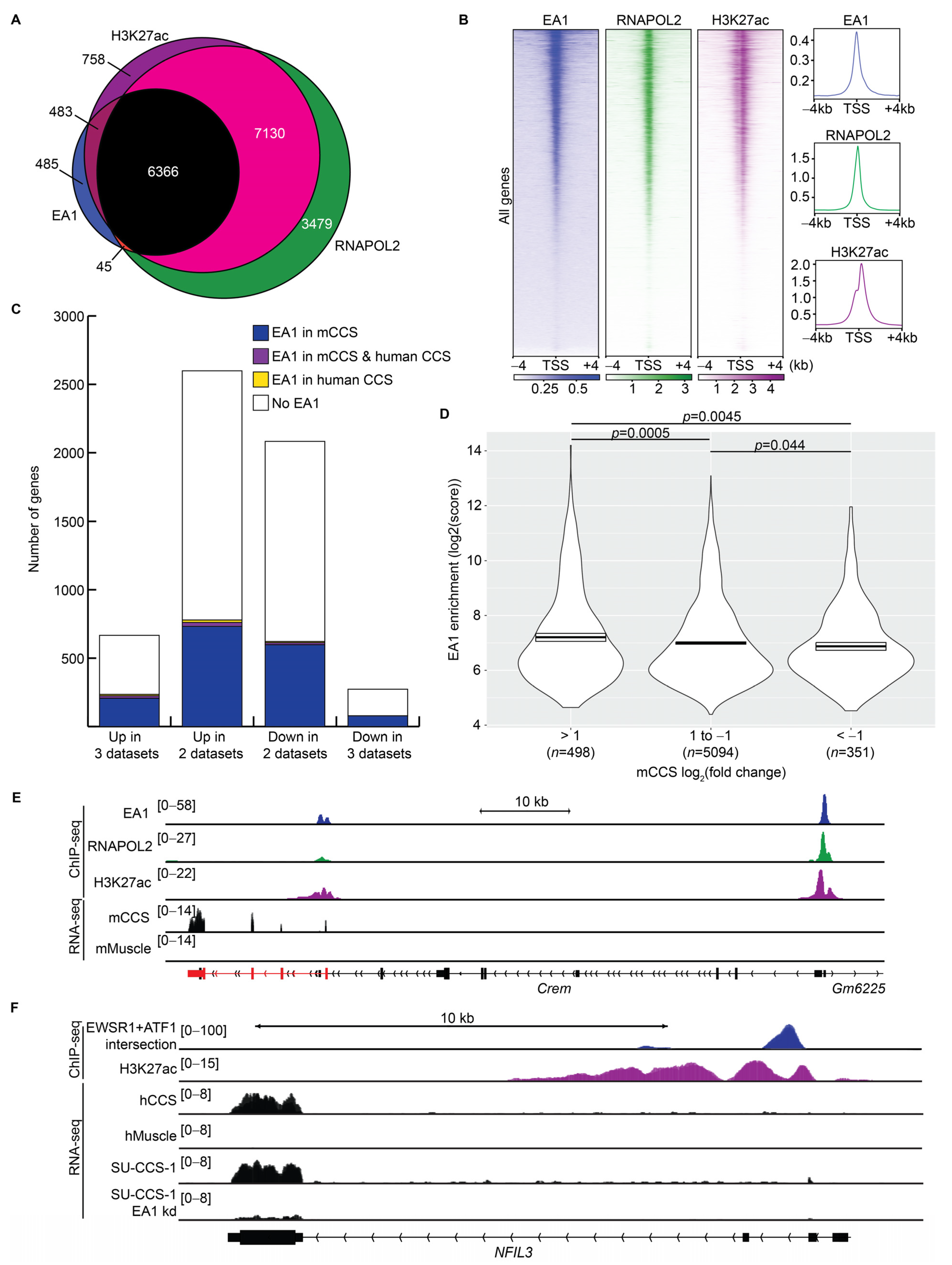
3.5. Promoter-Associated EA1 Binding Sites Promoted Transcription of Target Genes
3.6. Distal EA1 Binding Sites Demarcated Homologous Enhancer Regions in Mouse and Human CCS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Rabbitts, T.H. Chromosomal translocations in human cancer. Nature 1994, 372, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, R.M.; Steenstrup Jensen, S.; Juel, J. Clear cell sarcoma-A review. J. Orthop. 2018, 15, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga, M.I.; Grant, L.; Curtin, C.; Gootee, J.; Silberstein, P.; Voth, E. The epidemiology and survivorship of clear cell sarcoma: A National Cancer Database (NCDB) review. J. Cancer Res. Clin. Oncol. 2018, 144, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Straessler, K.M.; Jones, K.B.; Hu, H.; Jin, H.; van de Rijn, M.; Capecchi, M.R. Modeling clear cell sarcomagenesis in the mouse: Cell of origin differentiation state impacts tumor characteristics. Cancer Cell 2013, 23, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ohno, T.; Aoki, H.; Semi, K.; Watanabe, A.; Moritake, H.; Shiozawa, S.; Kunisada, T.; Kobayashi, Y.; Toguchida, J.; et al. EWS/ATF1 expression induces sarcomas from neural crest-derived cells in mice. J. Clin. Investig. 2013, 123, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Fisher, C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: The current status. Am. J. Surg. Pathol. 2012, 36, e1–e11. [Google Scholar] [CrossRef]
- Komatsu, M.; Sakai, Y.; Nishikubo, M.; Tane, S.; Nishio, W.; Kajimoto, K.; Hirose, T. EWSR1-CREM fusion in pulmonary mesenchymal neoplasm showing distinctive clear cell morphology. Pathol. Int. 2020, 70, 1020–1026. [Google Scholar] [CrossRef]
- Rossi, S.; Szuhai, K.; Ijszenga, M.; Tanke, H.J.; Zanatta, L.; Sciot, R.; Fletcher, C.D.; Dei Tos, A.P.; Hogendoorn, P.C. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin. Cancer Res. 2007, 13, 7322–7328. [Google Scholar] [CrossRef]
- Stockman, D.L.; Miettinen, M.; Suster, S.; Spagnolo, D.; Dominguez-Malagon, H.; Hornick, J.L.; Adsay, V.; Chou, P.M.; Amanuel, B.; Vantuinen, P.; et al. Malignant gastrointestinal neuroectodermal tumor: Clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am. J. Surg. Pathol. 2012, 36, 857–868. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Ganim, R.B.; Zhang, L.; Antonescu, C.; Mueller, J. Primary Pulmonary Myxoid Sarcoma: A Newly Described Entity-Report of a Case and Review of the Literature. Int. J. Surg. Pathol. 2017, 25, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhang, G.; Massett, M.; Cheng, J.; Guo, Z.; Wang, L.; Gao, Y.; Li, R.; Huang, X.; Li, P.; et al. Loci-specific phase separation of FET fusion oncoproteins promotes gene transcription. Nat. Commun. 2021, 12, 1491. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Thompson, M.A.; Wagner, S.; Greenberg, M.E.; Green, M.R. Activating transcription factor-1 can mediate Ca2+- and cAMP-inducible transcriptional activation. J. Biol. Chem. 1993, 268, 6714–6720. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y.; Ohno, T.; Siddique, H.; Lee, L.; Rao, V.N.; Reddy, E.S. The EWS-ATF-1 gene involved in malignant melanoma of soft parts with t(12;22) chromosome translocation, encodes a constitutive transcriptional activator. Oncogene 1996, 12, 159–167. [Google Scholar] [PubMed]
- Kovar, H. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma 2011, 2011, 837474. [Google Scholar] [CrossRef]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e19. [Google Scholar] [CrossRef]
- Linden, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Luna Santamariotaa, M.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019, 20, e45766. [Google Scholar] [CrossRef]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.S.; Sanchez-Vega, F.; Huang, C.H.; Dancsok, A.R.; Hatzi, K.; Chen, C.C.; Tschaharganeh, D.F.; et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 527–541.e8. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, e41305. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.E.; Lim, E.L.; Heravi-Moussavi, A.; Saberi, S.; Mungall, K.L.; Bilenky, M.; Carles, A.; Tse, K.; Shlafman, I.; Zhu, K.; et al. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 2016, 29, 394–406. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Möller, E.; Praz, V.; Rajendran, S.; Dong, R.; Cauderay, A.; Xing, Y.-H.; Lee, L.; Fusco, C.; Broye, L.C.; Cironi, L.; et al. EWSR1-ATF1 dependent 3D connectivity regulates oncogenic and differentiation programs in Clear Cell Sarcoma. Nat. Commun. 2022, 13, 2267. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pages, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Li, J.; Mulvihill, T.S.; Li, L.; Barrott, J.J.; Nelson, M.L.; Wagner, L.; Lock, I.C.; Pozner, A.; Lambert, S.L.; Ozenberger, B.B.; et al. A Role for SMARCB1 in Synovial Sarcomagenesis Reveals That SS18-SSX Induces Canonical BAF Destruction. Cancer Discov. 2021, 11, 2620–2637. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.; Leroux, M.M.; Fleming, M.D.; Orkin, S.H. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002, 2, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M. Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 2009, 89, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Partridge, E.C.; Chhetri, S.B.; Prokop, J.W.; Ramaker, R.C.; Jansen, C.S.; Goh, S.T.; Mackiewicz, M.; Newberry, K.M.; Brandsmeier, L.A.; Meadows, S.K.; et al. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature 2020, 583, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, J.K.; Vanoli, F.; Herviou, L.; Sung, Y.-S.; Zhang, L.; Singer, S.; Tap, W.D.; Benayed, R.; Bale, T.A.; Benhamida, J.K.; et al. Comprehensive genomic profiling of EWSR1/FUS::CREB translocation-associated tumors uncovers prognostically significant recurrent genetic alterations and methylation-transcriptional correlates. Mod. Pathol. 2022, 35, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Montminy, M.R.; Bilezikjian, L.M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 1987, 328, 175–178. [Google Scholar] [CrossRef]
- Bohmann, D.; Bos, T.J.; Admon, A.; Nishimura, T.; Vogt, P.K.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 2017, 68, 1067–1082.e12. [Google Scholar] [CrossRef]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar] [CrossRef]
- Monument, M.J.; Johnson, K.M.; Grossmann, A.H.; Schiffman, J.D.; Randall, R.L.; Lessnick, S.L. Microsatellites with macro-influence in ewing sarcoma. Genes 2012, 3, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Algarra, D.; Souaid, C.; Singh, H.; Dao, L.T.M.; Hussain, S.; Medina-Rivera, A.; Ramirez-Navarro, L.; Castro-Mondragon, J.A.; Sadouni, N.; Charbonnier, G.; et al. Epromoters function as a hub to recruit key transcription factors required for the inflammatory response. Nat. Commun. 2021, 12, 6660. [Google Scholar] [CrossRef] [PubMed]
- Blocher, S.; Fink, L.; Bohle, R.M.; Bergmann, M.; Steger, K. CREM activator and repressor isoform expression in human male germ cells. Int. J. Androl. 2005, 28, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozenberger, B.B.; Li, L.; Wilson, E.R.; Lazar, A.J.; Barrott, J.J.; Jones, K.B. EWSR1::ATF1 Orchestrates the Clear Cell Sarcoma Transcriptome in Human Tumors and a Mouse Genetic Model. Cancers 2023, 15, 5750. https://doi.org/10.3390/cancers15245750
Ozenberger BB, Li L, Wilson ER, Lazar AJ, Barrott JJ, Jones KB. EWSR1::ATF1 Orchestrates the Clear Cell Sarcoma Transcriptome in Human Tumors and a Mouse Genetic Model. Cancers. 2023; 15(24):5750. https://doi.org/10.3390/cancers15245750
Chicago/Turabian StyleOzenberger, Benjamin B., Li Li, Emily R. Wilson, Alexander J. Lazar, Jared J. Barrott, and Kevin B. Jones. 2023. "EWSR1::ATF1 Orchestrates the Clear Cell Sarcoma Transcriptome in Human Tumors and a Mouse Genetic Model" Cancers 15, no. 24: 5750. https://doi.org/10.3390/cancers15245750
APA StyleOzenberger, B. B., Li, L., Wilson, E. R., Lazar, A. J., Barrott, J. J., & Jones, K. B. (2023). EWSR1::ATF1 Orchestrates the Clear Cell Sarcoma Transcriptome in Human Tumors and a Mouse Genetic Model. Cancers, 15(24), 5750. https://doi.org/10.3390/cancers15245750