
The Nitric Oxide Donor [Zn(PipNONO)Cl] Exhibits Antitumor Activity through Inhibition of Epithelial and Endothelial Mesenchymal Transitions

 , , and
, , and 
Abstract
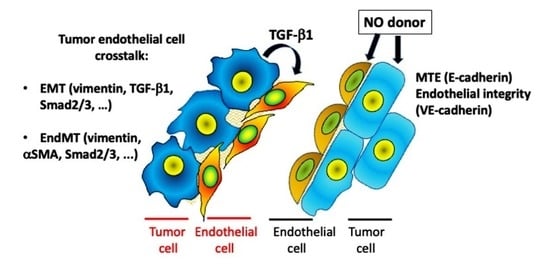
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Survival Assay
2.3. HUVEC Proliferation Assay
2.4. Co-Culturing of HUVECs with Cancer Cells and EndMT Protein Detection
2.4.1. Co-Culture Setup
2.4.2. Western Blot
2.4.3. Immunofluorescence Analysis
2.5. Tumor Cell Migration
2.6. Tumor Cell Invasion Assay
2.7. EMT Protein Expression in Tumor Cells
2.8. RNA Isolation and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-q PCR)
2.9. Data Analysis and Statistical Procedures
3. Results
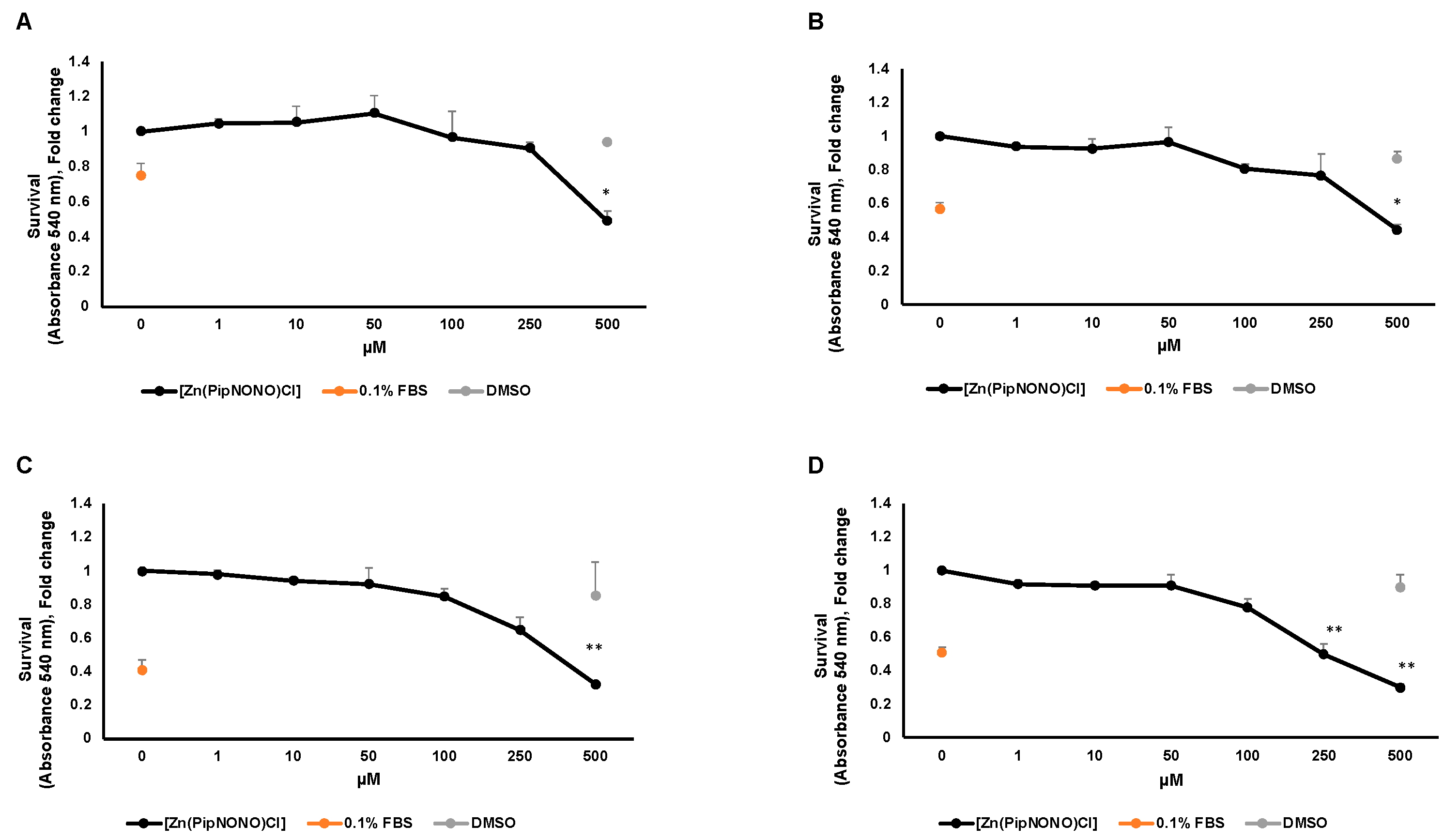
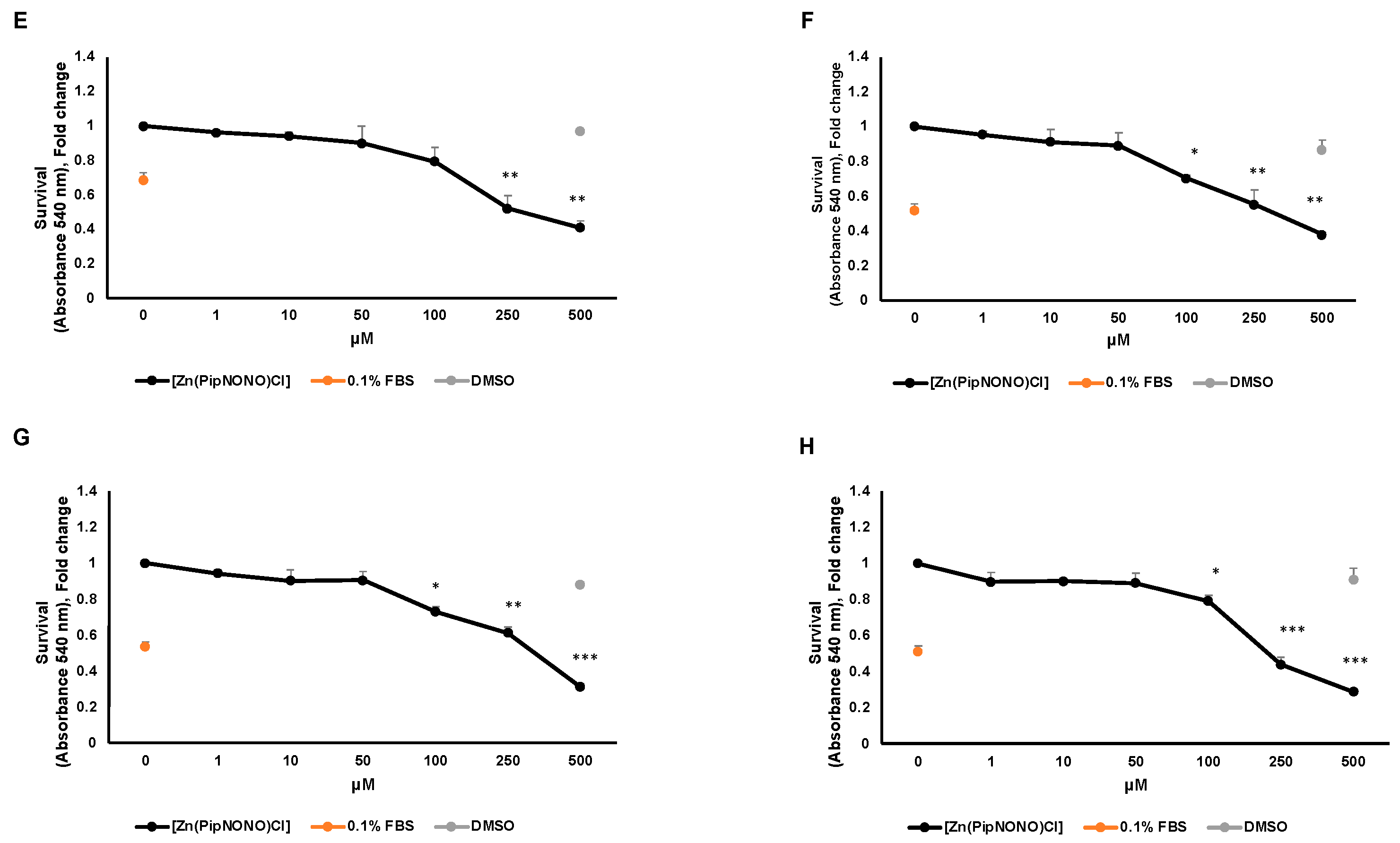
3.1. Effect of [Zn(PipNONO)Cl] on Tumor Cell Viability
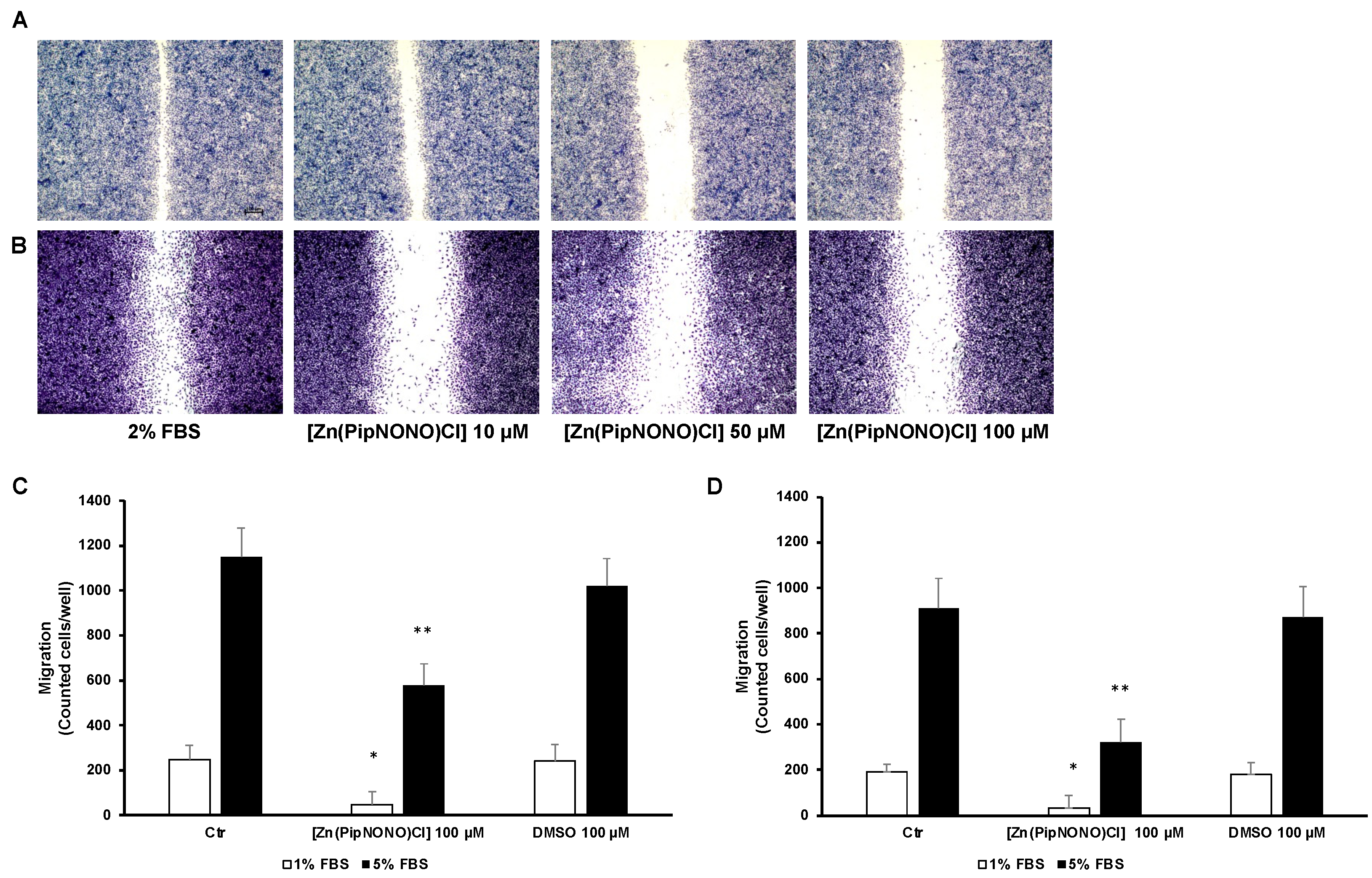
3.2. [Zn(PipNONO)Cl] Inhibited the Migration and Invasion of Tumor Cells
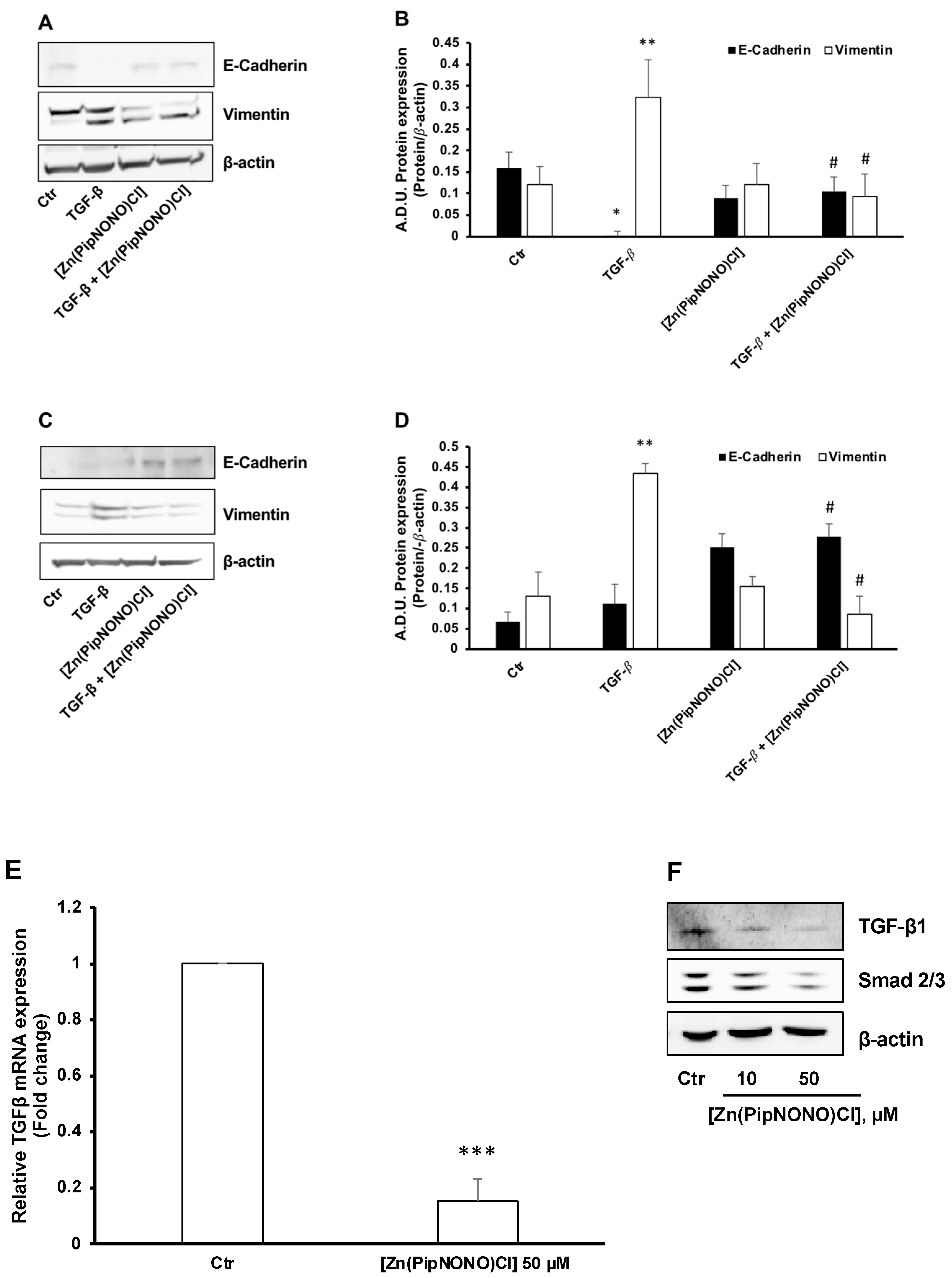
3.3. Effect of [Zn(PipNONO)Cl] on EMT Markers
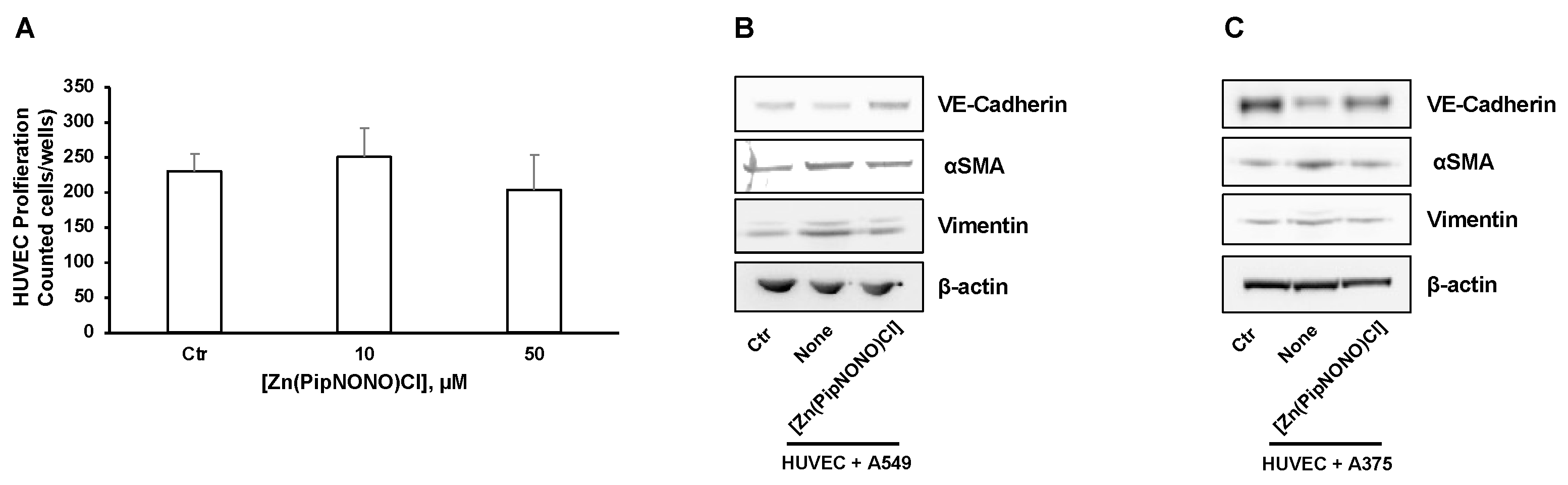
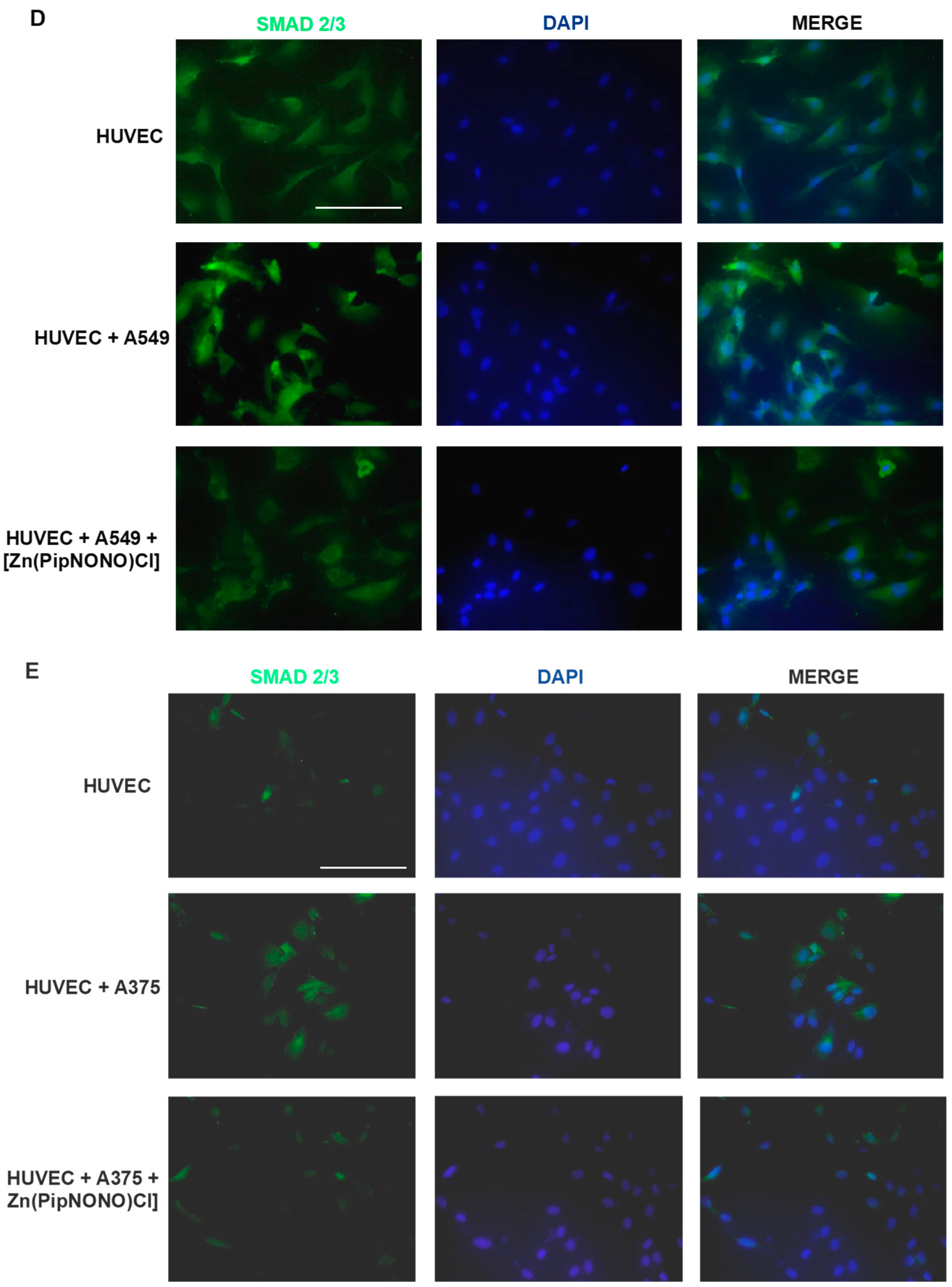
3.4. Effect of [Zn(PipNONO)Cl] on EndMT in Endothelial Cells Co-Cultured with Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Albina, J.E.; Reichner, J.S. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 1998, 17, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Diers, A.R.; Hogg, N. Cancer cell metabolism and the modulating effects of nitric oxide. Free Radic. Biol. Med. 2015, 79, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.T.; Pae, H.O.; Choi, B.M.; Billiar, T.R.; Kim, Y.M. Nitric oxide as a bioregulator of apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef]
- Huerta, S.; Chilka, S.; Bonavida, B. Nitric oxide donors: Novel cancer therapeutics (review). Int. J. Oncol. 2008, 33, 909–927. [Google Scholar] [CrossRef]
- Alimoradi, H.; Greish, K.; Gamble, A.B.; Giles, G.I. Controlled Delivery of Nitric Oxide for Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 279–303. [Google Scholar] [CrossRef]
- Xu, L.; Xie, K.; Fidler, I.J. Therapy of human ovarian cancer by transfection with the murine interferon beta gene: Role of macrophage-inducible nitric oxide synthase. Hum. Gene Ther. 1998, 9, 2699–2708. [Google Scholar] [CrossRef]
- Qiu, M.; Chen, L.; Tan, G.; Ke, L.; Zhang, S.; Chen, H.; Liu, J. A reactive oxygen species activation mechanism contributes to JS-K-induced apoptosis in human bladder cancer cells. Sci. Rep. 2015, 5, 15104. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S.; Huerta-Yepez, S.; Vega, M.I.; Chatterjee, D.; Yeung, K. Novel therapeutic applications of nitric oxide donors in cancer: Roles in chemo- and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide 2008, 19, 152–157. [Google Scholar] [CrossRef]
- Bonavida, B. Sensitizing activities of nitric oxide donors for cancer resistance to anticancer therapeutic drugs. Biochem. Pharmacol. 2020, 176, 113913. [Google Scholar] [CrossRef] [PubMed]
- Hirst, D.; Robson, T. Targeting nitric oxide for cancer therapy. J. Pharm. Pharmacol. 2007, 59, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Miraglia, E.; Viarisio, D.; Costamagna, C.; Pescarmona, G.; Ghigo, D.; Bosia, A. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005, 65, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ming, Y.; Liu, Y.; Xing, H.; Fu, R.; Li, Z.; Ni, R.; Li, L.; Duan, D.; Xu, J.; et al. Recent Developments in Pharmacological Effect, Mechanism and Application Prospect of Diazeniumdiolates. Front. Pharmacol. 2020, 11, 923. [Google Scholar] [CrossRef]
- Ziche, M.; Donnini, S.; Morbidelli, L.; Monzani, E.; Roncone, R.; Gabbini, R.; Casella, L. Nitric oxide releasing metal-diazeniumdiolate complexes strongly induce vasorelaxation and endothelial cell proliferation. ChemMedChem 2008, 3, 1039–1047. [Google Scholar] [CrossRef]
- Monti, M.; Ciccone, V.; Pacini, A.; Roggeri, R.; Monzani, E.; Casella, L.; Morbidelli, L. Anti-hypertensive property of a nickel-piperazine/NO donor in spontaneously hypertensive rats. Pharmacol. Res. 2016, 107, 352–359. [Google Scholar] [CrossRef]
- Monti, M.; Hyseni, I.; Pacini, A.; Monzani, E.; Casella, L.; Morbidelli, L. Cross-talk between endogenous H2S and NO accounts for vascular protective activity of the metal-nonoate Zn(PipNONO)Cl. Biochem. Pharmacol. 2018, 152, 143–152. [Google Scholar] [CrossRef]
- Ciccone, V.; Monti, M.; Monzani, E.; Casella, L.; Morbidelli, L. The metal-nonoate Ni(SalPipNONO) inhibits in vitro tumor growth, invasiveness and angiogenesis. Oncotarget 2018, 9, 13353–13365. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell. Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Zhang, Y.E. Transforming Growth Factor-β: An Agent of Change in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 764727. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: Downregulation of the NF-κB/Snail/YY1/RKIP circuitry. Nitric Oxide 2011, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wang, X.; Lei, W.; Min, L.; Yang, Y.; Wang, X.; Song, J. Nitric oxide suppresses transforming growth factor-β1-induced epithelial-to mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2009, 50, 1577–1587. [Google Scholar] [CrossRef]
- Powan, P.; Chanvorachote, P. Nitric oxide mediates cell aggregation and mesenchymal to epithelial transition in anoikis-resistant lung cancer cells. Mol. Cell. Biochem. 2014, 393, 237–245. [Google Scholar] [CrossRef]
- Maleszewska, M.; Moonen, J.R.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef]
- Bischoff, J. Endothelial-to-Mesenchymal Transition Purposeful Versus Maladaptive Differentiation. Circ. Res. 2019, 124, 1163–1165. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Krenning, G.; Moonen, J.R.; van Luyn, M.J.; Harmsen, M.C. Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials 2008, 29, 3703–3711. [Google Scholar] [CrossRef]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Gasparics, Á.; Rosivall, L.; Krizbai, I.A.; Sebe, A. When the endothelium scores an own goal: Endothelial cells actively augment metastatic extravasation through endothelial-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1055–H1063. [Google Scholar] [CrossRef]
- Anderberg, C.; Cunha, S.I.; Zhai, Z.; Cortez, E.; Pardali, E.; Johnson, J.R.; Franco, M.; Páez-Ribes, M.; Cordiner, R.; Fuxe, J.; et al. Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J. Exp. Med. 2013, 210, 563–579. [Google Scholar] [CrossRef]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef]
- Hutchinson, B.D.; Shroff, G.S.; Truong, M.T.; Ko, J.P. Spectrum of Lung Adenocarcinoma. Semin. Ultrasound CT MRI 2019, 40, 255–264. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef]
- Zafeiriadou, A.; Kollias, I.; Londra, T.; Tsaroucha, E.; Georgoulias, V.; Kotsakis, A.; Lianidou, E.; Markou, A. Metabolism-Related Gene Expression in Circulating Tumor Cells from Patients with Early Stage Non-Small Cell Lung Cancer. Cancers 2022, 14, 3237. [Google Scholar] [CrossRef] [PubMed]
- Murtas, D.; Maxia, C.; Diana, A.; Pilloni, L.; Corda, C.; Minerba, L.; Tomei, S.; Piras, F.; Ferreli, C.; Perra, M.T. Role of epithelial-mesenchymal transition involved molecules in the progression of cutaneous melanoma. Histochem. Cell Biol. 2017, 148, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Song, Y.; Seo, H.R. GSK-3β regulates the endothelial-to-mesenchymal transition via reciprocal crosstalk between NSCLC cells and HUVECs in multicellular tumor spheroid models. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, S.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153. [Google Scholar] [CrossRef]
- Munaweera, I.; Shi, Y.; Koneru, B.; Patel, A.; Dang, M.H.; Di Pasqua, A.J.; Balkus, K.J., Jr. Nitric oxide- and cisplatin-releasing silica nanoparticles for use against non-small cell lung cancer. J. Inorg. Biochem. 2015, 153, 23–31. [Google Scholar] [CrossRef]
- Arrieta, O.; Blake, M.; de la Mata-Moya, M.D.; Corona, F.; Turcott, J.; Orta, D.; Alexander-Alatorre, J.; Gallardo-Rincón, D. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014, 111, 311–315. [Google Scholar] [CrossRef]
- Ferraz, L.S.; Watashi, C.M.; Colturato-Kido, C.; Pelegrino, M.T.; Paredes-Gamero, E.J.; Weller, R.B.; Seabra, A.B.; Rodrigues, T. Antitumor Potential of S-Nitrosothiol-Containing Polymeric Nanoparticles against Melanoma. Mol. Pharm. 2018, 15, 1160–1168. [Google Scholar] [CrossRef]
- Bai, C.; Xue, R.; Wu, J.; Lv, T.; Luo, X.; Huang, Y.; Gong, Y.; Zhang, H.; Zhang, Y.; Huang, Z. O2-(6-Oxocyclohex-1-en-1-yl)methyl diazen-1-ium-1,2-diolates: A new class of nitric oxide donors activatable by GSH/GSTπ with both anti-proliferative and anti-metastatic activities against melanoma. Chem. Commun. 2017, 53, 5059–5062. [Google Scholar] [CrossRef]
- Ciccone, V.; Zazzetta, M.; Morbidelli, L. Comparison of the Effect of Two Hyaluronic Acid Preparations on Fibroblast and Endothelial Cell Functions Related to Angiogenesis. Cells 2019, 8, 1479. [Google Scholar] [CrossRef]
- Ciccone, V.; Terzuoli, E.; Ristori, E.; Filippelli, A.; Ziche, M.; Morbidelli, L.; Donnini, S. ALDH1A1 overexpression in melanoma cells promotes tumor angiogenesis by activating the IL-8/Notch signaling cascade. Int. J. Mol. Med. 2022, 50, 99. [Google Scholar] [CrossRef]
- Martelli, A.; Piragine, E.; Gorica, E.; Citi, V.; Testai, L.; Pagnotta, E.; Lazzeri, L.; Pecchioni, N.; Ciccone, V.; Montanaro, R.; et al. The H2S-Donor Erucin Exhibits Protective Effects against Vascular Inflammation in Human Endothelial and Smooth Muscle Cells. Antioxidants 2021, 10, 961. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Filippelli, A.; Angeli, A.; Supuran, C.T.; Morbidelli, L. Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. Int. J. Mol. Sci. 2020, 21, 2983. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Elmansuri, A.Z.; Tanino, M.A.; Mahabir, R.; Wang, L.; Kimura, T.; Nishihara, H.; Kinoshita, I.; Dosaka-Akita, H.; Tsuda, M.; Tanaka, S. Novel signaling collaboration between TGF-β and adaptor protein Crk facilitates EMT in human lung cancer. Oncotarget 2016, 7, 27094–27107. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L. Therapeutic Potential of Nitric Oxide Donors in Cancer: Focus on Angiogenesis. Crit. Rev. Oncog. 2016, 21, 447–458. [Google Scholar] [CrossRef]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef]
- Monti, M.; Solito, R.; Puccetti, L.; Pasotti, L.; Roggeri, R.; Monzani, E.; Casella, L.; Morbidelli, L. Protective effects of novel metal-nonoates on the cellular components of the vascular system. J. Pharmacol. Exp. Ther. 2014, 351, 500–509. [Google Scholar] [CrossRef]
- Smeda, M.; Kieronska, A.; Adamski, M.G.; Proniewski, B.; Sternak, M.; Mohaissen, T.; Przyborowski, K.; Derszniak, K.; Kaczor, D.; Stojak, M.; et al. Nitric oxide deficiency and endothelial-mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res. 2018, 20, 86. [Google Scholar] [CrossRef]
- Waheed, S.; Cheng, R.Y.; Casablanca, Y.; Maxwell, G.L.; Wink, D.A.; Syed, V. Nitric Oxide Donor DETA/NO Inhibits the Growth of Endometrial Cancer Cells by Upregulating the Expression of RASSF1 and CDKN1A. Molecules 2019, 24, 3722. [Google Scholar] [CrossRef] [PubMed]
- Gallo, O.; Masini, E.; Morbidelli, L.; Franchi, A.; Fini-Storchi, I.; Vergari, W.A.; Ziche, M. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J. Natl. Cancer Inst. 1998, 90, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Drehmer, D.; Luiz, J.P.M.; Hernandez, C.A.S.; Alves-Filho, J.C.; Hussell, T.; Townsend, P.A.; Moncada, S. Nitric oxide favours tumour-promoting inflammation through mitochondria-dependent and -independent actions on macrophages. Redox Biol. 2022, 54, 102350. [Google Scholar] [CrossRef]
- Puglisi, M.A.; Cenciarelli, C.; Tesori, V.; Cappellari, M.; Martini, M.; Di Francesco, A.M.; Giorda, E.; Carsetti, R.; Ricci-Vitiani, L.; Gasbarrini, A. High nitric oxide production, secondary to inducible nitric oxide synthase expression, is essential for regulation of the tumour-initiating properties of colon cancer stem cells. J. Pathol. 2015, 236, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Baritaki, S.; Vivarelli, S.; Falzone, L.; Scalisi, A.; Libra, M.; Bonavida, B. The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis. Antioxidants 2022, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- D’Este, F.; Della Pietra, E.; Badillo Pazmay, G.V.; Xodo, L.E.; Rapozzi, V. Role of nitric oxide in the response to photooxidative stress in prostate cancer cells. Biochem. Pharmacol. 2020, 182, 114205. [Google Scholar] [CrossRef]
- Pacova, H.; Astl, J.; Martinek, J. The pathogenesis of chronic inflammation and malignant transformation in the human upper airways: The role of beta-defensins, eNOS, cell proliferation and apoptosis. Histol. Histopathol. 2009, 24, 815–820. [Google Scholar] [CrossRef]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef]
- Lee, J.; Hlaing, S.P.; Hasan, N.; Kwak, D.; Kim, H.; Cao, J.; Yoon, I.S.; Yun, H.; Jung, Y.; Yoo, J.W. Tumor-Penetrable Nitric Oxide-Releasing Nanoparticles Potentiate Local Antimelanoma Therapy. ACS Appl. Mater. Interfaces 2021, 13, 30383–30396, Erratum in ACS Appl Mater. Interfaces 2022, 14, 26314. [Google Scholar] [CrossRef]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist. Updates 2020, 50, 100682. [Google Scholar] [CrossRef]
- Chegaev, K.; Riganti, C.; Lazzarato, L.; Rolando, B.; Guglielmo, S.; Campia, I.; Fruttero, R.; Bosia, A.; Gasco, A. Nitric oxide donor doxorubicins accumulate into Doxorubicin-resistant human colon cancer cells inducing cytotoxicity. ACS Med. Chem. Lett. 2011, 2, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, Y.; Luo, X.; Ma, Z.; Xu, Y.; Zhang, X.; Lv, T.; Zhang, Y.; Wang, M.; Huang, Z.; et al. Anti-CD24 Antibody-Nitric Oxide Conjugate Selectively and Potently Suppresses Hepatic Carcinoma. Cancer Res. 2019, 79, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y. Antiangiogenesis: Vessel Regression, Vessel Normalization, or Both? Cancer Res. 2022, 82, 15–17. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccone, V.; Filippelli, A.; Bacchella, C.; Monzani, E.; Morbidelli, L. The Nitric Oxide Donor [Zn(PipNONO)Cl] Exhibits Antitumor Activity through Inhibition of Epithelial and Endothelial Mesenchymal Transitions. Cancers 2022, 14, 4240. https://doi.org/10.3390/cancers14174240
Ciccone V, Filippelli A, Bacchella C, Monzani E, Morbidelli L. The Nitric Oxide Donor [Zn(PipNONO)Cl] Exhibits Antitumor Activity through Inhibition of Epithelial and Endothelial Mesenchymal Transitions. Cancers. 2022; 14(17):4240. https://doi.org/10.3390/cancers14174240
Chicago/Turabian StyleCiccone, Valerio, Arianna Filippelli, Chiara Bacchella, Enrico Monzani, and Lucia Morbidelli. 2022. "The Nitric Oxide Donor [Zn(PipNONO)Cl] Exhibits Antitumor Activity through Inhibition of Epithelial and Endothelial Mesenchymal Transitions" Cancers 14, no. 17: 4240. https://doi.org/10.3390/cancers14174240
APA StyleCiccone, V., Filippelli, A., Bacchella, C., Monzani, E., & Morbidelli, L. (2022). The Nitric Oxide Donor [Zn(PipNONO)Cl] Exhibits Antitumor Activity through Inhibition of Epithelial and Endothelial Mesenchymal Transitions. Cancers, 14(17), 4240. https://doi.org/10.3390/cancers14174240