Combined Methylome and Transcriptome Analyses Reveals Potential Therapeutic Targets for EGFR Wild Type Lung Cancers with Low PD-L1 Expression


Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Sample Datasets and Data Preprocessing
2.2. Immune Profile Analysis
2.3. Analysis of DNA Methylation Data
2.4. Gene Expression Data Analysis
2.5. Analysis of DMGs and DEGs in Different Regions
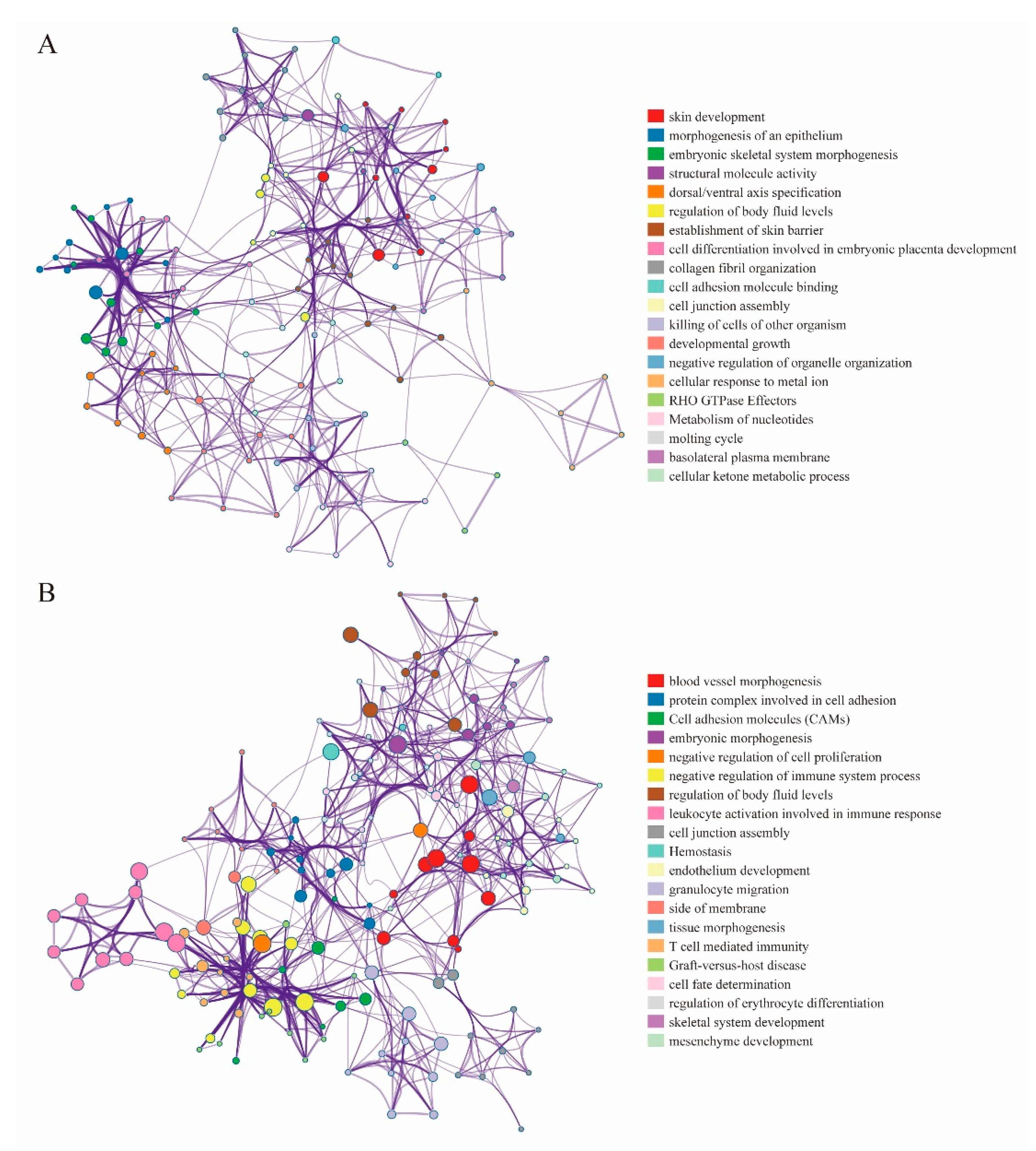
2.6. Functional Enrichment Analysis
2.7. Evaluation of Expression and Methylation Biomarkers
2.8. Construction of DMEGs-Based Prognostic Signature
2.9. Identification of Potential Drug Targets
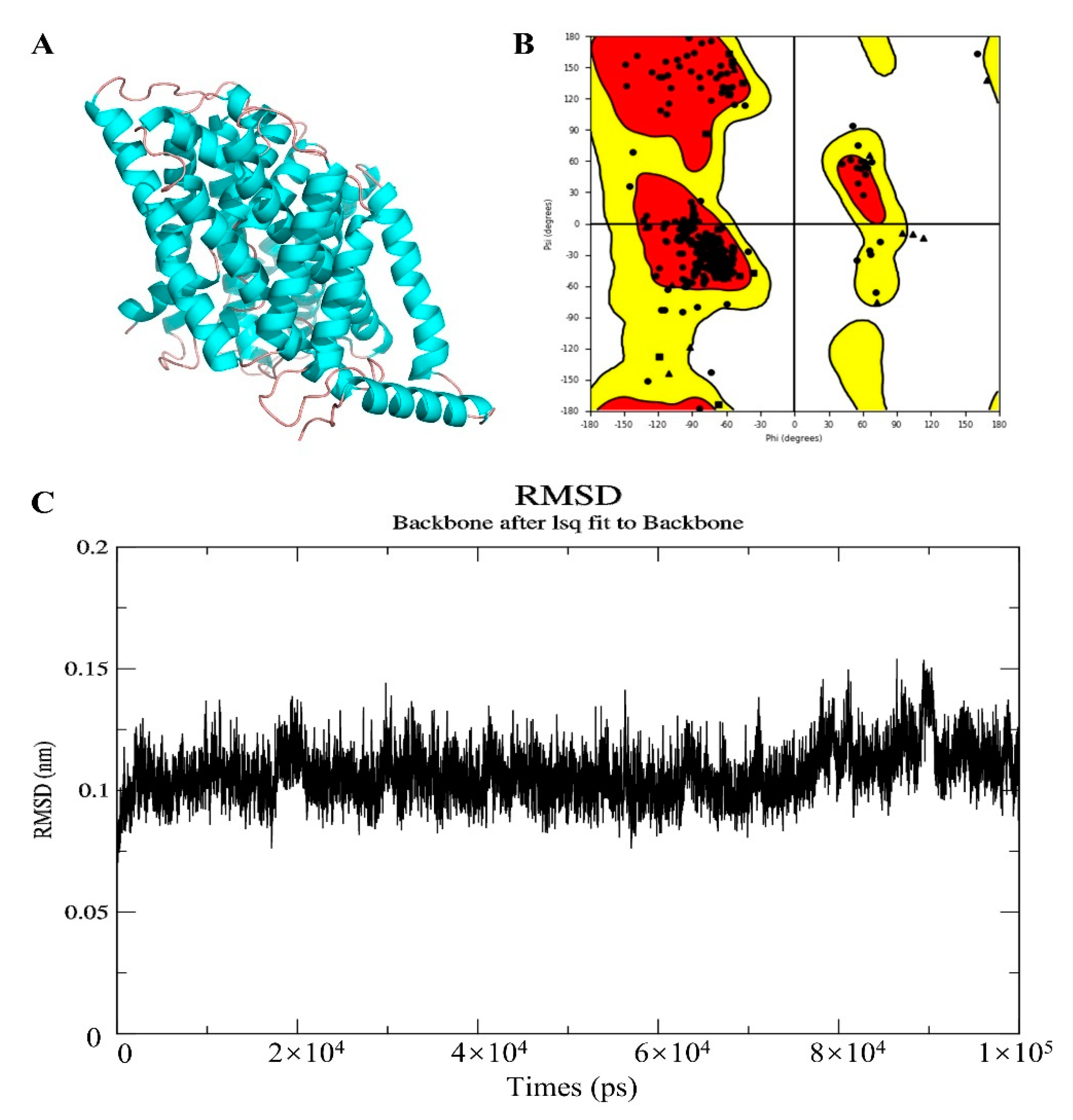
2.10. Homologous Modeling
2.11. Molecular Docking
3. Results
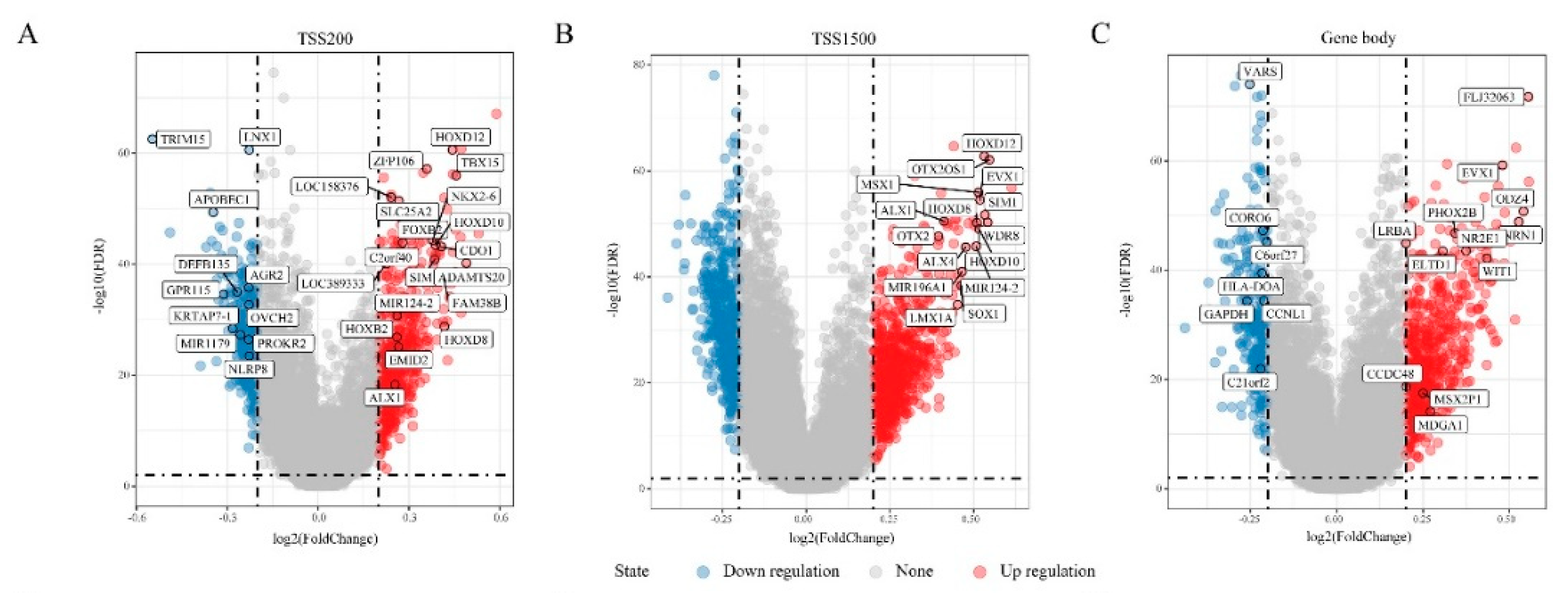
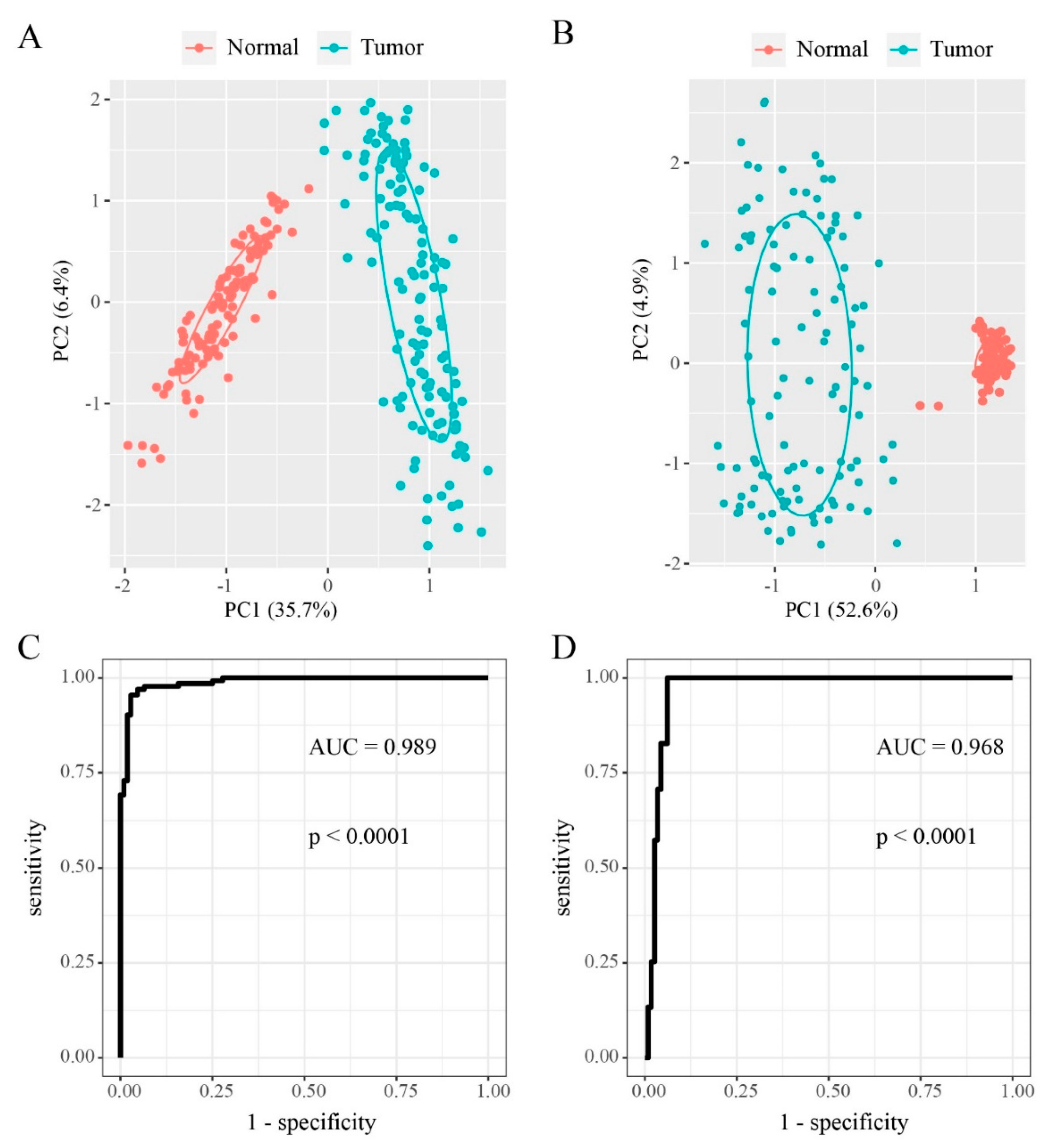
3.1. DMGs in “EGFR Wild Type/Low PD-L1 Expression” NSCLC
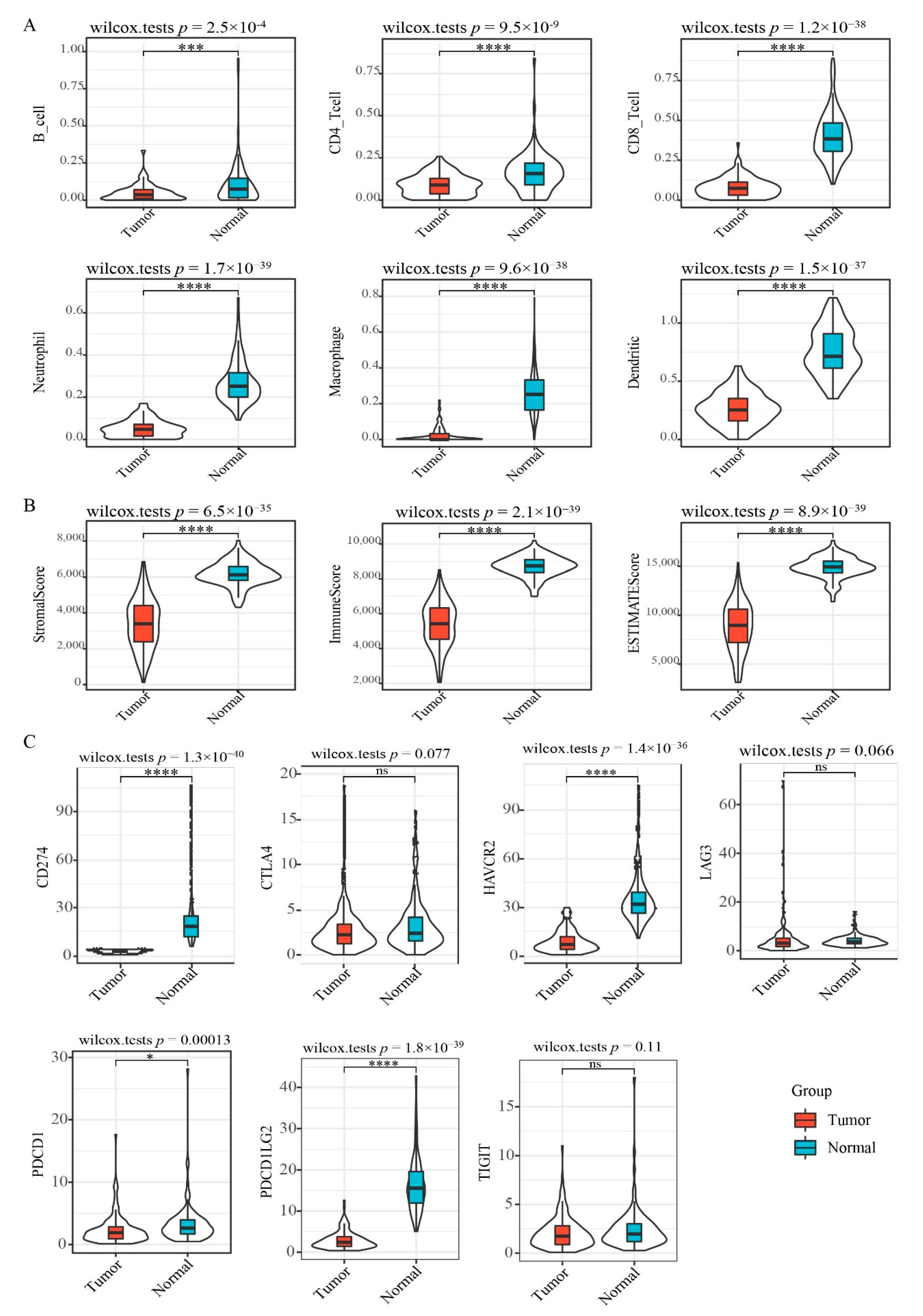
3.2. Immune Profile Analysis
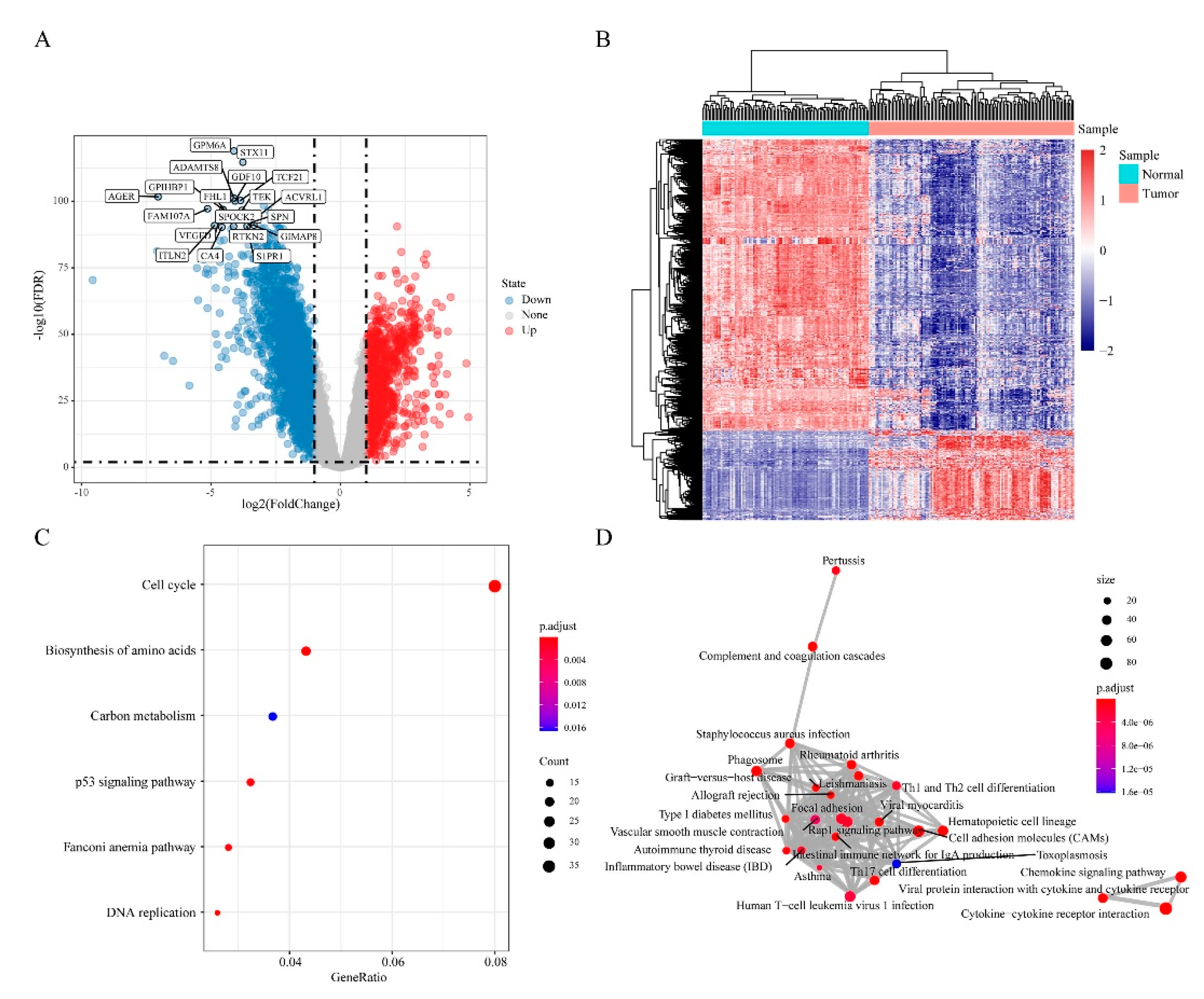
3.3. Differentially Expressed Genes (DEGs) in “EGFR Wild Type/Low PD-L1 Expression” NSCLC
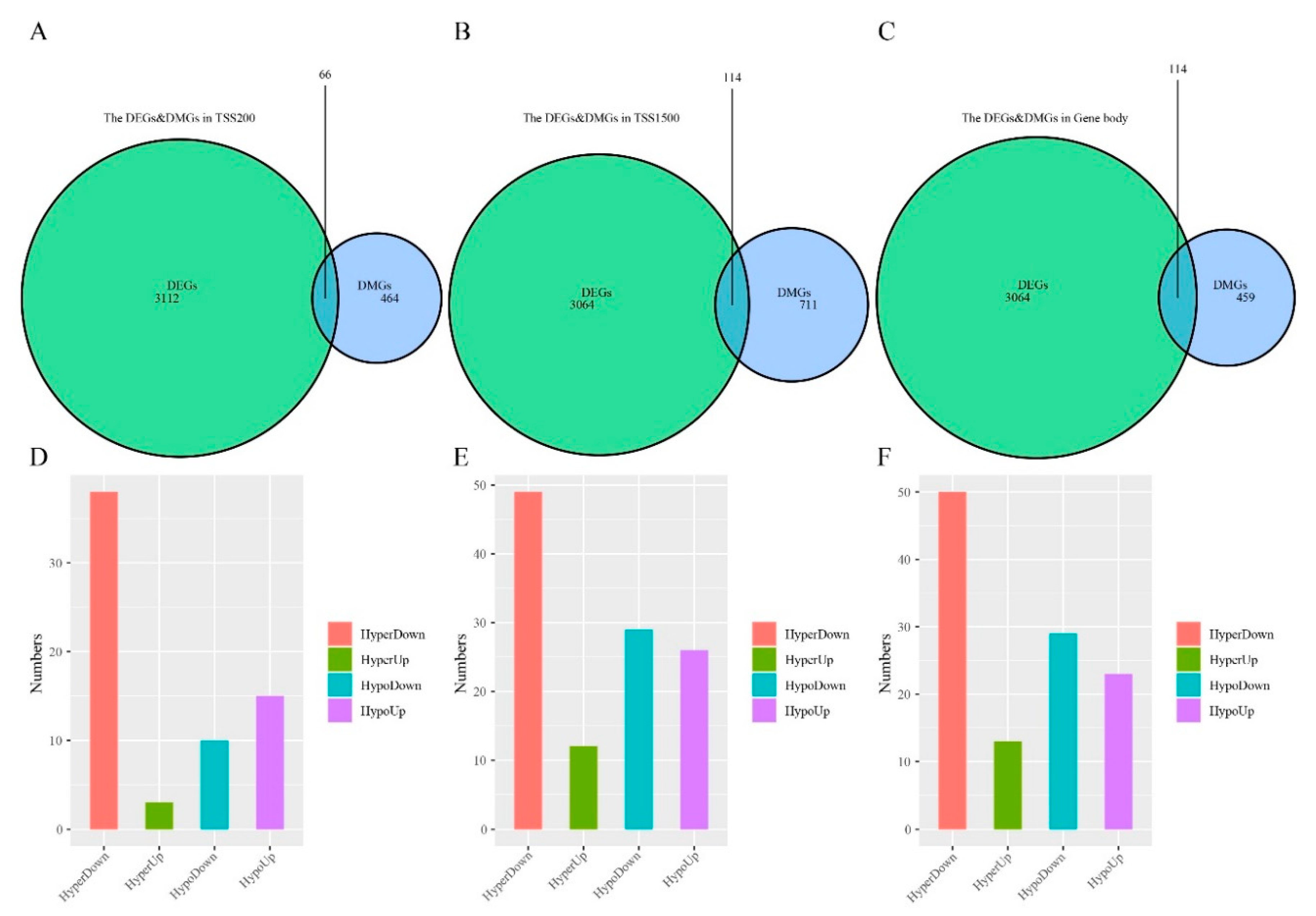
3.4. Differentially Methylated and Expressed Genes (DMEGs) in “EGFR Wild Type/Low PD-L1 Expression” NSCLC
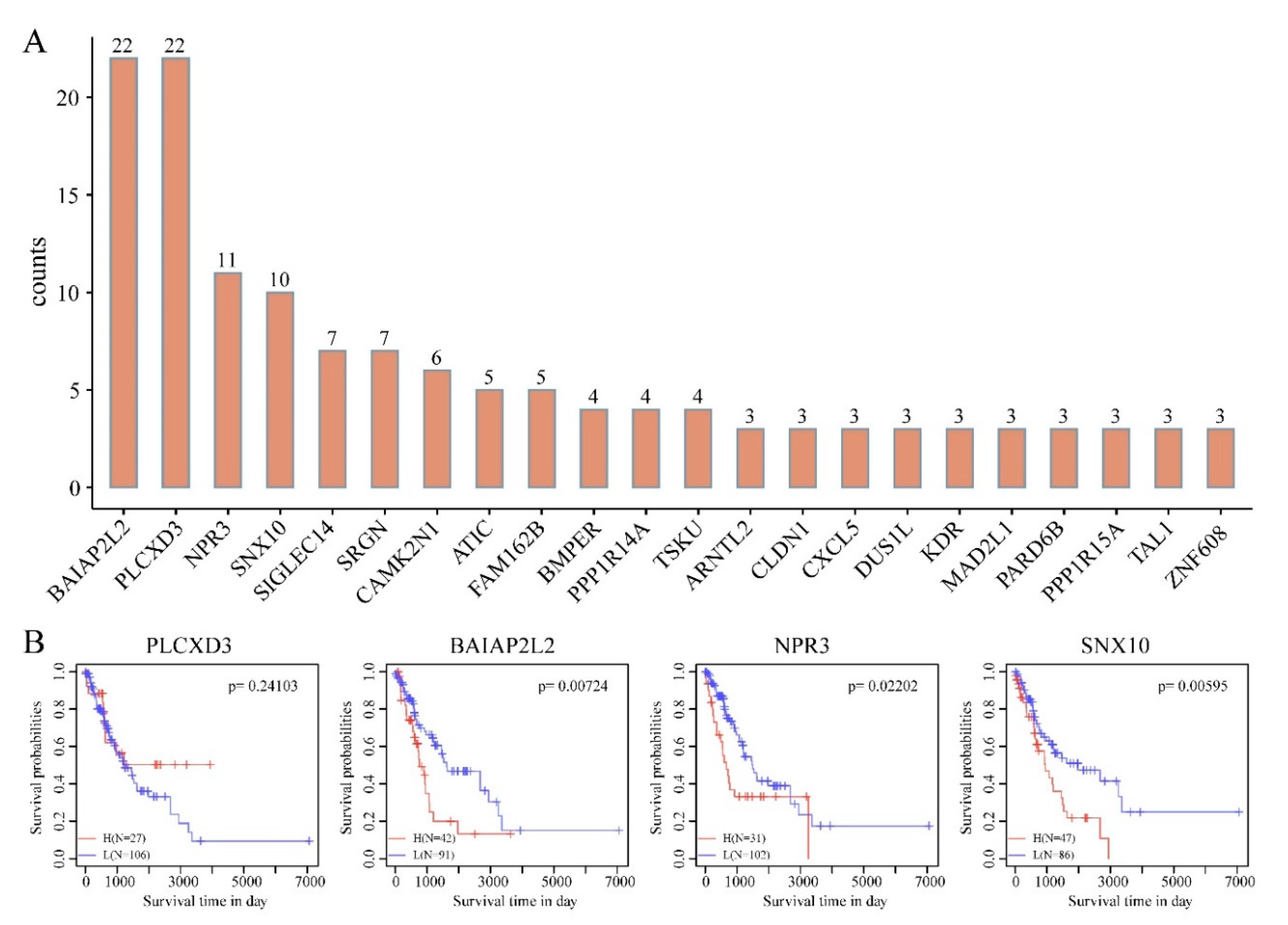
3.5. DMEGs Analysis
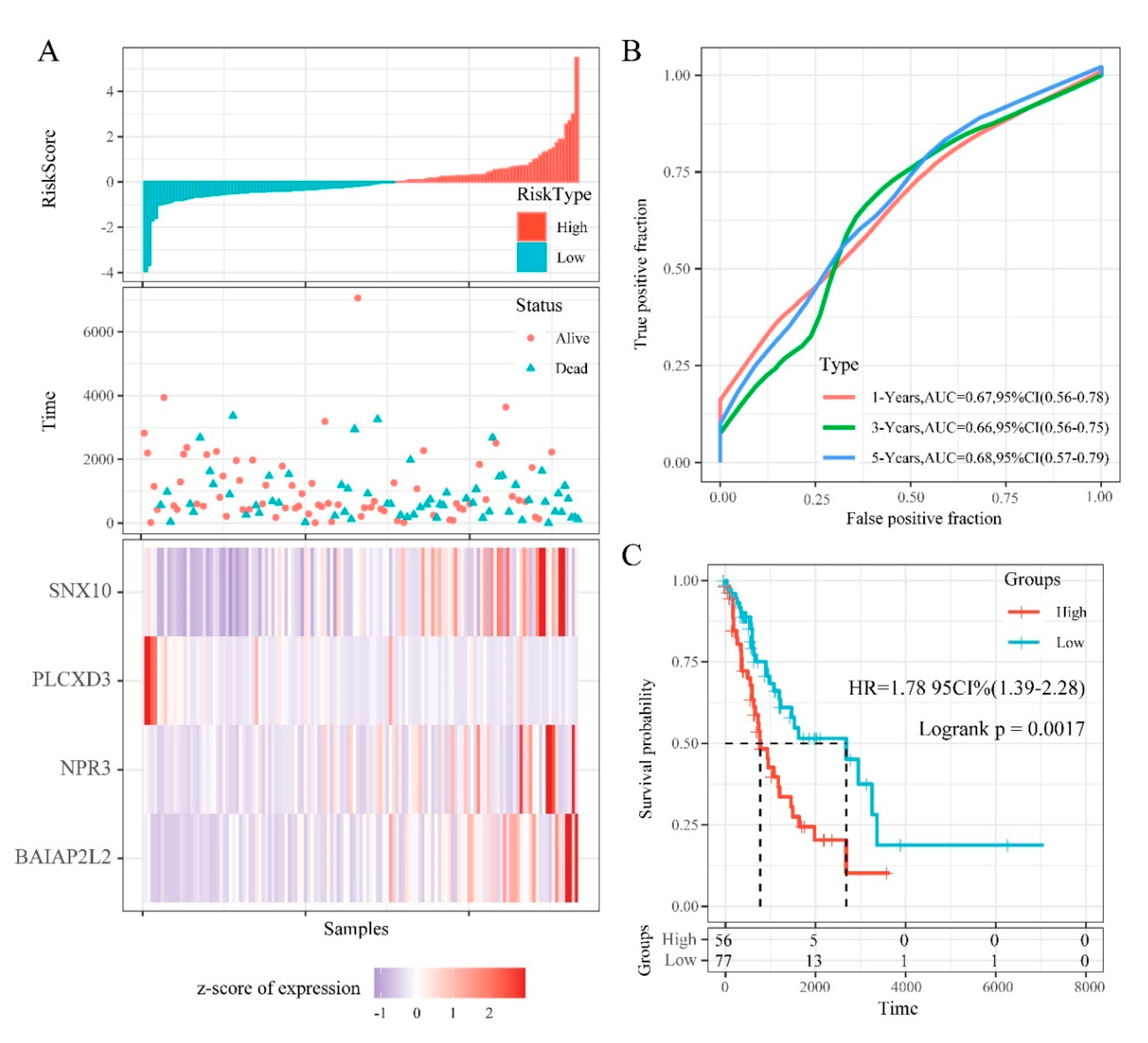
3.6. Construction and Evaluation of DMEGs-Based Prognostic Signature
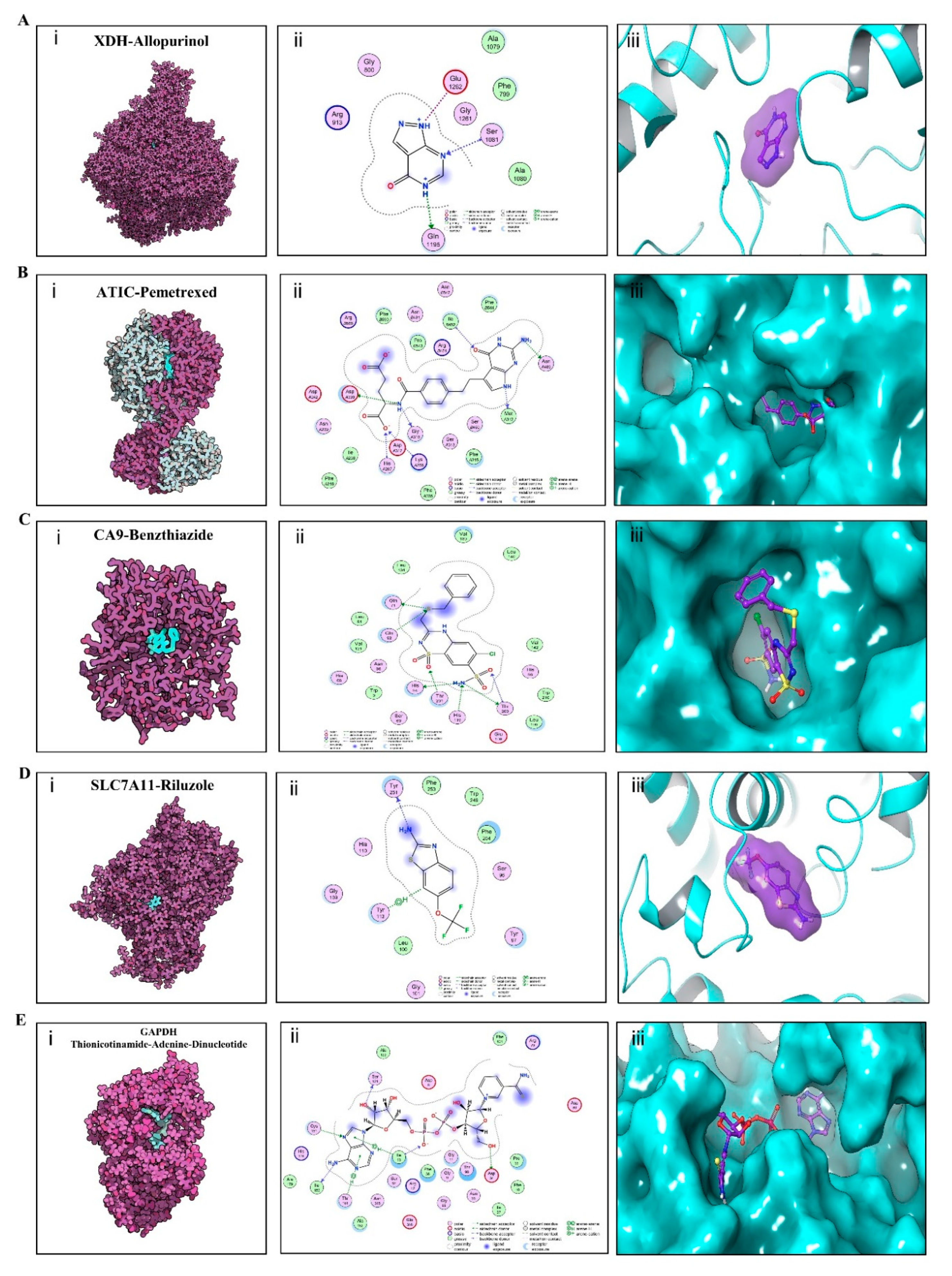
3.7. Multiple DMEGs Are Potential Druggable Targets
3.8. Validation of Affinity of the Candidate Drugs by Molecular Docking Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Supporting Data
Abbreviations
| ICIs: | Immune checkpoint inhibitors; |
| NSCLC: | Non-small cell lung cancer; |
| EGFR: | Epidermal growth factor receptor; |
| PD-L1: | programmed death-ligand 1; |
| DMRs: | differentially methylated regions; |
| PLCXD3: | Phosphatidylinositol-Specific Phospholipase C, X Domain Containing 3; |
| BAIAP2L2: | BAR/IMD Domain Containing Adaptor Protein 2 Like 2; |
| NPR3: | Natriuretic Peptide Receptor 3; |
| SNX10: | Sorting Nexin 10; |
| XDH: | Xanthine Dehydrogenase; |
| ATIC: | 5-Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase/IMP Cyclohydrolase; |
| CA9: | Carbonic Anhydrase 9; |
| SLC7A11: | Solute Carrier Family 7 Member 11; |
| GAPDH: | Glyceraldehyde-3-Phosphate Dehydrogenase; |
| TKI: | Tyrosine kinase inhibitors; |
| PD1: | Programmed death 1; |
| ALK: | ALK receptor tyrosine kinase; |
| OS: | Overall survival; |
| IDO: | Indoleamine 2,3-dioxygenase; |
| VEGF: | Vascular endothelial growth factor; |
| CTLA-4: | Cytotoxic T-lymphocyte-associated protein 4; |
| LAG-3: | Lymphocyte-activation gene 3; |
| Tim-3: | T-cell immunoglobulin mucin 3; |
| TCGA: | The Cancer Genome Atlas; |
| DMEGs: | differentially methylated and expressed genes; |
| GEO: | Gene Expression Omnibus; |
| DMSs: | Differentially methylated CpG sites; |
| DMRs: | Differentially methylated regions; |
| DMGs: | Differentially methylated genes; |
| FDR: | False discovery rate; |
| BH: | Benjamini and Hochberg; |
| DEGs: | Differentially expressed genes; |
| GO: | Gene Ontology; |
| KEGG: | Kyoto Encyclopedia of Genes and Genomes; |
| BP: | Biological process; |
| CC: | Cellular component; |
| MF: | Molecular functions; |
| PCA: | Principal Component Analysis; |
| LOOCV: | Leave-one-out cross-validation; |
| ROC: | Receiver operating characteristic; |
| AUC: | Area under the curve; |
| LASSO: | Least Absolute Shrinkage and Selection Operator; |
| TIMER: | Tumor immune estimation resource; |
| ESTIMATE: | Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data; |
| RMSD: | Root-mean-square deviation; |
| TSS | Transcription start sites |
| CD274: | CD274 molecule, also known as PD-L1; |
| HAVCR2: | Hepatitis A Virus Cellular Receptor 2, also known as TIM3; |
| PDCD1: | Programmed Cell Death 1, also known as PD1; |
| PDCD1LG2: | Programmed Cell Death 1 Ligand 2; |
| CDO1: | Cysteine Dioxygenase Type 1; |
| IRF8: | Interferon Regulatory Factor 8; |
| STAT5A: | Signal Transducer and Activator of Transcription 5A; |
| CFTR: | CF Transmembrane Conductance Regulator; |
| ADAMTS8: | ADAM Metallopeptidase with Thrombospondin Type 1 Motif 8; |
| WIF1: | WNT Inhibitory Factor 1; |
| GATA5: | GATA Binding Protein 5; |
| FOXA2: | FOXA2; |
| SHISA3: | Shisa Family Member 3; |
| AXIN2: | Axin 2; |
| DIRAS3: | DIRAS family GTPase 3; |
| IRX1: | Iroquois Homeobox 1; |
| ITGA5: | Integrin Subunit Alpha 5; |
| CAMK2N1: | Calcium/Calmodulin Dependent Protein Kinase II Inhibitor 1; |
| CRC: | Colorectal cancer; |
| SNX10: | Sorting nexin 10; |
| ALS: | Amyotrophic lateral sclerosis; |
| IBD: | Inflammatory bowel disease |
References
- Seliger, B.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.S.-K.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR- Mutated Non–Small Cell Lung Cancer—A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.S.-K.; Yang, J.C.-H. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ma, L.; Adame, C.; Lim, C.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.-K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Langer, C.; Gadgeel, S.M.; Borghaei, H.; A Papadimitrakopoulou, V.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Dómine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.G.; Luft, A.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Senler, F.C.; Fülöp, A.; Rodriguez-Cid, J.; Sugawara, S.; et al. Phase 3 study of carboplatin-paclitaxel/nab-paclitaxel (Chemo) with or without pembrolizumab (Pembro) for patients (Pts) with metastatic squamous (Sq) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 105. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Caro, R.B.; Zurawski, B.; Kim, S.-W.; Costa, E.C.; Park, K.; Alexandru, A.; Lupinacci, L.; Jimenez, E.D.L.M.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Ciuleanu, T.-E.; Dols, M.C.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. J. Clin. Oncol. 2020, 38, 9501. [Google Scholar] [CrossRef]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Zhou, C.; Hou, L.; Wu, C.; Ren, S.; et al. LAG-3 Protein Expression in Non-Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumor-Infiltrating Lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Liu, J.-F.; Ma, S.-R.; Mao, L.; Bu, L.-L.; Yu, G.-T.; Li, Y.-C.; Huang, C.-F.; Deng, W.-W.; Kulkarni, A.B.; Zhang, W.-F.; et al. T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol. Oncol. 2017, 11, 235–247. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Bird, A.P. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Moazed, D. Mechanisms for the Inheritance of Chromatin States. Cell 2011, 146, 510–518. [Google Scholar] [CrossRef]
- Lee, P.P.; Fitzpatrick, D.R.; Beard, C.; Jessup, H.K.; Lehar, S.; Makar, K.W.; Pérez-Melgosa, M.; Sweetser, M.T.; Schlissel, M.S.; Nguyen, S.; et al. A Critical Role for Dnmt1 and DNA Methylation in T Cell Development, Function, and Survival. Immunity 2001, 15, 763–774. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gamper, C.J.; Ladle, B.H.; Powell, J.D.; Wells, A.D. De Novo DNA Methylation Is Required to Restrict T Helper Lineage Plasticity. J. Boil. Chem. 2012, 287, 22900–22909. [Google Scholar] [CrossRef]
- Delacher, M.; Imbusch, C.D.; Weichenhan, D.; Breiling, A.; Hotz-Wagenblatt, A.; Träger, U.; Christoph, P.; Wang, Q.; Benedikt, B.; Carl, H.; et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017, 18, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Chappell, C.; Beard, C.; Altman, J.; Jaenisch, R.; Jacob, J. DNA Methylation by DNA Methyltransferase 1 Is Critical for Effector CD8 T Cell Expansion. J. Immunol. 2006, 176, 4562–4572. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; De Moura, M.C.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: A multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Esteller, M.; Lee, S.-H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-García, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, U.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Guo, M.; Sinha, S.; Wang, S.M. Coupled Genome-Wide DNA Methylation and Transcription Analysis Identified Rich Biomarkers and Drug Targets in Triple-Negative Breast Cancer. Cancers 2019, 11, 1724. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.W.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinform. 2008, 24. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 2, 19–25. [Google Scholar] [CrossRef]
- Ramachandran, G.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Boil. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P. 2D Depiction of Protein−Ligand Complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Minamiya, Y.; Saito, H.; Imai, K.; Kawaharada, Y.; Motoyama, S.; Ogawa, J.-I. Acquired xanthine dehydrogenase expression shortens survival in patients with resected adenocarcinoma of lung. Tumor Boil. 2012, 33, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Tavassoly, I.; Hu, Y.; Zhao, S.; Mariottini, C.; Boran, A.; Chen, Y.; Li, L.; Tolentino, R.E.; Jayaraman, G.; Goldfarb, J.; et al. Genomic signatures defining responsiveness to allopurinol and combination therapy for lung cancer identified by systems therapeutics analyses. Mol. Oncol. 2019, 13, 1725–1743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-C.; Zhang, S.; Yang, X.-N.; Yang, J.-J.; Zhou, Q.; Yin, X.; An, S.-J.; Lin, J.; Chen, S.; Xie, Z.; et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol. Cancer 2010, 9, 188. [Google Scholar] [CrossRef]
- Yu, Y.-Y.; Chiou, H.-L.; Tsao, S.-M.; Huang, C.-C.; Lin, C.-Y.; Lee, C.-Y.; Tsao, T.C.-Y.; Yang, S.-F.; Huang, Y.-W. Association of Carbonic Anhydrase 9 Polymorphism and the Epithelial Growth Factor Receptor Mutations in Lung Adenocarcinoma Patients. Diagnostics 2020, 10, 266. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245.e1–261.e1. [Google Scholar] [CrossRef]
- De Lima, G.L.; Dicksey, J.S.; Peters, W.P.; Palalay, M.; Chang, A.Y. Final results of a prematurely discontinued Phase 1/2 study of eniluracil with escalating doses of 5-fluorouracil administered orally in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2011, 68, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.T.; Muthu, V.; Biswas, B.; Malik, P.S.; Dabkara, D.; Ganguly, S.; Ghosh, J.; Kataria, B.; Khurana, S.; Verma, S.; et al. Utility and safety of maintenance chemotherapy in advanced non–small cell lung cancer across various performance status categories: Real-world experience. Curr. Probl. Cancer 2020, 44, 100565. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Bae, T.-J.; Lee, J.; Kim, D.G.; Oh, Y.S.; Jang, Y.; Kim, J.-T.; Lee, J.-J.; Innocenti, A.; Supuran, C.T.; et al. Rational drug repositioning guided by an integrated pharmacological network of protein, disease and drug. BMC Syst. Biol. 2012, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Turanli, B.; Zhang, C.; Kim, W.; Benfeitas, R.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Discovery of therapeutic agents for prostate cancer using genome-scale metabolic modeling and drug repositioning. EBioMedicine 2019, 42, 386–396. [Google Scholar] [CrossRef]
- Jacobsohn, K.M.; Wood, C.G. Adjuvant Therapy for Renal Cell Carcinoma. Semin. Oncol. 2006, 33, 576–582. [Google Scholar] [CrossRef]
- Zhao, J.; Li, G.; Wei, J.; Dang, S.; Yu, X.; Ding, L.; Shang, C.; Zhang, H.; Chen, H.; Liu, M. Ellagic acid induces cell cycle arrest and apoptosis via the TGF-β1/Smad3 signaling pathway in human colon cancer HCT-116 cells. Oncol. Rep. 2020, 44, 768–776. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Lemieszek, M.; Stepulak, A.; Sawa-Wejksza, K.; Czerwonka, A.; Ikonomidou, C.; Rzeski, W. Riluzole Inhibits Proliferation, Migration and Cell Cycle Progression and Induces Apoptosis in Tumor Cells of Various Origins. Anti-Cancer Agents Med. Chem. 2018, 18, 565–572. [Google Scholar] [CrossRef]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The xc−cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: Use of sulfasalazine. Cancer Chemother. Pharmacol. 2008, 64, 463. [Google Scholar] [CrossRef]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.-L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Tedeschi, P.; Lawal, R.A.; Banerjee, D.; Scotto, K.; Kerrigan, J.E.; Lee, K.-C.; Johnson-Farley, N.; Bertino, J.R.; Abali, E.E. Enhanced degradation of dihydrofolate reductase through inhibition of NAD kinase by nicotinamide analogs. Mol. Pharmacol. 2012, 83, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z. Advances in homology protein structure modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L.; DeMaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef]
- Wu, H.-X.; Chen, Y.-X.; Wang, Z.-X.; Zhao, Q.; He, M.-M.; Wang, Y.-N.; Wang, F.; Xu, R.-H. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J. Immunother. Cancer 2019, 7, 264–313. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Jones, P.A. DNA methylation and cancer. Oncogene 2002, 21, 5358–5360. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, H.; Shi, R.; Liu, X.; Liu, J.; Ren, F.; Zhao, Q.; Zhang, H.; Li, Y.; Liu, H.; et al. The Role of Plasma CDO1 Methylation in the Early Diagnosis of Lung Cancer. Zhongguo Fei Ai Za Zhi 2020, 23, 314–320. [Google Scholar]
- Liu, B.; Ricarte-Filho, J.; Mallisetty, A.; Villani, C.; Kottorou, A.E.; Rodgers, K.P.; Chen, C.; Ito, T.; Holmes, K.; Gastala, N.; et al. Detection of promoter DNA methylation in urine and plasma aids the detection of non-small cell lung cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Ye, L.; Xiang, T.-X.; Zhu, J.; Li, D.; Shao, Q.; Peng, W.; Tang, J.; Li, L.; Ren, G. Interferon Consensus Sequence-Binding Protein 8, a Tumor Suppressor, Suppresses Tumor Growth and Invasion of Non-Small Cell Lung Cancer by Interacting with the Wnt/β-Catenin Pathway. Cell. Physiol. Biochem. 2018, 51, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Son, J.W.; Kim, Y.J.; Cho, H.M.; Lee, S.Y.; Lee, S.M.; Kang, J.; Lee, J.U.; Lee, Y.M.; Kwon, S.J.; Choi, E.; et al. Promoter hypermethylation of the CFTR gene and clinical/pathological features associated with non-small cell lung cancer. Respirology 2011, 16, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.C.; Li, L.; Wang, Y.; Zhong, L.; Ma, B.; Su, X.; Ying, J.; Xiang, T.-X.; Rha, S.Y.; Yu, J.; et al. The Metalloprotease ADAMTS8 Displays Antitumor Properties through Antagonizing EGFR–MEK–ERK Signaling and Is Silenced in Carcinomas by CpG Methylation. Mol. Cancer Res. 2013, 12, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ye, S.; Jiang, Q.; Gong, Y.; Yuan, Y.; Hu, X.; Su, X.; Zhu, W. Wnt inhibitory factor-1-mediated autophagy inhibits Wnt/β-catenin signaling by downregulating dishevelled-2 expression in non-small cell lung cancer cells. Int. J. Oncol. 2018, 53, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, X.; Yin, W.; Peng, M.; Wu, F.; Wu, X.; Tang, J.; Chen, M.; Wang, X.; Hulbert, A.; et al. Ultrasensitive DNA hypermethylation detection using plasma for early detection of NSCLC: A study in Chinese patients with very small nodules. Clin. Epigenetics 2020, 12, 39. [Google Scholar] [CrossRef]
- Bassères, D.; D’Alò, F.; Yeap, B.Y.; Löwenberg, E.C.; González, D.A.; Yasuda, H.; Dayaram, T.; Kocher, O.N.; Godleski, J.J.; Richards, W.G.; et al. Frequent downregulation of the transcription factor Foxa2 in lung cancer through epigenetic silencing. Lung Cancer 2012, 77, 31–37. [Google Scholar] [CrossRef]
- Chen, C.-C.; Chen, H.-Y.; Su, K.-Y.; Hong, Q.-S.; Yan, B.-S.; Chen, C.-H.; Pan, S.-H.; Chang, Y.; Wang, C.-J.; Hung, P.-F.; et al. Shisa3 Is Associated with Prolonged Survival through Promoting β-Catenin Degradation in Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 190, 433–444. [Google Scholar] [CrossRef]
- Ji, P.; Zhou, Y.; Yang, Y.; Wu, J.; Zhou, H.; Quan, W.; Sun, J.; Yao, Y.; Shang, A.; Gu, C.; et al. Myeloid cell-derived LL-37 promotes lung cancer growth by activating Wnt/β-catenin signaling. Theranostics 2019, 9, 2209–2223. [Google Scholar] [CrossRef]
- Ye, K.; Wang, S.; Wang, J.; Han, H.; Ma, B.; Yang, Y. Zebularine enhances apoptosis of human osteosarcoma cells by suppressing methylation ofARHI. Cancer Sci. 2016, 107, 1851–1857. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, W.K.; Park, J.Y.; Kim, D.S. Prognostic value of Iroquois homeobox 1 methylation in non-small cell lung cancers. Genes Genom. 2020, 42, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Um, S.-W.; Kim, H.K.; Kim, Y.; Bin Lee, B.; Kim, D.; Han, J.; Kim, H.; Shim, Y.M.; Kim, D.-H. Bronchial biopsy specimen as a surrogate for DNA methylation analysis in inoperable lung cancer. Clin. Epigenetics 2017, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Miao, C.; Tang, Q.; Zhou, X.; Xi, P.; Chang, P.; Hua, L.; Ni, H. MiR-129-5p promotes docetaxel resistance in prostate cancer by down-regulating CAMK2N1 expression. J. Cell. Mol. Med. 2019, 24, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Aljaibeji, H.; Mukhopadhyay, D.; Mohammed, A.K.; Dhaiban, S.; Hachim, M.Y.; Elemam, N.M.; Sulaiman, N.; Salehi, A.; Taneera, J. Reduced Expression of PLCXD3 Associates With Disruption of Glucose Sensing and Insulin Signaling in Pancreatic β-Cells. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Xu, L.; Du, H.; Zhang, Q.; Wang, C.; Yan, L.; Tian, G.; Fu, X. BAI1-associated protein 2-like 2 is a potential biomarker in lung cancer. Oncol. Rep. 2018, 41. [Google Scholar] [CrossRef]
- Martinez-Romero, J.; Bueno-Fortes, S.; Martín-Merino, M.; De Molina, A.R.; Rivas, J.D.L. Survival marker genes of colorectal cancer derived from consistent transcriptomic profiling. BMC Genom. 2018, 19, 857. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, B.; You, Y.; Yang, Z.; Liu, L.; Tang, H.; Bao, W.; Guan, Y.; Shen, X. Sorting nexin 10 acts as a tumor suppressor in tumorigenesis and progression of colorectal cancer through regulating chaperone mediated autophagy degradation of p21(Cip1/WAF1). Cancer Lett. 2018, 419, 116–127. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Z.; Bao, W.; Liu, L.; You, Y.; Wang, X.; Shao, L.; Fu, W.; Kou, X.; Shen, W.; et al. SNX10 (sorting nexin 10) inhibits colorectal cancer initiation and progression by controlling autophagic degradation of SRC. Autophagy 2019, 16, 735–749. [Google Scholar] [CrossRef]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2015, 17, 2–12. [Google Scholar] [CrossRef]
- Shah, R.R.; Stonier, P.D. Repurposing old drugs in oncology: Opportunities with clinical and regulatory challenges ahead. J. Clin. Pharm. Ther. 2018, 44, 6–22. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2015, 76, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Methylation Cut-Off | Expression Cut-Off |
|---|---|---|
| HypoUp | FDR < 0.01 and delta β-value < −0.2 | FDR < 0.01 and log2FC > 1 |
| HypoDown | FDR < 0.01 and delta β-value < −0.2 | FDR < 0.01 and log2FC < −1 |
| HyperUp | FDR < 0.01 and delta β-value > 0.2 | FDR < 0.01 and log2FC > 1 |
| HyperDown | FDR < 0.01 and delta β-value > 0.2 | FDR < 0.01 and log2FC < −1 |
| Symbol | Coef | HR | p Value | Low 95% CI | High 95% CI |
|---|---|---|---|---|---|
| BAIAP2L2 | 0.022 | 1.023 | 0.000 | 1.011 | 1.035 |
| NPR3 | 0.011 | 1.011 | 0.042 | 1.000 | 1.023 |
| PLCXD3 | −0.102 | 0.903 | 0.222 | 0.768 | 1.063 |
| SNX10 | 0.017 | 1.017 | 0.013 | 1.004 | 1.031 |
| RefGene | Region | Relation to Island | Pattern | Drugs | Drug Example |
|---|---|---|---|---|---|
| XDH | TSS200 | OpenSea | HypoUp | 9 | Allopurinol, Eniluracil |
| Body | OpenSea | HypoUp | 9 | Allopurinol, Eniluracil | |
| ATIC | Body | S_Shore | HypoUp | 7 | Pemetrexed |
| CA9 | Body | Island | HyperUp | 6 | Benzthiazide, Hydroflumethiazide, Ellagic Acid |
| SLC7A11 | Body | OpenSea | HypoUp | 5 | Riluzole, Sulfasalazine |
| GAPDH | Body | S_Shore | HypoUp | 4 | Thionicotinamide-Adenine-Dinucleotide |
| PPIF | Body | S_Shore | HypoUp | 4 | Cyclosporine, L-Proline |
| AKR1B10 | Body | OpenSea | HypoUp | 3 | Tolrestat |
| MMP11 | Body | S_Shore | HypoUp | 2 | Marimastat |
| GMDS | Body | Island | HyperUp | 2 | Guanosine-5′-Diphosphate-Rhamnose, Guanosine-5′-Diphosphate |
| Target | Drug | Binding Energy (kcal/mol) |
|---|---|---|
| XDH | Allopurinol | −6.0 |
| XDH | Eniluracil | −5.8 |
| CA9 | Benzthiazide | −7.0 |
| CA9 | Ellagic-Acid | −7.1 |
| CA9 | Hydroflumethiazide | −6.2 |
| ATIC | Pemetrexed | −9.1 |
| GAPDH | Thionicotinamide-Adenine-Dinucleotide | −9.6 |
| SLC7A11 | Sulfasalazine | −9.8 |
| SLC7A11 | Riluzole | −7.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Wang, G.; Yarmus, L.B.; Wan, Y. Combined Methylome and Transcriptome Analyses Reveals Potential Therapeutic Targets for EGFR Wild Type Lung Cancers with Low PD-L1 Expression. Cancers 2020, 12, 2496. https://doi.org/10.3390/cancers12092496
Hu W, Wang G, Yarmus LB, Wan Y. Combined Methylome and Transcriptome Analyses Reveals Potential Therapeutic Targets for EGFR Wild Type Lung Cancers with Low PD-L1 Expression. Cancers. 2020; 12(9):2496. https://doi.org/10.3390/cancers12092496
Chicago/Turabian StyleHu, Weilei, Guosheng Wang, Lonny B. Yarmus, and Yuan Wan. 2020. "Combined Methylome and Transcriptome Analyses Reveals Potential Therapeutic Targets for EGFR Wild Type Lung Cancers with Low PD-L1 Expression" Cancers 12, no. 9: 2496. https://doi.org/10.3390/cancers12092496
APA StyleHu, W., Wang, G., Yarmus, L. B., & Wan, Y. (2020). Combined Methylome and Transcriptome Analyses Reveals Potential Therapeutic Targets for EGFR Wild Type Lung Cancers with Low PD-L1 Expression. Cancers, 12(9), 2496. https://doi.org/10.3390/cancers12092496