NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis

Simple Summary
Abstract
1. Introduction
2. Historical Perspective on NRF2 and Cancer Chemoprevention
3. Global Knockout of NRF2 in the Mouse Results in Diminished Intrinsic Resistance to Chemical Carcinogenesis and Impairs the Efficacy of Cancer Chemoprevention
4. Cancer Chemopreventive Blocking Agents Possess Thiol Reactivity
5. Repression of NRF2 by KEAP1 Requires Two Binding Motifs in the Transcription Factor
6. The Triggering of Multiple Thiol-Based Sensors in KEAP1 by Thiol-Reactive Small Molecules Results in De-repression of NRF2 and Protection against Oxidative Stress
7. NRF2 is Frequently Upregulated in Malignant Disease
8. Constitutive Activation of NRF2 Supports Post-Initiation Stages of Cancer
9. Mechanisms by Which Upregulation of NRF2 Supports Post-Initiation Stages of Cancer
10. Evidence of Dysregulation of NRF2 in Human Cancers and Segregation with Activated Oncogenes
10.1. Upregulation of NRF2 in Lung Tumours
10.2. Upregulation of NRF2 in Oesophageal Tumours
10.3. Upregulation of NRF2 in Liver Tumours
10.4. Upregulation of NRF2 in Head and Neck Tumours
10.5. Upregulation of NRF2 in Gastric Tumours
10.6. Upregulation of NRF2 in Bladder Tumours
10.7. Upregulation of NRF2 in Colorectal Tumours
11. Therapeutic Approaches to Treat Tumours in Which NRF2 Is Upregulated
12. Concluding Comments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. 2018, 73, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar]
- Friling, R.S.; Bergelson, S.; Daniel, V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc. Natl. Acad. Sci. USA 1992, 89, 668–672. [Google Scholar] [CrossRef]
- Kataoka, K.; Fujiwara, K.T.; Noda, M.; Nishizawa, M. MafB, a new Maf family transcription activator that can associate with Maf and Fos but not with Jun. Mol. Cell. Biol. 1994, 14, 7581–7591. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. J. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans. 2000, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap ‘n’ Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef]
- Chanas, S.A.; Jiang, Q.; McMahon, M.; McWalter, G.K.; McLellan, L.I.; Elcombe, C.R.; Henderson, C.J.; Wolf, C.R.; Moffat, G.J.; Itoh, K.; et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002, 365, 405–416. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer 2007, 6, 154. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An exceptionally potent inducer of cytoprotective enzymes: Elucidation of the structural features that determine inducer potency and reactivity with Keap1. J. Biol. Chem. 2010, 285, 33747–33755. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Soriano, F.X.; Baxter, P.; Murray, L.M.; Sporn, M.B.; Gillingwater, T.H.; Hardingham, G.E. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cells 2009, 27, 279–282. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.-R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, A.; Tsukamoto, S.; Nishikawa, K.; Yoshida, A.; Harada, N.; Motojima, K.; Ishii, T.; Nakane, A.; Yamamoto, M.; Itoh, K. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch. Biochem. Biophys. 2008, 477, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe Á, J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.W.; Gustafson, D.L.; Thorburn, A. Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition. Dev. Cell 2019, 50, 690–703.e6. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Bin, H.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Transcriptional Regulation of Activating Transcription Factor 4 under Oxidative Stress in Retinal Pigment Epithelial ARPE-19/HPV-16 Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1226–1234. [Google Scholar] [CrossRef]
- Dziunycz, P.J.; Lefort, K.; Wu, X.; Freiberger, S.N.; Neu, J.; Djerbi, N.; Iotzowa-Weiss, G.; French, L.E.; Dotto, G.P.; Hofbauer, G.F.L. The oncogene ATF3 is potentiated by cyclosporine A and ultraviolet light A. J. Investig. Dermatol. 2014, 134, 1998–2004. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.-Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e1007. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Chartoumpekis, D.V.; Kensler, T.W. Crosstalk between Nrf2 and Notch signaling. Free Radic. Biol. Med. 2015, 88, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hurtado, A.; Martin-Morales, R.; Robledinos-Antón, N.; Blanco, R.; Palacios-Blanco, I.; Lastres-Becker, I.; Cuadrado, A.; Garcia-Gonzalo, F.R. NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling. Sci. Rep. 2019, 9, 13896. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Dodson, M.; Fang, D.; Chapman, E.; Zhang, D.D. NRF2 negatively regulates primary ciliogenesis and hedgehog signaling. PLoS Biol. 2020, 18, e3000620. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, L.W. Chemoprevention of cancer. Cancer Res. 1985, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, L.W. Chemoprophylaxis of carcinogenesis: A review. Cancer Res. 1966, 26, 1520–1526. [Google Scholar] [PubMed]
- Berenblum, I. Experimental inhibition of tumour induction by mustard gas and other compounds. J. Pathol. Bacteriol. 1935, 40, 549–558. [Google Scholar] [CrossRef]
- Berenblum, I.; Smith, R. The anti-carcinogenic action of dichlorodiethylsulphide (mustard gas). J. Pathol. Bacteriol. 1931, 34, 731–746. [Google Scholar] [CrossRef]
- Crabtree, H.G. Influence of Unsaturated Dibasic Acids on the Induction of Skin Tumors by Chemical Carcinogens. Cancer Res. 1945, 5, 346–351. [Google Scholar]
- Crabtree, H.G. Retardation of the Rate of Tumor Induction by Hydrolyzing Chlor-Compounds. Cancer Res. 1941, 1, 39. [Google Scholar]
- Richardson, H.L.; Stier, A.R.; Borsos-Nachtnebel, E. Liver tumor inhibition and adrenal histologic responses in rats to which 3′-methyl-4-dimethylaminoazobenzene and 20-methylcholanthrene were simultaneously administered. Cancer Res. 1952, 12, 356–361. [Google Scholar]
- Meechan, R.J.; McCafferty, D.E.; Jones, R.S. 3-Methylcholanthrene as an inhibitor of hepatic cancer induced by 3′-methyl-4-dimethylaminoazobenzene in the diet of the rat: A determination of the time relationships. Cancer Res. 1953, 13, 802–806. [Google Scholar] [PubMed]
- Miller, E.C.; Miller, J.A.; Brown, R.R.; Macdonald, J.C. On the protective action of certain polycyclic aromatic hydrocarbons against carcinogenesis by aminoazo dyes and 2-acetylaminofluorene. Cancer Res. 1958, 18, 469–477. [Google Scholar] [PubMed]
- Miller, E.C.; Miller, J.A. Mechanisms of chemical carcinogenesis: Nature of proximate carcinogens and interactions with macromolecules. Pharmacol. Rev. 1966, 18, 805–838. [Google Scholar] [PubMed]
- Thakker, D.R.; Yagi, H.; Lu, A.Y.; Levin, W.; Conney, A.H. Metabolism of benzo[a]pyrene: Conversion of (+/−)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to highly mutagenic 7,8-diol-9,10-epoxides. Proc. Natl. Acad. Sci. USA 1976, 73, 3381–3385. [Google Scholar] [CrossRef]
- Levin, W.; Wood, A.W.; Yagi, H.; Jerina, D.M.; Conney, A.H. (+/−)-trans-7,8-dihydroxy-7,8-dihydrobenzo (a)pyrene: A potent skin carcinogen when applied topically to mice. Proc. Natl. Acad. Sci. USA 1976, 73, 3867–3871. [Google Scholar] [CrossRef]
- Wattenberg, L.W.; Page, M.A.; Leong, J.L. Induction of increased benzpyrene hydroxylase activity by flavones and related compounds. Cancer Res. 1968, 28, 934–937. [Google Scholar]
- Wattenberg, L.W.; Leong, J.L. Inhibition of the carcinogenic action of benzo(a)pyrene by flavones. Cancer Res. 1970, 30, 1922–1925. [Google Scholar]
- Wattenberg, L.W.; Bueding, E. Inhibitory effects of 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (Oltipraz) on carcinogenesis induced by benzo[a]pyrene, diethylnitrosamine and uracil mustard. Carcinogenesis 1986, 7, 1379–1381. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of carcinogenic and toxic effects of polycyclic hydrocarbons by phenolic antioxidants and ethoxyquin. J. Natl. Cancer Inst. 1972, 48, 1425–1430. [Google Scholar]
- Ulland, B.M.; Weisburger, J.H.; Yamamoto, R.S.; Weisburger, E.K. Antioxidants and carcinogenesis: Butylated hydroxytoluene, but not diphenyl-p-phenylenediamine, inhibits cancer induction by N-2-fluorenylacetamide and by N-hydroxy-N-2-fluorenylacetamide in rats. Food Cosmet. Toxicol. 1973, 11, 199–207. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Whysner, J. Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 1999, 37, 1027–1038. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of carcinogenic and toxic effects of polycyclic hydrocarbons by several sulfur-containing compounds. J. Natl. Cancer Inst. 1974, 52, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, L.W.; Loub, W.D. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978, 38, 1410–1413. [Google Scholar] [PubMed]
- Grantham, P.H.; Weisburger, J.H.; Weisburger, E.K. Effect of the antioxidant butylated hydroxytoluene (BHT) on the metabolism of the carcinogens N-2-fluorenylacetamide and N-hydroxy-N-2-fluorenylacetamide. Food Cosmet. Toxicol. 1973, 11, 209–217. [Google Scholar] [CrossRef]
- Cha, Y.N.; Martz, F.; Bueding, E. Enhancement of liver microsome epoxide hydratase activity in rodents by treatment with 2(3)-tert-butyl-4-hydroxyanisole. Cancer Res. 1978, 38, 4496–4498. [Google Scholar]
- Benson, A.M.; Batzinger, R.P.; Ou, S.Y.; Bueding, E.; Cha, Y.N.; Talalay, P. Elevation of hepatic glutathione S-transferase activities and protection against mutagenic metabolites of benzo(a)pyrene by dietary antioxidants. Cancer Res. 1978, 38, 4486–4495. [Google Scholar]
- Benson, A.M.; Cha, Y.N.; Bueding, E.; Heine, H.S.; Talalay, P. Elevation of extrahepatic glutathione S-transferase and epoxide hydratase activities by 2(3)-tert-butyl-4-hydroxyanisole. Cancer Res. 1979, 39, 2971–2977. [Google Scholar]
- Benson, A.M.; Hunkeler, M.J.; Talalay, P. Increase of NAD(P)H:quinone reductase by dietary antioxidants: Possible role in protection against carcinogenesis and toxicity. Proc. Natl. Acad. Sci. USA 1980, 77, 5216–5220. [Google Scholar] [CrossRef]
- Sparnins, V.L.; Chuan, J.; Wattenberg, L.W. Enhancement of glutathione S-transferase activity of the esophagus by phenols, lactones, and benzyl isothiocyanate. Cancer Res. 1982, 42, 1205–1207. [Google Scholar]
- Sparnins, V.L.; Venegas, P.L.; Wattenberg, L.W. Glutathione S-transferase activity: Enhancement by compounds inhibiting chemical carcinogenesis and by dietary constituents. J. Natl. Cancer Inst. 1982, 68, 493–496. [Google Scholar]
- Judah, D.J.; Hayes, J.D.; Yang, J.C.; Lian, L.Y.; Roberts, G.C.; Farmer, P.B.; Lamb, J.H.; Neal, G.E. A novel aldehyde reductase with activity towards a metabolite of aflatoxin B1 is expressed in rat liver during carcinogenesis and following the administration of an anti-oxidant. Biochem. J. 1993, 292, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Reinhart, J.; Sisk, S.C.; Anderson, K.S.; Adler, P.N. Tissue-specific induction of murine glutathione transferase mRNAs by butylated hydroxyanisole. J. Biol. Chem. 1988, 263, 13324–13332. [Google Scholar] [PubMed]
- Pearson, W.R.; Windle, J.J.; Morrow, J.F.; Benson, A.M.; Talalay, P. Increased synthesis of glutathione S-transferases in response to anticarcinogenic antioxidants. Cloning and measurement of messenger RNA. J. Biol. Chem. 1983, 258, 2052–2062. [Google Scholar] [PubMed]
- Cha, Y.N.; Heine, H.S. Comparative effects of dietary administration of 2(3)-tert-butyl-4-hydroxyanisole and 3,5-di-tert-butyl-4-hydroxytoluene on several hepatic enzyme activities in mice and rats. Cancer Res. 1982, 42, 2609–2615. [Google Scholar] [PubMed]
- Dock, L.; Martinez, M.; Jernström, B. Induction of hepatic glutathione S-transferase activity by butylated hydroxyanisole and conjugation of benzo[a]pyrene diol-epoxide. Carcinogenesis 1984, 5, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Human aldo-keto reductases and the metabolic activation of polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 2014, 27, 1901–1917. [Google Scholar] [CrossRef]
- Newberne, P.M.; Butler, W.H. Acute and chronic effects of aflatoxin on the liver of domestic and laboratory animals: A review. Cancer Res. 1969, 29, 236–250. [Google Scholar]
- Kensler, T.W.; Egner, P.A.; Davidson, N.E.; Roebuck, B.D.; Pikul, A.; Groopman, J.D. Modulation of aflatoxin metabolism, aflatoxin-N7-guanine formation, and hepatic tumorigenesis in rats fed ethoxyquin: Role of induction of glutathione S-transferases. Cancer Res. 1986, 46, 3924–3931. [Google Scholar]
- Kensler, T.W.; Egner, P.A.; Dolan, P.M.; Groopman, J.D.; Roebuck, B.D. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 1987, 47, 4271–4277. [Google Scholar]
- Hayes, J.D.; Judah, D.J.; McLellan, L.I.; Kerr, L.A.; Peacock, S.D.; Neal, G.E. Ethoxyquin-induced resistance to aflatoxin B1 in the rat is associated with the expression of a novel alpha-class glutathione S-transferase subunit, Yc2, which possesses high catalytic activity for aflatoxin B1-8,9-epoxide. Biochem. J. 1991, 279, 385–398. [Google Scholar] [CrossRef]
- Hayes, J.D.; Judah, D.J.; Neal, G.E. Resistance to aflatoxin B1 is associated with the expression of a novel aldo-keto reductase which has catalytic activity towards a cytotoxic aldehyde-containing metabolite of the toxin. Cancer Res. 1993, 53, 3887–3894. [Google Scholar] [PubMed]
- Ellis, E.M.; Judah, D.J.; Neal, G.E.; Hayes, J.D. An ethoxyquin-inducible aldehyde reductase from rat liver that metabolizes aflatoxin B1 defines a subfamily of aldo-keto reductases. Proc. Natl. Acad. Sci. USA 1993, 90, 10350–10354. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.W.; Ueng, Y.F.; Widersten, M.; Mannervik, B.; Hayes, J.D.; Sherratt, P.J.; Ketterer, B.; Guengerich, F.P. Conjugation of highly reactive aflatoxin B1 exo-8,9-epoxide catalyzed by rat and human glutathione transferases: Estimation of kinetic parameters. Biochemistry 1997, 36, 3056–3060. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Pulford, D.J.; Ellis, E.M.; McLeod, R.; James, R.F.; Seidegård, J.; Mosialou, E.; Jernström, B.; Neal, G.E. Regulation of rat glutathione S-transferase A5 by cancer chemopreventive agents: Mechanisms of inducible resistance to aflatoxin B1. Chem.-Biol. Interact. 1998, 111–112, 51–67. [Google Scholar] [CrossRef]
- Kelly, V.P.; Ellis, E.M.; Manson, M.M.; Chanas, S.A.; Moffat, G.J.; McLeod, R.; Judah, D.J.; Neal, G.E.; Hayes, J.D. Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits, and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res. 2000, 60, 957–969. [Google Scholar]
- Guengerich, F.P.; Cai, H.; McMahon, M.; Hayes, J.D.; Sutter, T.R.; Groopman, J.D.; Deng, Z.; Harris, T.M. Reduction of aflatoxin B1 dialdehyde by rat and human aldo-keto reductases. Chem. Res. Toxicol. 2001, 14, 727–737. [Google Scholar] [CrossRef]
- Hayes, J.D.; Judah, D.J.; Neal, G.E.; Nguyen, T. Molecular cloning and heterologous expression of a cDNA encoding a mouse glutathione S-transferase Yc subunit possessing high catalytic activity for aflatoxin B1-8,9-epoxide. Biochem. J. 1992, 285, 173–180. [Google Scholar] [CrossRef]
- Crawford, D.R.; Ilic, Z.; Guest, I.; Milne, G.L.; Hayes, J.D.; Sell, S. Characterization of liver injury, oval cell proliferation and cholangiocarcinogenesis in glutathione S-transferase A3 knockout mice. Carcinogenesis 2017, 38, 717–727. [Google Scholar] [CrossRef]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol. 2011, 85, 273–284. [Google Scholar] [CrossRef]
- Taguchi, K.; Takaku, M.; Egner, P.A.; Morita, M.; Kaneko, T.; Mashimo, T.; Kensler, T.W.; Yamamoto, M. Generation of a New Model Rat: Nrf2 Knockout Rats Are Sensitive to Aflatoxin B1 Toxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 152, 40–52. [Google Scholar] [CrossRef]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 Is Essential for the Chemopreventive Efficacy of Oltipraz against Urinary Bladder Carcinogenesis. Cancer Res. 2004, 64, 6424. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Jeffery, E.H.; Matusheski, N.V.; Moon, R.C.; Lantvit, D.D.; Pezzuto, J.M. Sulforaphane prevents mouse skin tumorigenesis during the stage of promotion. Cancer Lett. 2006, 236, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Jenkins, S.N.; Fahey, J.W.; Ye, L.; Wehage, S.L.; Liby, K.T.; Stephenson, K.K.; Wade, K.L.; Talalay, P. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006, 240, 243–252. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Benedict, A.L.; Jenkins, S.N.; Ye, L.; Wehage, S.L.; Talalay, P. Dietary glucoraphanin-rich broccoli sprout extracts protect against UV radiation-induced skin carcinogenesis in SKH-1 hairless mice. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2010, 9, 597–600. [Google Scholar] [CrossRef]
- Long, M.; Tao, S.; Rojo de la Vega, M.; Jiang, T.; Wen, Q.; Park, S.L.; Zhang, D.D.; Wondrak, G.T. Nrf2-dependent suppression of azoxymethane/dextran sulfate sodium-induced colon carcinogenesis by the cinnamon-derived dietary factor cinnamaldehyde. Cancer Prev. Res. 2015, 8, 444–454. [Google Scholar] [CrossRef]
- Lan, A.; Li, W.; Liu, Y.; Xiong, Z.; Zhang, X.; Zhou, S.; Palko, O.; Chen, H.; Kapita, M.; Prigge, J.R.; et al. Chemoprevention of oxidative stress-associated oral carcinogenesis by sulforaphane depends on NRF2 and the isothiocyanate moiety. Oncotarget 2016, 7, 53502–53514. [Google Scholar] [CrossRef]
- Ohkoshi, A.; Suzuki, T.; Ono, M.; Kobayashi, T.; Yamamoto, M. Roles of Keap1-Nrf2 system in upper aerodigestive tract carcinogenesis. Cancer Prev. Res. 2013, 6, 149–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Becks, L.; Prince, M.; Burson, H.; Christophe, C.; Broadway, M.; Itoh, K.; Yamamoto, M.; Mathis, M.; Orchard, E.; Shi, R.; et al. Aggressive mammary carcinoma progression in Nrf2 knockout mice treated with 7,12-dimethylbenz[a]anthracene. BMC Cancer 2010, 10, 540. [Google Scholar] [CrossRef]
- Kasala, E.R.; Bodduluru, L.N.; Barua, C.C.; Madhana, R.M.; Dahiya, V.; Budhani, M.K.; Mallugari, R.R.; Maramreddy, S.R.; Gogoi, R. Chemopreventive effect of chrysin, a dietary flavone against benzo(a)pyrene induced lung carcinogenesis in Swiss albino mice. Pharmacol. Rep. 2016, 68, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Jiang, T.; Long, M.; Chen, J.; Ren, D.M.; Wong, P.K.; Chapman, E.; Zhou, B.; Zhang, D.D. A Curcumin Derivative That Inhibits Vinyl Carbamate-Induced Lung Carcinogenesis via Activation of the Nrf2 Protective Response. Antioxid. Redox Signal. 2015, 23, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Rennhack, J.; Andrechek, E.R.; Rockwell, C.E.; Liby, K.T. Identification of an Unfavorable Immune Signature in Advanced Lung Tumors from Nrf2-Deficient Mice. Antioxid. Redox Signal. 2018, 29, 1535–1552. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Saigusa, D.; Baird, L.; Yu, L.; Rokutan, H.; Igarashi, K.; Ebina, M.; Shibata, T.; Yamamoto, M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016, 76, 3088–3096. [Google Scholar] [CrossRef]
- Tao, S.; Rojo de la Vega, M.; Chapman, E.; Ooi, A.; Zhang, D.D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. [Google Scholar] [CrossRef]
- Kombairaju, P.; Ma, J.; Thimmulappa, R.K.; Yan, S.G.; Gabrielson, E.; Singh, A.; Biswal, S. Prolonged sulforaphane treatment does not enhance tumorigenesis in oncogenic K-ras and xenograft mouse models of lung cancer. J. Carcinog. 2012, 11, 8. [Google Scholar] [CrossRef]
- Liu, J.; Wu, K.C.; Lu, Y.F.; Ekuase, E.; Klaassen, C.D. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid. Med. Cell. Longev. 2013, 2013, 305861. [Google Scholar] [CrossRef]
- Luo, L.; Chen, Y.; Wang, H.; Wang, S.; Liu, K.; Li, X.; Wang, X.J.; Tang, X. Mkp-1 protects mice against toxin-induced liver damage by promoting the Nrf2 cytoprotective response. Free Radic. Biol. Med. 2018, 115, 361–370. [Google Scholar] [CrossRef]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef]
- Prochaska, H.J.; De Long, M.J.; Talalay, P. On the mechanisms of induction of cancer-protective enzymes: A unifying proposal. Proc. Natl. Acad. Sci. USA 1985, 82, 8232–8236. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; De Long, M.J.; Prochaska, H.J. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Holtzclaw, W.D.; Zhang, Y.; Talalay, P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc. Natl. Acad. Sci. USA 1993, 90, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Chasseaud, L.F. The role of glutathione and glutathione S-transferases in the metabolism of chemical carcinogens and other electrophilic agents. Adv. Cancer Res.. 1979, 29, 175–274. [Google Scholar] [CrossRef] [PubMed]
- Coles, B. Effects of modifying structure on electrophilic reactions with biological nucleophiles. Drug Metab. Rev. 1984, 15, 1307–1334. [Google Scholar] [CrossRef]
- Spencer, S.R.; Xue, L.A.; Klenz, E.M.; Talalay, P. The potency of inducers of NAD(P)H:(quinone-acceptor) oxidoreductase parallels their efficiency as substrates for glutathione transferases. Structural and electronic correlations. Biochem. J. 1991, 273, 711–717. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef]
- Prestera, T.; Zhang, Y.; Spencer, S.R.; Wilczak, C.A.; Talalay, P. The electrophile counterattack response: Protection against neoplasia and toxicity. Adv. Enzym. Regul. 1993, 33, 281–296. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Hannink, M.; Beamer, L.J. Crystal structure of the Kelch domain of human Keap1. J. Biol. Chem. 2004, 279, 54750–54758. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e744. [Google Scholar] [CrossRef]
- Stöcker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Caggiano, M.; Schröder, E.; Cho, H.J.; Burgoyne, J.; Barallobre-Barreiro, J.; Mayr, M.; Eaton, P. Oxidant-induced Interprotein Disulfide Formation in Cardiac Protein DJ-1 Occurs via an Interaction with Peroxiredoxin 2. J. Biol. Chem. 2016, 291, 10399–10410. [Google Scholar] [CrossRef]
- Farber, E. Cellular biochemistry of the stepwise development of cancer with chemicals: G. H. A. Clowes memorial lecture. Cancer Res. 1984, 44, 5463–5474. [Google Scholar] [PubMed]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef]
- Siegel, D.; Franklin, W.A.; Ross, D. Immunohistochemical detection of NAD(P)H:quinone oxidoreductase in human lung and lung tumors. Clin. Cancer Res. 1998, 4, 2065–2070. [Google Scholar]
- Seo, D.C.; Sung, J.M.; Cho, H.J.; Yi, H.; Seo, K.H.; Choi, I.S.; Kim, D.K.; Kim, J.S.; El-Aty, A.A.; Shin, H.C. Gene expression profiling of cancer stem cell in human lung adenocarcinoma A549 cells. Mol. Cancer 2007, 6, 75. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Res. 2008, 68, 1303. [Google Scholar] [CrossRef]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. MCR 2017, 15, 1570–1578. [Google Scholar] [CrossRef]
- Duan, F.G.; Wang, M.F.; Cao, Y.B.; Dan, L.; Li, R.Z.; Fan, X.X.; Khan, I.; Lai, H.L.; Zhang, Y.Z.; Hsiao, W.W.; et al. MicroRNA-421 confers paclitaxel resistance by binding to the KEAP1 3’UTR and predicts poor survival in non-small cell lung cancer. Cell Death Dis. 2019, 10, 821. [Google Scholar] [CrossRef]
- Song, S.; Nguyen, V.; Schrank, T.; Mulvaney, K.; Walter, V.; Wei, D.; Orvis, T.; Desai, N.; Zhang, J.; Hayes, D.N.; et al. Loss of SWI/SNF Chromatin Remodeling Alters NRF2 Signaling in Non-Small Cell Lung Carcinoma. Mol. Cancer Res. MCR 2020. [Google Scholar] [CrossRef]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ1-1 Deficiency Activates NRF2/IGF1R Axis in HCC via Accumulation of Oncometabolite Succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. Oncogene-Stimulated Congestion at the KEAP1 Stress Signaling Hub Allows Bypass of NRF2 and Induction of NRF2-Target Genes that Promote Tumor Survival. Cancer Cell 2017, 32, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Pölönen, P.; Jawahar Deen, A.; Leinonen, H.M.; Jyrkkänen, H.-K.; Kuosmanen, S.; Mononen, M.; Jain, A.; Tuomainen, T.; Pasonen-Seppänen, S.; Hartikainen, J.M.; et al. Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene 2019, 38, 7473–7490. [Google Scholar] [CrossRef]
- Escoll, M.; Lastra, D.; Pajares, M.; Robledinos-Antón, N.; Rojo, A.I.; Fernández-Ginés, R.; Mendiola, M.; Martínez-Marín, V.; Esteban, I.; López-Larrubia, P.; et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol. 2020, 30, 101425. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- Taguchi, K.; Maher, J.M.; Suzuki, T.; Kawatani, Y.; Motohashi, H.; Yamamoto, M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 2010, 30, 3016–3026. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Anzawa, H.; Liu, Z.; Ota, N.; Kitamura, H.; Onodera, Y.; Alam, M.M.; Matsumaru, D.; Suzuki, T.; Katsuoka, F.; et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 2020, 11, 5911. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.M.; Montgomery, S.A.; Schrank, T.P.; Simon, J.M.; Ptacek, T.S.; Tamir, T.Y.; Mulvaney, K.M.; Weir, S.J.; Nguyen, T.T.; Murphy, R.M.; et al. A conditional mouse expressing an activating mutation in NRF2 displays hyperplasia of the upper gastrointestinal tract and decreased white adipose tissue. J. Pathol. 2020, 252, e5504. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef]
- Schäfer, M.; Willrodt, A.H.; Kurinna, S.; Link, A.S.; Farwanah, H.; Geusau, A.; Gruber, F.; Sorg, O.; Huebner, A.J.; Roop, D.R.; et al. Activation of Nrf2 in keratinocytes causes chloracne (MADISH)-like skin disease in mice. EMBO Mol. Med. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Rolfs, F.; Huber, M.; Kuehne, A.; Kramer, S.; Haertel, E.; Muzumdar, S.; Wagner, J.; Tanner, Y.; Böhm, F.; Smola, S.; et al. Nrf2 Activation Promotes Keratinocyte Survival during Early Skin Carcinogenesis via Metabolic Alterations. Cancer Res. 2015, 75, 4817–4829. [Google Scholar] [CrossRef]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e110. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental Activation of Nrf2 Restricts the Progression of Nrf2-Activated Malignant Tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef]
- Nabeshima, T.; Hamada, S.; Taguchi, K.; Tanaka, Y.; Matsumoto, R.; Yamamoto, M.; Masamune, A. Keap1 deletion accelerates mutant K-ras/p53-driven cholangiocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G419–G427. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.-P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.-Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef]
- Kudo, Y.; Sugimoto, M.; Arias, E.; Kasashima, H.; Cordes, T.; Linares, J.F.; Duran, A.; Nakanishi, Y.; Nakanishi, N.; L’Hermitte, A.; et al. PKCλ/ι Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 2020, 38, 247–262.e11. [Google Scholar] [CrossRef]
- Jessen, C.; Kreß, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Saverio, V.; Monzani, R.; Ferrari, E.; Piacentini, M.; Corazzari, M. Ferroptosis: A new unexpected chance to treat metastatic melanoma? Cell Cycle 2020, 19, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Hensing, T.; Chawla, A.; Batra, R.; Salgia, R. A personalized treatment for lung cancer: Molecular pathways, targeted therapies, and genomic characterization. Adv. Exp. Med. Biol. 2014, 799, 85–117. [Google Scholar]
- Evans, M. Lung cancer: Needs assessment, treatment and therapies. Br. J. Nurs. 2013, 22, S15–S22. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal. 2020, 18, 98. [Google Scholar] [CrossRef]
- Liu, Z.; Deng, M.; Wu, L.; Zhang, S. An integrative investigation on significant mutations and their down-stream pathways in lung squamous cell carcinoma reveals CUL3/KEAP1/NRF2 relevant subtypes. Mol. Med. 2020, 26, 48. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Hammerman, P.S.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; Gabriel, S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Baeissa, H.M.; Benstead-Hume, G.; Richardson, C.J.; Pearl, F.M. Mutational patterns in oncogenes and tumour suppressors. Biochem. Soc. Trans. 2016, 44, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Hast, B.E.; Cloer, E.W.; Goldfarb, D.; Li, H.; Siesser, P.F.; Yan, F.; Walter, V.; Zheng, N.; Hayes, D.N.; Major, M.B. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014, 74, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Bos, M.; Gardizi, M.; König, K.; Michels, S.; Fassunke, J.; Heydt, C.; Künstlinger, H.; Ihle, M.; Ueckeroth, F.; et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): Genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget 2015, 6, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Suzuki, A.; Shitara, M.; Okuda, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Keap1 mutations in lung cancer patients. Oncol. Lett. 2013, 6, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Ahrendt, S.A.; Decker, P.A.; Alawi, E.A.; Zhu, Y.R.; Sanchez-Cespedes, M.; Yang, S.C.; Haasler, G.B.; Kajdacsy-Balla, A.; Demeure, M.J.; Sidransky, D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 2001, 92, 1525–1530. [Google Scholar] [CrossRef]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889. [Google Scholar] [CrossRef]
- Wimuttisuk, W.; West, M.; Davidge, B.; Yu, K.; Salomon, A.; Singer, J.D. Novel Cul3 binding proteins function to remodel E3 ligase complexes. BMC Cell Biol. 2014, 15, 28. [Google Scholar] [CrossRef]
- Pintard, L.; Willems, A.; Peter, M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004, 23, 1681–1687. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of co-occurring Genomic alterations on outcomes in patients with KRAS-mutant non-“small cell lung cancer. Clin. Cancer Res. 2018, 24, 334. [Google Scholar] [CrossRef]
- Nadal, E.; Palmero, R.; Muñoz-Pinedo, C. Mutations in the Antioxidant KEAP1/NRF2 Pathway Define an Aggressive Subset of NSCLC Resistant to Conventional Treatments. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1881–1883. [Google Scholar] [CrossRef]
- Barrera-Rodriguez, R. Importance of the Keap1-Nrf2 pathway in NSCLC: Is it a possible biomarker? Biomed. Rep. 2018, 9, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Tamura, M.; Koyama, R.; Nakagaki, T.; Adachi, Y.; Tokino, T. Genomic characterization of esophageal squamous cell carcinoma: Insights from next-generation sequencing. World J. Gastroenterol. 2016, 22, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiao, Q.; Kong, L.; Yu, J.; Fang, A.; Li, M.; Yu, J. Nrf2 and Keap1 abnormalities in esophageal squamous cell carcinoma and association with the effect of chemoradiotherapy. Thorac. Cancer 2018, 9, 726–735. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Lin, D.-C.; Hao, J.-J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.-W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Kitano, Y.; Baba, Y.; Nakagawa, S.; Miyake, K.; Iwatsuki, M.; Ishimoto, T.; Yamashita, Y.I.; Yoshida, N.; Watanabe, M.; Nakao, M.; et al. Nrf2 promotes oesophageal cancer cell proliferation via metabolic reprogramming and detoxification of reactive oxygen species. J. Pathol. 2018, 244, 346–357. [Google Scholar] [CrossRef]
- Zuo, J.; Zhao, M.; Fan, Z.; Liu, B.; Wang, Y.; Li, Y.; Lv, P.; Xing, L.; Zhang, X.; Shen, H. MicroRNA-153-3p regulates cell proliferation and cisplatin resistance via Nrf-2 in esophageal squamous cell carcinoma. Thorac. Cancer 2020, 11, 738–747. [Google Scholar] [CrossRef]
- Liu, M.; Hu, C.; Xu, Q.; Chen, L.; Ma, K.; Xu, N.; Zhu, H. Methylseleninic acid activates Keap1/Nrf2 pathway via up-regulating miR-200a in human oesophageal squamous cell carcinoma cells. Biosci. Rep. 2015, 35, e00256. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Shin, S.M.; Yang, J.H.; Ki, S.H. Role of the Nrf2-ARE Pathway in Liver Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 763257. [Google Scholar] [CrossRef]
- Haque, E.; Karim, M.R.; Salam Teeli, A.; Åšmiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular Mechanisms Underlying Hepatocellular Carcinoma Induction by Aberrant NRF2 Activation-Mediated Transcription Networks: Interaction of NRF2-KEAP1 Controls the Fate of Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [CrossRef] [PubMed]
- Orru, C.; Szydlowska, M.; Taguchi, K.; Zavattari, P.; Perra, A.; Yamamoto, M.; Columbano, A. Genetic inactivation of Nrf2 prevents clonal expansion of initiated cells in a nutritional model of rat hepatocarcinogenesis. J. Hepatol. 2018, 69, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.L.; Degos, F.O.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.F.; Berres, M.L.; Fujiwara, N.; Goossens, N.; Chou, H.I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 2018, 9, 4962. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Orru, C.; Perra, A.; Kowalik, M.A.; Rizzolio, S.; Puliga, E.; Cabras, L.; Giordano, S.; Columbano, A. Distinct Mechanisms Are Responsible for Nrf2-Keap1 Pathway Activation at Different Stages of Rat Hepatocarcinogenesis. Cancers 2020, 12, 2305. [Google Scholar] [CrossRef] [PubMed]
- Stacy, D.R.; Ely, K.; Massion, P.P.; Yarbrough, W.G.; Hallahan, D.E.; Sekhar, K.R.; Freeman, M.L. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck 2006, 28, 813–818. [Google Scholar] [CrossRef]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef]
- Liu, R.; Peng, J.; Wang, H.; Li, L.; Wen, X.; Tan, Y.; Zhang, L.; Wan, H.; Chen, F.; Nie, X. Oxysophocarpine Retards the Growth and Metastasis of Oral Squamous Cell Carcinoma by Targeting the Nrf2/HO-1 Axis. Cell. Physiol. Biochem. 2018, 49, 1717–1733. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Pikor, L.A.; Lam, S.; Lam, W.L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: Association with poor prognosis in head and neck cancer. Head Neck 2015, 37, 727–734. [Google Scholar] [CrossRef]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef]
- Lu, B.C.; Li, J.; Yu, W.F.; Zhang, G.Z.; Wang, H.M.; Ma, H.M. Elevated expression of Nrf2 mediates multidrug resistance in CD133+ head and neck squamous cell carcinoma stem cells. Oncol. Lett. 2016, 12, 4333–4338. [Google Scholar] [CrossRef][Green Version]
- Kawasaki, Y.; Ishigami, S.; Arigami, T.; Uenosono, Y.; Yanagita, S.; Uchikado, Y.; Kita, Y.; Nishizono, Y.; Okumura, H.; Nakajo, A.; et al. Clinicopathological significance of nuclear factor (erythroid-2)-related factor 2 (Nrf2) expression in gastric cancer. BMC Cancer 2015, 15, 5. [Google Scholar] [CrossRef]
- Richters, A.; Aben, K.K.H.; Kiemeney, L.A.L.M. The global burden of urinary bladder cancer: An update. World J. Urol. 2020, 38, 1895–1904. [Google Scholar] [CrossRef]
- Reszka, E.; Jablonowski, Z.; Wieczorek, E.; Jablonska, E.; Krol, M.B.; Gromadzinska, J.; Grzegorczyk, A.; Sosnowski, M.; Wasowicz, W. Polymorphisms of NRF2 and NRF2 target genes in urinary bladder cancer patients. J. Cancer Res. Clin. Oncol. 2014, 140, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Ochoa, A.; McConkey, D.J.; Aine, M.; Höglund, M.; Kim, W.Y.; Real, F.X.; Kiltie, A.E.; Milsom, I.; Dyrskjøt, L.; et al. Genetic Alterations in the Molecular Subtypes of Bladder Cancer: Illustration in the Cancer Genome Atlas Dataset. Eur. Urol. 2017, 72, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Akbani, R.; Creighton, C.J.; Lerner, S.P.; Weinstein, J.N.; Getz, G.; Kwiatkowski, D.J. Invasive Bladder Cancer: Genomic Insights and Therapeutic Promise. Clin. Cancer Res. 2015, 21, 4514–4524. [Google Scholar] [CrossRef]
- Li, T.A.-O.; Jiang, D.; Wu, K. p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci. 2020, 111, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Guida, E.; Inokawa, Y.; Goldberg, R.; Reis, L.O.; Ooki, A.; Pilli, M.; Sadhukhan, P.; Woo, J.; Choi, W.; et al. GULP1 regulates the NRF2-KEAP1 signaling axis in urothelial carcinoma. Sci. Sig. 2020, 13, aba0443. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Wu, C.H.; Lewis, A.; Holmes, C.; Hawkins, M.A.; Maughan, T. NRF2 metagene signature is a novel prognostic biomarker in colorectal cancer. Cancer Genet. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Torrente, L.; Maan, G.; Oumkaltoum Rezig, A.; Quinn, J.; Jackson, A.; Grilli, A.; Casares, L.; Zhang, Y.; Kulesskiy, E.; Saarela, J.; et al. High NRF2 Levels Correlate with Poor Prognosis in Colorectal Cancer Patients and with Sensitivity to the Kinase Inhibitor AT9283 In Vitro. Biomolecules 2020, 10, 1365. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef]
- Taheri, Z.; Asadzadeh Aghdaei, H.; Irani, S.; Modarressi, M.H.; Zahra, N. Evaluation of the Epigenetic Demethylation of NRF2, a Master Transcription Factor for Antioxidant Enzymes, in Colorectal Cancer. Rep. Biochem. Mol. Biol. 2020, 9, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Talens-Visconti, R.; Rius-Perez, S.; Finamor, I.; Sastre, J. Redox signaling in the gastrointestinal tract. Free Radic. Biol. Med. 2017, 104, 75–103. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Q.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Wei, Y.; Jiang, T.; Wang, S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int. J. Clin. Exp. Pathol. 2014, 7, 1124–1131. [Google Scholar] [PubMed]
- Gonzalez-Donquiles, C.; Alonso-Molero, J.; Fernandez-Villa, T.; Vilorio-Marqués, L.; Molina, A.J.; Martín, V. The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS ONE 2017, 12, e0177549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Nat. Prod. Rep. 2020, 37, 797–826. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, H.; Xia, H.; Xu, M.; Pan, G.; Mao, J.; Tao, S.; Yamanaka, K.; An, Y. High NF-E2-related factor 2 expression predicts poor prognosis in patients with lung cancer: A meta-analysis of cohort studies. Free Radic. Res. 2019, 1–9. [Google Scholar] [CrossRef]
- Yu, S.; Wu, T.; Wang, J.; Cheng, C.; Wang, J.; Sun, L.; Liu, C.; Cao, G.; Hu, T. Combined Evaluation of Expression of CXCR4 and Nrf2 as Prognostic Factor for Patients with Gastric Carcinoma. Anti-Cancer Agents Med. Chem. 2018, 18, 388–393. [Google Scholar] [CrossRef]
- Lee, K.; Kim, S.; Lee, Y.; Lee, H.; Lee, Y.; Park, H.; Nahm, J.H.; Ahn, S.; Yu, S.J.; Lee, K.; et al. The Clinicopathological and Prognostic Significance of Nrf2 and Keap1 Expression in Hepatocellular Carcinoma. Cancers 2020, 12, 2128. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharm. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6, e28083. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, F.; Sun, Z.; Zhou, W.; Li, Z.Y.; You, Q.D.; Guo, Q.L.; Hu, R. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol. Carcinog. 2013, 52, 824–834. [Google Scholar] [CrossRef]
- Vartanian, S.; Ma, T.P.; Lee, J.; Haverty, P.M.; Kirkpatrick, D.S.; Yu, K.; Stokoe, D. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol. Cell. Proteom. MCP 2016, 15, 1220–1231. [Google Scholar] [CrossRef]
- Lee, S.; Lim, M.J.; Kim, M.H.; Yu, C.H.; Yun, Y.S.; Ahn, J.; Song, J.Y. An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic. Biol. Med. 2012, 53, 807–816. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Jung, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Siegel, D.; Ross, D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [CrossRef]
- Parkinson, E.I.; Hergenrother, P.J. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. Acc. Chem. Res. 2015, 48, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.J.; Boland, M.P.; Friedlos, F.; Coles, B.; Southan, C.; Roberts, J.J. The nitroreductase enzyme in Walker cells that activates 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) to 5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide is a form of NAD(P)H dehydrogenase (quinone) (EC 1.6.99.2). Biochem. Pharm. 1988, 37, 4671–4677. [Google Scholar] [CrossRef]
- Knox, R.J.; Friedlos, F.; Jarman, M.; Roberts, J.J. A new cytotoxic, DNA interstrand crosslinking agent, 5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide, is formed from 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by a nitroreductase enzyme in Walker carcinoma cells. Biochem. Pharm. 1988, 37, 4661–4669. [Google Scholar] [CrossRef]
- Siegel, D.; Beall, H.; Senekowitsch, C.; Kasai, M.; Arai, H.; Gibson, N.W.; Ross, D. Bioreductive activation of mitomycin C by DT-diaphorase. Biochemistry 1992, 31, 7879–7885. [Google Scholar] [CrossRef]
- Siegel, D.; Gibson, N.W.; Preusch, P.C.; Ross, D. Metabolism of mitomycin C by DT-diaphorase: Role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990, 50, 7483–7489. [Google Scholar]
- Chen, S.; Knox, R.; Wu, K.; Deng, P.S.; Zhou, D.; Bianchet, M.A.; Amzel, L.M. Molecular basis of the catalytic differences among DT-diaphorase of human, rat, and mouse. J. Biol. Chem. 1997, 272, 1437–1439. [Google Scholar] [CrossRef]
- Knox, R.J.; Jenkins, T.C.; Hobbs, S.M.; Chen, S.; Melton, R.G.; Burke, P.J. Bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by human NAD(P)H quinone oxidoreductase 2: A novel co-substrate-mediated antitumor prodrug therapy. Cancer Res. 2000, 60, 4179–4186. [Google Scholar] [PubMed]
- Beall, H.D.; Mulcahy, R.T.; Siegel, D.; Traver, R.D.; Gibson, N.W.; Ross, D. Metabolism of bioreductive antitumor compounds by purified rat and human DT-diaphorases. Cancer Res. 1994, 54, 3196–3201. [Google Scholar] [PubMed]
- Beall, H.D.; Murphy, A.M.; Siegel, D.; Hargreaves, R.H.; Butler, J.; Ross, D. Nicotinamide adenine dinucleotide (phosphate): Quinone oxidoreductase (DT-diaphorase) as a target for bioreductive antitumor quinones: Quinone cytotoxicity and selectivity in human lung and breast cancer cell lines. Mol. Pharmacol. 1995, 48, 499–504. [Google Scholar] [PubMed]
- Danson, S.J.; Johnson, P.; Ward, T.H.; Dawson, M.; Denneny, O.; Dickinson, G.; Aarons, L.; Watson, A.; Jowle, D.; Cummings, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the bioreductive drug RH1. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 1653–1660. [Google Scholar] [CrossRef]
- Phillips, R.M.; Loadman, P.M.; Reddy, G. Inactivation of apaziquone by haematuria: Implications for the design of phase III clinical trials against non-muscle invasive bladder cancer. Cancer Chemother. Pharmacol. 2019, 83, 1183–1189. [Google Scholar] [CrossRef]
- Starcher, C.L.; Pay, S.L.; Singh, N.; Yeh, I.J.; Bhandare, S.B.; Su, X.; Huang, X.; Bey, E.A.; Motea, E.A.; Boothman, D.A. Targeting Base Excision Repair in Cancer: NQO1-Bioactivatable Drugs Improve Tumor Selectivity and Reduce Treatment Toxicity Through Radiosensitization of Human Cancer. Front. Oncol. 2020, 10, 1575. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef]
- Huang, X.; Dong, Y.; Bey, E.A.; Kilgore, J.A.; Bair, J.S.; Li, L.S.; Patel, M.; Parkinson, E.I.; Wang, Y.; Williams, N.S.; et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012, 72, 3038–3047. [Google Scholar] [CrossRef]
- Guo, W.; Reigan, P.; Siegel, D.; Zirrolli, J.; Gustafson, D.; Ross, D. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: Role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res. 2005, 65, 10006–10015. [Google Scholar] [CrossRef]
- Guo, W.; Reigan, P.; Siegel, D.; Zirrolli, J.; Gustafson, D.; Ross, D. The bioreduction of a series of benzoquinone ansamycins by NAD(P)H:quinone oxidoreductase 1 to more potent heat shock protein 90 inhibitors, the hydroquinone ansamycins. Mol. Pharmacol. 2006, 70, 1194–1203. [Google Scholar] [CrossRef]
- Baird, L.; Suzuki, T.; Takahashi, Y.; Hishinuma, E.; Saigusa, D.; Yamamoto, M. Geldanamycin-derived HSP90 Inhibitors are Synthetic Lethal with NRF2. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. NRF2-dependent bioactivation of mitomycin C as a novel strategy to target KEAP1-NRF2 pathway activation in human cancer. Mol. Cell. Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.; Chang, C.H.; Xiong, R.; Williams, H.E.; Davis, A.L.; Lewis, W.; Dehn, D.L.; Siegel, D.; Roe, S.M.; Prodromou, C.; et al. Synthesis of 19-substituted geldanamycins with altered conformations and their binding to heat shock protein Hsp90. Nat. Chem. 2013, 5, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Drechsel, D.A.; Kitson, R.R.; Siegel, D.; You, Q.; Backos, D.S.; Ju, C.; Moody, C.J.; Ross, D. 19-substituted benzoquinone ansamycin heat shock protein-90 inhibitors: Biological activity and decreased off-target toxicity. Mol. Pharmacol. 2014, 85, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]







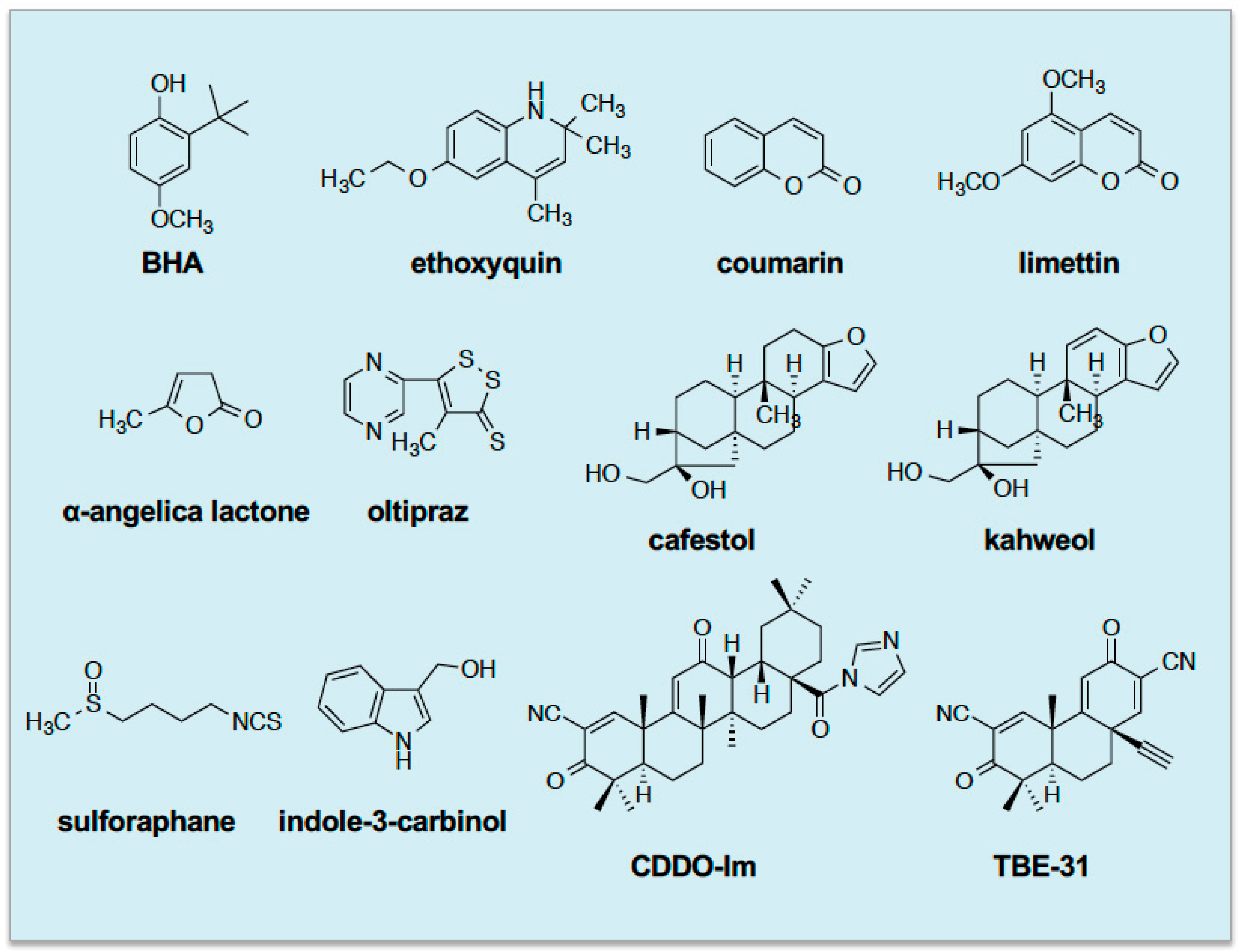{kind=link}
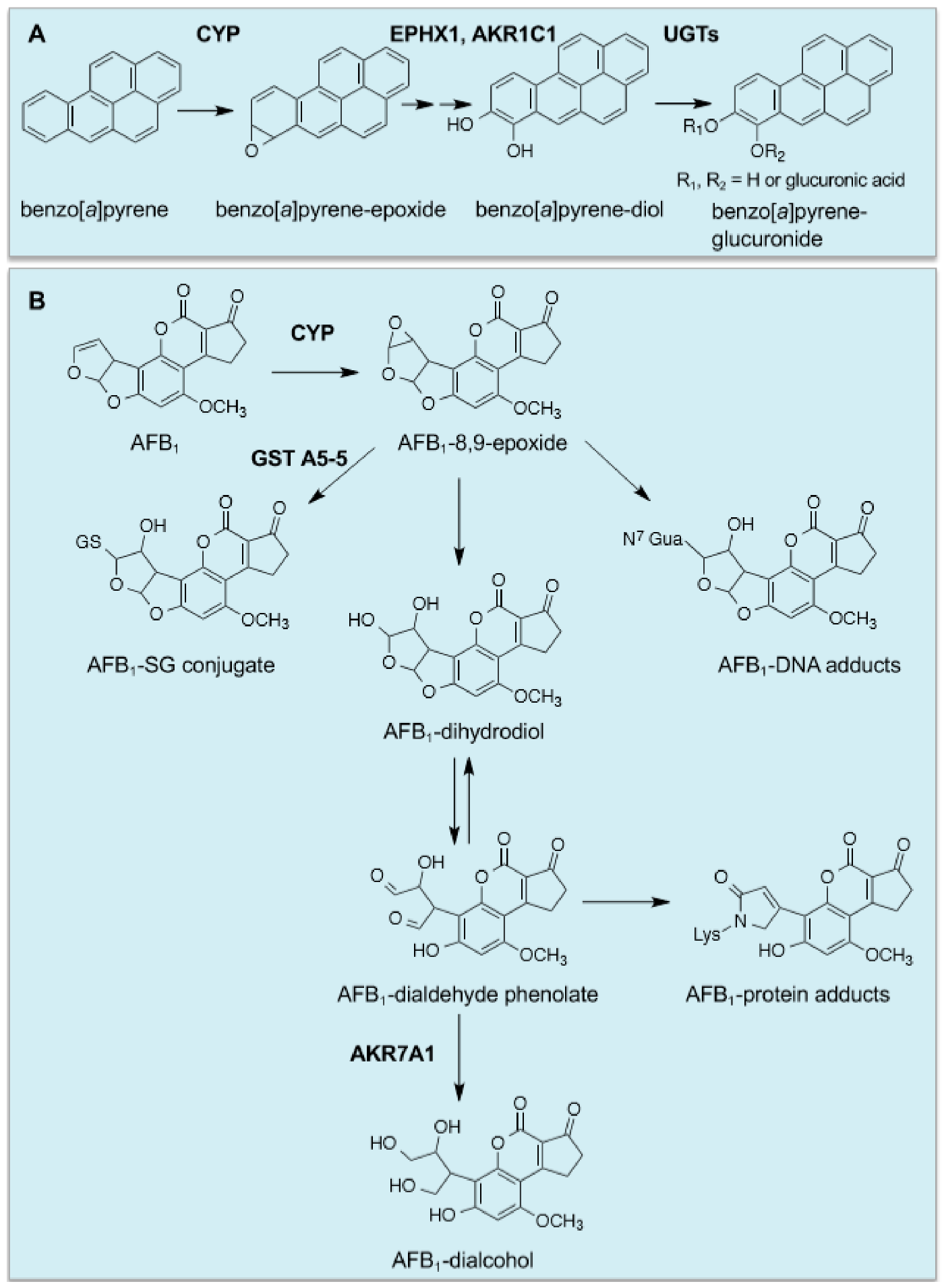{kind=link}
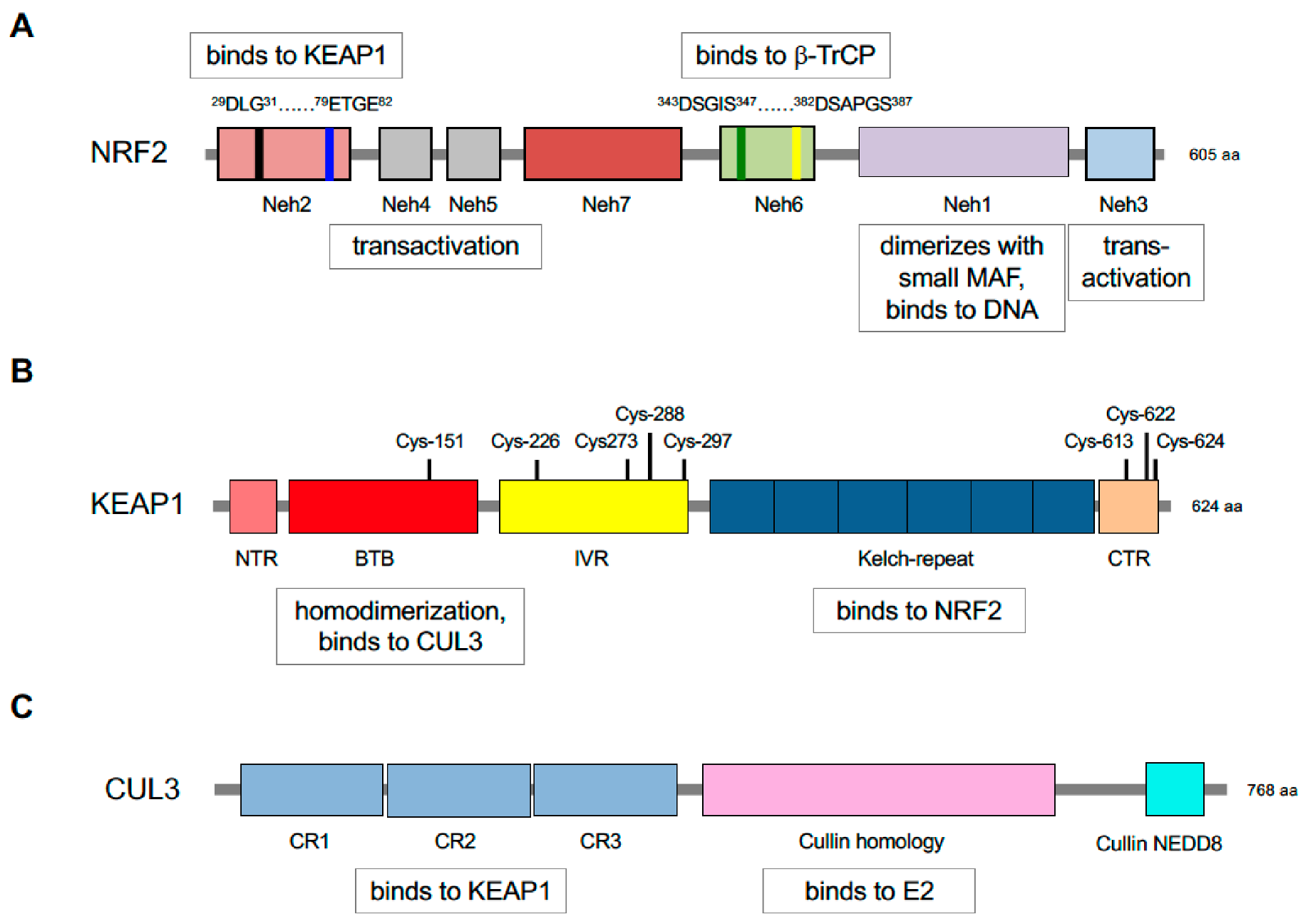{kind=link}
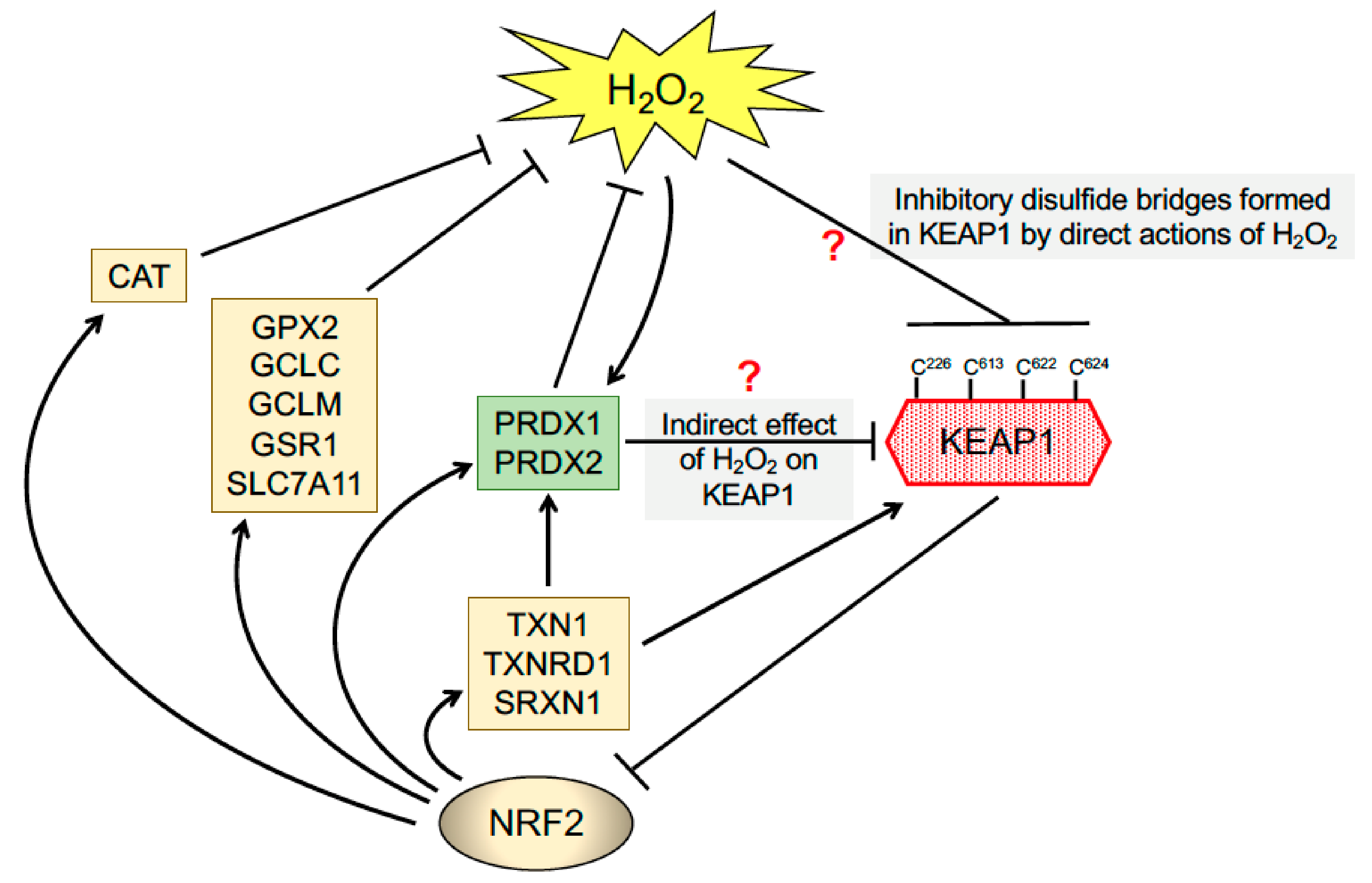{kind=link}
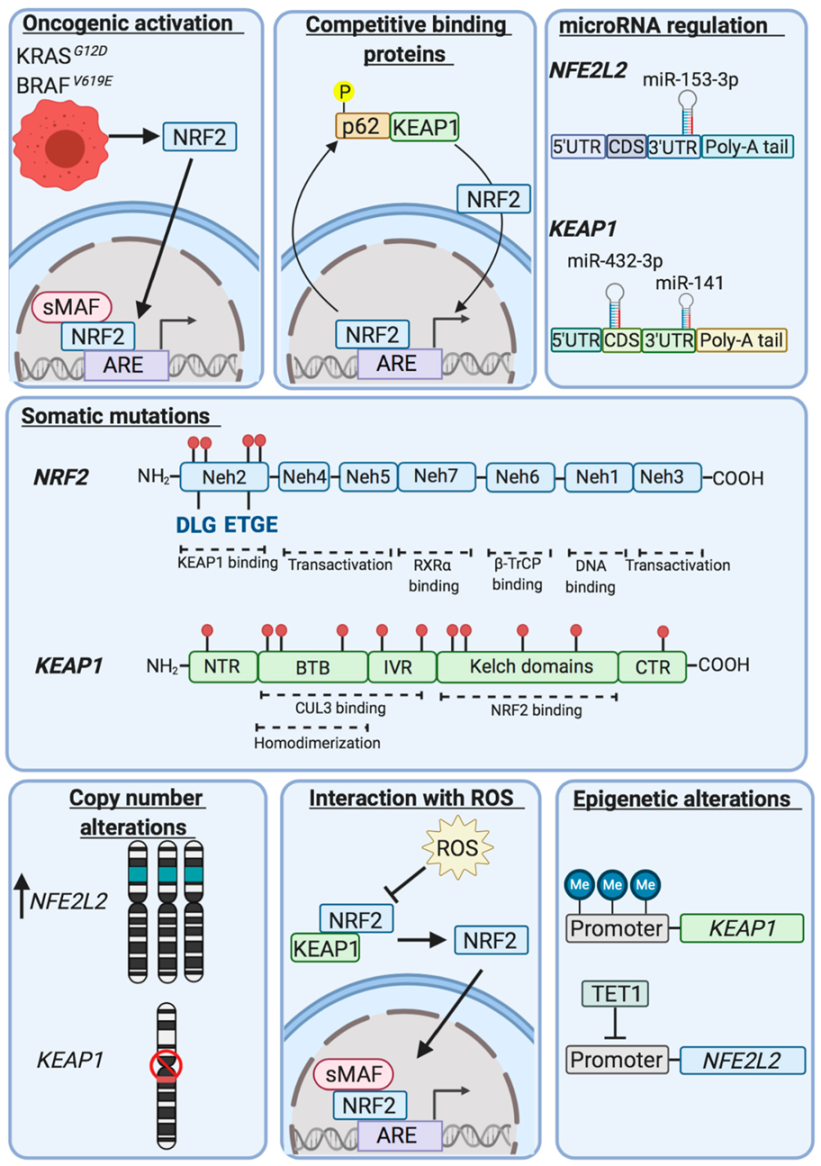{kind=link}
{kind=link}
| Function | Gene |
|---|---|
| Phase I drug oxidation, reduction and hydrolysis | AKR1B10, AKR1C1, AKR1C2, AKR1C3, ALDH1A1, ALDH3A1, ALDH7A1, CBR1, CES1G, CES1H, EPHX1, NQO1, PTGR1 |
| Phase II drug conjugation * | GSTA1, GSTA3, GSTM1, GSTP1, MGST1, SULT1A1, UGT1A1, UGT1A6, UGT2B7 |
| Phase III drug efflux | ABCB6, ABCC1, ABCC2, ABCC3 |
| Scavenging of H2O2 | CAT, GPX2, GPX4, PRDX1, PRDX6 |
| Iron/heme metabolism | BLVRB, FECH, FPN1 (also called SLC40A1), FTH1, FTL1, HMOX1 |
| Contribution to maintenance of the GSH-based antioxidant system | GCLC, GCLM, GGT1, GSR1, SLC7A11 |
| Contribution to maintenance of the TXN1-based antioxidant system | TXN1, TXNRD1, SRXN1 |
| NADPH generation to provide reducing equivalents for GSH and TXN1 systems | G6PD, PGD, IDH1, ME1 |
| Non-oxidative arm of the pentose phosphate pathway ** | TALDO1, TKT |
| Import of fatty acids | CD36 |
| Scavenger receptor | MARCO |
| Proteasomal subunits | PSMA1, PSMA4, PSMA7, PSMB1, PSMB2, PSMB3, PSMB4, PSMB5, PSMB6, PSMC1, PSMC3, PSMD1, PSMD5, PSMD7, PSMD11, PSMD12, PSMD13, PSME1 (encoding PA28αβ) |
| Autophagy-related proteins | SQSTM1 (encoding p62), CALCOCO2, ULK1, ATG2, ATG5, ATG7, GABARAPL1, LAMP2 |
| Transcription factors and associated factors | AHR, ATF3, ATF4, CEBPB (encoding C/EBPβ), MAFG, PPARG (encoding PPARγ, or NR1C3), PPARGC1B, RXRA (encoding RXRα, or NR2B1) |
| Ubiquitin ligase substrate adaptor | KEAP1 |
| Positive regulation of response to oxidative stress | NFE2L2 |
| Activation of Ca+2 channel signalling | TRPA1 |
| Activation of Notch signalling | NOTCH1 |
| Attenuation of hedgehog signalling *** | PTCH1 |
| Cancer Type | Percentage of Somatic Mutation | Reference | Samples Analysed | ||
|---|---|---|---|---|---|
| KEAP1 | NFE2L2 | CUL3 | |||
| Lung | 14.2% | 7.3% | 3.4% | [153] | Whole-exome sequencing of 660 LUAD and 484 LUSC tumour/normal pairs |
| Oesophageal | 3.4% | 4.5% | 1.1% | [154] | Whole-genome or whole-exon sequencing of 88 ESCC tumour/normal pairs |
| Liver | 5.1% | 4.0% | 0% | [155] | Hepatocellular carcinomas 369 and 3 fibrolamellar carcinoma |
| Head and neck | 4.3% | 6.1% | 3.6% | [156] | Whole exome sequencing and/or whole genome sequencing of 279 head and neck squamous cell carcinoma tumour/normal pairs |
| Gastric | 3.1% | 0.3% | 2.0% | [157] | Whole exome sequencing of 295 primary gastric adenocarcinomas tumours with matched normal |
| Bladder | 3.1% | 8.7% | 0% | [158] | Whole exome sequencing of 131 high grade muscle invasive urothelial bladder carcinomas |
| Colorectal | 1.4% | 0.9% | 0% | [159] | Whole exome sequencing in 224 of the 276 colorectal carcinoma tumour/normal pairs |
| Pan-Lung Cancer | ||
|---|---|---|
| Alteration | ||
| Gene Name | Number of Samples with the Mutation | Number of Samples without the Mutation |
| NFE2L2 | 105 (9%) | 1039 (91%) |
| KEAP1 | 170 (15%) | 974 (85%) |
| CUL3 | 51 (5%) | 1093 (96%) |
| KRAS | 259 (23%) | 885 (77%) |
| TP53 | 776 (68%) | 368 (32%) |
| PIK3CA | 276(24%) | 868 (76%) |
| PTEN | 102 (9%) | 1042 (91%) |
| STK11 | 118 (10%) | 1026 (90%) |
| KEAP1 Mutant Samples | |||
|---|---|---|---|
| Gene | Alteration | Co-Occurrence or Mutual Exclusivity with KEAP1 Mutation | |
| Number of KEAP1 Mutant Samples with the Alteration | Number of KEAP1 Wildtype Samples with the Alteration | ||
| NFE2L2 | 4(2.4%) | 80 (8.2%) | Mutual exclusivity |
| CUL3 | 6 (3.5%) | 33(3.4%) | Co-occurrence |
| KRAS | 41 (24.1%) | 181 (18.6%) | Co-occurrence |
| TP53 | 103 (60.6%) | 672 (69%) | Mutual exclusivity |
| PIK3CA | 12 (7.1%) | 82(8.4%) | Mutual exclusivity |
| PTEN | 6 (3.5%) | 61(6.3%) | Mutual exclusivity |
| STK11 | 39 (22.9%) | 72 (7.4%) | Co-occurrence |
| NFE2L2 Mutant Samples | |||
|---|---|---|---|
| Gene | Alteration | Co-Occurrence or Mutual Exclusivity with NFE2L2 Mutation | |
| Number of NFE2L2 Mutant Samples with the Alteration | Number of NFE2L2 Wildtype Samples with the Alteration | ||
| KEAP1 | 6(6.7%) | 155(15%) | Mutual exclusivity |
| CUL3 | 5 (4.8%) | 34(3.3%) | Co-occurrence |
| KRAS | 5 (4.8%) | 217 (20.9%) | Mutual exclusivity |
| TP53 | 88 (83.8%) | 687 (66.1%) | Co-occurrence |
| PIK3CA | 12 (11.4%) | 82 (7.9%) | Co-occurrence |
| PTEN | 5 (4.8%) | 62 (6%) | Mutual exclusivity |
| STK11 | 3 (2.9%) | 108 (10.4%) | Mutual exclusivity |
| CUL3 Mutant Samples | |||
|---|---|---|---|
| Gene | Alteration | Co-Occurrence or Mutual Exclusivity with CUL3 Mutation | |
| Number of CUL3 Mutant Samples with the Alteration | Number of CUL3 Wildtype Samples with the Alteration | ||
| NFE2L2 | 5 (9.8%) | 79 (7.2%) | Co-occurrence |
| KEAP1 | 6 (11.8%) | 156 (14.3%) | Mutual exclusivity |
| KRAS | 4 (7.8%) | 218 (20%) | Mutual exclusivity |
| TP53 | 42 (82.4%) | 733 (67.1%) | Co-occurrence |
| PIK3CA | 6 (11.8%) | 88 (8.1%) | Co-occurrence |
| PTEN | 4 (7.8%) | 63 (5.8%) | Co-occurrence |
| STK11 | 3 (5.9%) | 108 (9.9%) | Mutual exclusivity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers 2020, 12, 3609. https://doi.org/10.3390/cancers12123609
Robertson H, Dinkova-Kostova AT, Hayes JD. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers. 2020; 12(12):3609. https://doi.org/10.3390/cancers12123609
Chicago/Turabian StyleRobertson, Holly, Albena T. Dinkova-Kostova, and John D. Hayes. 2020. "NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis" Cancers 12, no. 12: 3609. https://doi.org/10.3390/cancers12123609
APA StyleRobertson, H., Dinkova-Kostova, A. T., & Hayes, J. D. (2020). NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers, 12(12), 3609. https://doi.org/10.3390/cancers12123609