Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma

Abstract
Simple Summary
Abstract
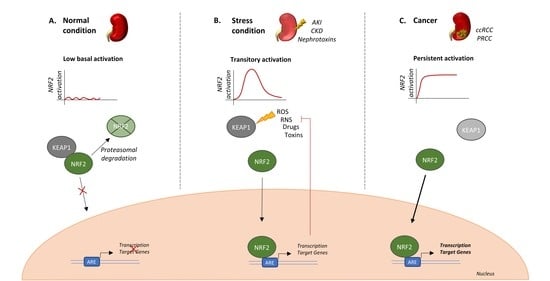
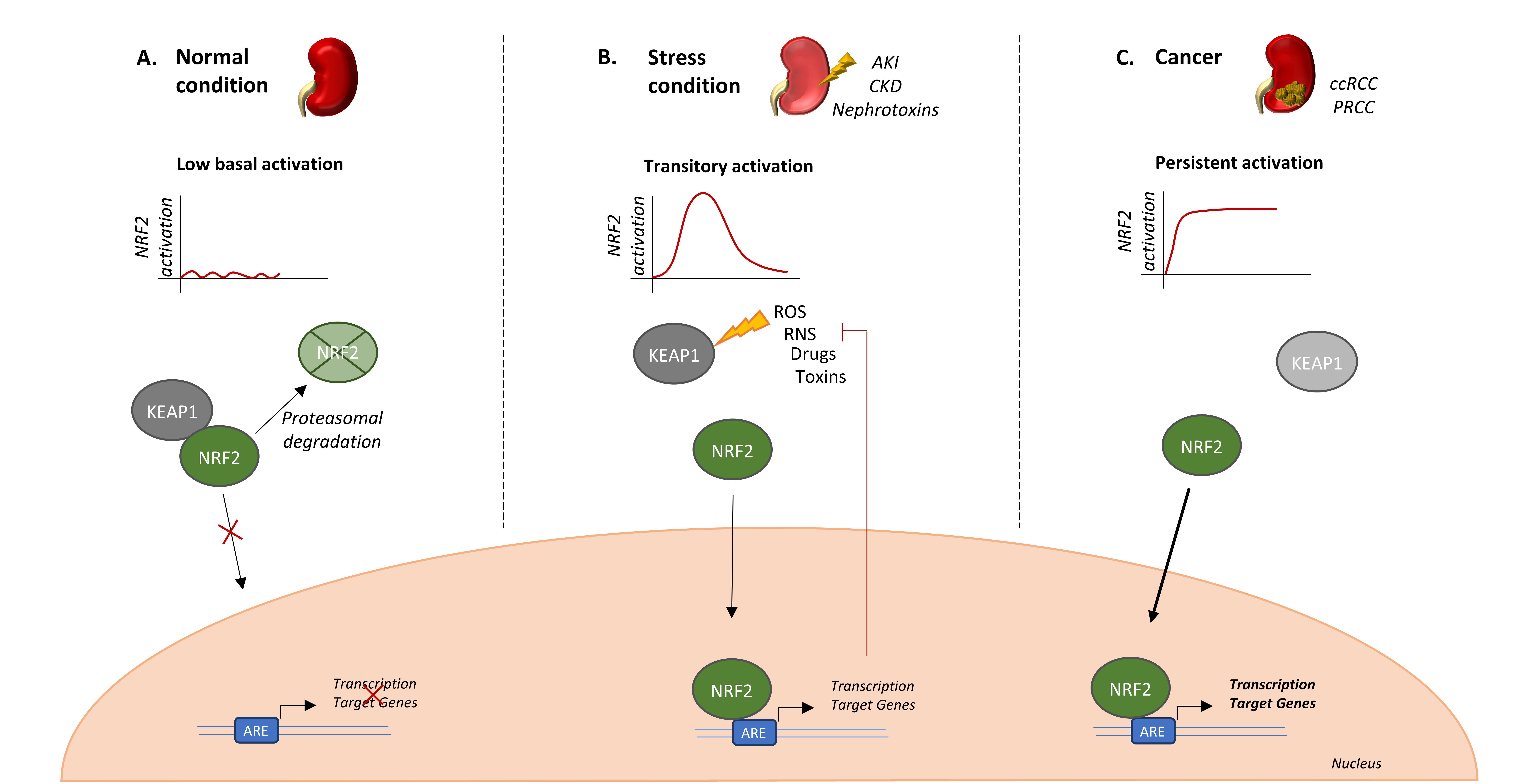
1. Introduction
2. NRF2 Expression in PRCC and ccRCC
3. NRF Hyperactivation Supports RCC
3.1. Balancing Cytoprotection and Chemoresistance
3.2. Epithelial-to-Mesenchymal Transition
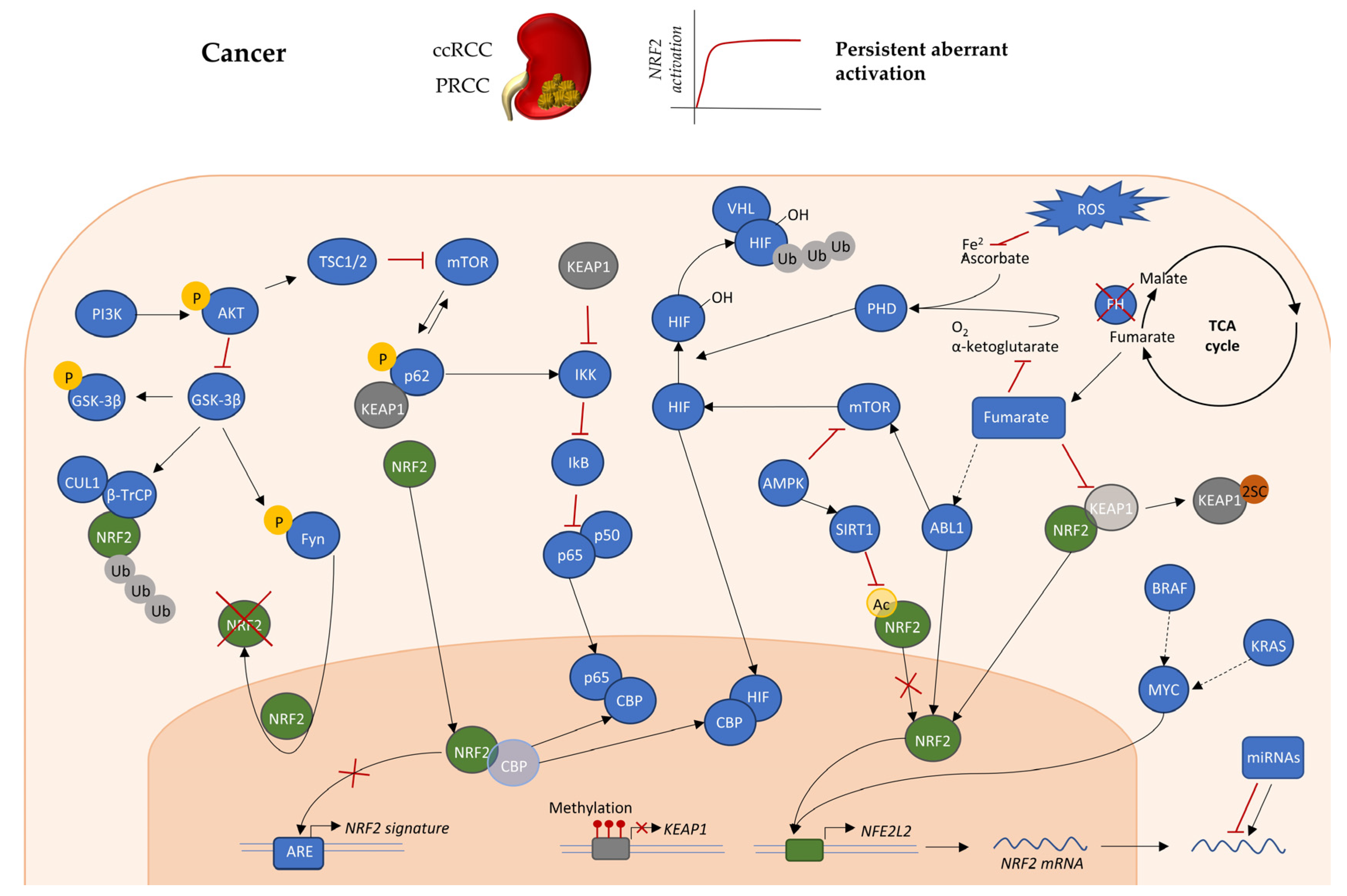
4. NRF2 Regulation in RCC
4.1. Mutations in Genes that Directly or Indirectly Affect KEAP1-NRF2
4.2. Epigenetic Regulation of KEAP1-NRF2 Axis
4.3. Oncogenes and Transcriptional Regulation of KEAP1-NRF2 Axis
4.4. Post-Translational Modifications Affecting the KEAP1-NRF2 Axis
4.5. The Role of Fumarate in Regulation of KEAP1
5. Conclusions and Highlights about Therapeutical Strategies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements erythroid transcription factor NF-E2 is a hetero. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2013, 6, 371–379. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N. Roles of NRF2 in protecting the kidney from oxidative damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Zhang, D.D. Filtering through the role of NRF2 in kidney disease. Arch. Pharm. Res. 2020, 43, 361–369. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Liu, Z.; Lu, Y.; Zhu, X.; Lan, B.; Mi, Z.; Dang, L.; Li, N.; Zhan, W.; et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci. Transl. Med. 2020, 12, eaba3613. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Linehan, W.M.; Ricketts, C.J. The cancer genome atlas of renal cell carcinoma: Findings and clinical implications. Nat. Rev. Urol. 2019, 16, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Robertson, G.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensivemolecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Vocke, C.D.; et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; de Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, Y.; Şenbabaoğlu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; Shuch, B.; Micevic, G.; de Velasco, G.; Shinbrot, E.; et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016, 14, 2476–2489. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx Renal. Cell 2018, 173, 611–623.e17. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef]
- Pal, S.K.; Ali, S.M.; Yakirevich, E.; Geynisman, D.M.; Karam, J.A.; Elvin, J.A.; Frampton, G.M.; Huang, X.; Lin, D.I.; Rosenzweig, M.; et al. Characterization of clinical cases of advanced papillary renal cell carcinoma via comprehensive genomic profiling. Eur. Urol. 2018, 73, 71–78. [Google Scholar] [CrossRef]
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer the multiple leiomyoma consortium. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.H.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An Antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Sourbier, C.; Ricketts, C.J.; Matsumoto, S.; Crooks, D.R.; Liao, P.J.; Mannes, P.Z.; Yang, Y.; Wei, M.H.; Srivastava, G.; Ghosh, S.; et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 2014, 26, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- Yuki, H.; Kamai, T.; Murakami, S.; Higashi, S.; Narimatsu, T.; Kambara, T.; Betsunoh, H.; Abe, H.; Arai, K.; Shirataki, H.; et al. Increased Nrf2 expression by renal cell carcinoma is associated with postoperative chronic kidney disease and an unfavorable prognosis. Oncotarget 2018, 9, 28351–28363. [Google Scholar] [CrossRef][Green Version]
- Yamaguchi, Y.; Kamai, T.; Higashi, S.; Murakami, S.; Arai, K.; Shirataki, H.; Yoshida, K.I. Nrf2 gene mutation and single nucleotide polymorphism rs6721961 of the Nrf2 promoter region in renal cell cancer. BMC Cancer 2019, 19, 1137. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Costantini, M.; Copetti, M.; la Torre, A.; Sparaneo, A.; Fontana, A.; Poeta, L.; Gallucci, M.; Sentinelli, S.; Graziano, P.; et al. Keap1/Nrf2 pathway in kidney cancer: Frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 2017, 8, 11187–11198. [Google Scholar] [CrossRef]
- Huang, H.; Wu, Y.; Fu, W.; Wang, X.; Zhou, L.; Xu, X.; Huang, F.; Wu, Y. Downregulation of Keap1 contributes to poor prognosis and Axitinib resistance of renal cell carcinoma via upregulation of Nrf2 expression. Int. J. Mol. Med. 2019, 43, 2044–2054. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, Y.; Zhao, P.; Weng, W.; Ye, M.; Sun, H.; Xu, M.; Wang, C. The NRF2/HO-1 axis can be a prognostic factor in clear cell renal cell carcinoma. Cancer Manag. Res. 2019, 11, 1221–1230. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ricketts, C.J.; Sourbier, C.; Linehan, W.M. New strategies in renal cell carcinoma: Targeting the genetic and metabolic basis of disease. Clin. Cancer Res. 2015, 21, 10–17. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Noé, G.; Cerles, O.; Cabel, L.; Kavian-Tessler, N.; Chouzenoux, S.; Bahuaud, M.; Chéreau, C.; Nicco, C.; Leroy, K.; et al. Dimethyl fumarate controls the NRF2/DJ-1 Axis in cancer cells: Therapeutic applications. Mol. Cancer Ther. 2017, 16, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Meierhofer, D. Glutathione metabolism in renal cell carcinoma progression and implications for therapies. Int. J. Mol. Sci. 2019, 20, 3672. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef]
- Levings, D.C.; Wang, X.; Kohlhase, D.; Bell, D.A.; Slattery, M. A distinct class of antioxidant response elements is consistently activated in tumors with NRF2 mutations. Redox Biol. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef]
- Joo, M.S.; Lee, C.G.; Koo, J.H.; Kim, S.G. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013, 4, e899. [Google Scholar] [CrossRef]
- Cao, X.; Nie, X.; Xiong, S.; Cao, L.; Wu, Z.; Moore, P.K.; Bian, J.S. Renal protective effect of polysulfide in cisplatin-induced nephrotoxicity. Redox Biol. 2018, 15, 513–521. [Google Scholar] [CrossRef]
- Li, F.; Yao, Y.; Huang, H.; Hao, H.; Ying, M. Xanthohumol attenuates cisplatin-induced nephrotoxicity through inhibiting NF-κB and activating Nrf2 signaling pathways. Int. Immunopharmacol. 2018, 61, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 97, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Xiong, Y.; Zhao, X.; Liu, Y.; Yu, L.Q. Effect of the nrf2-are signaling pathway on biological characteristics and sensitivity to sunitinib in renal cell carcinoma. Oncol. Lett. 2019, 17, 5175–5186. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Cai, L.C.; Wang, X.Y.; Cheng, S.Y.; Zhang, D.M.; Jian, W.G.; Wang, T.D.; Yang, J.K.; Yang, K.B.; Zhang, C. BMP8A promotes survival and drug resistance via Nrf2/TRIM24 signaling pathway in clear cell renal cell carcinoma. Cancer Sci. 2020, 111, 1555–1566. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Santoni, M.; Occhipinti, G.; Scarpelli, M.; Lopez-Beltran, A.; Cheng, L.; Principato, G.; Montironi, R. Epithelial to mesenchymal transition in renal cell carcinoma: Implications for cancer therapy. Mol. Diagn. Ther. 2016, 20, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef]
- Shibata, T.; Saito, S.; Kokubu, A.; Suzuki, T.; Yamamoto, M.; Hirohashi, S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2—Related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010, 70, 9095–9105. [Google Scholar] [CrossRef]
- Sampson, V.B.; David, J.M.; Puig, I.; Patil, P.U.; de Herreros, A.G.; Thomas, G.V.; Rajasekaran, A.K. Wilms’ tumor protein induces an epithelial-mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS ONE 2014, 9, e102041. [Google Scholar] [CrossRef]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. 2019, 11, 251–263. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Onuchic, J.N.; Levine, H.; Ben-Jacob, E. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.D.; Kim, Y.W.; Cho, I.J.; Lee, C.H.; Kim, S.G. E-cadherin inhibits nuclear accumulation of Nrf2: Implications for chemoresistance of cancer cells. J. Cell Sci. 2012, 125, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to mesenchymal transition: A mechanism that fuels cancer radio/chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Müller, G.A.; Colasanti, G.; D’amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef]
- Ivanova, L.; Butt, M.J.; Matsell, D.G. Mesenchymal transition in kidney collecting duct epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F1238–F1248. [Google Scholar] [CrossRef]
- Zhou, P.; Yu, J.F.; Zhao, C.G.; Sui, F.X.; Teng, X.; Wu, Y.B. Therapeutic potential of EGCG on acute renal damage in a rat model of obstructive nephropathy. Mol. Med. Rep. 2013, 7, 1096–1102. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Ha, H.; Kwak, M.K. Inhibitory role of the KEAP1-NRF2 pathway in TGFbβ1-stimulated renal epithelial transition to fibroblastic cells: A modulatory effect on SMAD signaling. PLoS ONE 2014, 9, e93265. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Shin, D.H.; Kang, K.S.; Kwak, M.K. Involvement of Nrf2-GSH signaling in TGFβ1-stimulated epithelial-to-mesenchymal transition changes in rat renal tubular cells. Arch. Pharm. Res. 2015, 38, 272–281. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Su, X.; Zhou, G.; Sun, G.; Du, F.; Bian, X.; Wang, B. Epigallocatechin-3-gallate attenuates transforming growth factor-β1 induced epithelial-mesenchymal transition via Nrf2 regulation in renal tubular epithelial cells. Biomed. Pharmacother. 2015, 70, 260–267. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, D.; Guo, L.; Liang, W.; Jiang, Y.; Li, H.; Zhao, Y.; Lu, S.; Chi, Z.H. Curcumin protects renal tubular epithelial cells from high glucose-induced epithelial-to-mesenchymal transition through Nrf2-mediated upregulation of heme oxygenase-1. Mol. Med. Rep. 2015, 12, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.; Arend, L.J.; Bandapalle, S.; Reddy, S.P.; Rabb, H. Kidney epithelium specific deletion of kelch-like ECH-associated protein 1 (Keap1) causes hydronephrosis in mice. BMC Nephrol. 2016, 17, 110. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Seki, S.; Hiramoto, K.; Naganuma, E.; Kobayashi, E.H.; Yamaoka, A.; Baird, L.; Takahashi, N.; Sato, H.; Yamamoto, M. Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat. Commun. 2017, 8, 14577. [Google Scholar] [CrossRef] [PubMed]
- Teshiba, R.; Tajiri, T.; Sumitomo, K.; Masumoto, K.; Taguchi, T.; Yamamoto, K. Identification of a KEAP1 germline mutation in a family with multinodular goitre. PLoS ONE 2013, 8, e65141. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef]
- Kovac, M.; Navas, C.; Horswell, S.; Salm, M.; Bardella, C.; Rowan, A.; Stares, M.; Castro-Giner, F.; Fisher, R.; de Bruin, E.C.; et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat. Commun. 2015, 6, 6336. [Google Scholar] [CrossRef]
- Thiesen, H.J.; Steinbeck, F.; Maruschke, M.; Koczan, D.; Ziems, B.; Hakenberg, O.W. Stratification of clear cell renal cell carcinoma (ccRCC) genomes by gene-directed copy number alteration (CNA) analysis. PLoS ONE 2017, 12, e0176659. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoh, K.; Kobayashi, A.; Ishii, Y.; Kure, S.; Koyama, A.; Sakamoto, T.; Sekizawa, K.; Motohashi, H.; Yamamoto, M. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 2004, 321, 72–79. [Google Scholar] [CrossRef]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.-Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Dykema, K.; Ansari, A.; Petillo, D.; Snider, J.; Kahnoski, R.; Anema, J.; Craig, D.; Carpten, J.; Teh, B.T.; et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013, 73, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Jeh, S.U.; Park, J.J.; Lee, J.S.; Kim, D.C.; Do, J.; Lee, S.W.; Choi, S.M.; Hyun, J.S.; Seo, D.H.; Lee, C.; et al. Differential expression of the sirtuin family in renal cell carcinoma: Aspects of carcinogenesis and prognostic significance. Urol. Oncol. 2017, 35, 675.e9–675.e15. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Y.; Sheng, Y.; Xiao, J.; Xiao, Y.; Cheng, N.; Chai, Y.; Wu, X.; Zhang, S.; Xiang, T. SIRT1 downregulated FGB expression to inhibit RCC tumorigenesis by destabilizing STAT3. Exp. Cell Res. 2019, 382, 111466. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Li, B.; Peng, F.; Gong, G.; Li, N. Integrative analysis of sirtuins and their prognostic significance in clear cell renal cell carcinoma. Front. Oncol. 2020, 10, 218. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Arai, E.; Sakamoto, H.; Ichikawa, H.; Totsuka, H.; Chiku, S.; Gotoh, M.; Mori, T.; Nakatani, T.; Ohnami, S.; Nakagawa, T.; et al. Multilayer-omics analysis of renal cell carcinoma, including the whole exome, methylome and transcriptome. Int. J. Cancer 2014, 135, 1330–1342. [Google Scholar] [CrossRef]
- Durinck, S.; Stawiski, E.W.; Pavía-Jiménez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Trombetta, D.; Muscarella, L.A. Epigenetic versus genetic deregulation of the KEAP1/NRF2 axis in solid tumors: Focus on methylation and noncoding RNAs. Oxidative Med. Cell. Longev. 2018, 2018, 2492063. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Zhang, Y.F.; Xia, Y.F.; Zhou, Z.M.; Cao, Y.Q. Promoter demethylation of nuclear factor-erythroid 2-related factor 2 gene in drug-resistant colon cancer cells. Oncol. Lett. 2015, 10, 1287–1292. [Google Scholar] [CrossRef]
- Ah Kang, K.; Piao, M.J.; Ryu, Y.S.; Kang, H.K.; Chang, W.Y.; Keum, Y.S.; Hyun, J.W. Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5-fluorouracil-resistant colon cancer cells. Oncotarget 2016, 7, 40594–40620. [Google Scholar] [CrossRef]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Wu, R.; Xia, M.; Fei, X.; He, Q.E.; Zha, C.; Wu, F. Identification of eight key miRNAs associated with renal cell carcinoma: A meta-analysis. Oncol. Lett. 2018, 16, 5847–5855. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Saleeb, R.; Kim, S.S.; Ding, Q.; Scorilas, A.; Lin, S.; Khella, H.W.; Boulos, C.; Ibrahim, G.; Yousef, G.M. The miR-200 family as prognostic markers in clear cell renal cell carcinoma. Urol. Oncol. 2019, 37, 955–963. [Google Scholar] [CrossRef]
- Gilyazova, I.R.; Klimentova, E.A.; Bulygin, K.V.; Izmailov, A.A.; Bermisheva, M.A.; Galimova, E.F.; Safiullin, R.I.; Galimov, S.N.; Pavlov, V.N.; Khusnutdinova, E.K. MicroRNA-200 family expression analysis in metastatic clear cell renal cell carcinoma patients. Cancer Gene Ther. 2010, 27, 768–772. [Google Scholar] [CrossRef]
- Li, Y.; Guan, B.; Liu, J.; Zhang, Z.; He, S.; Zhan, Y.; Su, B.; Han, H.; Zhang, X.; Wang, B.; et al. MicroRNA-200b is downregulated and suppresses metastasis by targeting LAMA4 in renal cell carcinoma. EBioMedicine 2019, 44, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Peng, F.H.; Peng, L.K. MiR-200c sensitizes clear-cell renal cell carcinoma cells to sorafenib and imatinib by targeting heme oxygenase-1. Neoplasma 2014, 61, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Yao, Y.; Yang, M.; Zhang, Y.; Chumsri, S.; Zhou, Q. miR-200a regulates SIRT1 expression and Epithelial to Mesenchymal Transition (EMT)-like transformation in mammary epithelial cells. J. Biol. Chem. 2011, 286, 25992–26002. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating Human Hepatocellular Carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.S.; Lee, D.K.; Kim, J.; Kwak, S.N.; Ha, K.S.; Choe, J.; Won, M.H.; Cho, B.R.; Jeoung, D.; et al. Hypoxia-responsive MicroRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid. Redox Signal. 2014, 21, 2469–2482. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Kurozumi, A.; Nohata, N.; Kojima, S.; Matsushita, R.; Yoshino, H.; Yamazaki, K.; Ishida, Y.; Ichikawa, T.; Naya, Y.; et al. The microRNA signature of patients with sunitinib failure: Regulation of UHRF1 pathways by microRNA-101 in renal cell carcinoma. Oncotarget 2016, 7, 59070–59086. [Google Scholar] [CrossRef]
- Yamada, Y.; Nohata, N.; Uchida, A.; Kato, M.; Arai, T.; Moriya, S.; Mizuno, K.; Kojima, S.; Yamazaki, K.; Naya, Y.; et al. Replisome genes regulation by antitumor miR-101-5p in clear cell renal cell carcinoma. Cancer Sci. 2020, 111, 1392–1406. [Google Scholar] [CrossRef]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef]
- Tang, S.W.; Chang, W.H.; Su, Y.C.; Chen, Y.C.; Lai, Y.H.; Wu, P.T.; Hsu, C.I.; Lin, W.C.; Lai, M.K.; Lin, J.Y. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009, 273, 35–43. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Trand, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef]
- Kozma, L.; Kiss, I.; Nagy, A.; Szakáll, S.; Ember, I. Investigation of c-myc and K-ras amplification in renal clear cell adenocarcinoma. Cancer Lett. 1997, 111, 127–131. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.K.; Baker, A.C.; DeBiasi, R.M.; Winckler, W.; LaFramboise, T.; Lin, W.M.; Wang, M.; Feng, W.; Zander, T.; MacConnaill, L.E.; et al. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 2007, 39, 347–351. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Castiglione, F.; Martignoni, G.; Cheng, L.; Montironi, R.; Lopez-Beltran, A. Unlike in clear cell renal cell carcinoma, KRAS is not mutated in multilocular cystic clear cell renal cell neoplasm of low potential. Virchows Arch. 2015, 467, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Gattenlöhner, S.; Etschmann, B.; Riedmiller, H.; Müller-Hermelink, H.K. Lack of KRAS and BRAF mutation in renal cell carcinoma. Eur. Urol. 2009, 55, 1490–1491. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Basu, A.; Datta, D.; Gasser, M.; Waaga-Gasser, A.M.; Pal, S. The heme oxygenase-1 protein is overexpressed in human renal cancer cells following activation of the Ras-Raf-ERK pathway and mediates anti-apoptotic signal. J. Biol. Chem. 2011, 286, 33580–33590. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediat. Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 Ligase-mediated downregulation of NF-κB signaling by targeting IKKβ. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-κB signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- An, J.; Rettig, M.B. Mechanism of von hippel-lindau protein-mediated suppression of nuclear factor kappa B activity. Mol. Cell. Biol. 2005, 25, 7546–7556. [Google Scholar] [CrossRef]
- An, J.; Fisher, M.; Rettig, M.B. VHL expression in renal cell carcinoma sensitizes to bortezomib (PS-341) through an NF-κB-dependent mechanism. Oncogene 2005, 24, 1563–1570. [Google Scholar] [CrossRef]
- Oya, M.; Takayanagi, A.; Horiguchi, A.; Mizuno, R.; Ohtsubo, M.; Marumo, K.; Shimizu, N.; Murai, M. Increased nuclear factor-κB activation is related to the tumor development of renal cell carcinoma. Carcinogenesis 2003, 24, 377–384. [Google Scholar] [CrossRef]
- Ng, K.L.; Yap, N.Y.; Rajandram, R.; Small, D.; Pailoor, J.; Ong, T.A.; Razack, A.H.; Wood, S.T.; Morais, C.; Gobe, G.C. Nuclear factor-kappa B subunits and their prognostic cancer-specific survival value in renal cell carcinoma patients. Pathology 2018, 50, 511–518. [Google Scholar] [CrossRef]
- Morais, C.; Gobe, G.; Johnson, D.W.; Healy, H. The emerging role of nuclear factor kappa B in renal cell carcinoma. Int. J. Biochem. Cell Biol. 2011, 43, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the Nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT pathway and renal cell carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; Ruiz De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-Kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef]
- Harrison, E.M.; McNally, S.J.; Devey, L.; Garden, O.J.; Ross, J.A.; Wigmore, S.J. Insulin induces heme oxygenase-1 through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in renal cells. FEBS J. 2006, 273, 2345–2356. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011, 25, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Zhang, H.F.; Wang, J.H.; Wang, Y.L.; Gao, C.; Gu, Y.T.; Huang, J.; Zhang, Z. Salvianolic acid A protects the kidney against oxidative stress by activating the Akt/GSK-3 β /Nrf2 signaling pathway and inhibiting the NF- B signaling pathway in 5/6 nephrectomized rats. Oxidative Med. Cell. Longev. 2019, 2019, 2853534. [Google Scholar] [CrossRef]
- Anraku, T.; Kuroki, H.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.; Giles, F.J.; Ugolkov, A.; Tomita, Y. Clinically relevant GSK 3β inhibitor 9 ING 41 is active as a single agent and in combination with other antitumor therapies in human renal cancer. Int. J. Mol. Med. 2020, 45, 315–323. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE 2012, 7, e41831. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef]
- Lam, H.C.; Baglini, C.V.; Lope, A.L.; Parkhitko, A.A.; Liu, H.J.; Alesi, N.; Malinowska, I.A.; Ebrahimi-Fakhari, D.; Saffari, A.; Yu, J.J.; et al. P62/SQSTM1 cooperates with hyperactive mTORC1 to regulate glutathione production, maintain mitochondrial integrity, and promote tumorigenesis. Cancer Res. 2017, 77, 3255–3267. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shen, C.; Nakamura, E.; Ando, K.; Signoretti, S.; Beroukhim, R.; Cowley, G.S.; Lizotte, P.; Liberzon, E.; Bair, S.; et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 2013, 24, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Nakajima, M.; Yuasa, A.; Obata, R.; Takahashi, K.; Ohe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Imamura, R.; et al. Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorg. Med. Chem. Lett. 2016, 26, 5956–5959. [Google Scholar] [CrossRef]
- Alam, N.A.; Rowan, A.J.; Wortham, N.C.; Pollard, P.J.; Mitchell, M.; Tyrer, J.P.; Barclay, E.; Calonje, E.; Manek, S.; Adams, S.J.; et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum. Mol. Genet. 2003, 12, 1241–1252. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Yun, J.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Pollard, P.J.; Ratcliffe, P.J. Cancer: Puzzling patterns of predisposition. Science 2009, 324, 192–194. [Google Scholar] [CrossRef]
- O’Flaherty, L.; Adam, J.; Heather, L.C.; Zhdanov, A.V.; Chung, Y.L.; Miranda, M.X.; Croft, J.; Olpin, S.; Clarke, K.; Pugh, C.W.; et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum. Mol. Genet. 2010, 19, 3844–3851. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; El-Bahrawy, M.; Frizzell, N.; Adam, J.; Ternette, N.; Hatipoglu, E.; Howarth, K.; O’Flaherty, L.; Roberts, I.; Turner, G.; et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 2011, 225, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Drusian, L.; Nigro, E.A.; Mannella, V.; Pagliarini, R.; Pema, M.; Costa, A.S.H.; Benigni, F.; Larcher, A.; Chiaravalli, M.; Gaude, E.; et al. mTORC1 upregulation leads to accumulation of the oncometabolite fumarate in a mouse model of renal cell carcinoma. Cell Rep. 2018, 24, 1093–1104.e6. [Google Scholar] [CrossRef]
- Drusian, L.; Boletta, A. mTORC1-driven accumulation of the oncometabolite fumarate as a potential critical step in renal cancer progression. Mol. Cell. Oncol. 2019, 6, 1537709. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Bretagne, M.; Mansuet, A.L.; Just, P.A.; Leroy, K.; Cerles, O.; Chouzenoux, S.; Nicco, C.; Damotte, D.; Alifano, M.; et al. Dimethyl fumarate is highly cytotoxic in KRAS mutated cancer cells but spares non-tumorigenic cells. Oncotarget 2018, 9, 9088–9099. [Google Scholar] [CrossRef]
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the dna damage response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.Y.; et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Leshets, M.; Silas, Y.B.H.; Lehming, N.; Pines, O. Fumarase: From the TCA Cycle to DNA damage response and tumor suppression. Front. Mol. Biosci. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Rhoades Smith, K.E.; Bilen, M.A. A review of papillary renal cell carcinoma and MET inhibitors. Kidney Cancer 2019, 3, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonenn, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Panieri, E.; Buha, A.; Telkoparan-akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential applications of NRF2 modulators in cancer therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef]
- Harder, B.; Tian, W.; la Clair, J.J.; Tan, A.C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef]
- Kweon, M.H.; Adhami, V.M.; Lee, J.S.; Mukhtar, H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J. Biol. Chem. 2006, 281, 33761–33772. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Zhu, J.; Yu, L.; Jiang, X.; Zhang, J.; Dong, X.; Ma, B.; Zhang, Q. Oridonin exerts anticancer effect on osteosarcoma by activating PPAR-γ and inhibiting Nrf2 pathway article. Cell Death Dis. 2018, 9, 15. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Zhang, Y.; Yang, L.; Liu, Y.; Huang, S.; Lu, L.; Kong, L.; Li, Z.; Guo, Q.; et al. Wogonin reversed resistant human myelogenous leukemia cells via inhibiting Nrf2 signaling by Stat3/NF-κB inactivation. Sci. Rep. 2017, 7, 39950. [Google Scholar] [CrossRef]
- Gao, A.M.; Ke, Z.P.; Shi, F.; Sun, G.C.; Chen, H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem. Biol. Interact. 2013, 206, 100–108. [Google Scholar] [CrossRef]
- Tian, D.; Shi, Y.; Chen, D.; Liu, Q.; Fan, F. The Wnt inhibitor LGK-974 enhances radiosensitivity of HepG2 cells by modulating Nrf2 signaling. Int. J. Oncol. 2017, 51, 545–554. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Function | Genes | Mutations and Alterations | |
|---|---|---|---|
| PRCC | ccRCC | ||
| Antioxidant response | HMOX1 | TM | MM |
| GPX1 | Del | MM | |
| TXN | MM | Del | |
| Drug metabolism and disposition | NQO1 | Amp | Amp |
| AKR1B10 | MM, Del | Amp | |
| AKR1C1 | MM, Amp | MM | |
| AKR1C3 | MM, Amp | - | |
| Autophagy | SQSTM1 | MM, Amp | Amp |
| Mitochondrial apoptosis | PARK7 | Amp, Del | Del |
| Xenobiotic response and metabolism | AHR | MM | TM, Amp |
| NADPH generation and pentose synthesis | G6PD | Amp | MM, Amp |
| IDH1 | Del | MM, TM, Amp, Del | |
| NFE2L2 mutations | KEAP1 mutations | ||||||
|---|---|---|---|---|---|---|---|
| ccRCC | ccRCC | ||||||
| CDS mutation | AA mutation | Mutation | Ref. | CDS mutation | AA mutation | Mutation | Ref. |
| c.70T>C | p.W24R | MM | [76] | c.160T>G | p.Y54D | MM | [12] |
| c.85C>G | p.D29H | MM | [76] TCGA | c.761A>C | p.K254T | MM | [23] |
| c.86A>T | p.D29V | MM | [77] | c.779G>A | p.R260Q | MM | [23] |
| c.89T>A | p.L30H | MM | [12] | c.779G>T | p.R260L | MM | [23] |
| c.92G>C | p.G31A | MM | [77] | c.1226T>C | p.M409T | MM | [12] |
| c.100C>G | p.R34G | MM | [12] | c.1330G>T | p.E444 | NsM | [23] |
| c.239C>A | p.T80K | MM | [23] | c.1630T>C | p.W544R | MM | [12] |
| c.242G>A | p.G81D | MM | [12] | c.1735G>T | p.D759Y | MM | [23] |
| c.246A>C | p.E82D | MM | [12] | c.1752del | p.Y584 | Del | [23] |
| c.739C>T | p.L247F | MM | [12] | ||||
| c.1279G>T | p.E427 | NsM | [12] | ||||
| PRCC | PRCC | ||||||
| CDS mutation | AA mutation | Mutation | Ref. | CDS mutation | AA mutation | Mutation | Ref. |
| c.70T>C | p.W24R | MM | [78] | c.532C>T | p.Q178 | NsM | TCGA |
| c.85G>T | p.D29Y | MM | TCGA | ||||
| c.88C>T | L30F | MM | [68] | ||||
| c.89T>G | p.L30R | MM | TCGA | Copy number alterations | |||
| c.106_108del | p.V36del | Del | [72] | ccRCC | |||
| c.230A>C | p.D77A | MM | [68] | Gene | Cytoband | Alteration | Ref. |
| c.239C>A | p.T80K | MM | TCGA | NFE2L2 | 2q31.2 | Amp | [12] |
| c.242G>T | p.G81V | MM | TCGA | KEAP1 | 19p13.2 | Del | [12] |
| c.245A>G | p.E82G | MM | [72] TCGA | ||||
| c.246A>C | p.E82D | MM | TCGA | PRCC | |||
| p.F339L | MM | TCGA | Gene | Cytoband | Alteration | Ref. | |
| p.P174T | MM | TCGA | NFE2L2 | 2q31.2 | Amp | [12] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerici, S.; Boletta, A. Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma. Cancers 2020, 12, 3458. https://doi.org/10.3390/cancers12113458
Clerici S, Boletta A. Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma. Cancers. 2020; 12(11):3458. https://doi.org/10.3390/cancers12113458
Chicago/Turabian StyleClerici, Sara, and Alessandra Boletta. 2020. "Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma" Cancers 12, no. 11: 3458. https://doi.org/10.3390/cancers12113458
APA StyleClerici, S., & Boletta, A. (2020). Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma. Cancers, 12(11), 3458. https://doi.org/10.3390/cancers12113458