
p53-Dependent Repression: DREAM or Reality?


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Factors Which Confer the Diversity of p53 Responses
3. Identification of the Core Transcriptional Program of p53
4. Controversy over the Mechanisms of p53-Mediated Repression
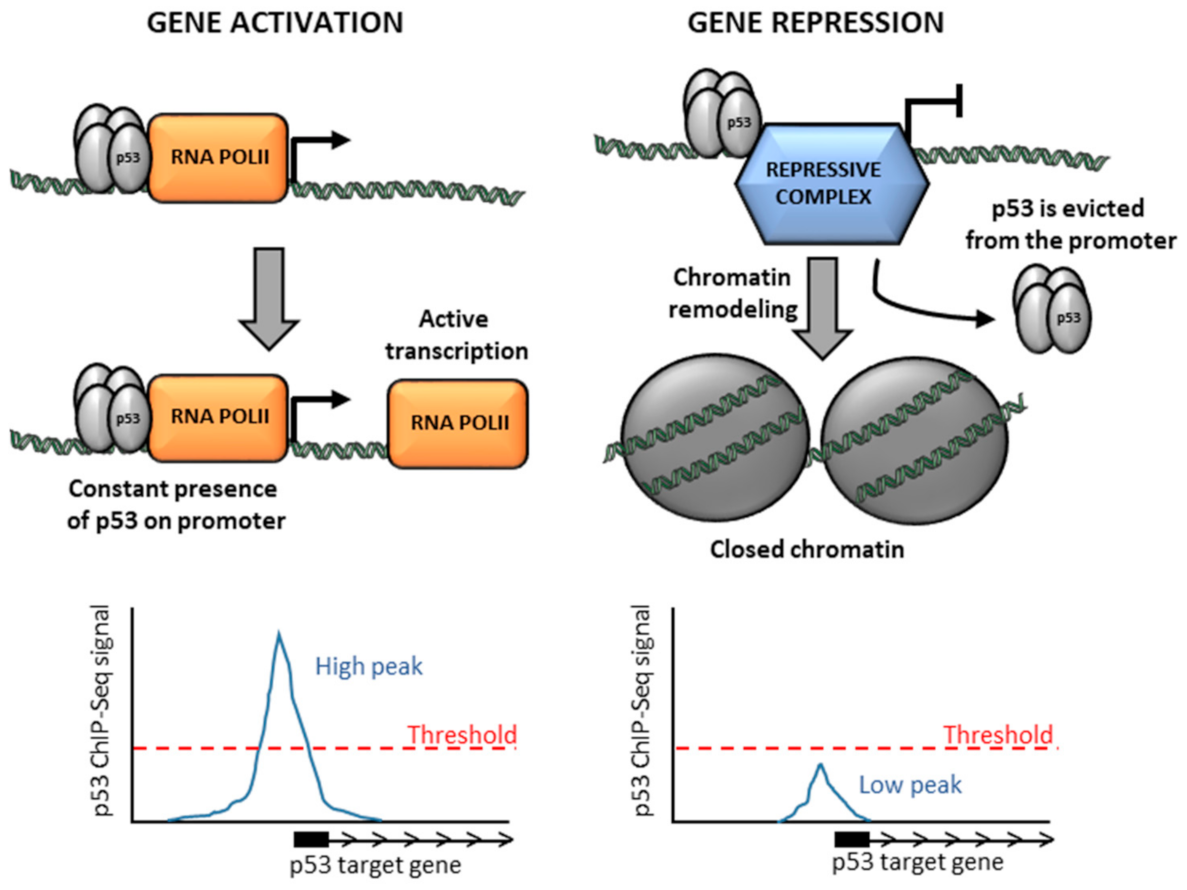
5. Low p53 Occupancy on Repressed Genes Might Be Due to a Short Time of Residence
6. Low Affinity p53 REs
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, G. Wild Type P53 Reactivation: From Lab Bench to Clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of P53 Inhibitors: Progress, Challenges and Perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. Dissecting P53-Dependent Apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Steiner, L.; Engeland, K. The Transcription Factor P53: Not a Repressor, Solely an Activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M. Census and Evaluation of P53 Target Genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrysik, Z.; Galbraith, M.D.; Guarnieri, A.L.; Zaccara, S.; Sullivan, K.D.; Pandey, A.; MacBeth, M.; Inga, A.; Espinosa, J.M. Identification of a Core TP53 Transcriptional Program with Highly Distributed Tumor Suppressive Activity. Genome Res. 2017, 27, 1645–1657. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional Control of Human P53-Regulated Genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging Roles of P53 and Other Tumour-Suppressor Genes in Immune Regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of P53 to Metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. P53-Mediated Inhibition of Angiogenesis through up-Regulation of a Collagen Prolyl Hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Jin, L.; Huang, X.; Geng, S.; He, C.; Hu, X. P53 Signaling and Autophagy in Cancer: A Revolutionary Strategy Could Be Developed for Cancer Treatment. Autophagy 2011, 7, 565–571. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Harris, C.C. P53: Traffic Cop at the Crossroads of DNA Repair and Recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The Antioxidant Function of the P53 Tumor Suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Menendez, S.; Camus, S.; Izpisua Belmonte, J.C. P53: Guardian of Reprogramming. Cell Cycle 2010, 9, 3887–3891. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. P53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Ablain, J.; Rice, K.; Soilihi, H.; De Reynies, A.; Minucci, S.; De Thé, H. Activation of a Promyelocytic Leukemia-Tumor Protein 53 Axis Underlies Acute Promyelocytic Leukemia Cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. SnapShot: P53 Posttranslational Modifications. Cell 2008, 133, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of P53-Mediated Cell Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for P53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, J.P.; Gu, W. Modes of P53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-Interacting Protein Kinase-2 Phosphorylates P53 at Ser 46 and Mediates Apoptosis. Nat. Cell Biol. 2002, 4, 11–20. [Google Scholar] [CrossRef]
- Sullivan, A.; Lu, X. ASPP: A New Family of Oncogenes and Tumour Suppressor Genes. Br. J. Cancer 2007, 96, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. HnRNP K: An HDM2 Target and Transcriptional Coactivator of P53 in Response to DNA Damage. Cell 2005, 123, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 Directs a Selective P53-Dependent Transcriptional Response towards Growth Arrest and DNA Repair Targets. Mol. Cell. Biol. 2002, 22, 4280–4292. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.R.; Kelley, M.R.; Smith, M.L. Selenomethionine Regulation of P53 by a Ref1-Dependent Redox Mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 14548–14553. [Google Scholar] [CrossRef] [Green Version]
- Lu, X. P53: A Heavily Dictated Dictator of Life and Death. Curr. Opin. Genet. Dev. 2005, 15, 27–33. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.-C. P53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieler, M.; Sanyal, S. P53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.J.; Hrstka, R.; Candeias, M.; Bourougaa, K.; Vojtesek, B.; Fåhraeus, R. Stress-Dependent Changes in the Properties of P53 Complexes by the Alternative Translation Product P53/47. Cell Cycle 2008, 7, 950–959. [Google Scholar] [CrossRef]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.-C.; Lane, D.P.; Bourdon, J.-C. P53 Directly Transactivates Δ133p53α, Regulating Cell Fate Outcome in Response to DNA Damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, I.; Park, K.-Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133p53 Represses P53-Inducible Senescence Genes and Enhances the Generation of Human Induced Pluripotent Stem Cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. P53 Dynamics Control Cell Fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in P53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Stewart-Ornstein, J.; Purvis, J.E.; Forrester, W.C.; Bulyk, M.L.; Lahav, G. P53 Pulses Lead to Distinct Patterns of Gene Expression Albeit Similar DNA Binding Dynamics. Nat. Struct Mol. Biol. 2017, 24, 840–847. [Google Scholar] [CrossRef]
- Nikulenkov, F.; Spinnler, C.; Li, H.; Tonelli, C.; Shi, Y.; Turunen, M.; Kivioja, T.; Ignatiev, I.; Kel, A.; Taipale, J.; et al. Insights into P53 Transcriptional Function via Genome-Wide Chromatin Occupancy and Gene Expression Analysis. Cell Death Differ. 2012, 19, 1992–2002. [Google Scholar] [CrossRef] [Green Version]
- Moyer, S.M.; Wasylishen, A.R.; Qi, Y.; Fowlkes, N.; Su, X.; Lozano, G. P53 Drives a Transcriptional Program That Elicits a Non-Cell-Autonomous Response and Alters Cell State in Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 23663–23673. [Google Scholar] [CrossRef]
- Menendez, D.; Nguyen, T.-A.; Freudenberg, J.M.; Mathew, V.J.; Anderson, C.W.; Jothi, R.; Resnick, M.A. Diverse Stresses Dramatically Alter Genome-Wide P53 Binding and Transactivation Landscape in Human Cancer Cells. Nucleic Acids Res. 2013, 41, 7286–7301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botcheva, K.; McCorkle, S.R. Cell Context Dependent P53 Genome-Wide Binding Patterns and Enrichment at Repeats. PLoS ONE 2014, 9, e113492. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Huarte, M. To Repress or Not to Repress: This Is the Guardian’s Question. Trends Cell Biol. 2011, 21, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-Y.; Teng, C.-L.J.; Lin, T.-C.C.; Chen, K.-C.; Hsu, S.-L.; Wu, C.-C. Transcriptional Repression of Aurora-A Gene by Wild-Type P53 through Directly Binding to Its Promoter with Histone Deacetylase 1 and MSin3a. Int. J. Cancer 2018, 142, 92–108. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Wang, Q.; Gruber, A.; Björkholm, M.; Chen, Z.; Zaid, A.; Selivanova, G.; Peterson, C.; Wiman, K.G.; Pisa, P. Downregulation of Telomerase Reverse Transcriptase MRNA Expression by Wild Type P53 in Human Tumor Cells. Oncogene 2000, 19, 5123–5133. [Google Scholar] [CrossRef] [Green Version]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. P53-Dependent Nestin Regulation Links Tumor Suppression to Cellular Plasticity in Liver Cancer. Cell 2016, 165, 1546–1547. [Google Scholar] [CrossRef] [Green Version]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M.; et al. P53 Is Essential for DNA Methylation Homeostasis in Naïve Embryonic Stem Cells, and Its Loss Promotes Clonal Heterogeneity. Genes Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef]
- Leonova, K.I.; Brodsky, L.; Lipchick, B.; Pal, M.; Novototskaya, L.; Chenchik, A.A.; Sen, G.C.; Komarova, E.A.; Gudkov, A.V. P53 Cooperates with DNA Methylation and a Suicidal Interferon Response to Maintain Epigenetic Silencing of Repeats and Noncoding RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E89–E98. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zeng, J.; Lowe, C.B.; Sellers, R.G.; Salama, S.R.; Yang, M.; Burgess, S.M.; Brachmann, R.K.; Haussler, D. Species-Specific Endogenous Retroviruses Shape the Transcriptional Network of the Human Tumor Suppressor Protein P53. Proc. Natl. Acad. Sci. USA 2007, 104, 18613–18618. [Google Scholar] [CrossRef] [Green Version]
- Estève, P.-O.; Chin, H.G.; Pradhan, S. Human Maintenance DNA (Cytosine-5)-Methyltransferase and P53 Modulate Expression of P53-Repressed Promoters. Proc. Natl. Acad. Sci. USA 2005, 102, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.A.; Kamarova, Y.; Shen, K.C.; Jiang, Z.; Hahn, M.-J.; Wang, Y.; Brooks, S.C. DNA Methyltransferase-3a Interacts with P53 and Represses P53-Mediated Gene Expression. Cancer Biol. Ther. 2005, 4, 1138–1143. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.-P.; Solano-Gonzàlez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. P53 Acts as a Safeguard of Translational Control by Regulating Fibrillarin and RRNA Methylation in Cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, K.; Samarajiwa, S.A.; Cairns, J.M.; Menon, S.; Pérez-Mancera, P.A.; Tomimatsu, K.; Bermejo-Rodriguez, C.; Ito, Y.; Chandra, T.; Narita, M.; et al. Phenotype Specific Analyses Reveal Distinct Regulatory Mechanism for Chronically Activated P53. PLoS Genet. 2015, 11, e1005053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goiran, T.; Duplan, E.; Rouland, L.; Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Costa, C.A. da Nuclear P53-Mediated Repression of Autophagy Involves PINK1 Transcriptional down-Regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. P53-Dependent Transcriptional Repression of c-Myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Jin, R.; Gao, M.; Xu, H.; Zou, S.; Li, X.; Xing, C.; Wang, Q.; Wang, H.; Feng, J.; et al. Transcriptional Repression of IKKβ by P53 in Arsenite-Induced GADD45α Accumulation and Apoptosis. Oncogene 2019, 38, 731–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, B.; Jones, A.E.; Caillet, C.J.; Das, S.; Royer, S.K.; Abrams, J.M. P53 Directly Represses Human LINE1 Transposons. Genes Dev. 2020, 34, 1439–1451. [Google Scholar] [CrossRef]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.-J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. P53 Genes Function to Restrain Mobile Elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeland, K. Cell Cycle Arrest through Indirect Transcriptional Repression by P53: I Have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Ptashne, M. Principles of a Switch. Nat. Chem. Biol. 2011, 7, 484–487. [Google Scholar] [CrossRef]
- Crocker, J.; Noon, E.P.-B.; Stern, D.L. The Soft Touch: Low-Affinity Transcription Factor Binding Sites in Development and Evolution. Curr. Top. Dev. Biol. 2016, 117, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, K.; Heyl, C.; Krampitz, A.-M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the P53 Cistrome–DNA Binding Cooperativity Dissects P53′s Tumor Suppressor Functions. PLoS Genet. 2013, 9, e1003726. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xiao, Z.; Ren, E.C. Redefining the P53 Response Element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different Promoter Affinities Account for Specificity in MYC-Dependent Gene Regulation. eLife 2016, 5, e18871. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Lee Kraus, W.; et al. Global Analysis of P53-Regulated Transcription Identifies Its Direct Targets and Unexpected Regulatory Mechanisms. eLife 2014, 2014, e02200. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-A.T.; Grimm, S.A.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; Lavender, C.A.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a Human P53 Universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peuget, S.; Selivanova, G. p53-Dependent Repression: DREAM or Reality? Cancers 2021, 13, 4850. https://doi.org/10.3390/cancers13194850
Peuget S, Selivanova G. p53-Dependent Repression: DREAM or Reality? Cancers. 2021; 13(19):4850. https://doi.org/10.3390/cancers13194850
Chicago/Turabian StylePeuget, Sylvain, and Galina Selivanova. 2021. "p53-Dependent Repression: DREAM or Reality?" Cancers 13, no. 19: 4850. https://doi.org/10.3390/cancers13194850
APA StylePeuget, S., & Selivanova, G. (2021). p53-Dependent Repression: DREAM or Reality? Cancers, 13(19), 4850. https://doi.org/10.3390/cancers13194850