
Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. Analysis of Sensitivity Biomarkers
2.2.1. End-Point Calculation and Comparison
2.2.2. Data Availability
2.3. Gene Expression
2.3.1. Transcriptome Analysis
2.3.2. Quantitative PCR
2.3.3. Gene Silencing
2.4. Proliferation Assay
2.4.1. Acumen
2.4.2. Resazurin Proliferation Assay
2.4.3. Trypan Blue
2.5. Fluorocytometry
2.5.1. Quantification of Mitochondrial Membrane Potential and Cell Permeability
2.5.2. Cell Cycle Analysis
2.6. DNA Damage: Alkaline Comet Assay
2.7. Statistical Analysis and Synergy
3. Results
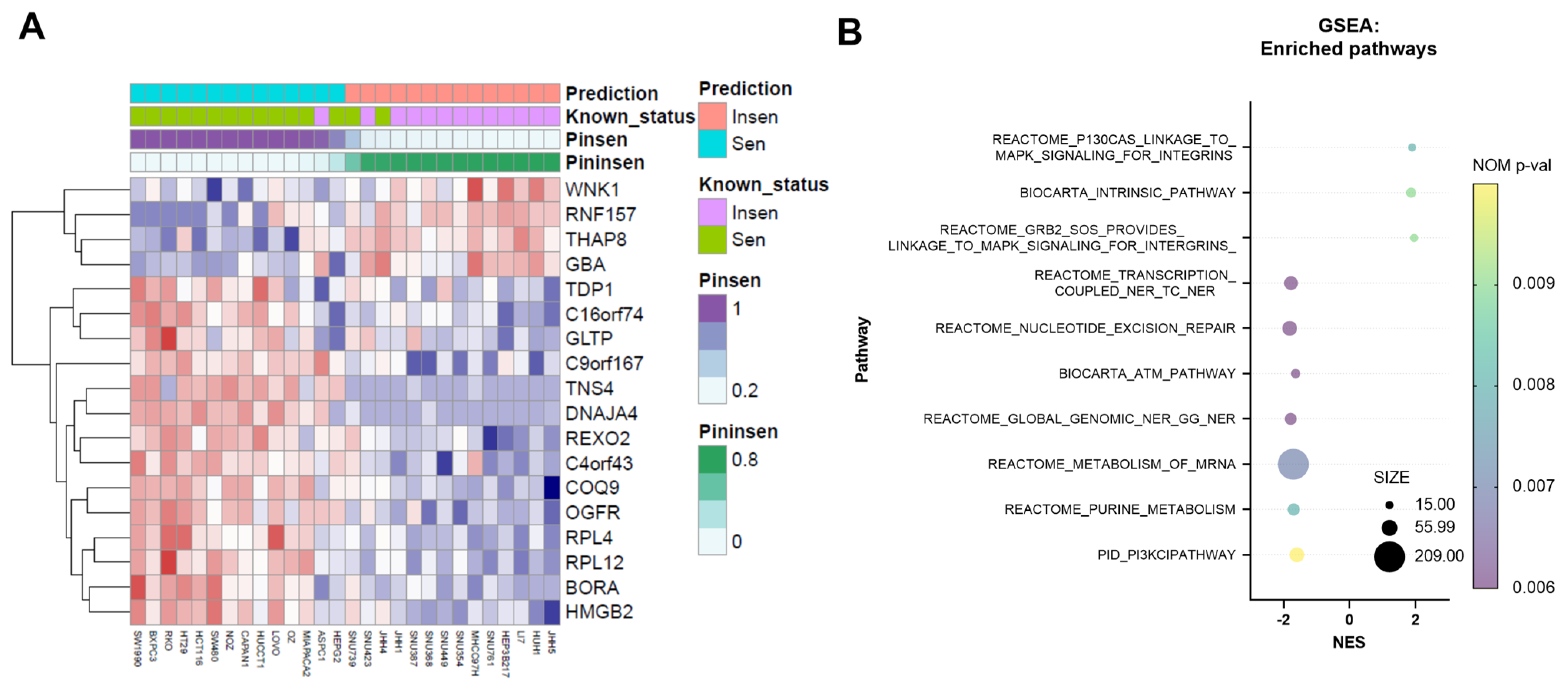
3.1. NV651 Decreases Proliferation in Colorectal, Liver and Pancreatic Cancer Cell Lines; Furthermore, 18 Genes Can Effectively Predict NV651 Sensitivity
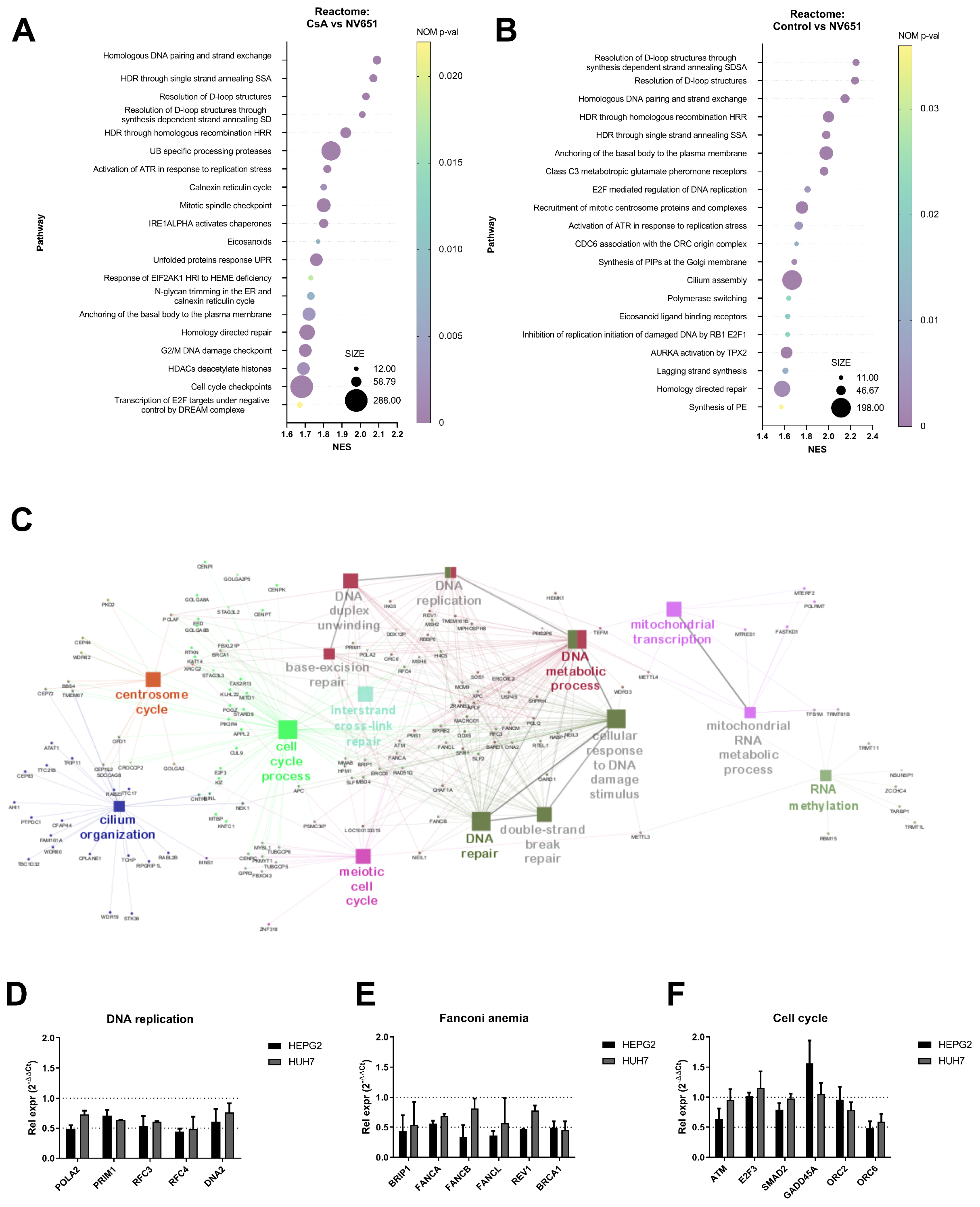
3.2. NV651 Affects DNA Damage Repair and Alters the Cell Cycle
3.3. Combination of NV651 and Cisplatin Results in a Synergistic Effect on Cell Viability in HCC Cell Lines
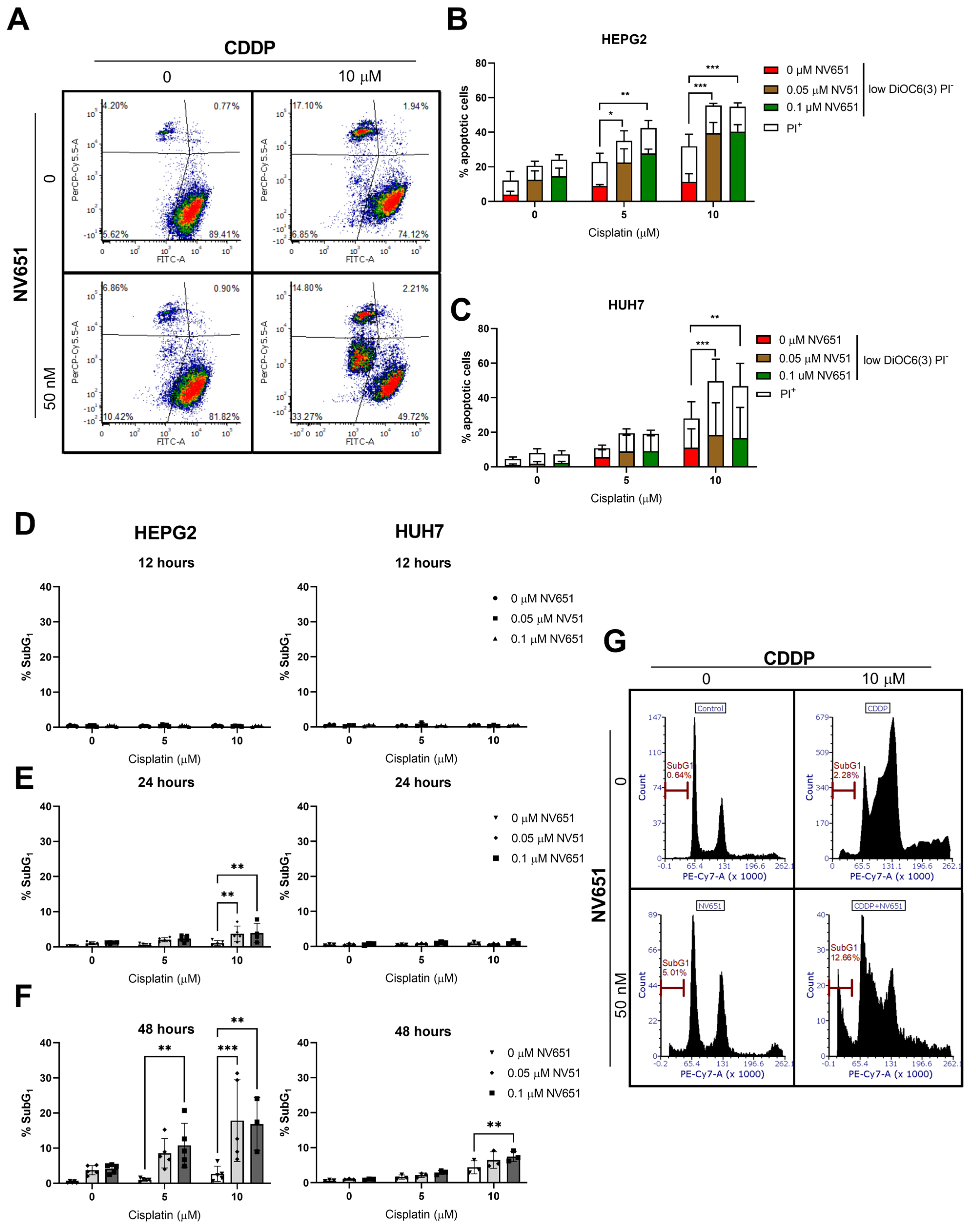
3.4. NV651 and Cisplatin Activate the Intrinsic Apoptotic Pathway in HCC Cells
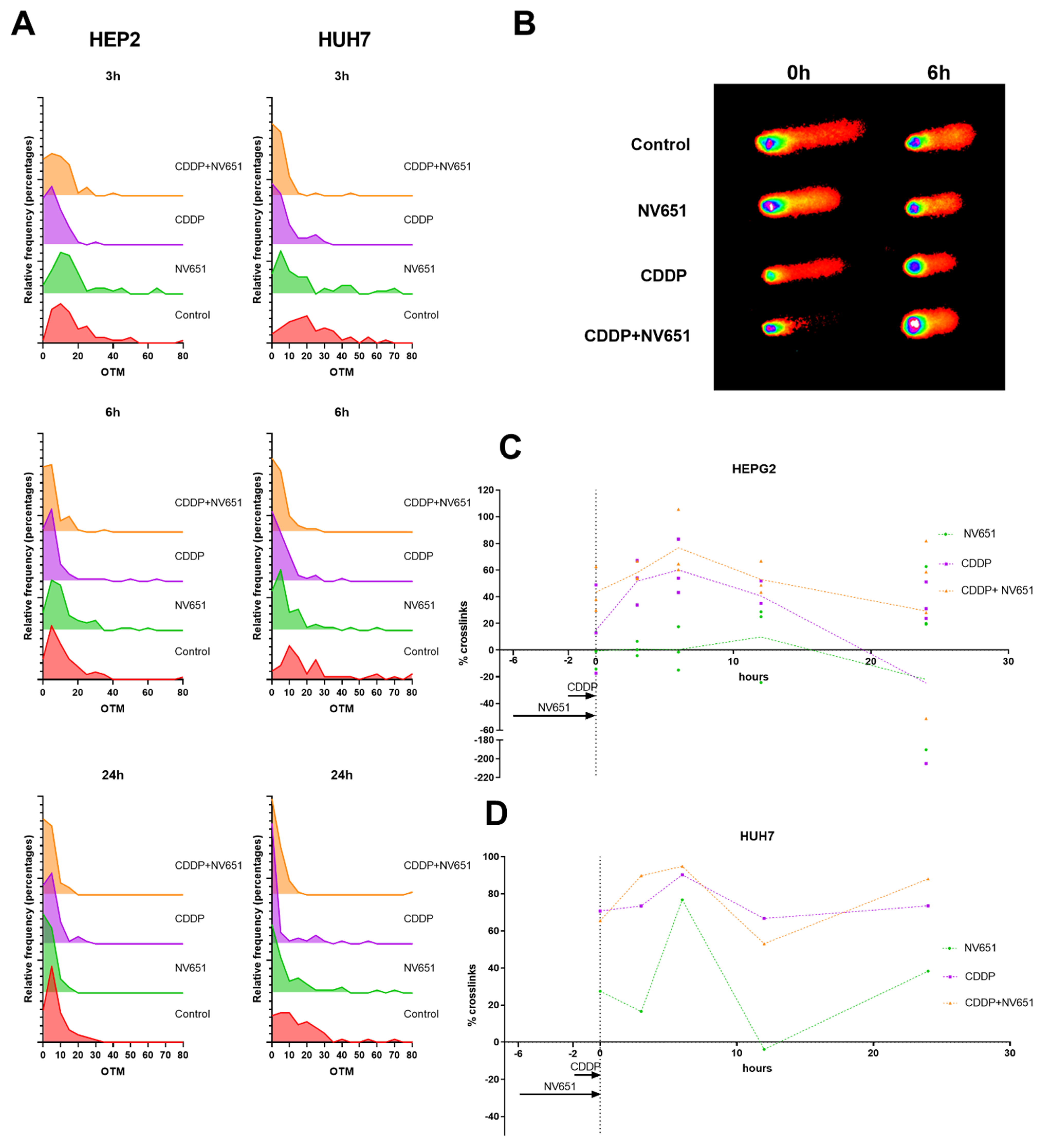
3.5. NV651 and Cisplatin Cause a Decrease in the DNA Repair Capacity of HCC Cells




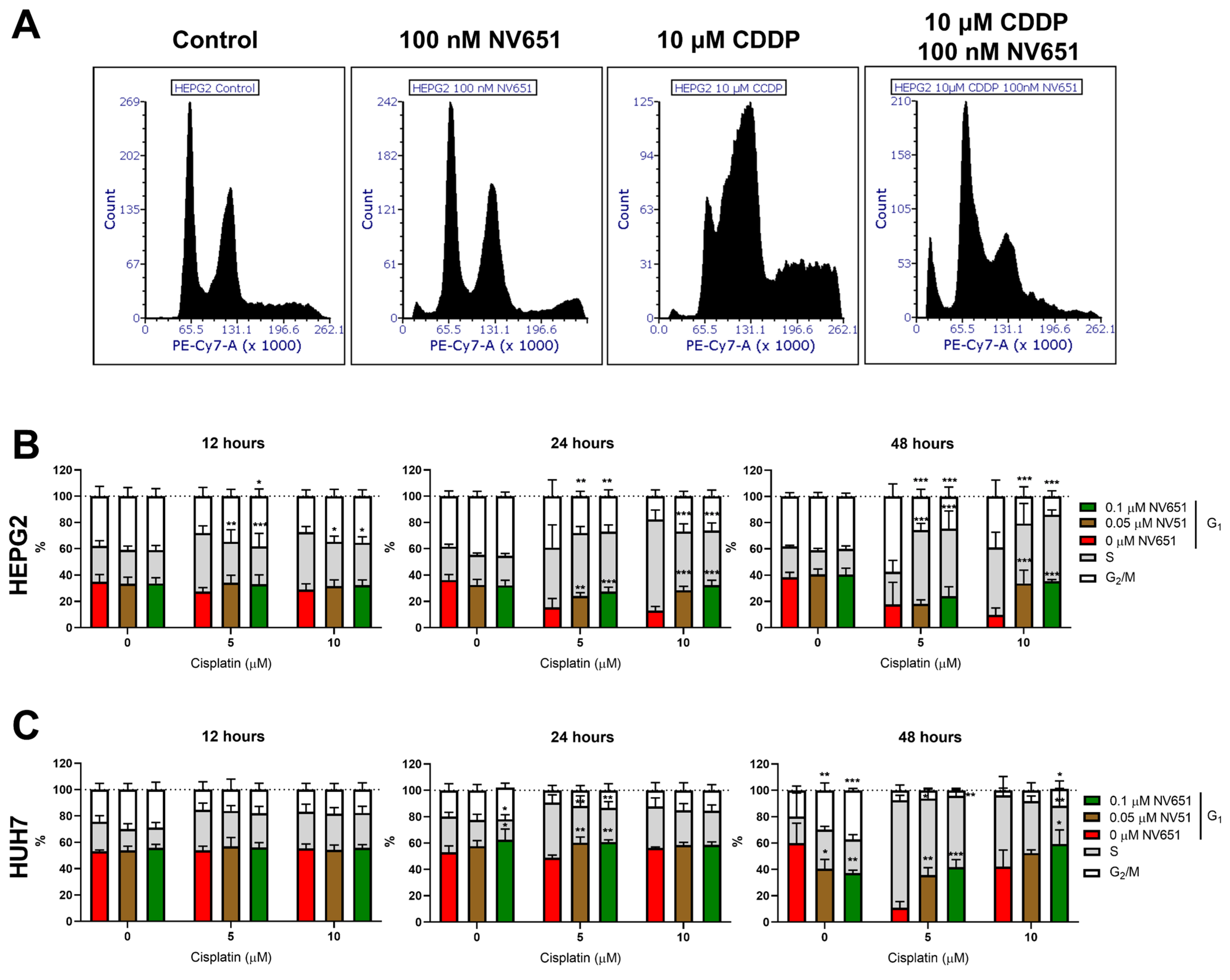
3.6. NV651 and Cisplatin Cause a Disturbance in the Cell Cycle
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Liver Fact Sheet. 2020. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 1 November 2021).
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular Carcinoma: Risk Factors, Diagnosis and Treatment. Open Access Maced. J. Med Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and Endpoints of Clinical Trials in Hepatocellular Carcinoma. JNCI J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef]
- Tsurusaki, M.; Murakami, T. Surgical and Locoregional Therapy of HCC: TACE. Liver Cancer 2015, 4, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73 (Suppl. S1), e478s. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Gadaleta-Caldarola, G.; Brandi, G. First-line immune checkpoint inhibitor-based combinations in unresectable hepatocellular carcinoma: Current management and future challenges. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 1245–1251. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Perrucci, G.L.; Gowran, A.; Zanobini, M.; Capogrossi, M.C.; Pompilio, G.; Nigro, P. Peptidyl-prolyl isomerases: A full cast of critical actors in cardiovascular diseases. Cardiovasc. Res. 2015, 106, 353–364. [Google Scholar] [CrossRef]
- Fischer, G.; Tradler, T.; Zarnt, T. The mode of action of peptidyl prolyl cis/trans isomerases in vivo: Binding vs. catalysis. FEBS Lett. 1998, 426, 17–20. [Google Scholar] [CrossRef]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef][Green Version]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and Biochemical Characterization of the Human Cyclophilin Family of Peptidyl-Prolyl Isomerases. PLOS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef]
- Kim, J.; Choi, T.G.; Ding, Y.; Kim, Y.; Ha, K.S.; Lee, K.H.; Kang, I.; Ha, J.; Kaufman, R.J.; Lee, J.; et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J. Cell Sci. 2008, 121, 3636–3648. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Matsuda, S.; Koyasu, S. Mechanisms of action of cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Sanglier, J.-J.; Quesniaux, V.; Fehr, T.; Hofmann, H.; Mahnke, M.; Memmert, K.; Schuler, W.; Zenke, G.; Gschwind, L.; Maurer, C.; et al. Sanglifehrins A, B, C and D, Novel Cyclophilin-binding Compounds Isolated from Streptomyces sp. A92-308110. I. Taxonomy, Fermentation, Isolation and Biological Activity. J. Antibiot. 1999, 52, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Wang, J.; Zhang, M.; Gong, Z.; Wei, Y.; Li, L.; Zhang, Y.; Zhao, X.; Jiang, S.; et al. Cyclophilin J Is a Novel Peptidyl-Prolyl Isomerase and Target for Repressing the Growth of Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0127668. [Google Scholar] [CrossRef]
- Ren, Y.-X.; Wang, S.-J.; Fan, J.-H.; Sun, S.-J.; Li, X.; Padhiar, A.A.; Zhang, J.-N. CD147 stimulates hepatoma cells escaping from immune surveillance of T cells by interaction with Cyclophilin A. Biomed. Pharmacother. 2016, 80, 289–297. [Google Scholar] [CrossRef]
- Gong, Z.; Chi, C.; Huang, X.; Chu, H.; Wang, J.; DU, F.; Jiang, L.; Chen, J. Cyclophilin A Is Overexpressed in Hepatocellular Carcinoma and Is Associated with the Cell Cycle. Anticancer Res. 2017, 37, 4443–4447. [Google Scholar]
- Feng, W.; Xin, Y.; Xiao, Y.; Li, W.; Sun, D. Cyclophilin A Enhances Cell Proliferation and Xenografted Tumor Growth of Early Gastric Cancer. Am. J. Dig. Dis. 2015, 60, 2700–2711. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, M.; Ma, H.; Saiyin, H.; Shen, S.; Xi, J.; Wan, B.; Yu, L. Oligo-microarray analysis reveals the role of cyclophilin A in drug resistance. Cancer Chemother. Pharmacol. 2007, 61, 459–469. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Choi, Y.H.; Choi, C.G.; Nam, T.-J. Effects of the cyclophilin-type peptidylprolyl cis-trans isomerase from Pyropia yezoensis against hydrogen peroxide-induced oxidative stress in HepG2 cells. Mol. Med. Rep. 2017, 15, 4132–4138. [Google Scholar] [CrossRef]
- Lee, J. Novel combinational treatment of cisplatin with cyclophilin a inhibitors in human heptocellular carcinomas. Arch. Pharmacal Res. 2010, 33, 1401–1409. [Google Scholar] [CrossRef]
- Scanlon, K.J.; Wang, W.; Han, H. Cyclosporin A suppresses cisplatin-induced oncogene expression in human cancer cells. Cancer Treat. Rev. 1990, 17 (Suppl. SA), 27–35. [Google Scholar] [CrossRef]
- Hansson, M.J.; Moss, S.J.; Bobardt, M.; Chatterji, U.; Coates, N.; Garcia-Rivera, J.A.; Elmér, E.; Kendrew, S.; Leyssen, P.; Neyts, J.; et al. Bioengineering and Semisynthesis of an Optimized Cyclophilin Inhibitor for Treatment of Chronic Viral Infection. Chem. Biol. 2015, 22, 285–292. [Google Scholar] [CrossRef][Green Version]
- Serrano, S.S.; Tavecchio, M.; Grönberg, A.; Sime, W.; Jemaà, M.; Moss, S.; Gregory, M.; Gallay, P.; Elmér, E.; Hansson, M.; et al. Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma. Cancers 2021, 13, 3041. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Piaggi, S.; Raggi, C.; Corti, A.; Pitzalis, E.; Mascherpa, M.C.; Saviozzi, M.; Pompella, A.; Casini, A.F. Glutathione transferase omega 1-1 (GSTO1-1) plays an anti-apoptotic role in cell resistance to cisplatin toxicity. Carcinogenesis 2010, 31, 804–811. [Google Scholar] [CrossRef]
- Benhusein, G.M.; Mutch, E.; Aburawi, S.; Williams, F.M. Genotoxic effect induced by hydrogen peroxide in human hepatoma cells using comet assay. Libyan J. Med. 2010, 5, 4637. [Google Scholar] [CrossRef]
- Wu, J.H.; Jones, N.J. Assessment of DNA Interstrand Crosslinks Using the Modified Alkaline Comet Assay. Methods Mol. Biol. 2011, 817, 165–181. [Google Scholar]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Bahman, A.A.; Abaza, M.S.I.; Khoushiash, S.I.; Al-Attiyah, R.J. Sequence-dependent effect of sorafenib in combination with natural phenolic compounds on hepatic cancer cells and the possible mechanism of action. Int. J. Mol. Med. 2018, 42, 1695–1715. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jang, M.; Lim, S.; Won, H.; Yoon, K.-S.; Park, J.-H.; Kim, H.J.; Kim, B.-H.; Park, W.-S.; Ha, J.; et al. Role of cyclophilin B in tumorigenesis and cisplatin resistance in hepatocellular carcinoma in humans. Hepatology 2011, 54, 1661–1678. [Google Scholar] [CrossRef]
- Huang, C.; Sun, Z.; Sun, Y.; Chen, X.; Zhu, X.; Fan, C.; Liu, B.; Zhao, Y.; Zhang, W. Association of increased ligand cyclophilin A and receptor CD147 with hypoxia, angiogenesis, metastasis and prognosis of tongue squamous cell carcinoma. Histopathology 2012, 60, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Soltani, K.; Ming, M.; He, Y.-Y. Deregulation of XPC and CypA by Cyclosporin A: An Immunosuppression-Independent Mechanism of Skin Carcinogenesis. Cancer Prev. Res. 2012, 5, 1155–1162. [Google Scholar] [CrossRef]
- Rebillard, A.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.-T. Cisplatin cytotoxicity: DNA and plasma membrane targets. Curr. Med. Chem. 2008, 15, 2656–2663. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; D’Andrea, A.D. How the Fanconi Anemia Pathway Guards the Genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-P.; Hsu, S.-H.; Wu, C.-K.; Chang, S.-B.; Lin, Y.-J.; Yang, W.-B.; Hung, J.-J.; Chiu, W.-T.; Tzeng, S.-F.; Tseng, Y.-L.; et al. Chronic treatment with cisplatin induces replication-dependent sister chromatid recombination to confer cisplatin-resistant phenotype in nasopharyngeal carcinoma. Oncotarget 2014, 5, 6323–6337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Furuta, T.; Ueda, T.; Aune, G.; Sarasin, A.; Kraemer, K.H.; Pommier, Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002, 62, 4899–4902. [Google Scholar]
- Hanawalt, P.C. Controlling the efficiency of excision repair. Mutat. Res. Repair 2001, 485, 3–13. [Google Scholar] [CrossRef]
- Köberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–276. [Google Scholar] [CrossRef]
- Enoiu, M.; Jiricny, J.; Scharer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef]
- Dabholkar, M.; Vionnet, J.; Bostick-Bruton, F.; Yu, J.J.; Reed, E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J. Clin. Investig. 1994, 94, 703–708. [Google Scholar] [CrossRef]
- Li, Q.; Tsang, B.; Bostick-Bruton, F.; Reed, E. Modulation of excision repair cross complementation group 1 (ercc-1) mrna expression by pharmacological agents in human ovarian carcinoma cells. Biochem. Pharmacol. 1999, 57, 347–353. [Google Scholar] [CrossRef]
- Colton, S.L.; Xu, X.S.; Wang, Y.A.; Wang, G. The Involvement of Ataxia-telangiectasia Mutated Protein Activation in Nucleotide Excision Repair-facilitated Cell Survival with Cisplatin Treatment. J. Biol. Chem. 2006, 281, 27117–27125. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and other potential predictors of response to immunotherapy for hepatocellular carcinoma: How can they assist drug clinical trials? Expert Opin. Investig. Drugs 2022, 31, 415–423. [Google Scholar] [CrossRef]
- Kloeckner, R.; Galle, P.R.; Bruix, J. Local and Regional Therapies for Hepatocellular Carcinoma. Hepatology 2020, 73 (Suppl. S1), 137–149. [Google Scholar] [CrossRef] [PubMed]
- Simon Serrano, S.; Grönberg, A.; Longato, L.; Rombouts, K.; Kuo, J.; Gregory, M.; Moss, S.; Elmér, E.; Mazza, G.; Gallay, P.; et al. Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells 2019, 8, 1409. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simón Serrano, S.; Tavecchio, M.; Mallik, J.; Grönberg, A.; Elmér, E.; Kifagi, C.; Gallay, P.; Hansson, M.J.; Massoumi, R. Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma. Cancers 2022, 14, 4553. https://doi.org/10.3390/cancers14194553
Simón Serrano S, Tavecchio M, Mallik J, Grönberg A, Elmér E, Kifagi C, Gallay P, Hansson MJ, Massoumi R. Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma. Cancers. 2022; 14(19):4553. https://doi.org/10.3390/cancers14194553
Chicago/Turabian StyleSimón Serrano, Sonia, Michele Tavecchio, Josef Mallik, Alvar Grönberg, Eskil Elmér, Chamseddine Kifagi, Philippe Gallay, Magnus Joakim Hansson, and Ramin Massoumi. 2022. "Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma" Cancers 14, no. 19: 4553. https://doi.org/10.3390/cancers14194553
APA StyleSimón Serrano, S., Tavecchio, M., Mallik, J., Grönberg, A., Elmér, E., Kifagi, C., Gallay, P., Hansson, M. J., & Massoumi, R. (2022). Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma. Cancers, 14(19), 4553. https://doi.org/10.3390/cancers14194553