Importance of Cullin4 Ubiquitin Ligase in Malignant Pleural Mesothelioma

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. CUL4A and CUL4B Expression Was Elevated in a Subset of MPM Patients, and High CUL4B Expression Was Associated with a Poor Outcome
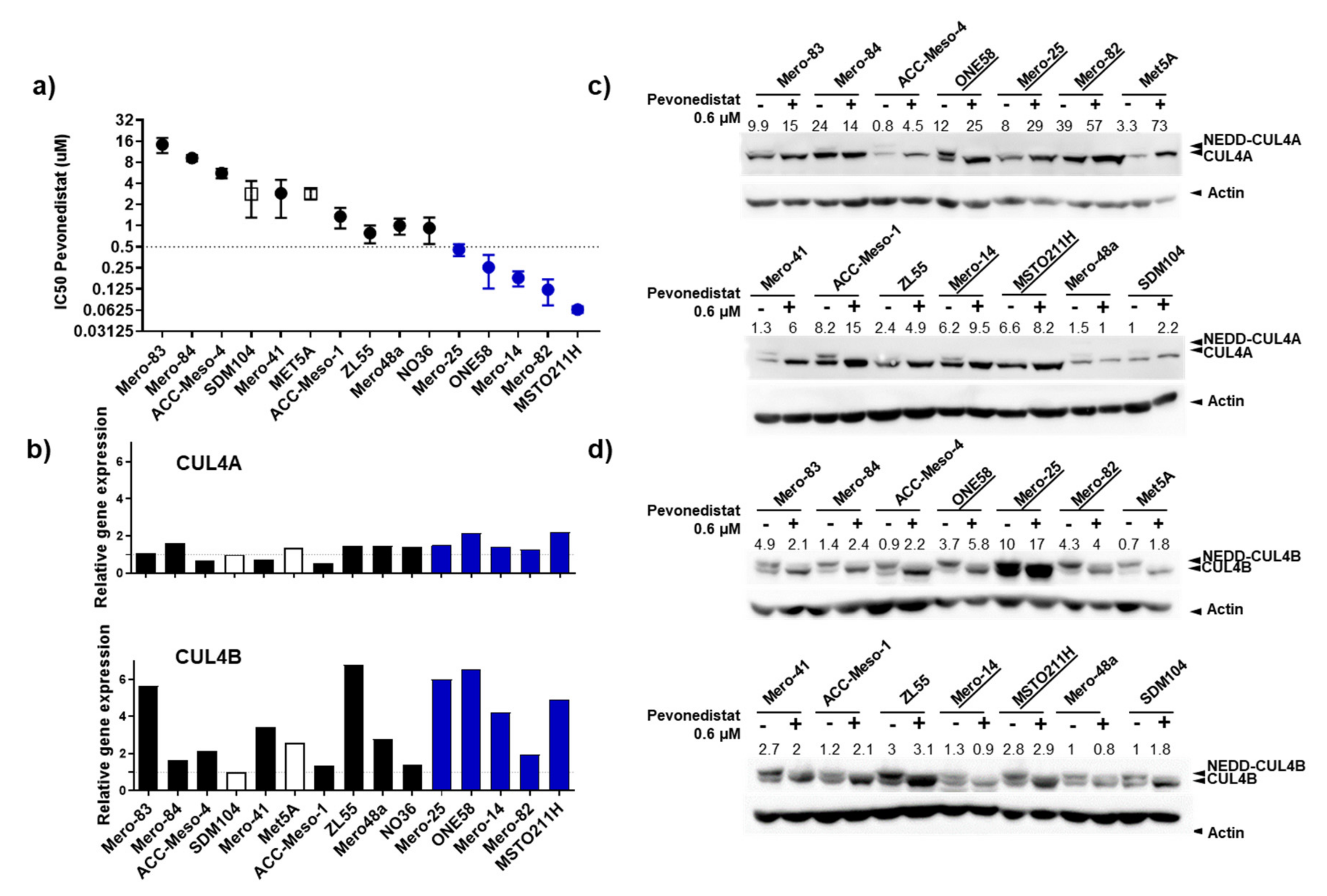
2.2. CU4LA and CUL4B Are Overexpressed in MPM Cell Lines, and a Subset of Them Were Susceptible to the Inhibition of NEDD8 Activating Enzyme (NAE) by Pevonedistat
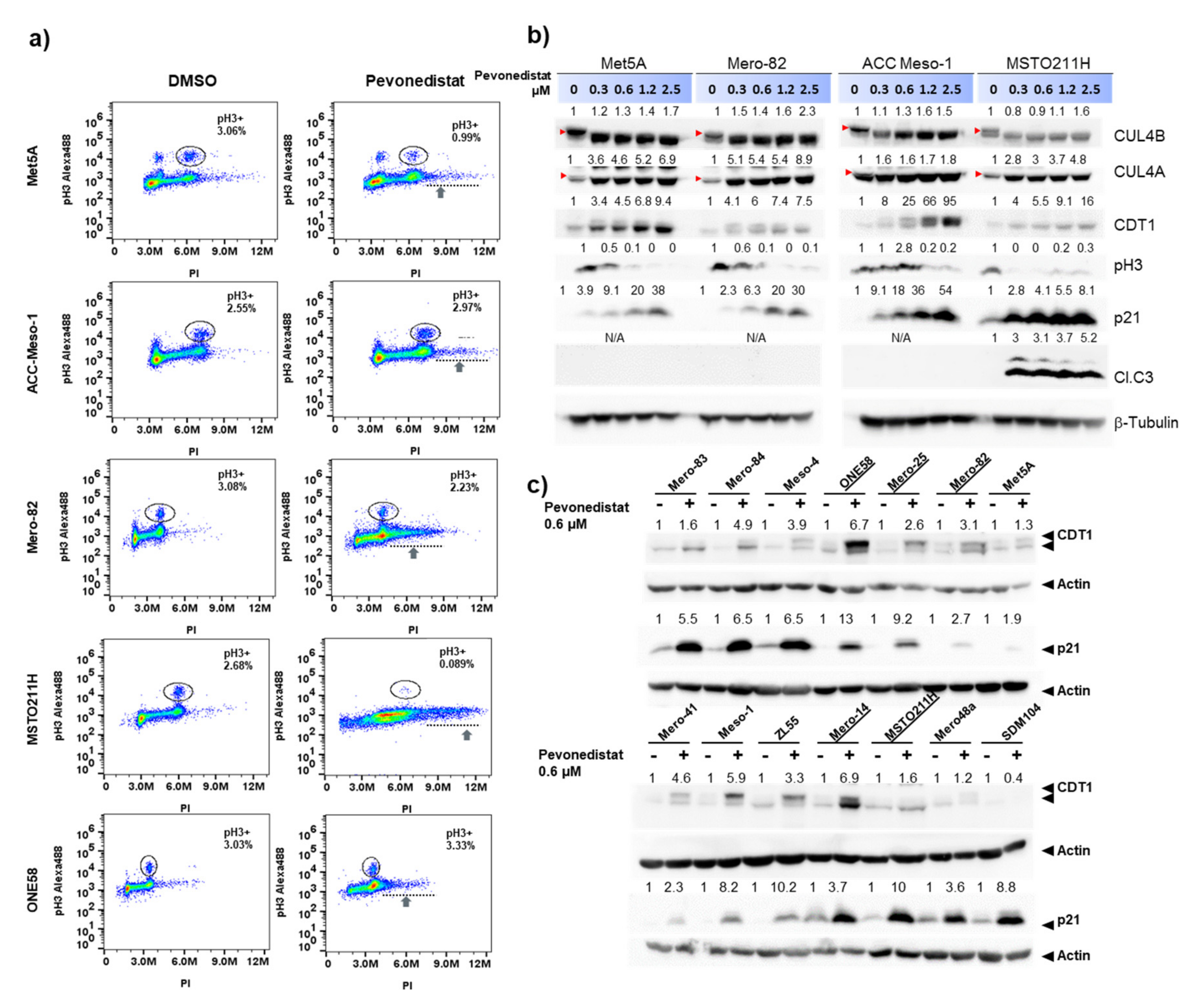
2.3. Increased Accumulation of Cells with DNA Rereplication in Pevonedistat-Sensitive Cell Lines
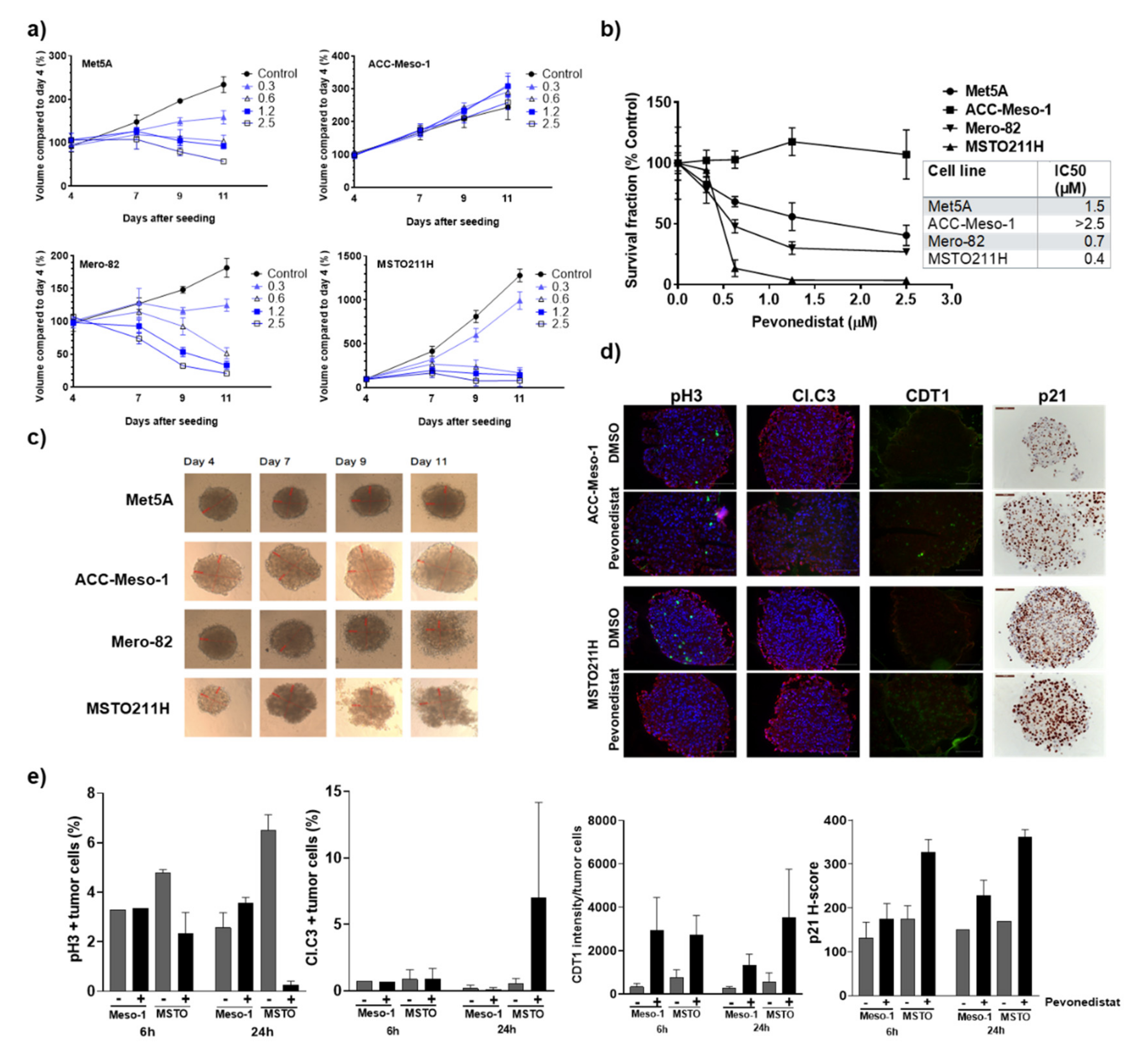
2.4. Three-Dimentional Culture Confirmed Differential Sensitivity of MPM Cell Lines to Pevonedistat
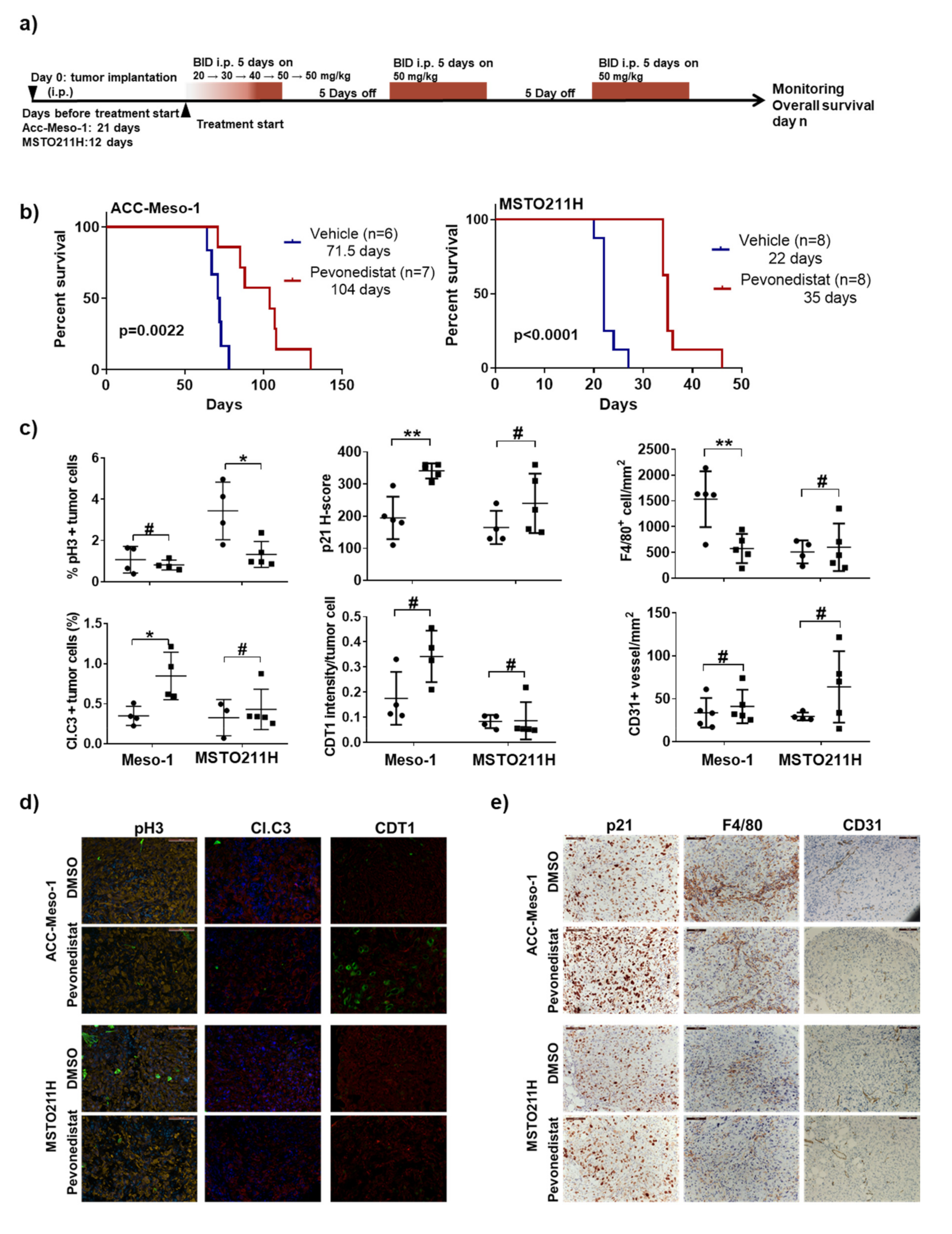
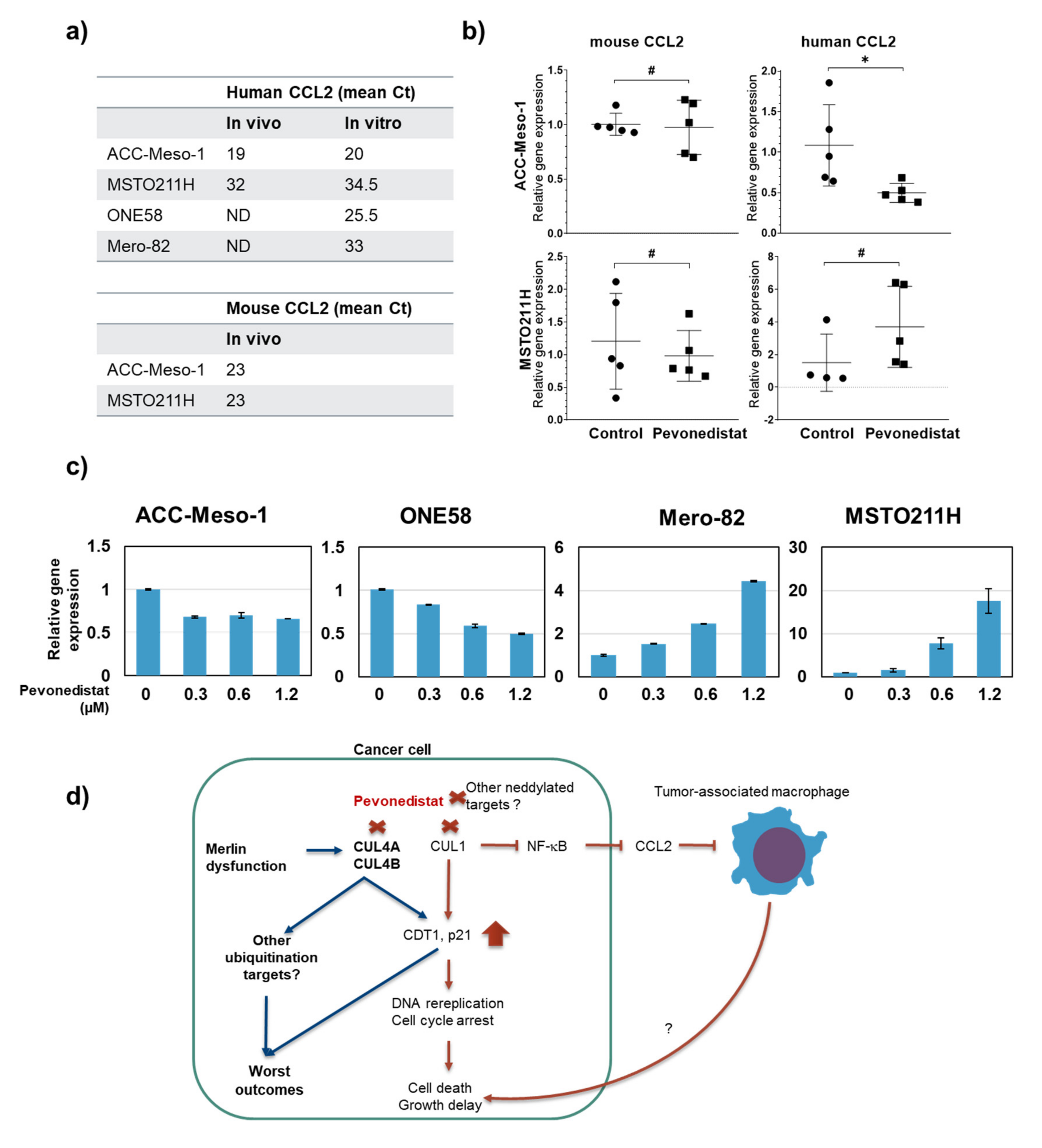
2.5. Efficacy of Pevonedistat In Vivo May Also Be Mediated by the Modulation of the Tumor Microenvironment in the Immune-Deficient Model
3. Discussion
4. Materials and Methods
4.1. MPM Tissue Microarrays (TMAs)
4.2. Immunohistochemical Staining and Quantification for Protein Expression
4.3. Tissue Samples for RNA Extraction
4.4. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR
4.5. Cell Lines
4.6. Treatment and Cell Survival Analysis for 2D and 3D Culture
4.7. Protein Extraction and Western Blot
4.8. Cell Cycle Analysis
4.9. MPM Spheriod Cultivation, Treatment and Viability Measurement
4.10. Immunofluorescence
4.11. Animal Experiment
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase iii study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Bjorkqvist, A.M.; Tammilehto, L.; Anttila, S.; Mattson, K.; Knuutila, S. Recurrent DNA copy number changes in 1q, 4q, 6q, 9p, 13q, 14q and 22q detected by comparative genomic hybridization in malignant mesothelioma. Br. J. Cancer 1997, 75, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Molecular insights into nf2/merlin tumor suppressor function. FEBS Lett. 2014, 588, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; You, L.; Giancotti, F.G. Shedding light on merlin’s wizardry. Trends Cell Biol. 2007, 17, 222–229. [Google Scholar] [CrossRef]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; Hanemann, C.O.; Long, S.B.; et al. Merlin/nf2 suppresses tumorigenesis by inhibiting the e3 ubiquitin ligase crl4(dcaf1) in the nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef]
- Meerang, M.; Berard, K.; Friess, M.; Bitanihirwe, B.K.; Soltermann, A.; Vrugt, B.; Felley-Bosco, E.; Bueno, R.; Richards, W.G.; Seifert, B.; et al. Low merlin expression and high survivin labeling index are indicators for poor prognosis in patients with malignant pleural mesothelioma. Mol. Oncol. 2016, 10, 1255–1265. [Google Scholar] [CrossRef]
- Lee, J.; Zhou, P. Cullins and cancer. Genes Cancer 2010, 1, 690–699. [Google Scholar] [CrossRef]
- Sharma, P.; Nag, A. Cul4a ubiquitin ligase: A promising drug target for cancer and other human diseases. Open Biol. 2014, 4, 130217. [Google Scholar] [CrossRef]
- Hung, M.S.; Mao, J.H.; Xu, Z.; Yang, C.T.; Yu, J.S.; Harvard, C.; Lin, Y.C.; Bravo, D.T.; Jablons, D.M.; You, L. Cul4a is an oncogene in malignant pleural mesothelioma. J. Cell. Mol. Med. 2011, 15, 350–358. [Google Scholar] [CrossRef]
- Yang, Y.L.; Hung, M.S.; Wang, Y.; Ni, J.; Mao, J.H.; Hsieh, D.; Au, A.; Kumar, A.; Quigley, D.; Fang, L.T.; et al. Lung tumourigenesis in a conditional cul4a transgenic mouse model. J. Pathol. 2014, 233, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tang, H.M.; Wu, Z.H.; Chen, J.; Lu, S.; Zhou, C.Z.; Yan, D.W.; Peng, Z.H. Cullin 4b is a novel prognostic marker that correlates with colon cancer progression and pathogenesis. Med. Oncol. 2013, 30, 534. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jiang, B.; Zhang, A.; Qian, Y.; Tan, H.; Gao, J.; Shao, C.; Gong, Y. Accelerated hepatocellular carcinoma development in cul4b transgenic mice. Oncotarget 2015, 6, 15209–15221. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. ’Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 969–976. [Google Scholar] [CrossRef]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The nedd8 e1 inhibitor mln4924 forms a nedd8-amp mimetic in situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. Mln4924: A novel first-in-class inhibitor of nedd8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. Gepia: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual pi3k/mtor inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef]
- Milhollen, M.A.; Narayanan, U.; Soucy, T.A.; Veiby, P.O.; Smith, P.G.; Amidon, B. Inhibition of nedd8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating cdt1 turnover. Cancer Res. 2011, 71, 3042–3051. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of nedd8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Liu, X.; Li, L.; Yang, X.; Dong, C.; Liu, X.; Lin, Y.; Li, Y.; Yu, J.; et al. Promotion of tumor-associated macrophages infiltration by elevated neddylation pathway via nf-kappab-ccl2 signaling in lung cancer. Oncogene 2019, 38, 5792–5804. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Xiong, Y. X-linked mental retardation gene cul4b targets ubiquitylation of h3k4 methyltransferase component wdr5 and regulates neuronal gene expression. Mol. Cell 2011, 43, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yang, Y.; Ji, Q.; Zhao, W.; Jiang, B.; Liu, R.; Yuan, J.; Liu, Q.; Li, X.; Zou, Y.; et al. Crl4b catalyzes h2ak119 monoubiquitination and coordinates with prc2 to promote tumorigenesis. Cancer Cell 2012, 22, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. Ezh2 generates a methyl degron that is recognized by the dcaf1/ddb1/cul4 e3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, W.; Jiang, K.; Chen, B.; Wang, K.; Lao, L.; Hou, C.; Wang, F.; Zhang, C.; Shen, H. Microrna-300 regulates the ubiquitination of pten through the crl4b(dcaf13) e3 ligase in osteosarcoma cells. Mol. Ther. Nucleic Acids 2018, 10, 254–268. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Hu, B.; Zhang, F.; Wei, H.; Li, L. Circular rna zfr accelerates non-small cell lung cancer progression by acting as a mir-101-3p sponge to enhance cul4b expression. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3410–3416. [Google Scholar] [CrossRef]
- Ramirez-Salazar, E.G.; Salinas-Silva, L.C.; Vazquez-Manriquez, M.E.; Gayosso-Gomez, L.V.; Negrete-Garcia, M.C.; Ramirez-Rodriguez, S.L.; Chavez, R.; Zenteno, E.; Santillan, P.; Kelly-Garcia, J.; et al. Analysis of microrna expression signatures in malignant pleural mesothelioma, pleural inflammation, and atypical mesothelial hyperplasia reveals common predictive tumorigenesis-related targets. Exp. Mol. Pathol. 2014, 97, 375–385. [Google Scholar] [CrossRef]
- Wong, K.M.; Micel, L.N.; Selby, H.M.; Tan, A.C.; Pitts, T.M.; Bagby, S.M.; Spreafico, A.; Klauck, P.J.; Blakemore, S.J.; Smith, P.F.; et al. Targeting the protein ubiquitination machinery in melanoma by the nedd8-activating enzyme inhibitor pevonedistat (mln4924). Investig. New Drugs 2017, 35, 11–25. [Google Scholar] [CrossRef]
- Cooper, J.; Xu, Q.; Zhou, L.; Pavlovic, M.; Ojeda, V.; Moulick, K.; de Stanchina, E.; Poirier, J.T.; Zauderer, M.; Rudin, C.M.; et al. Combined inhibition of nedd8-activating enzyme and mtor suppresses nf2 loss-driven tumorigenesis. Mol. Cancer Ther. 2017, 16, 1693–1704. [Google Scholar] [CrossRef]
- Vaziri, C.; Saxena, S.; Jeon, Y.; Lee, C.; Murata, K.; Machida, Y.; Wagle, N.; Hwang, D.S.; Dutta, A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 2003, 11, 997–1008. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Burt, B.M.; Rodig, S.J.; Tilleman, T.R.; Elbardissi, A.W.; Bueno, R.; Sugarbaker, D.J. Circulating and tumor-infiltrating myeloid cells predict survival in human pleural mesothelioma. Cancer 2011, 117, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.J.; van Zandwijk, N.; Rasko, J.E.J. The immune microenvironment in mesothelioma: Mechanisms of resistance to immunotherapy. Front. Oncol. 2019, 9, 1366. [Google Scholar] [CrossRef]
- Gueugnon, F.; Leclercq, S.; Blanquart, C.; Sagan, C.; Cellerin, L.; Padieu, M.; Perigaud, C.; Scherpereel, A.; Gregoire, M. Identification of novel markers for the diagnosis of malignant pleural mesothelioma. Am. J. Pathol. 2011, 178, 1033–1042. [Google Scholar] [CrossRef]
- Kishimoto, T.; Fujimoto, N.; Ebara, T.; Omori, T.; Oguri, T.; Niimi, A.; Yokoyama, T.; Kato, M.; Usami, I.; Nishio, M.; et al. Serum levels of the chemokine ccl2 are elevated in malignant pleural mesothelioma patients. BMC Cancer 2019, 19, 1204. [Google Scholar] [CrossRef]
- Hildebrand, D.G.; Alexander, E.; Horber, S.; Lehle, S.; Obermayer, K.; Munck, N.A.; Rothfuss, O.; Frick, J.S.; Morimatsu, M.; Schmitz, I.; et al. Ikappabzeta is a transcriptional key regulator of ccl2/mcp-1. J. Immunol. 2013, 190, 4812–4820. [Google Scholar] [CrossRef]
- Sidi, R.; Pasello, G.; Opitz, I.; Soltermann, A.; Tutic, M.; Rehrauer, H.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: An exploratory analysis. Eur. J. Cancer 2011, 47, 326–332. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Friedrich, J.; Eder, W.; Castaneda, J.; Doss, M.; Huber, E.; Ebner, R.; Kunz-Schughart, L.A. A reliable tool to determine cell viability in complex 3-d culture: The acid phosphatase assay. J. Biomol. Screen 2007, 12, 925–937. [Google Scholar] [CrossRef]
- Ullman-Cullere, M.H.; Foltz, C.J. Body condition scoring: A rapid and accurate method for assessing health status in mice. Comp. Med. 1999, 49, 319–323. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meerang, M.; Kreienbühl, J.; Orlowski, V.; Müller, S.L.C.; Kirschner, M.B.; Opitz, I. Importance of Cullin4 Ubiquitin Ligase in Malignant Pleural Mesothelioma. Cancers 2020, 12, 3460. https://doi.org/10.3390/cancers12113460
Meerang M, Kreienbühl J, Orlowski V, Müller SLC, Kirschner MB, Opitz I. Importance of Cullin4 Ubiquitin Ligase in Malignant Pleural Mesothelioma. Cancers. 2020; 12(11):3460. https://doi.org/10.3390/cancers12113460
Chicago/Turabian StyleMeerang, Mayura, Jessica Kreienbühl, Vanessa Orlowski, Seraina L. C. Müller, Michaela B. Kirschner, and Isabelle Opitz. 2020. "Importance of Cullin4 Ubiquitin Ligase in Malignant Pleural Mesothelioma" Cancers 12, no. 11: 3460. https://doi.org/10.3390/cancers12113460
APA StyleMeerang, M., Kreienbühl, J., Orlowski, V., Müller, S. L. C., Kirschner, M. B., & Opitz, I. (2020). Importance of Cullin4 Ubiquitin Ligase in Malignant Pleural Mesothelioma. Cancers, 12(11), 3460. https://doi.org/10.3390/cancers12113460