

PARP Inhibitors in Advanced Prostate Cancer in Tumors with DNA Damage Signatures
 and
and
Abstract
Simple Summary
Abstract
1. Introduction

{kind=link}
{kind=link}
| Year | Trial | Drug Class | Study Treatment | Control | (n) | Pretreated Chemo (c) ADT (h) (ARSi) | Sequence Approved by FDA | HR for Death (95% CI) | Biomarker |
|---|---|---|---|---|---|---|---|---|---|
| 2004 | Tax 327 [11] | Chemo | Docetaxel | Mito + P | 1006 | (h) | 1st line | 0.76 (0.62–0.94) | N/A |
| SWOG 9916 [33] | Chemo | Docetaxel + Estramustine | Mito + P | 674 | (h)(c) | 1st line | 0.80 (0.67–0.97) | N/A | |
| 2010 | TROPIC2 [34] | Chemo | Cabazitaxel | Mito + P | 755 | (h)(c) | 2nd line | 0.70 (0.59–0.83) | N/A |
| IMPACT [35] | IO | Sipuleucel-T | Placebo | 512 | (h)(c) | 1st line | 0.77 (0.61–0.98) | N/A | |
| 2011 | NCT00321620 [36] | BMA | Denosumab | ZA | 1904 | (h) | 1st line | 1.03 (0.91–1.17) | N/A |
| COU-AA-301 [7] | Chemo | Abiraterone + P | Placebo + P | 1195 | (h) | 2nd line | 0.65 (0.54–0.83) | N/A | |
| 2012 | AFFIRM [37] | ARSi | Enzalutamide | Placebo | 1199 | (h)(c) | 2nd line | 0.63 (0.53–0.75) | N/A |
| 2013 | COU-AA-302 [38] | ARSi | Abiraterone + P | Placebo + P | 1088 | (h) | 1st line | 0.75 (0.61–0.93) | N/A |
| ALSYMPCA [36] | Radio-pharmaceutical | Radium-223 + SOC | SOC | 921 | (h) | 1st line | 0.70 (0.58–0.83) | N/A | |
| 2014 | PREVAIL [8] | ARSi | Enzalutamide | Placebo | 1717 | (h) | 1st line | 0.71 (0.60–0.84) | N/A |
| 2017 | KEYNOTE 028 [16] | IO | Pembrolizumab | Placebo | 23 | (h)(c)(ARSi) | 3rd line | N/A | MMR/MSI |
| 2019 | TITAN [39] | ARSi | Apalutamide | Placebo | 1207 | (h)(c) | 2nd line | 0.67 (0.51–0.89) | N/A |
| ARAMIS [10] | ARSi | Darolutamide | Placebo | 1509 | (h) | 1st line | 0.71 (0.50–0.99) | N/A | |
| 2020 | PROFOUND [20] | PARPi | Olaparib | Placebo | 387 | (h)(c)(ARSi) | 2nd line | 0.55 (0.29–1.06) | HRD * |
| 2021 | TRITON2 [40] | PARPi | Rucaparib | Placebo | 115 | (h)(c)(ARSi) | 3rd line | N/A | BRCA1/2 Mutation |
2. DNA Repair, Cellular Pathways and Synthetic Lethality
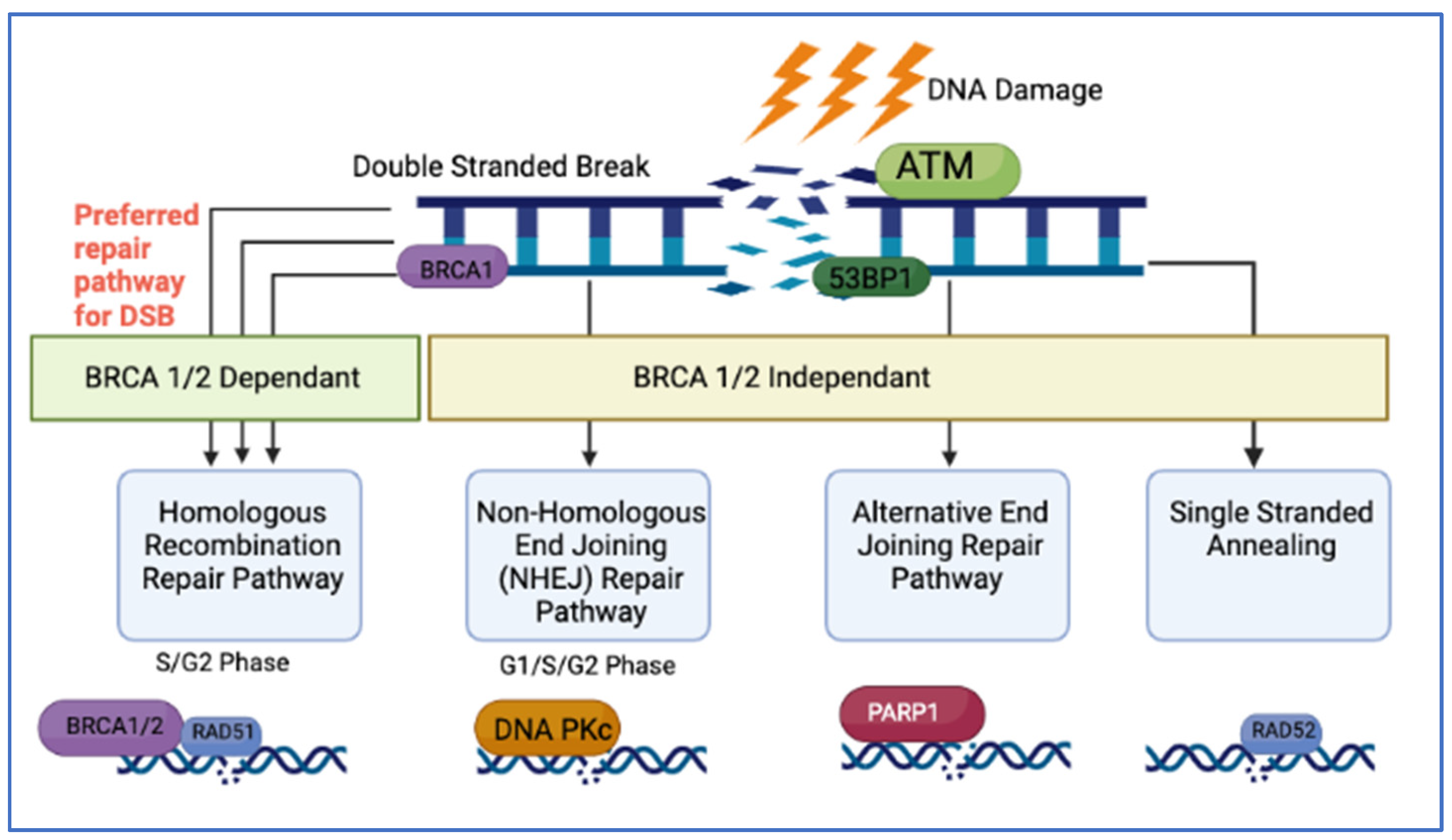
2.1. DNA Damage Repair Pathways
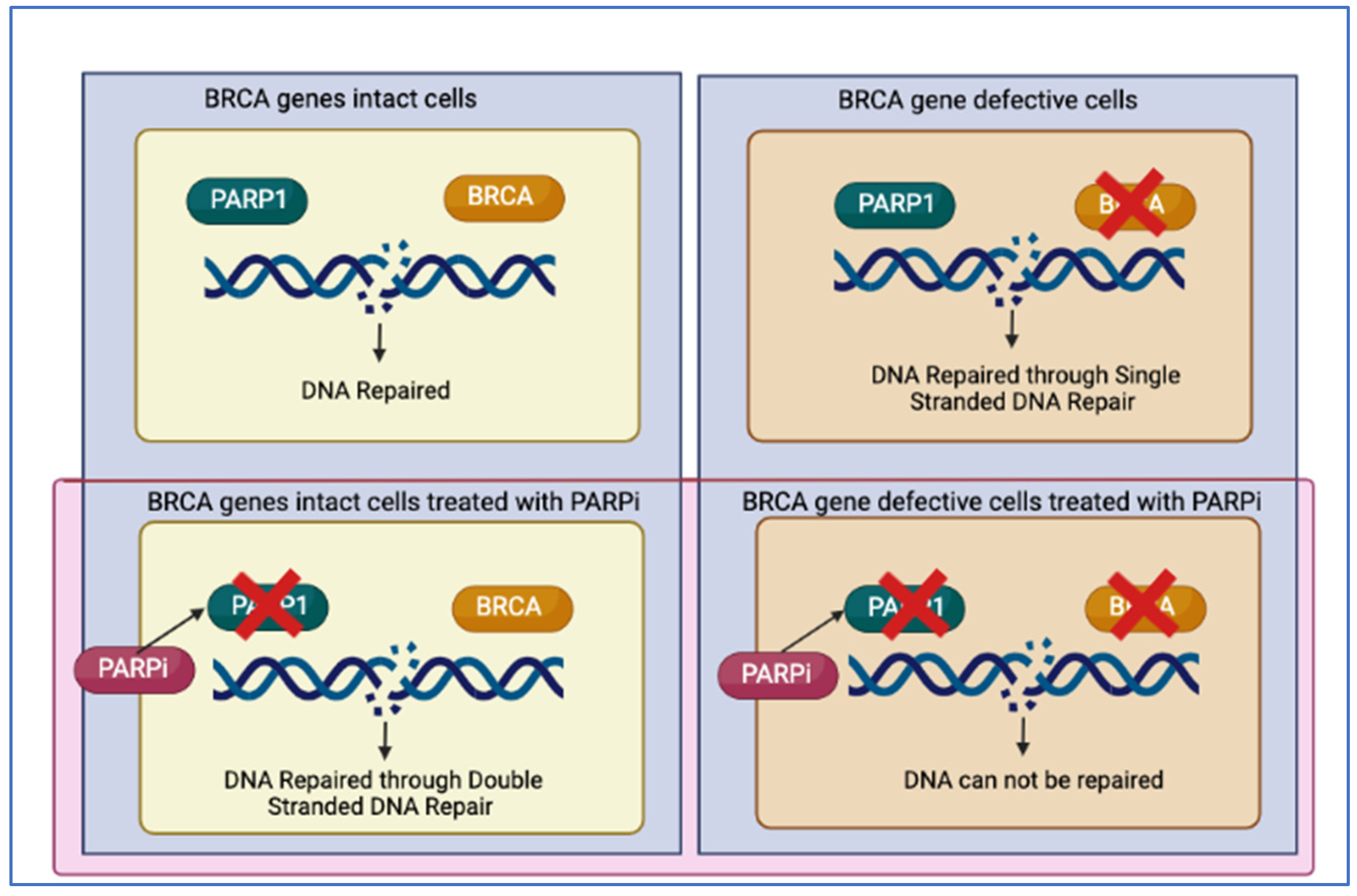
2.2. Synthetic Lethality and PARP
2.3. The Concept of BRCAness and Mutational Signatures
3. Genomic Heterogeneity in PCa
4. Clinical Impact of DNA Damage Signatures in mCRPC
4.1. DNA Repair Pathways and Clinical Trials with PARPi
4.2. Specific Non-BRCA Biomarkers in DNA Repair Pathways and Mutational Signatures
4.2.1. ATM Gene
4.2.2. PALB2
4.2.3. CHEK2
4.2.4. FANCA
4.2.5. RAD51
4.2.6. CDK12
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Society, A.C. Survival Rates for Prostate Cancer 2022. Available online: https://www.cancer.org/cancer/prostate-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 15 March 2022).
- ASCO.org. Prostate Cancer: Statistics 2021. Available online: https://www.cancer.net/cancer-types/prostate-cancer/statistics (accessed on 15 March 2022).
- Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2020 with a focus on prostate cancer. Ann. Oncol. 2020, 31, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Huggins, C.; Webster, W.O. Duality of Human Prostate in Response to Estrogen. J. Urol. 1948, 59, 258–266. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Faculty Opinions recommendation of Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juárez Soto, Á.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Cas-tration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Pred-nisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial—FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O′Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- (FDA) UFaDA. FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication: FDA.GOV. 2017. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 22 March 2022).
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.-P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Astrazeneca. LYNPARZA™ Approved by the Us Food and Drug Administration (Fda) for the Treatment of Advanced Ovarian Cancer in Patients with Germline Brca-Mutations 2014. Available online: https://www.astrazeneca.com/media-centre/press-releases/2014/lynparza-approved-us-fda-brca-mutated-ovarian-cancer-treatment-19122014.html# (accessed on 23 March 2022).
- FDA. FDA approves Olaparib for Germline BRCA-Mutated Metastatic Breast Cancer 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-Olaparib-germline-brca-mutated-metastatic-breast-cancer (accessed on 29 March 2022).
- FDA. FDA Approves Olaparib for gBRCAm Metastatic Pancreatic Adenocarcinoma 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 23 March 2022).
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Faculty Opinions recommendation of Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. 2019, 17, 515–521. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Borcsok, J.; Pomerantz, M.M.; Tisza, V.; Spisak, S.; Rusz, O.; Csabai, I.; Freedman, M.L.; et al. Detection of Molecular Signatures of Homologous Recombination Deficiency in Prostate Cancer with or without BRCA1/2 Mutations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2673–2680. [Google Scholar] [CrossRef]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Gallagher, D.J.; Cronin, A.M.; Milowsky, M.I.; Morris, M.J.; Bhatia, J.; Scardino, P.T.; Eastham, J.A.; Offit, K.; Robson, M.E. Germline BRCA mutation does not prevent response to taxane-based therapy for the treatment of castration-resistant prostate cancer. Br. J. Urol. 2012, 109, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.-M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Cheng, H.H.; Beltran, H.; Dolling, D.; Xu, W.; Pritchard, C.C.; Mossop, H.; Rescigno, P.; Perez-Lopez, R.; Sailer, V.; et al. Clinical Outcome of Prostate Cancer Patients with Germline DNA Repair Mutations: Retrospective Analysis from an International Study. Eur. Urol. 2018, 73, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Annala, M.; Struss, W.J.; Warner, E.W.; Beja, K.; Vandekerkhove, G.; Wong, A.; Khalaf, D.; Seppälä, I.L.; So, A.; Lo, G.; et al. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur Urol. 2017, 72, 34–42. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and Estramustine Compared with Mitoxantrone and Prednisone for Advanced Refractory Prostate Cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- NCT04030559. Niraparib Before Surgery in Treating Patients With High Risk Localized Prostate Cancer and DNA Damage Response Defects. 2021. Available online: https://www.clinicaltrials.gov/ (accessed on 3 April 2022).
- NCT00261456. The IMPACT Study—Identification of Men With a Genetic Predisposition to ProstAte Cancer. Available online: https://clinicaltrials.gov/ct2/show/record/NCT002614562021 (accessed on 3 April 2022).
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; De Bono, J.S.; Molina, A.; Logothetis, C.J.; De Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in Metastatic Prostate Cancer without Previous Chemotherapy. N. Engl. J. Med. 2012, 368, 138–148. [Google Scholar] [CrossRef]
- NCT03652493. Trial Evaluating the Efficacy of CARBOPLATIN in Metastatic Prostate Cancer With Gene Alterations in the Homologous Recombination Pathway (PRO-CARBO). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03652493 (accessed on 24 April 2022).
- NCT03432897. BrUOG 337: Olaparib Prior to Radical Prostatectomy For Patients With Locally Advanced Prostate Cancer and Defects in DNA Repair Genes (337). clinicaltrials.gov. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03432897 (accessed on 24 April 2022).
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 2012, 69, 1447–1473. [Google Scholar] [CrossRef] [PubMed]
- Stok, C.; Kok, Y.P.; Tempel, N.V.D.; Vugt, M.A.T.M.V. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef] [PubMed]
- Neiger, H.; Siegler, E.; Shi, Y. Breast Cancer Predisposition Genes and Synthetic Lethality. Int. J. Mol. Sci. 2021, 22, 5614. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Erixon, K.; Arnaudeau, C.; Schultz, N.; Jenssen, D.; Meuth, M.; Helleday, T. Different Roles for Nonhomologous End Joining and Homologous Recombination following Replication Arrest in Mammalian Cells. Mol. Cell. Biol. 2002, 22, 5869–5878. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I.; Alhmoud, J.F. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Castro, E.; Mahmud, N.; Guy, M.; Edwards, S.; O′Brien, L.; Sawyer, E.; et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br. J. Cancer 2011, 105, 1230–1234. [Google Scholar] [CrossRef]
- McNevin, C.S.; Cadoo, K.; Baird, A.-M.; Murchan, P.; Sheils, O.; McDermott, R.; Finn, S. Pathogenic BRCA Variants as Biomarkers for Risk in Prostate Cancer. Cancers 2021, 13, 5697. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA Mutations Are Associated With Higher Risk of Nodal Involvement, Distant Metastasis, and Poor Survival Outcomes in Prostate Cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef]
- Francica, P.; Rottenberg, S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018, 10, 101. [Google Scholar] [CrossRef]
- Rao, A.; Moka, N.; Hamstra, D.A.; Ryan, C.J. Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now? Cancers 2022, 14, 801. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O′Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Spisák, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O′Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [PubMed]
- De Sarkar, N.; Dasgupta, S.; Chatterjee, P.; Coleman, I.; Ha, G.; Ang, L.S.; Kohlbrenner, E.M.; Frank, S.B.; Nunez, T.A.; Salipante, S.J.; et al. Genomic attributes of homology-directed DNA repair deficiency in metastatic prostate cancer. JCI Insight. 2021, 6, e152789. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves Niraparib for First-Line Maintenance of Advanced Ovarian Cancer 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-first-line-maintenance-advanced-ovarian-cancer (accessed on 13 April 2022).
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Póti, A.; Gyergyák, H.; Németh, E.; Rusz, O.; Tóth, S.; Kovácsházi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike., T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef]
- NCCN. NCCN. NCCN Clinical Practice Guidelines in Oncology. In Prostate Cancer Version 2.2031; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- Cooney, K.A. Inherited Predisposition to Prostate Cancer: From Gene Discovery to Clinical Impact. Trans. Am. Clin. Clim. Assoc. 2017, 128, 14–23. [Google Scholar]
- Gallagher, D.J.; Gaudet, M.M.; Pal, P.; Kirchhoff, T.; Balistreri, L.; Vora, K.; Bhatia, J.; Stadler, Z.; Fine, S.W.; Reuter, V.; et al. Germline BRCA Mutations Denote a Clinicopathologic Subset of Prostate Cancer. Clin. Cancer Res. 2010, 16, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Warner, E.W.; Yip, S.M.; Chi, K.N.; Wyatt, A.W. DNA repair defects in prostate cancer: Impact for screening, prognostication and treatment. Br. J. Urol. 2019, 123, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Foye, A.; Wagle, N.; Kim, W.; Carter, S.L.; McKenna, A.; Simko, J.P.; Garraway, L.A.; Febbo, P.G. Successful whole-exome sequencing from a prostate cancer bone metastasis biopsy. Prostate Cancer Prostatic Dis. 2014, 17, 23–27. [Google Scholar] [CrossRef]
- Mehra, R.; Kumar-Sinha, C.; Shankar, S.; Lonigro, R.J.; Jing, X.; Philips, N.E.; Siddiqui, J.; Han, B.; Cao, X.; Smith, D.C.; et al. Characterization of Bone Metastases from Rapid Autopsies of Prostate Cancer Patients. Clin. Cancer Res. 2011, 17, 3924–3932. [Google Scholar] [CrossRef]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [PubMed]
- Dall’Era, M.A.; McPherson, J.D.; Gao, A.C.; White, R.W.D.; Gregg, J.P.; Lara, P.N. Germline and somatic DNA repair gene alterations in prostate cancer. Cancer 2020, 126, 2980–2985. [Google Scholar] [CrossRef]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Mul-ticlonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Altera-tions and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Carreira, S.; Porta, N.; Arce-Gallego, S.; Seed, G.; Llop-Guevara, A.; Bianchini, D.; Rescigno, P.; Paschalis, A.; Bertan, C.; Baker, C.; et al. Biomarkers Associating with PARP Inhibitor Benefit in Prostate Cancer in the TOPARP-B Trial. Cancer Discov. 2021, 11, 2812–2827. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [PubMed]


| Pathway | Gene | Robinson (n = 150) [15] | Quigley (n = 100) [22] | Abida (n = 444) [23] |
|---|---|---|---|---|
| AR | AR | 62.70% | 69.31% | |
| AR Enhancer | 80.20% | |||
| ASXL2 | 6.93% | |||
| FOXA1 | 12% | 18.81% | ||
| NCOR1 | 6.70% | 1.98% | ||
| NCOR2 | 5.30% | 0.99% | ||
| Cell Cycle | CCND1 | 4.70% | 7.92% | |
| CDKN1B | 4.00% | |||
| CDKN2A | 2.70% | 3.96% | ||
| RB1 | 9.30% | 1.98% | ||
| TP53 | 53.30% | 56.44% | ||
| Chromatin Modifier | CHD1 | 8.00% | 8.91% | |
| KDM6A | 3.30% | 2.97% | ||
| KMT2C | 12.70% | 7.92% | ||
| KMT2D | 2.70% | 1.98% | ||
| DNA Repair Pathway | ATM | 7.30% | 5.94% | 5.80% |
| ATR | <2% | |||
| BRCA1 | 0.70% | 0.99% | 1% | |
| BRCA2 | 13.30% | 9.90% | 11.40% | |
| BRIP1 | 4.70% | |||
| CDK12 | 2.97% | |||
| CHEK2 | <1% | |||
| ERCC2 | 2.97% | |||
| MLH1 | 0.70% | 0.99% | <2% | |
| MSH2 | 1.98% | 2.10% | ||
| MSH6 | 2.00% | 0.99% | 2.50% | |
| PALB2 | <1% | |||
| PRKDC | 7.92% | |||
| RAD51 | <1% | |||
| ETS | ETS fusions | 56.70% | ||
| ETV1 | 9.90% | |||
| ETV4 | 4.95% | |||
| ETV5 | 1.98% | |||
| ERG | 42.57% | |||
| PI3K Pathway | AKT1 | 1.30% | 1.98% | |
| PIK3CA | 5.30% | 0.99% | ||
| PIK3CB | 6.00% | |||
| PIK3R1 | 5.30% | |||
| PTEN | 40.70% | 44.55% | ||
| WNT Pathway | APC | 8.70% | 8.91% | |
| CTNNB1 | 4% | 5.94% | ||
| RNF43 | 2.70% | |||
| RSPO2 | 1.30% | |||
| ZNRF3 | 2% | 3.96% | ||
| RAS/RAS Fusions | RAF1 | 2.00% | ||
| BRAF | 2.70% | 3.96% | ||
| HRAS | 1.98% |
| Pathway | Gene | Pritchard (n = 82) [16] | Castro (n = 68) [24] |
|---|---|---|---|
| DNA Repair Pathway | ATM | 1.6% | 1.91% |
| ATR | 0.29% | ||
| BRCA1 | 0.9% | 0.95% | |
| BRCA2 | 5.3% | 3.34% | |
| BRIP1 | 0.18% | ||
| CDK12 | |||
| CHEK2 | 1.9% | 3.34% | |
| ERCC2 | 0.24% | ||
| MLH1 | |||
| MSH2 | 0.14% | ||
| MSH6 | 0.14% | ||
| PALB2 | 0.4% | 0.00% | |
| PRKDC | |||
| RAD51 | 0.4% |
| Author (Year) | Year | (n) | Disease Subtype | Somatic(s) V Germline(g) | Testing Adopted | % Clinically Actionable Aberration |
|---|---|---|---|---|---|---|
| Robinson [78] | 2015 | 150 | mCRPC | (s) | Panel of 38 genetic mutations | 89% |
| Pritchard [49] | 2016 | 82/692 | mPCa | (g) | Panel of 20 germline genetic mutations | 25% |
| Quigley [79] | 2018 | 100 | mCRPC | (g)(s) | WGS | n/a |
| Abida [48] | 2019 | 444 | mCRPC | (s) | WES | >20% |
| Castro [81] | 2019 | 68/419 | mCRPC | (g) | Germline DDR mutations in 107 gene | 16.2% |
| Mateo [82] | 2020 | 470 1/61 2 | PCa/mCRPC | (g)(s) | NGS 1/WGS 2 | 1 23% |
| Dall ‘Era [80] | 2020 | 154 | PCa/mCRPC | (g)(s) | Panel of 24 genetic mutations (NGS) | 16% |
| Year | Trial | Phase | PARPi | Primary End Point | Genes Included | Testing Method | DDR Gene Aberration Detected/Screened (%) | Key Finding |
|---|---|---|---|---|---|---|---|---|
| 2014 | TOPARP-A [83] NCT01682772 | II | Olaparib | Response Rate according to RECIST, PSA or CTC | BRCA2, ATM, BRCA1, FANCA, CHEK2, PALB2, HDAC2, RAD51, MLH3, ERCC3, MRE11, NBN | WES from tumor-biopsy samples; germline WES from saliva samples. | 16/50 (32%) | Overall RR: 33% (16/49) RR in HRR positive subgroup: 88% (14/16) PFS: HRR + ve: 9.8 vs. HRR − ve: 2.7 months; p < 0.001 OS: HRR + ve: 13.8 vs. HRR − ve: 7.5 months; p = 0.05 |
| 2019 | TOPARP-B [84] NCT01682772 | II | Olaparib | Response Rate according to RECIST, PSA or CTC | BRCA1/2, ATM, CDK12,PALB2, CHEK1,CHEK2, ARID1A, ATRX, FANCA, FANCF, FANCG, FANCI, FANCM, MSH2, NBN, RAD50, WRN | NGS of biopsies | 161/711 (27%) | RR: Olaparib 400 mg group: 54.3% vs. Olaparib 300 mg group: 39.1% PFS: Olaparib 400 mg 5.5 months vs. Olaparib 300 mg 5.4 months OS: Olaparib 400 mg 14.3 vs. Olaparib 300 mg 10.1 months |
| 2020 | PROFOUND [20] NCT02987543 | III | Olaparib | Imaging-based PFS | Cohort A: BRCA1, BRCA2, and ATM Cohort B: BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L | FoundationOne CDx NGS of archival or recent biopsy tissue | 778/2792 (28%) | Cohort A + B RR: Olaparib 22.0% vs. ADT 4.0% PFS: Olaparib 5.8 vs. ADT 3.5 months OS: Olaparib 17.5 vs. ADT 14.3 months Cohort A RR: Olaparib 33.0% vs. ADT 2.0% PFS: Olaparib 7.4 vs. ADT 3.6 months OS: Olaparib 18.5 vs. ADT 15.1 months |
| 2020 | TRITON2 [21,22] NCT02952534 | II | Rucaparib | Response Rate according to RECIST, PCWG3 criteria | First Analysis: BRCA1/BRCA2 Secondary Analysis: ATM, CHEK2, FANCA, PALB2, FANCA, BRIP1, and RAD51B | Foundation Medicine. Germline testing by Color Genomics. | 115/78 | RR: 44% in participants with BRCA1/2 mutations Confirmed PSA response in 51.1% es in BRCA1/2 group, 1 ntsent with a CDK12 alteration, 1 participant with a BRIP1 alteration, and 1 participant with a FANCA alteration |
| 2021 | TALAPRO-1 [85] NCT03148795 | II | Talazoparib | ORR | ATM, ATR, BRCA1, BRCA2, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, RAD51C | Foundation One CDx™ NGS gene panel test. Saliva sample collection for a germline comparator. | 127 | Objective RR 29·8% (31 of 104 participants) |
| 2021 | GALAHAD [86] NCT02854436 (preliminary results) | II | Niraparib | ORR | Total: 81; (BRCA1/2: 46; non-BRCA: 35) BRCA1/2 (BRCA), ATM, FANCA, PALB2, CHEK2, BRIP1, or HDAC2. | Plasma or tissue-based test | 127 | RR: BRCA ½ 41% vs. Non-BRCA 9% PFS: BRCA ½ 8.2 vs. Non-BRCA 5.3 months OS: BRCA ½ 12.6 vs. Non-BRCA 14 |
| Gene | (n) | Radiographic Responses (%) | PSA Responses (%) |
|---|---|---|---|
| BRCA2 + BRCA1 | 115 (102/13) | 43.5% | 54.8% |
| ATM | 49 | 10.5% | 4.1% |
| CDK12 | 15 | 0 | 6.7% |
| CHEK2 | 12 | 11.1% | 16.7% |
| FANCA, NBN, BRIP1, PALB2, RAD51, RAD51B, RAD54L ^^ | 14 | 28.6% | 35.7% |
| Gene | (n) | a,b ORR, % (Response/n) | b rPFS, Months (95% CI) | b,c Composite Response, % (Response/n) |
|---|---|---|---|---|
| BRCA2 + BRCA1 | 46 | 43.9 (18/41) | 9.3 (8.1–13.7) | 76.1 (35/46) |
| PALB2 | 4 | 33.3 (1/3) | 7.4 (2–7.4) | 50.0 (2/4) |
| ATM | 18 | 11.8 (2/17) | 5.5 (1.7–8.2) | 27.8 (5/18) |
| OTHER DDR Genes | 18 | 0 | 3.7 (1.7–3.9) | 11.1 (2/18) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McNevin, C.S.; Cadoo, K.; Baird, A.-M.; Finn, S.P.; McDermott, R. PARP Inhibitors in Advanced Prostate Cancer in Tumors with DNA Damage Signatures. Cancers 2022, 14, 4751. https://doi.org/10.3390/cancers14194751
McNevin CS, Cadoo K, Baird A-M, Finn SP, McDermott R. PARP Inhibitors in Advanced Prostate Cancer in Tumors with DNA Damage Signatures. Cancers. 2022; 14(19):4751. https://doi.org/10.3390/cancers14194751
Chicago/Turabian StyleMcNevin, Ciara S., Karen Cadoo, Anne-Marie Baird, Stephen P. Finn, and Ray McDermott. 2022. "PARP Inhibitors in Advanced Prostate Cancer in Tumors with DNA Damage Signatures" Cancers 14, no. 19: 4751. https://doi.org/10.3390/cancers14194751
APA StyleMcNevin, C. S., Cadoo, K., Baird, A.-M., Finn, S. P., & McDermott, R. (2022). PARP Inhibitors in Advanced Prostate Cancer in Tumors with DNA Damage Signatures. Cancers, 14(19), 4751. https://doi.org/10.3390/cancers14194751