A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent

 , ,
, ,
Abstract
1. Introduction
2. Results
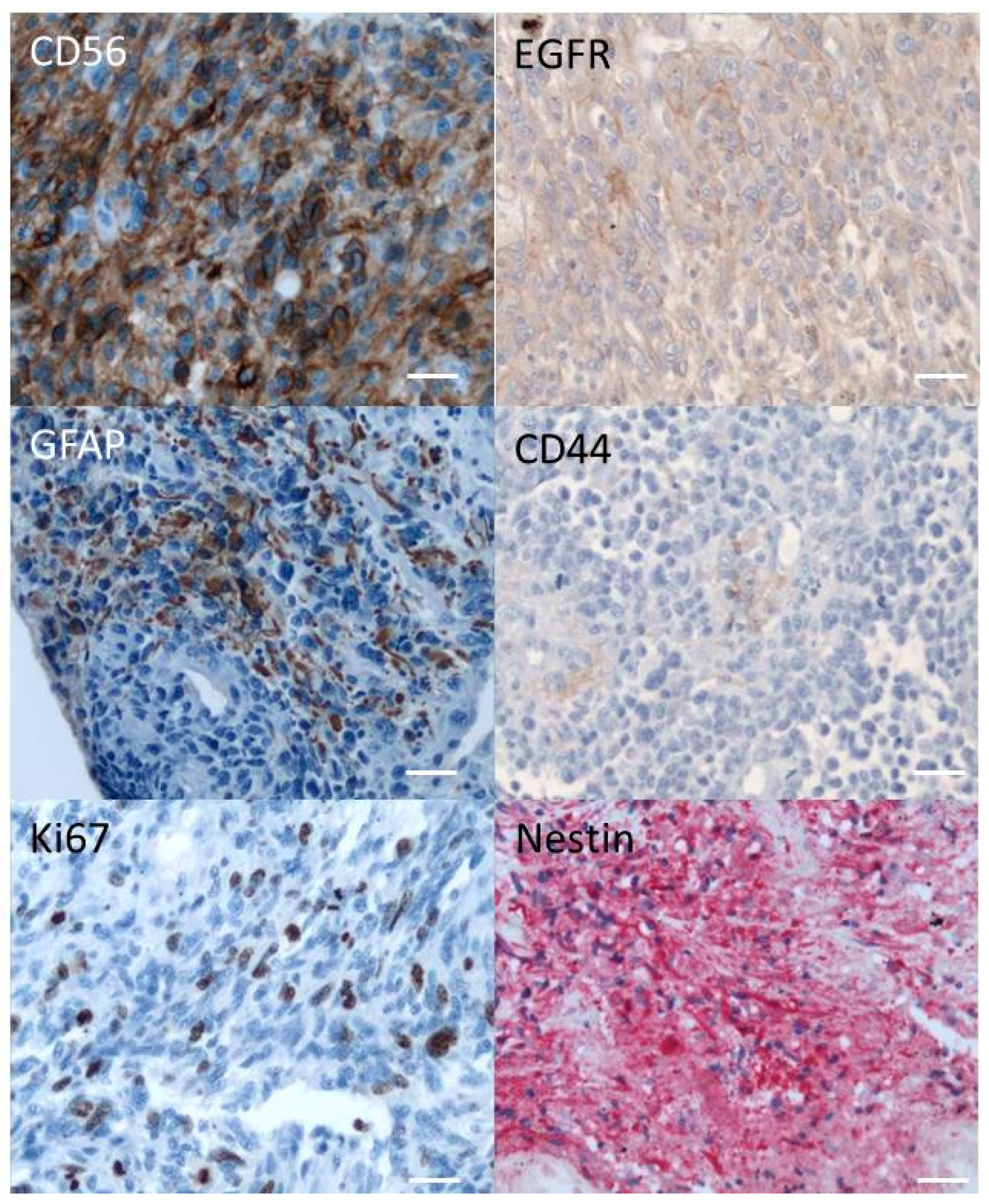
2.1. Immunohistochemistry of the Original Tumor
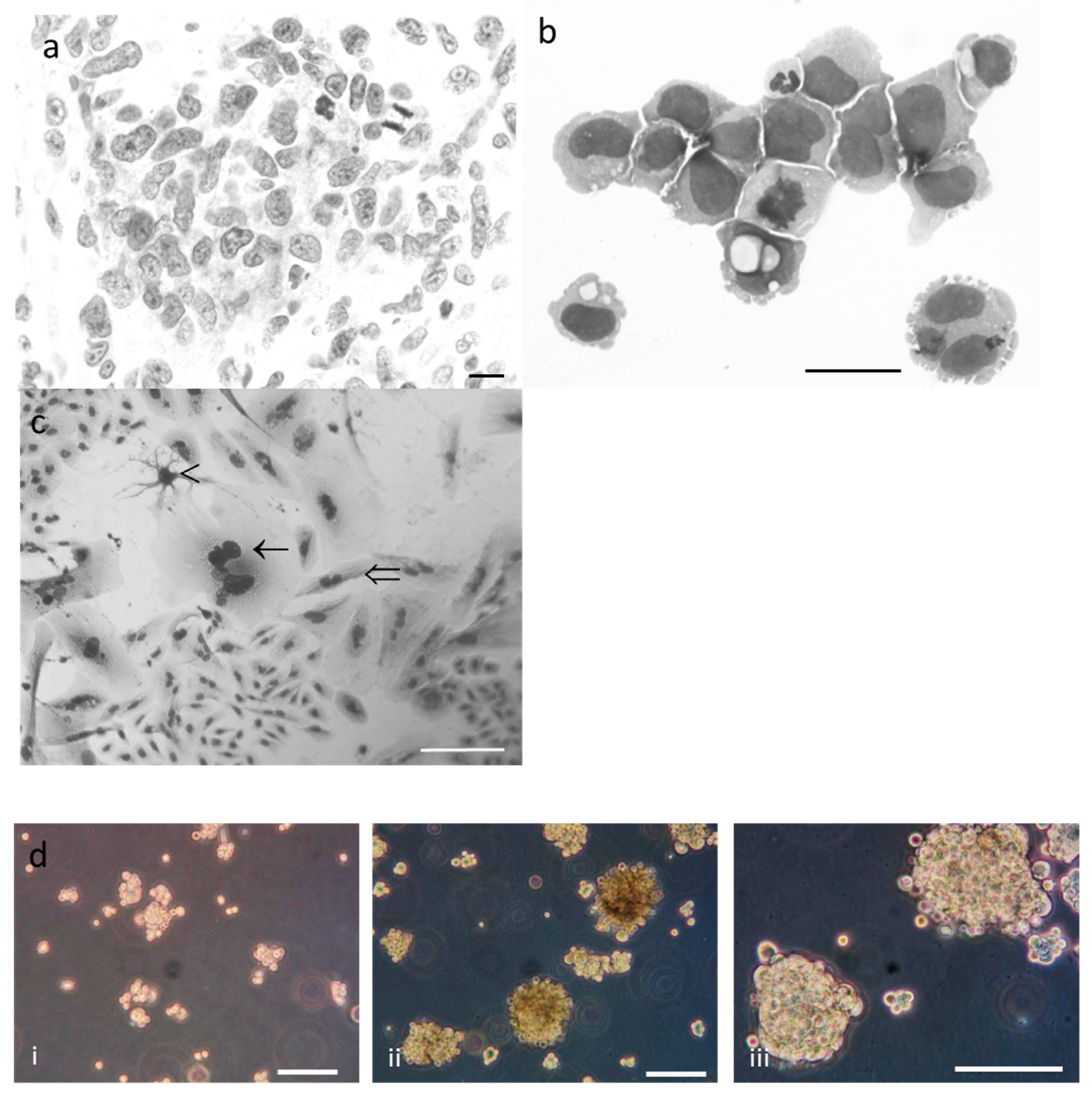
2.2. Phenotypic Characterisation of Tumor and R2J Cells
2.3. R2J Cells are Able to Form Gliospheres
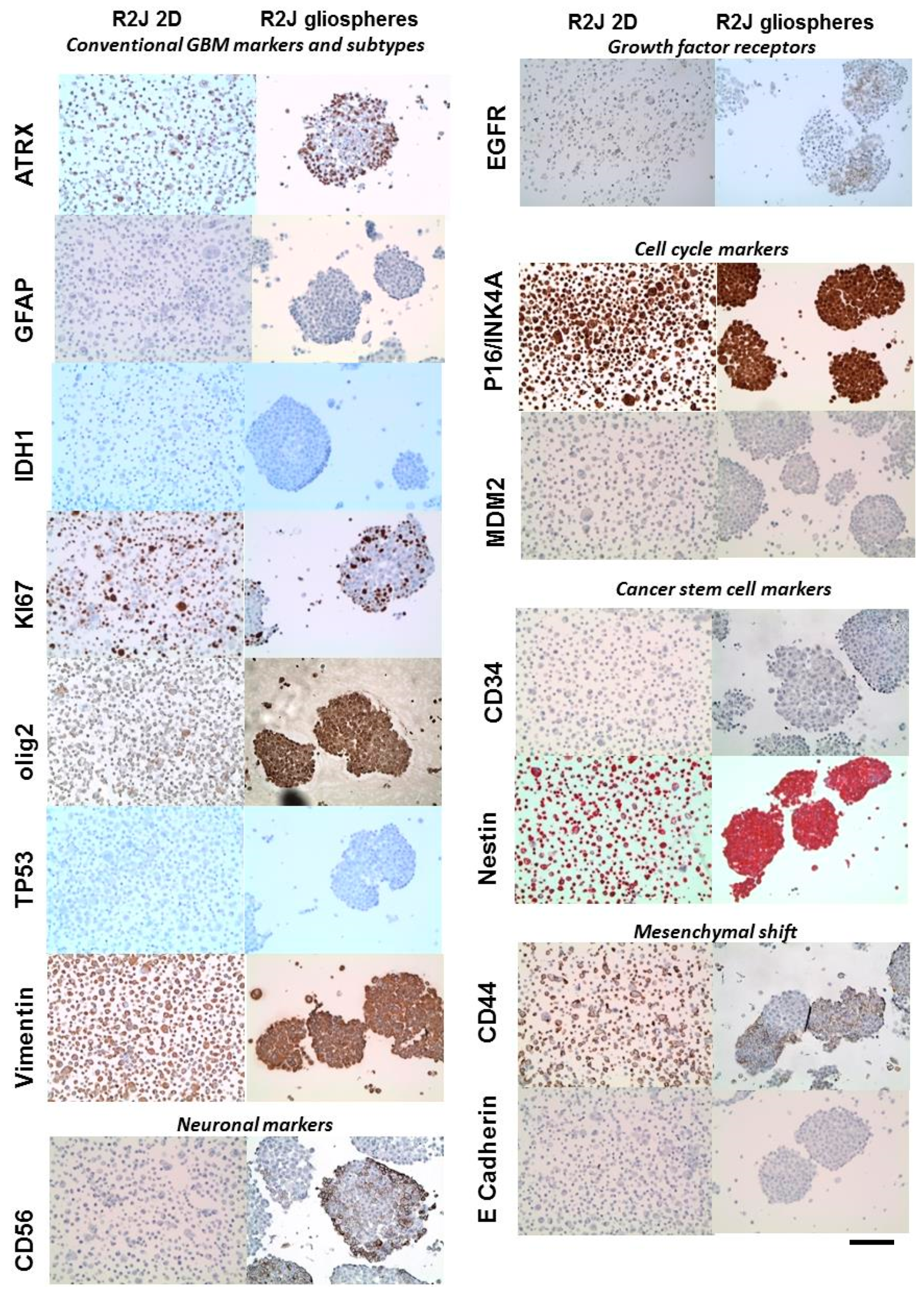
2.4. Immunohistochemistry of R2J Cells Cultured in 2D and in Gliospheres
2.5. MGMT Status of R2J Cells
2.6. Chromosome Analysis
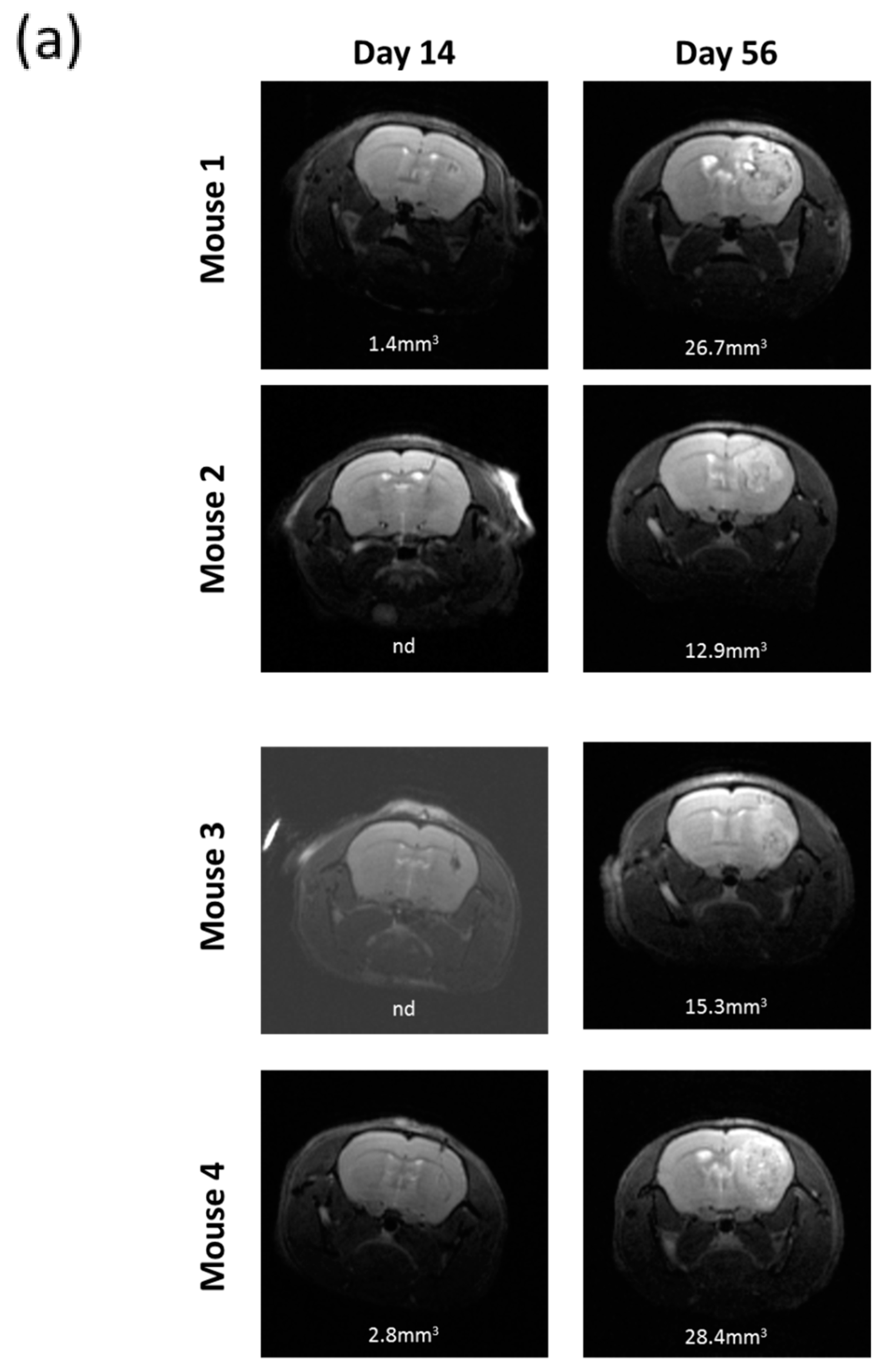
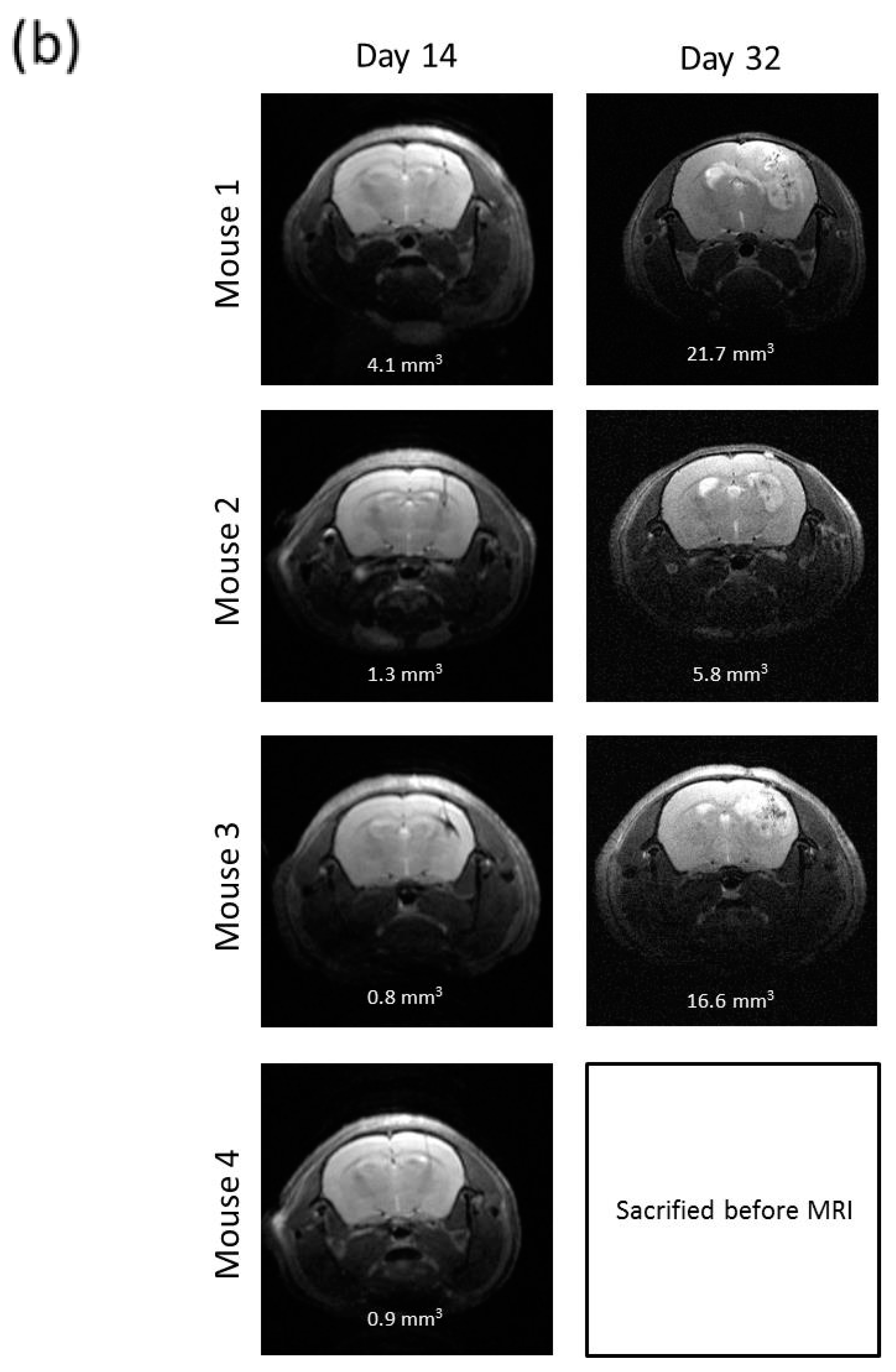
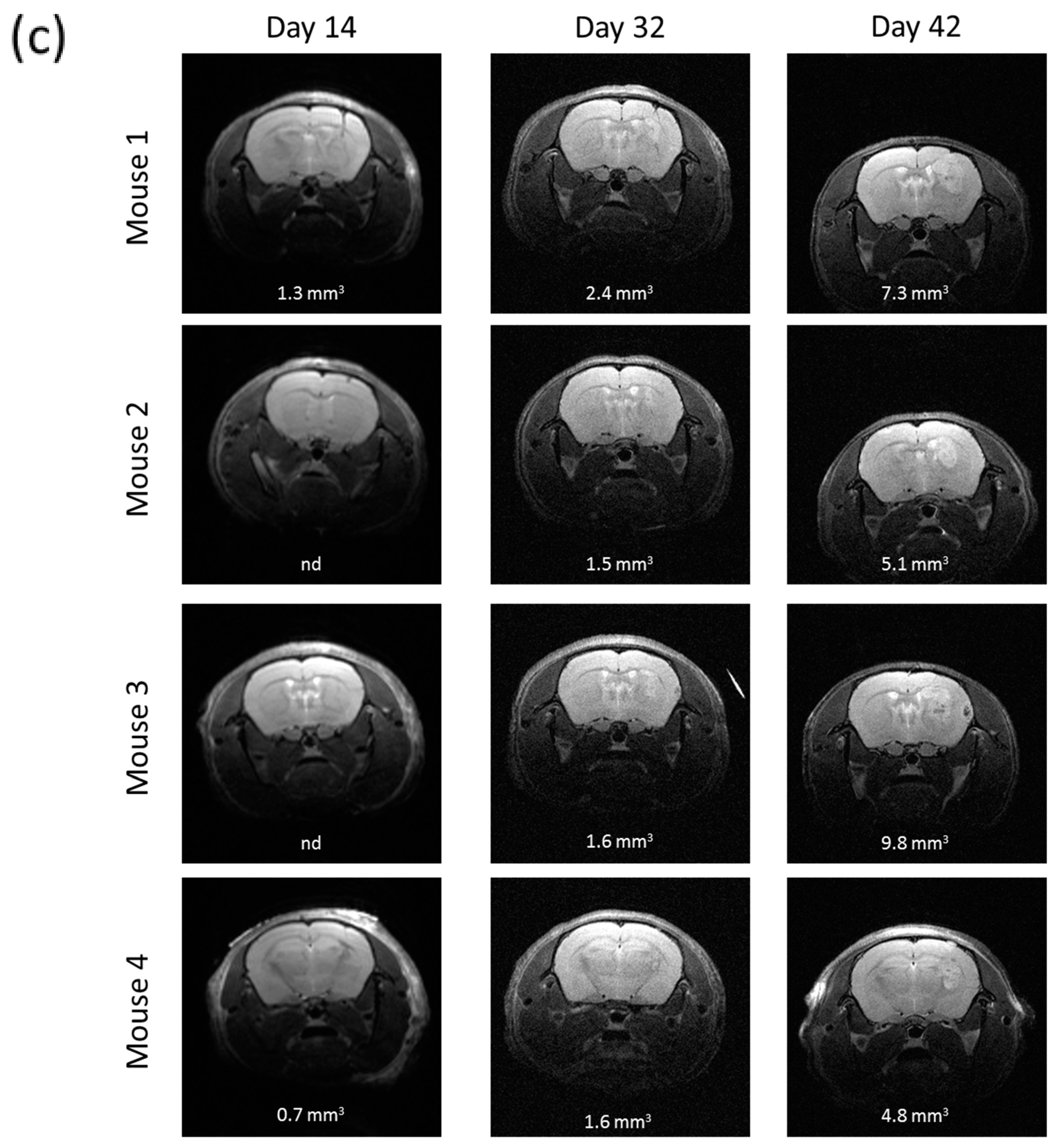
2.7. R2J Cells are Tumorigenic and Cancer Stem Cells
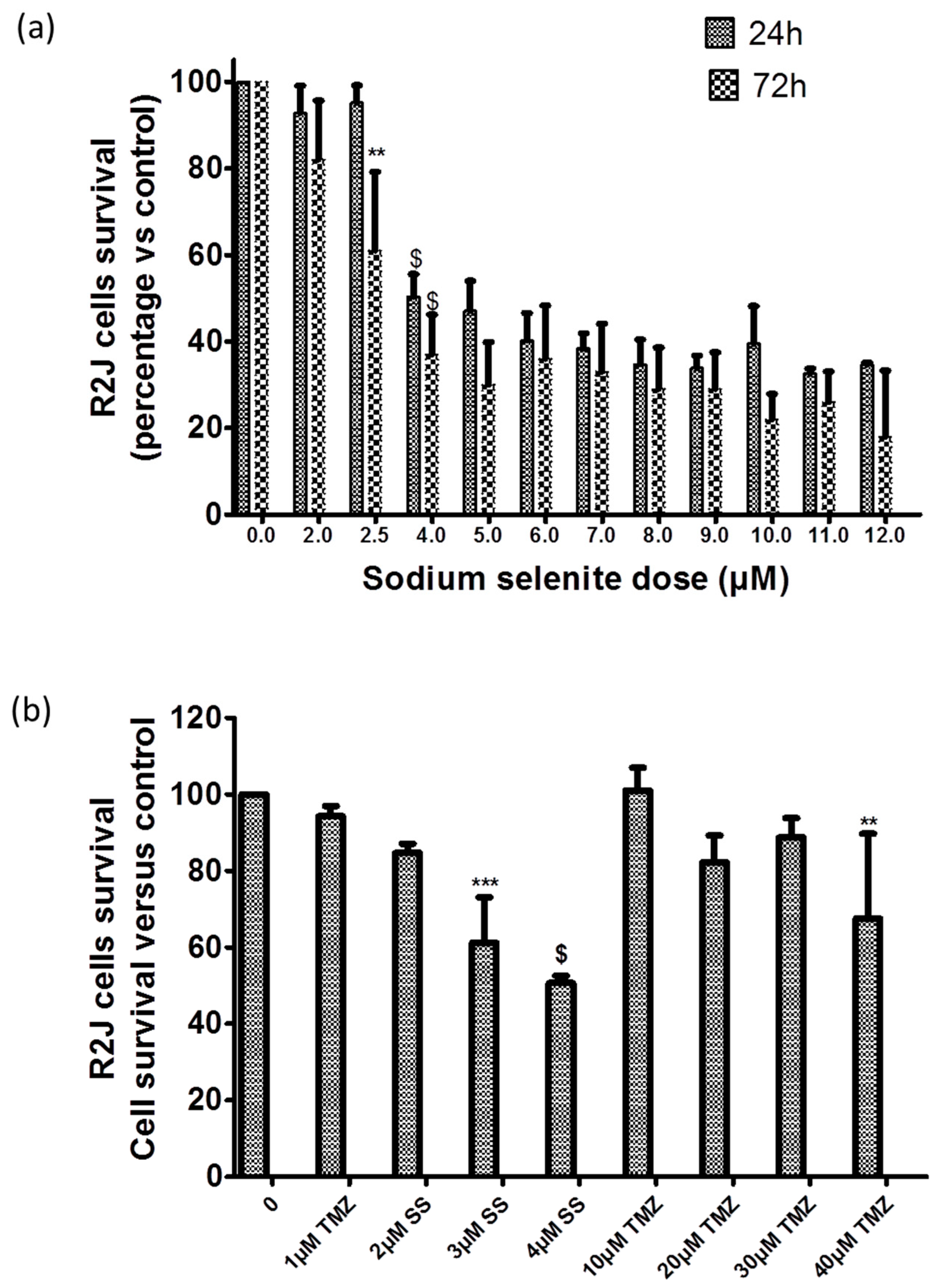
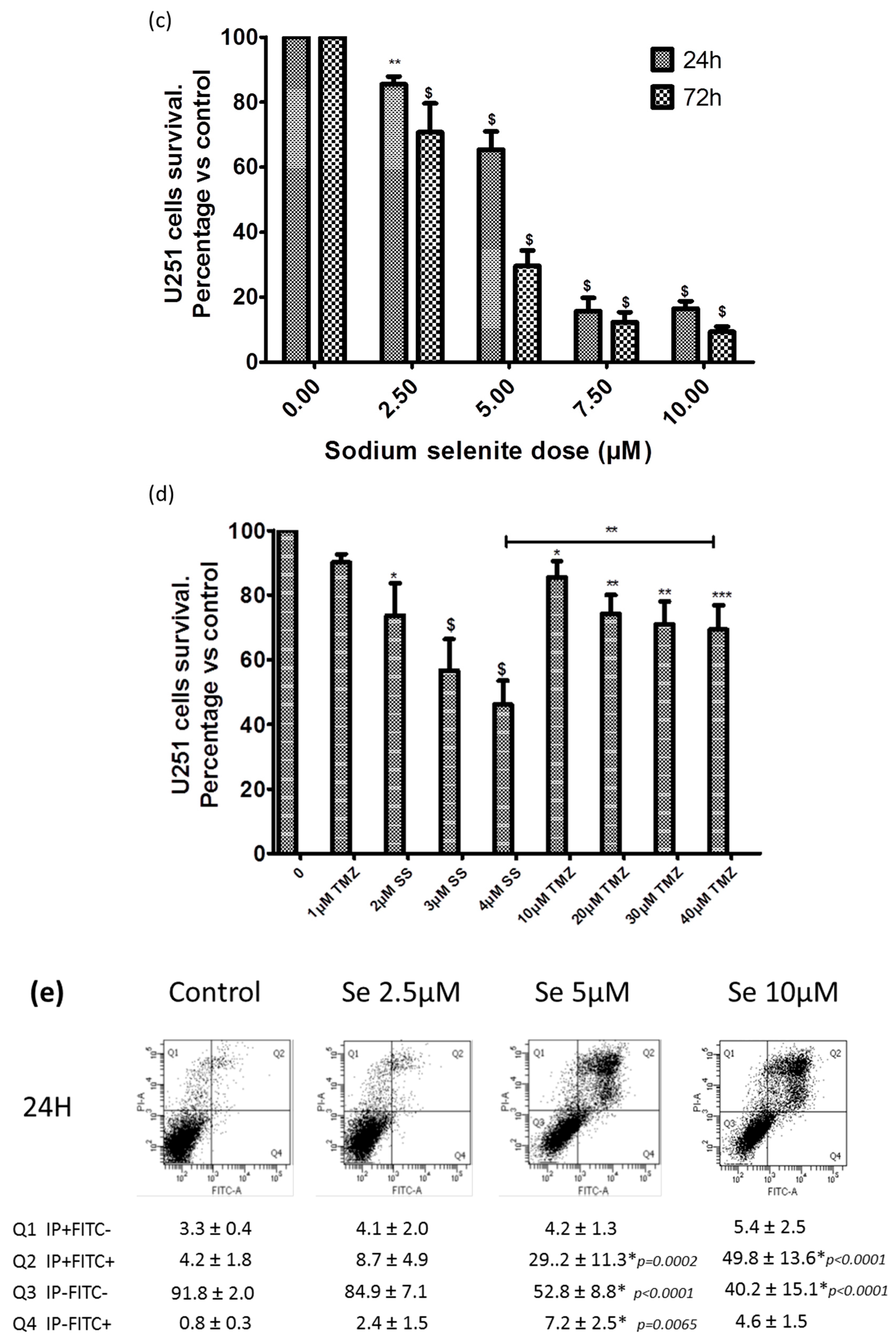
2.8. SS Absorption
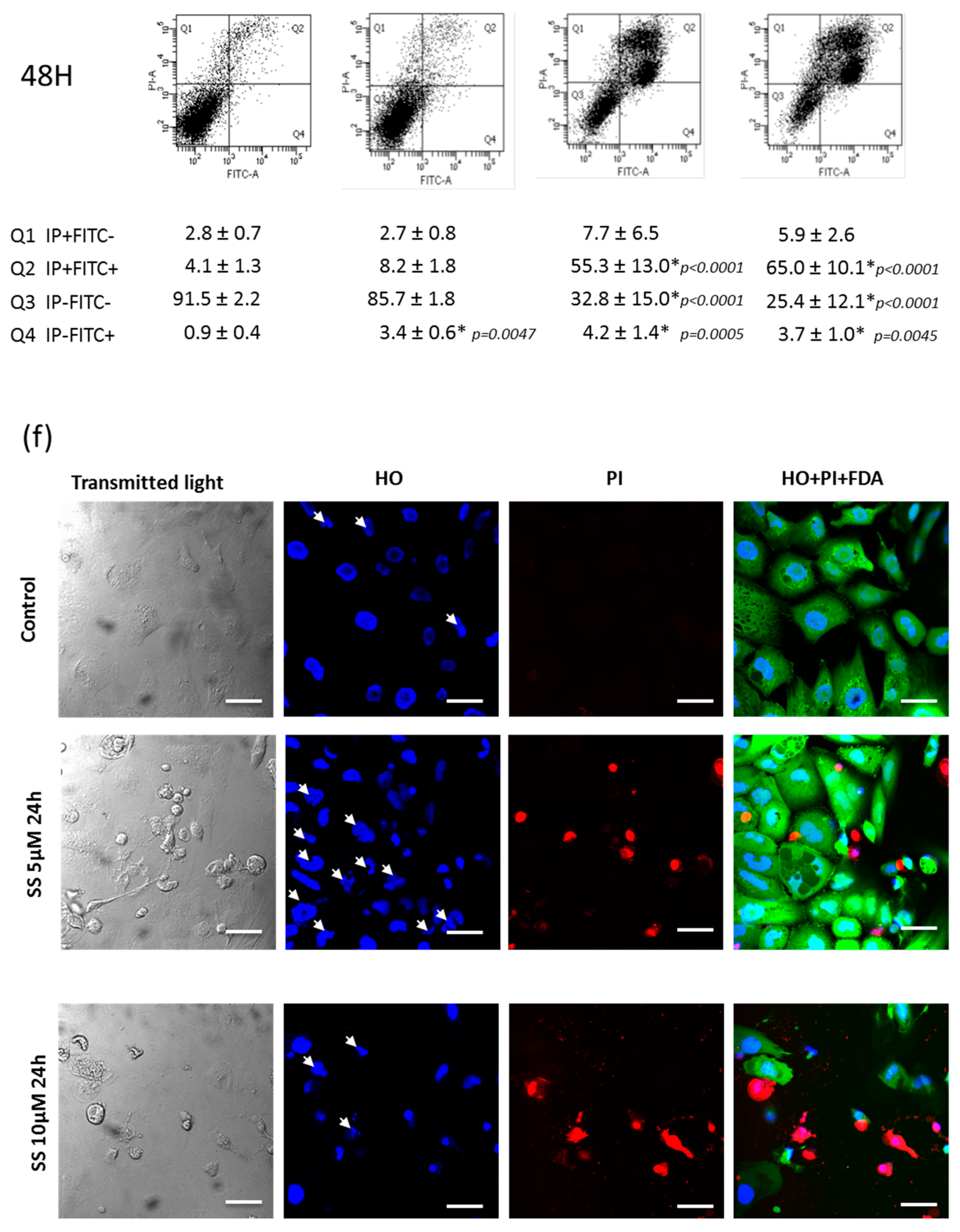
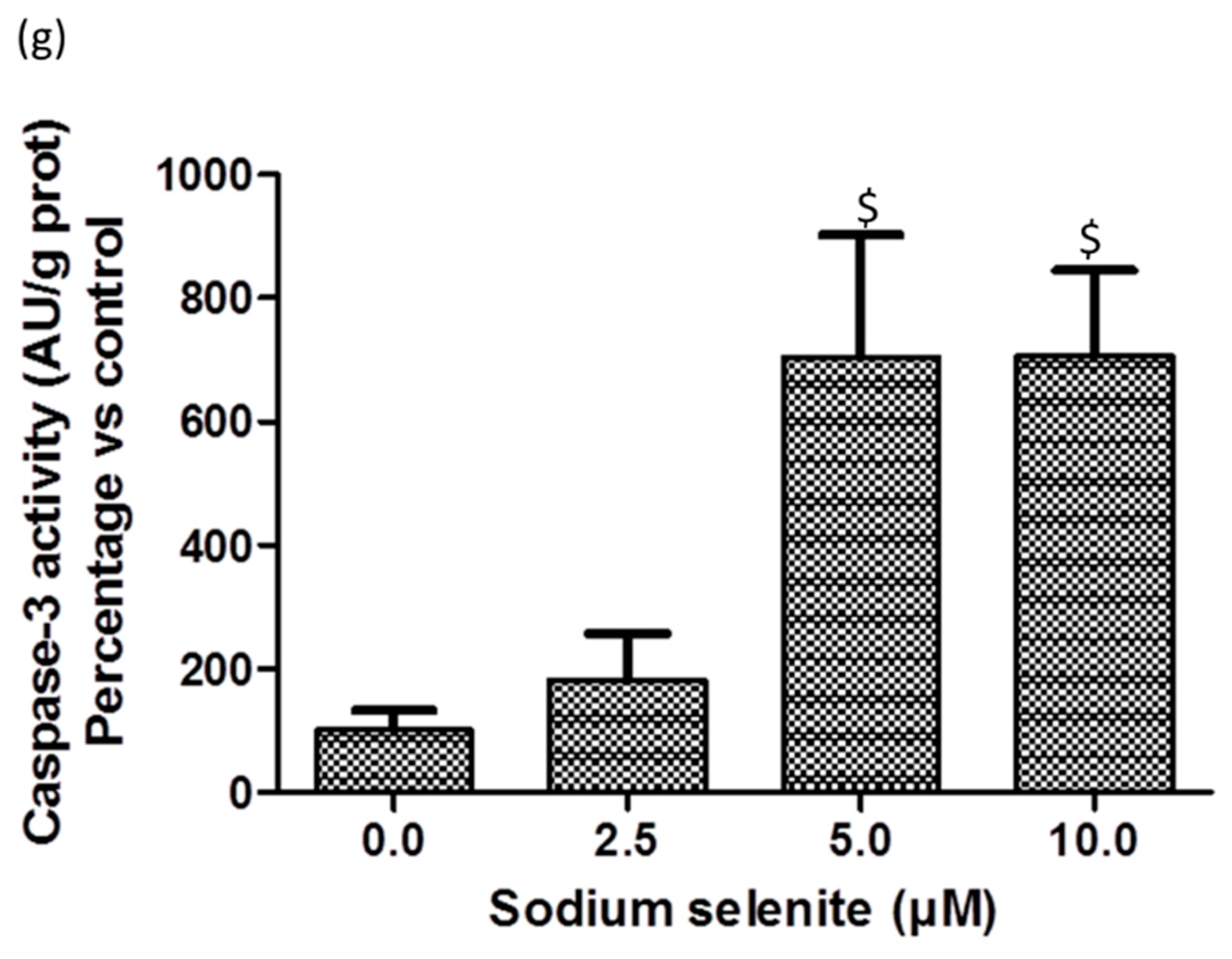
2.9. SS Triggered both Apoptosis and the Necrosis Cell Death Process
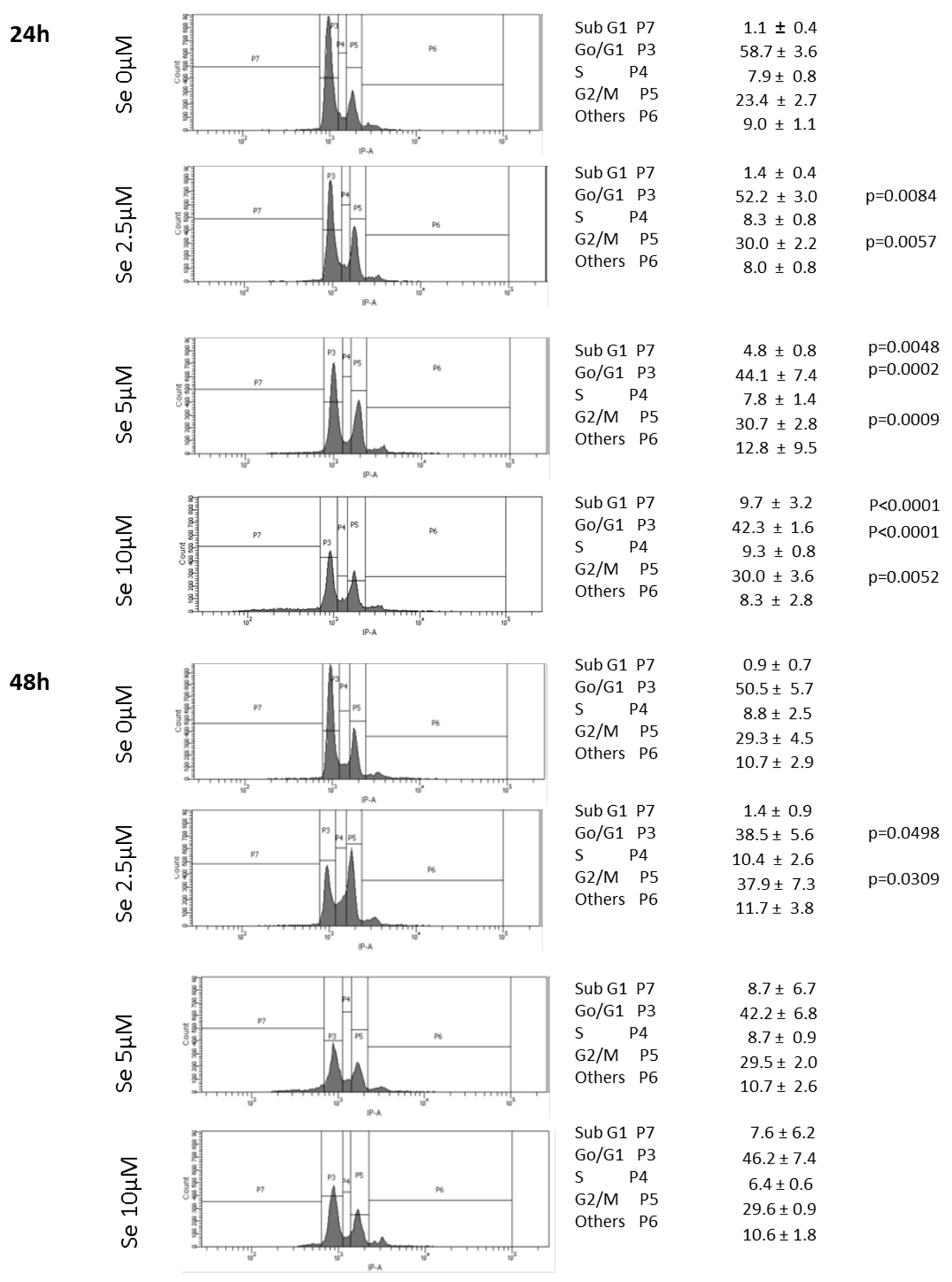
2.10. SS Induced DNA Fragmentation and Cell Cycle Arrest in G2
2.11. SS Acted at the Epigenetic Level
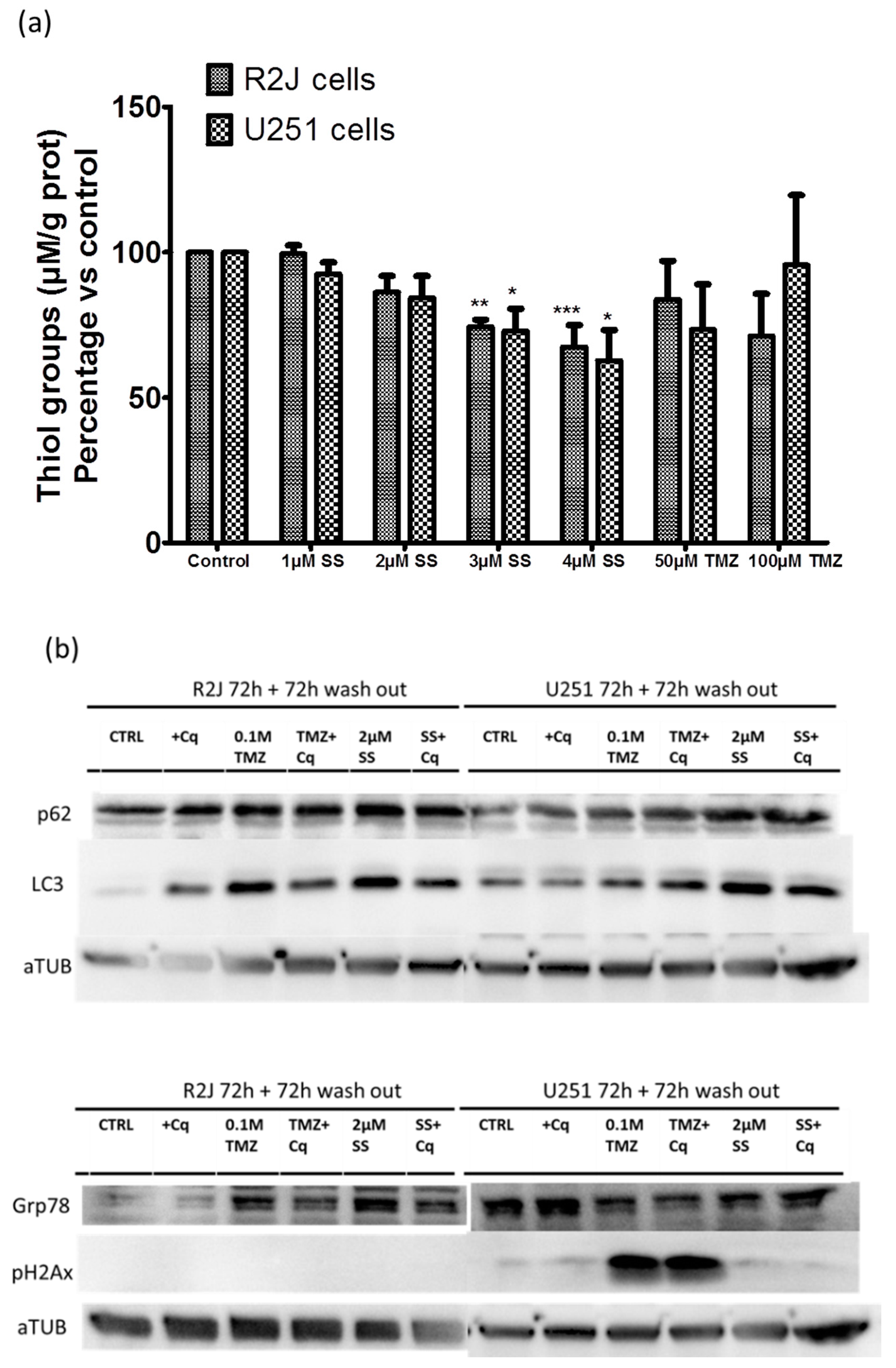
2.12. SS Induced Oxidative Stress, Autophagy, and Reticulum Endoplasmic Stress
3. Discussion
4. Materials and Methods
4.1. Clinical Data of the Patient
4.2. Origin of R2J Cells
4.3. Morphological Analysis
4.4. Cell Culture
4.5. Immunohistochemistry
4.6. Flow Cytometry Analysis
4.7. Chromosome Analysis
4.8. Limiting Dilution/Tumor Sphere Initiation Assay (LDA)
4.9. Intracranial Cell Transplantation into Immunocompromised Mice
4.10. Magnetic Resonance Imaging
4.11. Selenium Absorption
4.12. MTT Assay
4.13. Confocal Microscopy Analysis of Cell Viability
4.14. Caspase-3 Activity Evaluation
4.15. Determination of HDAC Activity
4.16. Real Time PCR
4.17. Thiol Group Determination
4.18. Western Blot Analysis
4.19. Protein Concentration Determination
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Monolayer |
| BSA | Bovine Serum Albumin |
| CSC | Cancer Stem Cell |
| CSF | Cerebro-Spinal Fluid |
| Ct | Cycle threshold |
| CT | ChemoTherapy |
| ECM | ExtraCellular Matrix |
| EGFR | Epithelial Growth Factor-Receptor |
| GMT | Glial to Mesenchymal Transition |
| FACS | Fluorescence Activated Cell Sorter |
| FCS | Foetal Calf Serum |
| FDA | Fluorescein DiAcetate |
| FISH | Fluorescence In Situ Hybridization |
| FITC | Fluorescein IsoThioCyanate |
| G-CSF-R | Granulocyte-Colony Stimulating Factor-Receptor |
| GFAP | Glial Fibrillary Acidic Protein |
| H3K9m2 | dimethyl-Histone-3-lysine-9 |
| HDAC | Histone Deacetylase |
| HES | Hematoxylin Eosin Safran |
| ICP-MS | Inductively Coupled Plasma Mass Spectrometry |
| IDH | Isocitrate DesHydrogenase |
| KDM | Histone Lysine Demethylase |
| MDM2 | Mouse Double Minute 2 homolog |
| MGG | May Grunwald Giemsa |
| MGMTt+ | positive transcript expression of O(6)-MethyGuanine-DNA-MethylTransferase |
| MMPs | Matrix Metallo-Proteases |
| MRI | Magnetic Resonance Imaging |
| N-CAM | Neural Cell Adhesion Molecule |
| neuN | NEUronal Nuclear antigen |
| p15/INK4B | Inhibitor of CDK4B |
| p16/INK4A | Inhibitor of CDK4A |
| p21/WAF1 | Cyclin-dependent kinase inhibitor 1 |
| p27/KIP1 | Cyclin-dependent kinase inhibitor 1B |
| PBS | Phosphate Buffer Saline |
| PI | Propidium iodide |
| PI3K | Phosphatidylinositol-3 kinase |
| PBS | Phosphate buffer salt |
| PFA | ParaFormAldehyde |
| ROS | Reactive Oxygen Species |
| RT | Radiation Therapy |
| TMZ | Temozolomide |
| SS | Sodium selenite |
Appendix A
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic alternative splicing switches: Role in cancer progression and prospects for therapy. Int. J. Cell Biol. 2013, 2013, 962038. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Hamoudi, R.A. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet. Cytogenet. 2000, 122, 87–92. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef]
- Jin, X.; Jin, X.; Jung, J.E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Molecular biomarkers of cancer stem/progenitor cells associated with progression, metastases, and treatment resistance of aggressive cancers. Cancer Epidemiol. Prev. Biomark. 2013, 23, 234–254. [Google Scholar] [CrossRef]
- Choi, S.A.; Wang, K.C.; Phi, J.H.; Lee, J.Y.; Park, C.K.; Park, S.H.; Kim, S.K. A distinct subpopulation within CD133 positive brain tumor cells shares characteristics with endothelial progenitor cells. Cancer Lett. 2012, 324, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma cell line-derived spheres in serumcontaining medium versus serum-free medium: A comparison of cancer stem cell properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2013, 16, 671–685. [Google Scholar] [CrossRef]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-mesenchymal transition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13, 202. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Harabin-Slowinska, M.; Slowinski, J.; Konecki, J.; Mrowka, R. Expression of adhesion molecule CD44 in metastatic brain tumors. Folia Neuropathol. 1998, 36, 179–184. [Google Scholar]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat. 2012, 15, 21–38. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Kondengaden, S.M.; Che, F.; Wang, L.; Heng, X. Potential Epigenetic-Based Therapeutic Targets for Glioma. Front. Mol. Neurosci. 2018, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, S.H.; Kim, T.M.; Choi, S.H.; Park, S.H.; Heo, D.S.; Kim, I.H.; Jung, H.W. The value of temozolomide in combination with radiotherapy during standard treatment for newly diagnosed glioblastoma. J. Neurooncol. 2013, 112, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Hazane-Puch, F.; Champelovier, P.; Arnaud, J.; Garrel, C.; Ballester, B.; Faure, P.; Laporte, F. Long-term selenium supplementation in HaCaT cells: Importance of chemical form for antagonist (protective versus toxic) activities. Biol. Trace Elem. Res. 2013, 154, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Hazane-Puch, F.; Champelovier, P.; Arnaud, J.; Trocme, C.; Garrel, C.; Faure, P.; Laporte, F. Six-day selenium supplementation led to either UVA-photoprotection or toxic effects in human fibroblasts depending on the chemical form and dose of Se. Met. Integr. Biomet. Sci. 2014, 6, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Hazane-Puch, F.; Arnaud, J.; Trocme, C.; Faure, P.; Laporte, F.; Champelovier, P. Sodium Selenite Decreased HDAC Activity, Cell Proliferation and Induced Apoptosis in Three Human Glioblastoma Cells. Anti-Cancer Agents Med. Chem. 2016, 16, 490–500. [Google Scholar] [CrossRef]
- Berthier, S.; Arnaud, J.; Champelovier, P.; Col, E.; Garrel, C.; Cottet, C.; Boutonnat, J.; Laporte, F.; Faure, P.; Hazane-Puch, F. Anticancer properties of sodium selenite in human glioblastoma cell cluster spheroids. J. Trace Elem. Med. Biol. 2017, 44, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Rooprai, H.K.; Kyriazis, I.; Nuttall, R.K.; Edwards, D.R.; Zicha, D.; Aubyn, D.; Davies, D.; Gullan, R.; Pilkington, G.J. Inhibition of invasion and induction of apoptosis by selenium in human malignant brain tumour cells in vitro. Int. J. Oncol. 2007, 30, 1263–1271. [Google Scholar] [CrossRef]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.J.; Lee, S.J.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chinen, Y.; Zhu, Z.; Kimura, M.; Itokawa, Y. Uptake and distribution of sodium selenite in rat brain tumor. Biol. Trace Elem. Res. 1995, 48, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, R.R.; Scott, K.G.; Sairenji, E. Selenite (75Se) as a tumor-localizing agent in man. J. Nucl. Med. 1966, 7, 197–208. [Google Scholar] [PubMed]
- Hazane-Puch, F.; Soldini, A. Unit Nutritional and Hormonal Biochemistry, Institute of Biology and Pathology, Grenoble Alpes Hospital, CS10217, 38043 Grenoble Cedex 9, France. Unpublished Work. 2017. [Google Scholar]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells—Much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Loja, T.; Chlapek, P.; Kuglik, P.; Pesakova, M.; Oltova, A.; Cejpek, P.; Veselska, R. Characterization of a GM7 glioblastoma cell line showing CD133 positivity and both cytoplasmic and nuclear localization of nestin. Oncol. Rep. 2009, 21, 119–127. [Google Scholar] [PubMed]
- Onda, K.; Nagai, S.; Tanaka, R.; Morii, K.; Yoshimura, J.I.; Tsumanuma, I.; Kumanishi, T. Establishment of two glioma cell lines from two surgical specimens obtained at different times from the same individual. J. Neuro-Oncol. 1999, 41, 247–254. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef]
- Therman, E.; Buchler, D.A.; Nieminen, U.; Timonen, S. Mitotic modifications and aberrations in human cervical cancer. Cancer Genet. Cytogenet. 1984, 11, 185–197. [Google Scholar] [CrossRef]
- Yu, S.C.; Ping, Y.F.; Yi, L.; Zhou, Z.H.; Chen, J.H.; Yao, X.H.; Gao, L.; Wang, J.M.; Bian, X.W. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett. 2008, 265, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Iacopino, F.; Angelucci, C.; Piacentini, R.; Biamonte, F.; Mangiola, A.; Maira, G.; Grassi, C.; Sica, G. Isolation of cancer stem cells from three human glioblastoma cell lines: Characterization of two selected clones. PLoS ONE 2014, 9, e105166. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Hansel, M.; Hamel, W.; Kunzmann, R.; Holzel, F. Karyotype analyses of 20 human glioma cell lines. Acta Neurochir. 1994, 126, 17–26. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Agasse, F.; Armstrong, D.; Peng, L.; Commins, D.; Wang, W.; Rosenstein-Sisson, R.; Vaikari, V.P.; Santiago, S.V.; Santos, T.; et al. A novel drug conjugate, NEO212, targeting proneural and mesenchymal subtypes of patient-derived glioma cancer stem cells. Cancer Lett. 2016, 371, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Takano, H.; Okamura, T.; Park, J.S.; Tanimoto, K.; Sekikawa, T.; Yamamoto, W.; Sparreboom, A.; Verweij, J.; Nishiyama, M. O(6)-methylguanine-DNA methyltransferase (MGMT) as a determinant of resistance to camptothecin derivatives. Jpn. J. Cancer Res. 2002, 93, 93–102. [Google Scholar] [CrossRef]
- Lunoe, K.; Gabel-Jensen, C.; Sturup, S.; Andresen, L.; Skov, S.; Gammelgaard, B. Investigation of the selenium metabolism in cancer cell lines. Met. Integr. Biomet. Sci. 2011, 3, 162–168. [Google Scholar] [CrossRef]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef]
- Weekley, C.M.; Aitken, J.B.; Vogt, S.; Finney, L.A.; Paterson, D.J.; de Jonge, M.D.; Howard, D.L.; Witting, P.K.; Musgrave, I.F.; Harris, H.H. Metabolism of selenite in human lung cancer cells: X-ray absorption and fluorescence studies. J. Am. Chem. Soc. 2011, 133, 18272–18279. [Google Scholar] [CrossRef]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlof, A.K.; Larsen, E.H.; Danielsson, O.; Bjornstedt, M. Extracellular thiol-assisted selenium uptake dependent on the x(c)-cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef] [PubMed]
- Coles-Takabe, B.L.; Brain, I.; Purpura, K.A.; Karpowicz, P.; Zandstra, P.W.; Morshead, C.M.; van der Kooy, D. Don’t look: Growing clonal versus nonclonal neural stem cell colonies. Stem Cells 2008, 26, 2938–2944. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Sehested, J. A simple method for R banding of human chromosomes, showing a pH-dependent connection between R and G bands. Humangenetik 1974, 21, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; Overbergh, L.; Valckx, D.; Decallonne, B.; Bouillon, R.; Mathieu, C. An overview of real-time quantitative PCR: Applications to quantify cytokine gene expression. Methods 2001, 25, 386–401. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Related Family | Proteins | Tumor | R2J Monolayer (2D) | R2J-Gliospheres |
|---|---|---|---|---|
| Conventional GBM markers and subtypes | ATRX | nd | + | + |
| GFAP | + | − | − | |
| IDH1 | nd | − | − | |
| Ki67 | + | + | + | |
| Olig2 | − | − | + rare cells | |
| TP53 mutated | nd | − | − | |
| Vimentin | + | + | + | |
| Neuronal markers | CD56 | + | − | + |
| neuN | − | − | − | |
| NF70 | − | − | − | |
| Growth factor receptors | EGFR | + low expression | + few cells | + few cells |
| Cell cycle markers | P16/INK4 | nd | + | + |
| MDM2 | nd | − | − | |
| Cancer stem cell markers | CD34 | − | − | − |
| Nestin | + | + | + | |
| Mesenchymal shift | CD44 | + rare cells | + (>90%) | + few cells |
| E-cadherin | nd | − | − |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berthier, S.; Larrouquère, L.; Champelovier, P.; Col, E.; Lefebvre, C.; Cottet-Rouselle, C.; Arnaud, J.; Garrel, C.; Laporte, F.; Boutonnat, J.; et al. A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent. Cancers 2019, 11, 12. https://doi.org/10.3390/cancers11010012
Berthier S, Larrouquère L, Champelovier P, Col E, Lefebvre C, Cottet-Rouselle C, Arnaud J, Garrel C, Laporte F, Boutonnat J, et al. A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent. Cancers. 2019; 11(1):12. https://doi.org/10.3390/cancers11010012
Chicago/Turabian StyleBerthier, Sylvie, Louis Larrouquère, Pierre Champelovier, Edwige Col, Christine Lefebvre, Cécile Cottet-Rouselle, Josiane Arnaud, Catherine Garrel, François Laporte, Jean Boutonnat, and et al. 2019. "A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent" Cancers 11, no. 1: 12. https://doi.org/10.3390/cancers11010012
APA StyleBerthier, S., Larrouquère, L., Champelovier, P., Col, E., Lefebvre, C., Cottet-Rouselle, C., Arnaud, J., Garrel, C., Laporte, F., Boutonnat, J., Faure, P., & Hazane-Puch, F. (2019). A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent. Cancers, 11(1), 12. https://doi.org/10.3390/cancers11010012