Comprehensive Genomic Profiling Reveals Diverse but Actionable Molecular Portfolios across Hematologic Malignancies: Implications for Next Generation Clinical Trials

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
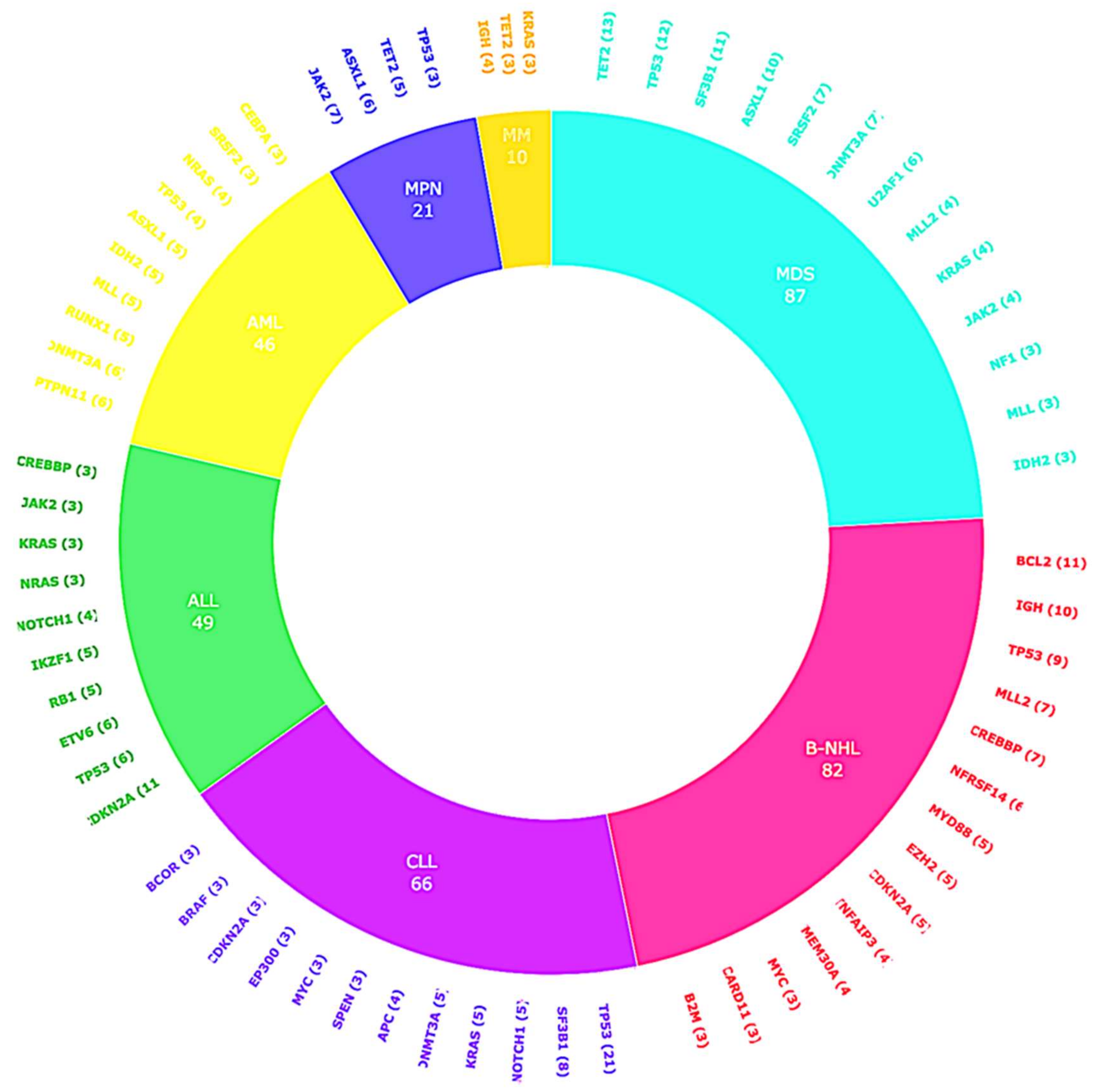
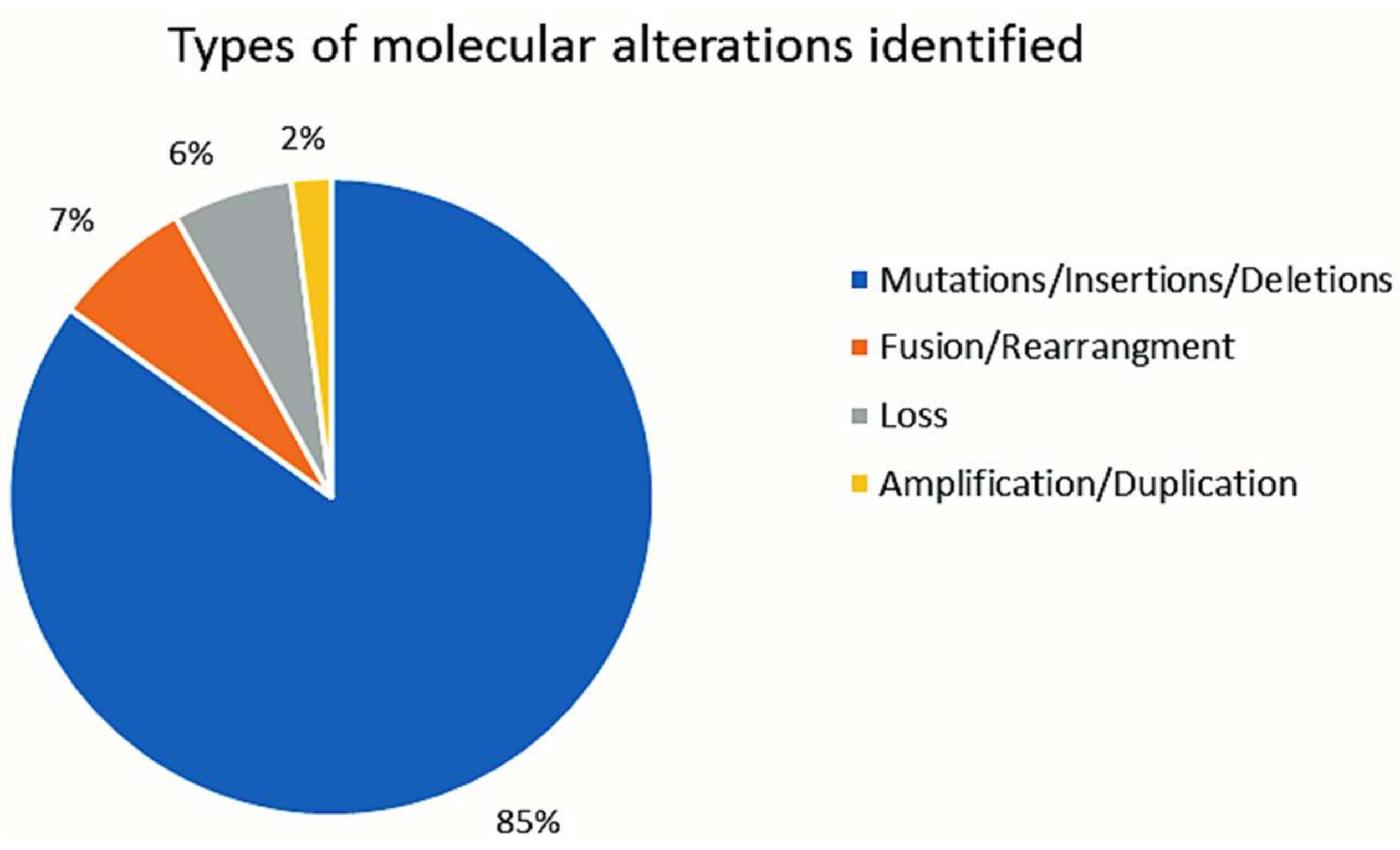
2.2. CGP Results
2.3. TMB
2.4. Actionable Alterations
3. Discussion
4. Patients and Methods
4.1. Patients
4.2. Comprehensive Genomic Profiling (CGP)
4.3. Tumor Mutational Burden (TMB)
4.4. Definition of a Potentially Actionable Alteration
4.5. Data Analysis and Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wheler, J.; Lee, J.J.; Kurzrock, R. Unique molecular landscapes in cancer: Implications for individualized, curated drug combinations. Cancer Res. 2014, 74, 7181–7184. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Gordon, L.I.; Wierda, W.G.; Abramson, J.S.; Advani, R.H.; Andreadis, C.B.; Bartlett, N.; Byrd, J.C.; Fayad, L.E.; Fisher, R.I.; et al. Diffuse Large B-Cell Lymphoma Version 1.2016. J. Natl. Compr. Cancer Netw. JNCCN 2016, 14, 196–231. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Hunter, Z. MYD88 Mutations and Response to Ibrutinib in Waldenstrom’s Macroglobulinemia. N. Engl. J. Med. 2015, 373, 584–586. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Schlette, E.; Kurzrock, R. Rapid response to vemurafenib in a heavily pretreated patient with hairy cell leukemia and a BRAF mutation. J. Clin. Oncol. 2013, 31, e351–e352. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Samadder, N.J.; Neklason, D.W.; Boucher, K.M.; Byrne, K.R.; Kanth, P.; Samowitz, W.; Jones, D.; Tavtigian, S.V.; Done, M.W.; Berry, T.; et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA 2016, 315, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Functional and cancer genomics of ASXL family members. Br. J. Cancer 2013, 109, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Wiestner, A. Targeting B-Cell receptor signaling for anticancer therapy: the Bruton’s tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J. Clin. Oncol. 2013, 31, 128–130. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B Cell Receptor Signaling in Diffuse Large B Cell Lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis-Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A Mutations and Response to the Hypomethylating Agent Decitabine in Acute Myeloid Leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Muller, C.; Gruning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. FLT3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [PubMed]
- Dias, S.; Choy, M.; Alitalo, K.; Rafii, S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood 2002, 99, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Faramand, R.; Carter-Cooper, B.; Tolu, S.; Ford, L.A.; Lapidus, R.G.; Wetzler, M.; Wang, E.S.; Etemadi, A.; Griffiths, E.A. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am. J. Hematol. 2015, 90, E77–E79. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Boyle, D.L.; Soma, K.; Hodge, J.; Kavanaugh, A.; Mandel, D.; Mease, P.; Shurmur, R.; Singhal, A.K.; Wei, N.; Rosengren, S.; et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1311–1316. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Gotlib, J.; Durocher, J.A.; Chao, M.P.; Mariappan, M.R.; Lay, M.; Jones, C.; Zehnder, J.L.; Lilleberg, S.L.; Weissman, I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 6224–6229. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet. Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Lindsay, Y.E.; Leslie, N.R. PTEN inhibitors: An evaluation of current compounds. Adv. Biol. Regul. 2015, 57, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.S.; Rosario, M.; Birchmeier, C.; Birchmeier, W. The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 2010, 106, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Siau, E.D.; Drilon, A. Targeting RET-rearranged lung cancers with multikinase inhibitors. Oncoscience 2017, 4, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Perez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Lazar, V.; Validire, P.; Hansson, J.; Lacroix, L.; Soria, J.C.; Pawitan, Y.; Kurzrock, R. VEGF-A Expression Correlates with TP53 Mutations in Non-Small Cell Lung Cancer: Implications for Antiangiogenesis Therapy. Cancer Res. 2015, 75, 1187–1190. [Google Scholar] [CrossRef]
- Schwaederle, M.; Daniels, G.A.; Piccioni, D.E.; Fanta, P.T.; Schwab, R.B.; Shimabukuro, K.A.; Parker, B.A.; Kurzrock, R. On the Road to Precision Cancer Medicine: Analysis of Genomic Biomarker Actionability in 439 Patients. Mol. Cancer Ther. 2015, 14, 1488–1494. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, A.; Curiel-Olmo, S.; Mollejo, M.; Cereceda, L.; Martinez, N.; Montes-Moreno, S.; Almaraz, C.; Revert, J.B.; Piris, M.A. MYD88 (L265P) somatic mutation in marginal zone B-cell lymphoma. Am. J. Surg. Pathol. 2015, 39, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Hadzidimitriou, A.; Agathangelidis, A.; Rossi, D.; Sutton, L.A.; Kminkova, J.; Scarfo, L.; Pospisilova, S.; Gaidano, G.; Stamatopoulos, K.; et al. Prognostic relevance of MYD88 mutations in CLL: The jury is still out. Blood 2015, 126, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, Y.; Lee, J.H.; Kim, Y.S. MYD88 expression and L265P mutation in diffuse large B-cell lymphoma. Hum. Pathol. 2013, 44, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Aguilar, A.; Idbaih, A.; Boisselier, B.; Habbita, N.; Rossetto, M.; Laurenge, A.; Bruno, A.; Jouvet, A.; Polivka, M.; Adam, C.; et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Grommes, C.; Kaley, T.J.; Nolan, C.; Omuro, A.M.P.; Wolfe, J.; Mellinghoff, I.K.; DeAngelis, L.M. Phase I study of single agent ibrutinib in recurrent/refractory primary (PCNSL) and secondary CNS lymphoma (SCNSL). J. Clin. Oncol. 2016, 34, 2046. [Google Scholar] [CrossRef]
- Tiacci, E.; Park, J.H.; De Carolis, L.; Chung, S.S.; Broccoli, A.; Scott, S.; Zaja, F.; Devlin, S.; Pulsoni, A.; Chung, Y.R.; et al. Targeting Mutant BRAF in Relapsed or Refractory Hairy-Cell Leukemia. N. Engl. J. Med. 2015, 373, 1733–1747. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef]
- Jebaraj, B.M.; Kienle, D.; Buhler, A.; Winkler, D.; Dohner, H.; Stilgenbauer, S.; Zenz, T. BRAF mutations in chronic lymphocytic leukemia. Leukemia Lymphoma 2013, 54, 1177–1182. [Google Scholar] [CrossRef]
- Flaherty, K.T.; McArthur, G. BRAF, a target in melanoma: Implications for solid tumor drug development. Cancer 2010, 116, 4902–4913. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Hasserjian, R.P.; Oduro, K.; Glomski, K.; Nardi, V.; Cote, G.M.; Graubert, T.A.; Brunner, A.M.; Chen, Y.B.A.; Fathi, A.T. Combined Targeted Therapy for BRAF-Mutant, Treatment-Related Acute Myeloid Leukemia. JCO Precis. Oncol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Turski, M.L.; Vidwans, S.J.; Janku, F.; Garrido-Laguna, I.; Munoz, J.; Schwab, R.; Subbiah, V.; Rodon, J.; Kurzrock, R. Genomically Driven Tumors and Actionability across Histologies: BRAF-Mutant Cancers as a Paradigm. Mol. Cancer Ther. 2016, 15, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Choi, M.; Wieduwilt, M.; Mulroney, C.; Costello, C.; Frampton, G.; Miller, V.; Kurzrock, R. Next Generation Sequencing Reveals Potentially Actionable Alterations in the Majority of Patients with Lymphoid Malignancies. JCO Precis. Oncol. 2017, 1. [Google Scholar] [CrossRef]
- Fashoyin-Aje, L.; Donoghue, M.; Chen, H.; He, K.; Veeraraghavan, J.; Goldberg, K.B.; Keegan, P.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Pembrolizumab for Recurrent Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Adenocarcinoma Expressing PD-L1. Oncologist 2018. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Fanta, P.T.; Boles, S.G.; Daniels, G.A.; Bazhenova, L.A.; Subramanian, R.; Coutinho, A.C.; Ojeda-Fournier, H.; et al. Molecular tumor board: The University of California-San Diego Moores Cancer Center experience. Oncologist 2014, 19, 631–636. [Google Scholar] [CrossRef]
- He, J.; Abdel-Wahab, O.; Nahas, M.K.; Wang, K.; Rampal, R.K.; Intlekofer, A.M.; Patel, J.; Krivstov, A.; Frampton, G.M.; Young, L.E.; et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016, 127, 3004–3014. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9. [Google Scholar] [CrossRef]
- Kowanetz, M.; Zou, W.; Shames, D.S.; Cummings, C.; Rizvi, N.; Spira, A.I.; Frampton, G.M.; Leveque, V.; Flynn, S.; Mocci, S.; et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Demographics and Baseline Characteristics | |
|---|---|
| Gender: Men (%); Women (%) | 133 (59%); 94 (41%) |
| Age: Median age (range) | 59 years (17–88) |
| Ethnicity: Caucasian (%) | 163 (71.8%) |
| Asian (%) | 18 (8%) |
| Hispanic (%) | 18 (8%) |
| African American (%) | 8 (3.5%) |
| Other (%) | 20 (8.8%) |
| Histologies: | |
| Myeloid Disorders | N = 124 (54.6%) |
| Myelodysplastic syndrome (MDS) | 52 (22.9%) |
| Acute myeloid leukemia (AML) | 25 (11%) |
| Myeloproliferative neoplasia (MPN) | 21 (9.2%) |
| Multiple Myeloma (MM) | 17 (7.5%) |
| Chronic myeloid leukemia (CML) | 4 (1.8%) |
| Other myeloid disorders | 5 (2.2%) |
| Lymphoid disorders | N = 103 (45.4%) |
| Chronic lymphocytic leukemia (CLL) | 39 (17.2%) |
| Acute lymphocytic leukemia (ALL) | 20 (8.8%) |
| Diffuse large B-cell lymphoma (DLBCL) | 18 (7.9%) |
| Follicular lymphoma (FL) | 6 (2.6%) |
| Marginal zone lymphoma (MZL) | 4 (1.8%) |
| Anaplastic large cell lymphoma (ALCL) | 2 (0.9%) |
| Castleman disease | 2 (0.9%) |
| Other lymphoid disorders | 12 (5.3%) |
| Summary of alterations | |
| Number of patients with alterations | 197 (87%) |
| Number of patients with potentially actionable alterations | 170 (75%) |
| Median number of alterations/patient (range) | 3 (0–14) |
| Median number of potentially actionable alterations/patient (range) | 1 (0–7) |
| Total alterations | 698 |
| Number of distinct alterations | 546 |
| Number of distinct potentially actionable alterations | 256 |
| Gene Alteration | Gene Function | Examples of Potential on/off-Label Therapy | Examples of Potential Experimental Therapy/Clinical Trial * | Ref. |
|---|---|---|---|---|
| ABL1/2 | ABL (Abelson tyrosine-protein) kinase regulates cell survival and division/differentiation | Imatinib, Dasatinib, Nilotinib, Bosutinib, Ponatinib | ||
| APC | APC (adenomatous polyposis coli) is a tumor suppressor, regulating cell division/adhesion, controls Wnt signaling pathway | Sulindac (Tankyrase inh) | [13] | |
| ARID1A | ARID1A (AT-rich interactive domain-containing protein 1A) regulates transcription | Dasatinib, EZH2 inh. | Talazoparib Tosylate NCT02286687 ** | |
| ASXL1 | ASXL1 (additional sex combs-like1) regulates transcription and ubiquitin-proteasome protein degradation via BAP pathway. | Cabozantinib | [14] | |
| ATM | ATM (ataxia telangiectasia mutated) regulates DNA damage response via the PI3K-like protein kinase pathway | Olaparib | [15] | |
| BCL2 | BCL2 (B-cell lymphoma 2) regulates apoptosis | Venetoclax | [16] | |
| BRAF | BRAF regulates cell growth via MAPK (RAF-MEK-ERK) signaling cascade | Dabrafenib, Regorafenib, Trametinib, Vemurafenib, Cobimetinib | [17] | |
| BRCA2 | BRCA2 (breast cancer 1/2) regulates DNA double-strand break repair | Olaparib, Niraparib, Rucaparib | ||
| BRIP1 | BRIP1 (BRCA1-interacting protein 1) functions in DNA repair | Olaparib | ||
| BTK | BTK (Bruton’s tyrosine kinase) regulates B-cell receptor signaling and B-cell development | Ibrutinib, Acalabrutinib | [18] | |
| CCND2 | CCND1/3 (cyclin D1/3) regulates cell cycle via CDK4/6 | Palbociclib | [19] | |
| CD274 | CD274 (cluster of differentiation 274) encodes immune inhibitory receptor B7-H1, also known as programmed cell death ligand-1 (PD-L1) | Atezolizumab, Avelumab, Durvalumab, Nivolumab, Pembrolizumab | ||
| CD79B | CD79A/B (cluster of differentiation 79) complexes with B-cell receptor, mediates downstream signaling to the NF-kB, PI3K, MAPK and NF-AT pathways | Ibrutinib | Polatuzumab vedotin | [20] |
| CDK4 | CDK4 (cyclin-dependent kinase 4) regulates cell cycle | Palbociclib, Ribociclib | [19] | |
| CDKN2A/B | CDKN2A (cyclin dependent kinase inhibitor encodes tumor suppressors and regulates cell cycle; loss results in increased CDK4/6 | Palbociclib, Ribociclib | [19] | |
| CSF1R | CSF1 (colony stimulating factor 1) regulates differentiation and survival | Chiauranib NCT03074825 ** | ||
| CXCR4 | CXCR4 (C-X-C chemokine receptor type 4) regulates hematopoiesis and CD20 expression | Plerixafor | BMS-936564 NCT01120457 ** | [21] |
| DNMT3A | DNMT3A (DNA methyltransferase 3A) regulates gene expression | Azacitidine, Decitabine | [22] | |
| EP300 | Histone acetyltransferase p300 regulates transcription via chromatin remodeling | Mocetinostat NCT02282358 ** | ||
| ERBB4 | Member of the EGFR (epidermal growth factor receptor) regulates proliferation | Trastuzumab, Pertuzumab Afatinib, Erlotinib, Lapatinib | ||
| EZH2 | EZH2 (enhancer of zeste-homolog 2) regulates DNA methylation and transcription repression | Tazemetostat (NCT02601950) ** | [23] | |
| FGFR3 | FGFR3 (fibroblast growth factor receptor 3) promotes cell cycle via activation of RAS/MAPK/AKT pathway | Lenvatinib, Pazopanib, Ponatinib, Regorafenib | ||
| FLT3 | FLT3 (FMS-like tyrosine kinase 3) activates signaling of Akt1, RAS, ERK, and mTOR. | Midostaurin, Gilteritinib Quizartinib | [9,24] | |
| FLT4 | FLT4 (FMS like tyrosine kinase 4), also known as VEGFR-3 (vascular endothelial growth factor receptor 3) | Sorafenib, Sunitinib, Pazopanib, Axitinib, Vandetanib, | [25] | |
| GNAS | GNAS (Guanine nucleotide binding protein, α stimulating) regulates adenylate cyclase via MAPK | Trametinib | ||
| IDH1 | IDH1 (isocitrate dehydrogenases 1) | Azacitidine, Decitabine | [26] | |
| IDH/2 | IDH2 (isocitrate dehydrogenases 2) regulates citric acid (Krebs) cycle and cell metabolism | Enasidenib | [27] | |
| IGF1R | IGF1R (insulin-like growth factor-1 receptor) mediates anti-apoptotic signals | Ganitumab NCT00562380 ** | ||
| JAK1 | JAK1 (Janus kinase 1) in involved in signal regulation | Tofacitinib | Fedratinib | [28] |
| JAK2 | JAK2 (Janus kinase 2) is involved in signal regulation | Ruxolitinib | [29] | |
| KIT | KIT (also known as c-Kit or CD117), activates PI3K/Akt and RAS/MAPK signaling pathway | Imatinib, Midastaurin | ||
| KRAS | KRAS (Kirsten rat sarcoma) regulates signal transduction via MAPK pathway | Cetuximab, Trametinib, Panitumumab, Regorafenib | [30] | |
| MAP2K1 | MAP2K1 (mitogen-activated protein kinase 1 (MKK1 or MEK1) mediates RAS/RAF/MAPK pathway | Cobimetinib, Selumetinib, Trametinib | ||
| MAP3K14 | MAP3K14 (mitogen-activated protein kinase 14) also known as NF-kappa-B-inducing kinase | Trametinib | ||
| MLL | MLL (mixed lineage leukemia) encodes a histone methyltransferase | EPZ-5676 NCT02141828 ** | ||
| MSH2 | MSH2 (MutS homolog2) is a tumor suppressor encodes DNA mismatch repair (MMR) protein 2 | Atezolizumab, Nivolumab Pembrolizumab | ||
| MSH6 | MSH6 (MutS homolog 6) encodes DNA mismatch repair (MMR) protein 6 involved in DNA repair | Atezolizumab, Nivolumab Pembrolizumab | ||
| MYC | MYC regulates cell cycle progression, apoptosis, proliferation | BET inhibitors NCT02431260 ** | ||
| MYD88 | MYD88 (myeloid differentiation primary response gene 88) activates transcription factor NFkB | Ibrutinib, acalabrutinib (IRAK1 inh) | zanubrutinib | [7] |
| NF1/2 | NF1 (neurofibromin 1/2) a GTPase-activating negative regulator of the RAS signaling pathway | Everolimus, Temsirolimus, Trametinib | ||
| NRAS | NRAS (neuroblastoma RAS) mediates signal transduction via RAF/MEK/ERK and PI3K | Trametinib, Panitumumab | [31] | |
| PALB2 | PALB2 (partner and localizer of BRCA2) | Olaparib | [32] | |
| PDCD1LG2 | Programmed cell death 1 ligand 2 (also known as CD273) essential for T-cell proliferation | Atezolizumab, Avelumab, Durvalumab, Nivolumab, Pembrolizumab | ||
| PIK3CA | PIK3CA (phosphatidylinositol 3-kinase (PI3K), which regulates the PI3K/AKT/MTOR axis | Everolimus, Temsirolimus, Copanlisib, Duvalisib, Idelalisib | Taselisib NCT02465060 ** | |
| PIK3R1 | PIK3R1 (PI3K regulatory subunit alpha) | Copanlisib | NCT02369016 ** | |
| PTCH1 | PTCH1 (Protein patched homolog 1) is a receptor for Sonic hedgehog (Shh) for gene transcription | Vismodegib, Sonidegib | [33] | |
| PTEN | PTEN (phosphatase and tensin homolog) is a tumor suppressor, functions via PI3K/AKT/mTOR pathway | Everolimus, Temsirolimus | [34] | |
| PTPN11 | PTPN11 (Tyrosine-protein phosphatase non-receptor type 11) activates PI3K, MEK axis | Trametinib, | [35] | |
| RET | RET (rearranged during transfection) is a proto-oncogene | Cabozantinib, Sorafenib, Vandetanib, Lenvatinib | [36] | |
| RUNX1 | RUNX1 (Runt-related transcription factor, also known as acute myeloid leukemia 1 protein (AML1), core-binding factor subunit alpha 2 (CBFA2) is a tumor suppressor | Mocetinostat (MGCD0103) or Sorafenib NCT00217646 ** | ||
| STAT3 | STAT3 (signal transducer and activator of transcription 3) encodes a transcription factor | AZD9150 (NCT01839604) ** | ||
| STK11 | STK11 (serine/threonine kinase 11) functions as a tumor suppressor gene | Dasatinib, Bosutinib, Everolimus, Temsirolimus | ||
| TET2 | TET2 (Tet methylcytosine dioxygenase 2) regulates DNA demethylation | Azacitidine, Decitabine | [37] | |
| TP53 | TP53 (Tumor protein p53) is a tumor suppressor; loss leads to overexpression of VEGF levels | Bevacizumab, Pazopanib | Wee-1 inh, MDM inh, PRIMA-1MET inhibitors. | [38] |
| VHL | VHL (von Hippel-Lindau) is a tumor suppressor activates the HIF/VEGF pathway | Axitinib, Bevacizumab, Everolimus, Pazopanib, Sorafenib, Sunitinib, Temsirolimus, Vandetanib, | ||
| XPO1 | XPO1 (exportin-1) regulates nuclear export of tumor suppressor genes | Selinexor NCT02227251 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galanina, N.; Bejar, R.; Choi, M.; Goodman, A.; Wieduwilt, M.; Mulroney, C.; Kim, L.; Yeerna, H.; Tamayo, P.; Vergilio, J.-A.; et al. Comprehensive Genomic Profiling Reveals Diverse but Actionable Molecular Portfolios across Hematologic Malignancies: Implications for Next Generation Clinical Trials. Cancers 2019, 11, 11. https://doi.org/10.3390/cancers11010011
Galanina N, Bejar R, Choi M, Goodman A, Wieduwilt M, Mulroney C, Kim L, Yeerna H, Tamayo P, Vergilio J-A, et al. Comprehensive Genomic Profiling Reveals Diverse but Actionable Molecular Portfolios across Hematologic Malignancies: Implications for Next Generation Clinical Trials. Cancers. 2019; 11(1):11. https://doi.org/10.3390/cancers11010011
Chicago/Turabian StyleGalanina, Natalie, Rafael Bejar, Michael Choi, Aaron Goodman, Matthew Wieduwilt, Carolyn Mulroney, Lisa Kim, Huwate Yeerna, Pablo Tamayo, Jo-Anne Vergilio, and et al. 2019. "Comprehensive Genomic Profiling Reveals Diverse but Actionable Molecular Portfolios across Hematologic Malignancies: Implications for Next Generation Clinical Trials" Cancers 11, no. 1: 11. https://doi.org/10.3390/cancers11010011
APA StyleGalanina, N., Bejar, R., Choi, M., Goodman, A., Wieduwilt, M., Mulroney, C., Kim, L., Yeerna, H., Tamayo, P., Vergilio, J.-A., Mughal, T. I., Miller, V., Jamieson, C., & Kurzrock, R. (2019). Comprehensive Genomic Profiling Reveals Diverse but Actionable Molecular Portfolios across Hematologic Malignancies: Implications for Next Generation Clinical Trials. Cancers, 11(1), 11. https://doi.org/10.3390/cancers11010011