
Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy



Highlights
- The ketogenic diet and its variants, including the modified Atkins and the medium-chain triglyceride diets, as well as low glycemic index treatment, show variable efficacy in genetically confirmed drug-resistant epilepsy.
- The clinical effects of these dietary therapies are influenced by underlying genetic and metabolic mechanisms, with disease-specific pathways shaping therapeutic outcomes.
- The safety and tolerability of the ketogenic diet and its applications are strongly dependent on genetic background, with certain metabolic disorders representing clear contraindications.
- A genetics-informed approach is essential for optimizing patient selection and guiding individualized dietary therapy use in epilepsy management.
Abstract
1. Introduction
2. Ketogenic Diet and Its Applications
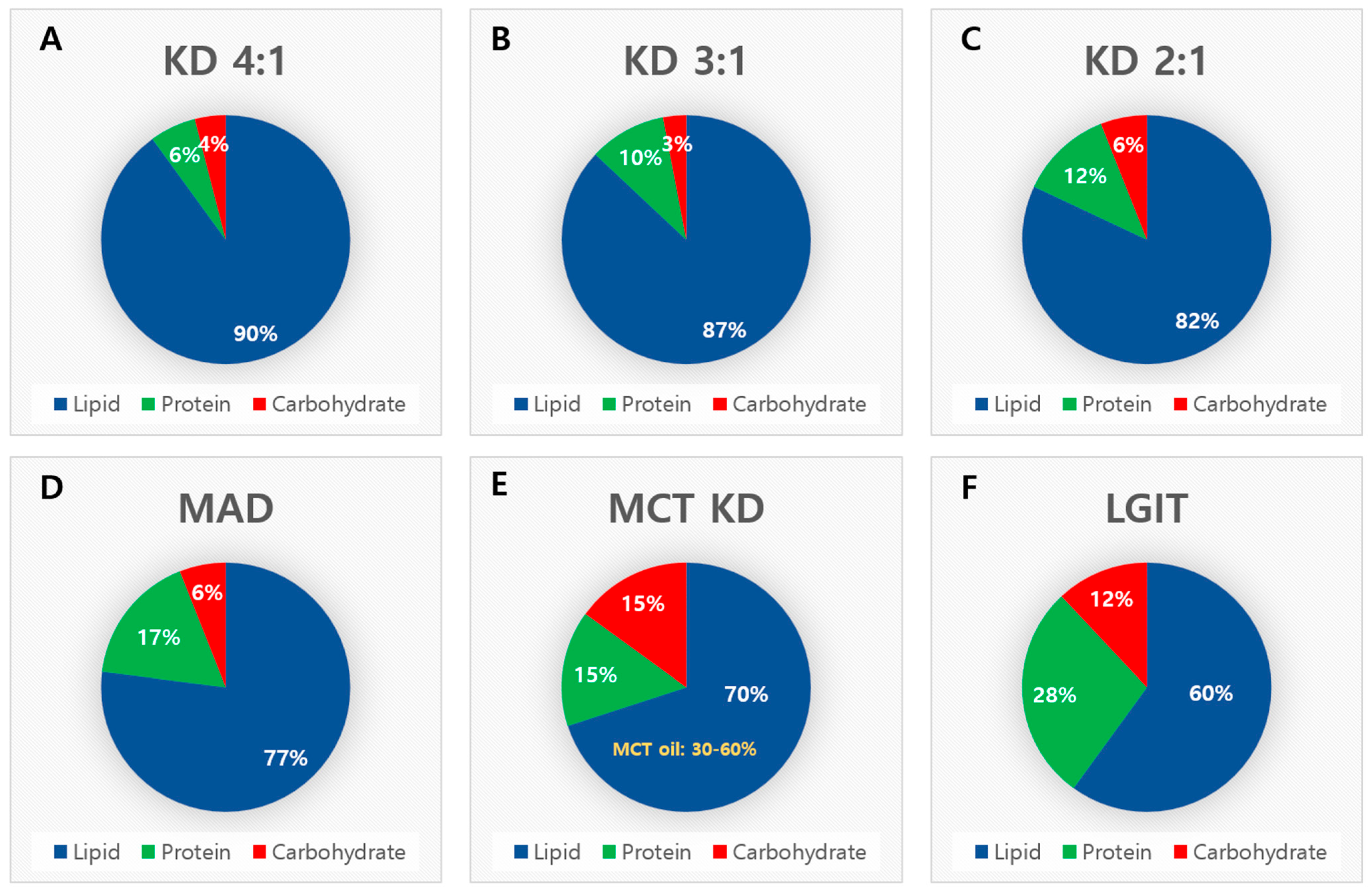
2.1. Classic Ketogenic Diet
2.2. Modified Atkins Diet
2.3. Medium-Chain Triglyceride Diet
2.4. Low Glycemic Index Treatment
3. Efficacy of the KD and Its Applications in Genetically Derived DRE
3.1. Pyruvate Dehydrogenase E1-Alpha Subunit 1
3.2. Solute Carrier Family 2 Member 1
3.3. Sodium Voltage-Gated Channel Alpha Subunit 1
3.4. Tuberous Sclerosis Complex 1 and 2
3.5. Cyclin-Dependent Kinase-like 5
3.6. Syntaxin-Binding Protein 1 (STXBP1)
3.7. Ubiquitin Protein Ligase E3A
3.8. Mitochondrial-Encoded tRNALeu (UUR) 1
3.9. Other Genes
4. Safety of Ketogenic Diet and Its Applications
5. Limitations and Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cKD | Classic ketogenic diet |
| SLC2A1 | Solute carrier family 2 member 1 |
| GLUT1DS | Glucose transporter type 1 deficiency syndrome |
| PDHA1 | Pyruvate dehydrogenase E1-alpha subunit 1 |
| PDCD | Pyruvate dehydrogenase complex deficiency |
| SCN1A | Sodium voltage-gated channel alpha subunit 1 |
| KCNQ2 | Potassium voltage-gated channel subfamily Q member 2 |
| KCNQ3 | Potassium voltage-gated channel subfamily Q member 3 |
| DEE | Developmental and epileptic encephalopathy |
| TSC1 | Tuberous sclerosis complex 1 |
| TSC2 | Tuberous sclerosis complex 2 |
| TSC | Tuberous sclerosis complex |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CDD | CDKL5 deficiency disorder |
| STXBP1 | Syntaxin-binding protein 1 |
| SCN8A | Sodium voltage-gated channel alpha subunit 8 |
| GRIN2A | Glutamate ionotropic receptor NMDA type subunit 2A |
| LKS | Landau–Kleffner syndrome |
| MT-TL1 | Mitochondrially encoded tRNA leucine 1 (UUR) |
| MELAS | Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes |
| PC | Pyruvate carboxylase |
| PCCA | Propionyl-CoA carboxylase subunit alpha |
| mtDNA | Mitochondrial deoxyribonucleic acid |
| POLG | Polymerase gamma |
| MCTD | Medium-chain triglyceride diet |
| LCHADD | Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency |
| PCCB | Propionyl-CoA carboxylase subunit beta |
| SLC22A5 | Solute carrier family 22 member 5 |
| ACADM | Acyl-CoA dehydrogenase medium-chain |
| MCADD | Medium-chain acyl-CoA dehydrogenase deficiency |
| ACADVL | Acyl-CoA dehydrogenase very long-chain |
| VLCADD | Very long-chain acyl-CoA dehydrogenase deficiency |
| HADHA | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha |
| HADHB | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta |
| LGIT | Low glycemic index treatment |
| LPL | Lipoprotein lipase |
| ECG | Electrocardiogram |
| CLCN5 | Chloride voltage-gated channel 5 |
| MCTD | medium-chain triglyceride diet |
| LCHAD | long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency |
References
- Hsieh, T.Y.; Su, T.Y.; Hung, K.Y.; Hsu, M.S.; Lin, Y.J.; Kuo, H.C.; Hung, P.L. Ketogenic diet effectiveness is superior for drug resistant epilepsy with causative genetic mutation than those without genetic etiology. Epilepsy Behav. 2024, 161, 110052. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 6, CD001903. [Google Scholar] [CrossRef]
- Perucca, E.; Perucca, P.; White, H.S.; Wirrell, E.C. Drug resistance in epilepsy. Lancet Neurol. 2023, 22, 723–734. [Google Scholar] [CrossRef]
- Samanta, D. Cognitive and behavioral impact of antiseizure medications, neuromodulation, ketogenic diet, and surgery in lennox-gastaut syndrome: A comprehensive review. Epilepsy Behav. 2025, 164, 110272. [Google Scholar] [CrossRef]
- Mesraoua, B.; Brigo, F.; Lattanzi, S.; Abou-Khalil, B.; Al Hail, H.; Asadi-Pooya, A.A. Drug-resistant epilepsy: Definition, pathophysiology, and management. J. Neurol. Sci. 2023, 452, 120766. [Google Scholar] [CrossRef]
- Armeno, M.; Calligaris, S.; Gagiulo, D.; Cresta, A.; Vaccarezza, M.M.; Diez, C.G.; Alberti, M.J.; Viollaz, R.; Vilavedra, F.; Caraballo, R.H. Use of ketogenic dietary therapy for drug-resistant epilepsy in early infancy. Epilepsia Open 2024, 9, 138–149. [Google Scholar] [CrossRef]
- Pizzo, F.; Collotta, A.D.; Di Nora, A.; Costanza, G.; Ruggieri, M.; Falsaperla, R. Ketogenic diet in pediatric seizures: A randomized controlled trial review and meta-analysis. Expert Rev. Neurother. 2022, 22, 169–177. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Ozturk, E.; Aslan Cin, N.N.; Cansu, A.; Akyol, A. Ketogenic diet as a therapeutic approach in autism spectrum disorder: A narrative review. Metab. Brain Dis. 2024, 40, 67. [Google Scholar] [CrossRef]
- Guerreiro, D.; Almeida, A.; Ramalho, R. Ketogenic Diet and Neuroinflammation: Implications for Neuroimmunometabolism and Therapeutic Approaches to Refractory Epilepsy. Nutrients 2024, 16, 3994. [Google Scholar] [CrossRef]
- Ko, A.; Jung, D.E.; Kim, S.H.; Kang, H.C.; Lee, J.S.; Lee, S.T.; Choi, J.R.; Kim, H.D. The Efficacy of Ketogenic Diet for Specific Genetic Mutation in Developmental and Epileptic Encephalopathy. Front. Neurol. 2018, 9, 530. [Google Scholar] [CrossRef] [PubMed]
- Muthaffar, O.Y.; Alyazidi, A.S.; Alsowat, D.; Alasiri, A.A.; Albaradie, R.; Jad, L.A.; Kayyali, H.; Jan, M.M.S.; Bamaga, A.K.; Alsubaie, M.A.; et al. Short-term effectiveness and side effects of ketogenic diet for drug-resistant epilepsy in children with genetic epilepsy syndromes. Front. Neurol. 2024, 15, 1484752. [Google Scholar] [CrossRef] [PubMed]
- Rastin, C.; Schenkel, L.C.; Sadikovic, B. Complexity in Genetic Epilepsies: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 14606. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, N.E.; Cross, J.H.; Sander, J.W.; Sisodiya, S.M. Can we predict a favourable response to Ketogenic Diet Therapies for drug-resistant epilepsy? Epilepsy Res. 2013, 106, 1–16. [Google Scholar] [CrossRef]
- Ulamek-Koziol, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Ketogenic Diet and Epilepsy. Nutrients 2019, 11, 2510. [Google Scholar] [CrossRef]
- Kang, H.C.; Lee, Y.M.; Kim, H.D.; Lee, J.S.; Slama, A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia 2007, 48, 82–88. [Google Scholar] [CrossRef]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy—A Review. Nutrients 2020, 12, 1809. [Google Scholar] [CrossRef]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef]
- Attaye, I.; van Oppenraaij, S.; Warmbrunn, M.V.; Nieuwdorp, M. The Role of the Gut Microbiota on the Beneficial Effects of Ketogenic Diets. Nutrients 2021, 14, 191. [Google Scholar] [CrossRef]
- McDonald, T.J.W.; Cervenka, M.C. Ketogenic Diets for Adult Neurological Disorders. Neurotherapeutics 2018, 15, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Borowicz-Reutt, K.; Krawczyk, M.; Czernia, J. Ketogenic Diet in the Treatment of Epilepsy. Nutrients 2024, 16, 1258. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Madaan, P.; Kandoth, N.; Bansal, D.; Sahu, J.K. Efficacy and Safety of Dietary Therapies for Childhood Drug-Resistant Epilepsy: A Systematic Review and Network Meta-analysis. JAMA Pediatr. 2023, 177, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Na, J.-H. Application and Effectiveness of Dietary Therapies for Pediatric Migraine. Headache Pain Res. 2024, 25, 34–41. [Google Scholar] [CrossRef]
- Sondhi, V.; Agarwala, A.; Pandey, R.M.; Chakrabarty, B.; Jauhari, P.; Lodha, R.; Toteja, G.S.; Sharma, S.; Paul, V.K.; Kossoff, E.; et al. Efficacy of Ketogenic Diet, Modified Atkins Diet, and Low Glycemic Index Therapy Diet Among Children With Drug-Resistant Epilepsy: A Randomized Clinical Trial. JAMA Pediatr. 2020, 174, 944–951. [Google Scholar] [CrossRef]
- Kim, D.W.; Kang, H.C.; Park, J.C.; Kim, H.D. Benefits of the nonfasting ketogenic diet compared with the initial fasting ketogenic diet. Pediatrics 2004, 114, 1627–1630. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Krauss, G.L.; McGrogan, J.R.; Freeman, J.M. Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurology 2003, 61, 1789–1791. [Google Scholar] [CrossRef]
- Griffen, C.; Schoeler, N.E.; Browne, R.; Cameron, T.; Kirkpatrick, M.; Thowfeek, S.; Munn, J.; Champion, H.; Mills, N.; Phillips, S.; et al. Tolerance, adherence, and acceptability of a ketogenic 2.5:1 ratio, nutritionally complete, medium chain triglyceride-containing liquid feed in children and adults with drug-resistant epilepsy following a ketogenic diet. Epilepsia Open 2024, 9, 727–738. [Google Scholar] [CrossRef]
- Lin, T.Y.; Liu, H.W.; Hung, T.M. The Ketogenic Effect of Medium-Chain Triacylglycerides. Front. Nutr. 2021, 8, 747284. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.C.; Lee, E.J.; Lee, J.S.; Kim, H.D. Low glycemic index treatment in patients with drug-resistant epilepsy. Brain Dev. 2017, 39, 687–692. [Google Scholar] [CrossRef]
- Pfeifer, H.H.; Thiele, E.A. Low-glycemic-index treatment: A liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 2005, 65, 1810–1812. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Deng, J.; Chen, C.; Wang, X.; Han, T.; Wang, X.; Fang, T.; Tian, X.; Fang, F. Long-term effectiveness and tolerability of ketogenic diet therapy in patients with genetic developmental and epileptic encephalopathy onset within the first 6 months of life. Epilepsia Open 2024, 9, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012, 105, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.; McCormick, E.M.; Falk, M.J. Primary Pyruvate Dehydrogenase Complex Deficiency Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Sofou, K.; Dahlin, M.; Hallbook, T.; Lindefeldt, M.; Viggedal, G.; Darin, N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef]
- Habarou, F.; Bahi-Buisson, N.; Lebigot, E.; Pontoizeau, C.; Abi-Warde, M.T.; Brassier, A.; Le Quan Sang, K.H.; Broissand, C.; Vuillaumier-Barrot, S.; Roubertie, A.; et al. Ketone Bodies as a Possible Adjuvant to Ketogenic Diet in PDHc Deficiency but Not in GLUT1 Deficiency. JIMD Rep. 2018, 38, 53–59. [Google Scholar] [CrossRef]
- Falsaperla, R.; Sortino, V.; Kluger, G.J.; Herberhold, T.; Ruegger, A.; Striano, P.; Ruggieri, M.; Klepper, J.; Ramantani, G. Exploring ketogenic diet resistance in glucose transporter type 1 deficiency syndrome: A comprehensive review and critical appraisal. Epilepsia Open 2025, 10, 31–39. [Google Scholar] [CrossRef]
- Klepper, J.; Akman, C.; Armeno, M.; Auvin, S.; Cervenka, M.; Cross, H.J.; De Giorgis, V.; Della Marina, A.; Engelstad, K.; Heussinger, N.; et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef]
- De Giorgis, V.; Masnada, S.; Varesio, C.; Chiappedi, M.A.; Zanaboni, M.; Pasca, L.; Filippini, M.; Macasaet, J.A.; Valente, M.; Ferraris, C.; et al. Overall cognitive profiles in patients with GLUT1 Deficiency Syndrome. Brain Behav. 2019, 9, e01224. [Google Scholar] [CrossRef]
- Amer, A.; Murrell, K.; Edmonds, L.; Bernhardt, I.; Akroyd, R.; Ryder, B.; Wilson, C.; Glamuzina, E. D,L-3-hydroxybutyrate in the treatment of glucose transporter 1 deficiency syndrome (Glut1DS). JIMD Rep. 2025, 66, e12461. [Google Scholar] [CrossRef]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The genetics of Dravet syndrome. Epilepsia 2011, 52 (Suppl. S2), 24–29. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.O.; Sotero de Menezes, M.A. SCN1A Seizure Disorders. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- van Hugte, E.J.H.; Lewerissa, E.I.; Wu, K.M.; Scheefhals, N.; Parodi, G.; van Voorst, T.W.; Puvogel, S.; Kogo, N.; Keller, J.M.; Frega, M.; et al. SCN1A-deficient excitatory neuronal networks display mutation-specific phenotypes. Brain 2023, 146, 5153–5167. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, X.; Tian, H.; Wang, L.; Guo, B.; Wang, Y.; Li, W.; Wang, F.; Sun, T. SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 2021, 12, 743726. [Google Scholar] [CrossRef] [PubMed]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Man, A.; Di Scipio, M.; Grewal, S.; Suk, Y.; Trinari, E.; Ejaz, R.; Whitney, R. The Genetics of Tuberous Sclerosis Complex and Related mTORopathies: Current Understanding and Future Directions. Genes 2024, 15, 332. [Google Scholar] [CrossRef]
- Curatolo, P.; Specchio, N.; Aronica, E. Advances in the genetics and neuropathology of tuberous sclerosis complex: Edging closer to targeted therapy. Lancet Neurol. 2022, 21, 843–856. [Google Scholar] [CrossRef]
- Nabavi Nouri, M.; Zak, M.; Jain, P.; Whitney, R. Epilepsy Management in Tuberous Sclerosis Complex: Existing and Evolving Therapies and Future Considerations. Pediatr. Neurol. 2022, 126, 11–19. [Google Scholar] [CrossRef]
- Youn, S.E.; Park, S.; Kim, S.H.; Lee, J.S.; Kim, H.D.; Kang, H.C. Long-term outcomes of ketogenic diet in patients with tuberous sclerosis complex-derived epilepsy. Epilepsy Res. 2020, 164, 106348. [Google Scholar] [CrossRef]
- Fang, Y.; Li, D.; Wang, M.; Zhao, X.; Duan, J.; Gu, Q.; Li, B.; Zha, J.; Mei, D.; Bian, G.; et al. Ketogenic Diet Therapy for Drug-Resistant Epilepsy and Cognitive Impairment in Children with Tuberous Sclerosis Complex. Front. Neurol. 2022, 13, 863826. [Google Scholar] [CrossRef]
- Leonard, H.; Downs, J.; Benke, T.A.; Swanson, L.; Olson, H.; Demarest, S. CDKL5 deficiency disorder: Clinical features, diagnosis, and management. Lancet Neurol. 2022, 21, 563–576. [Google Scholar] [CrossRef]
- Hong, W.; Haviland, I.; Pestana-Knight, E.; Weisenberg, J.L.; Demarest, S.; Marsh, E.D.; Olson, H.E. CDKL5 Deficiency Disorder-Related Epilepsy: A Review of Current and Emerging Treatment. CNS Drugs 2022, 36, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, J.; Chang, X.; Wu, P.; Li, S.; Wu, Y. Efficacy of ketogenic diet in CDKL5-related epilepsy: A single arm meta-analysis. Orphanet J. Rare Dis. 2022, 17, 385. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.; Parthasarathy, S.; Ruggiero, S.M.; Balagura, G.; Fitch, E.; Helbig, K.; Gan, J.; Ganesan, S.; Kaufman, M.C.; Ellis, C.A.; et al. Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain 2022, 145, 1668–1683. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.; Thalwitzer, K.M.; McKee, J.; Sullivan, K.R.; Brimble, E.; Fitch, E.; Toib, J.; Kaufman, M.C.; deCampo, D.; Cunningham, K.; et al. Delineating clinical and developmental outcomes in STXBP1-related disorders. Brain 2023, 146, 5182–5197. [Google Scholar] [CrossRef]
- Dagli, A.I.; Mathews, J.; Williams, C.A. Angelman Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Yang, L.; Shu, X.; Mao, S.; Wang, Y.; Du, X.; Zou, C. Genotype-Phenotype Correlations in Angelman Syndrome. Genes 2021, 12, 987. [Google Scholar] [CrossRef]
- Melikishvili, G.; Bienvenu, T.; Tabatadze, N.; Gachechiladze, T.; Kurua, E.; Gverdtsiteli, S.; Melikishvili, M.; Dulac, O. Novel UBE3A pathogenic variant in a large Georgian family produces non-convulsive status epilepticus responsive to ketogenic diet. Seizure 2022, 94, 70–73. [Google Scholar] [CrossRef]
- Duis, J.; Nespeca, M.; Summers, J.; Bird, L.; Bindels-de Heus, K.; Valstar, M.J.; de Wit, M.Y.; Navis, C.; Ten Hooven-Radstaake, M.; van Iperen-Kolk, B.M.; et al. A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol. Genet. Genom. Med. 2022, 10, e1843. [Google Scholar] [CrossRef]
- Grocott, O.R.; Herrington, K.S.; Pfeifer, H.H.; Thiele, E.A.; Thibert, R.L. Low glycemic index treatment for seizure control in Angelman syndrome: A case series from the Center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017, 68, 45–50. [Google Scholar] [CrossRef]
- Belal, S.; Goudenege, D.; Bocca, C.; Dumont, F.; Chao De La Barca, J.M.; Desquiret-Dumas, V.; Gueguen, N.; Geffroy, G.; Benyahia, R.; Kane, S.; et al. Glutamate-Induced Deregulation of Krebs Cycle in Mitochondrial Encephalopathy Lactic Acidosis Syndrome Stroke-Like Episodes (MELAS) Syndrome Is Alleviated by Ketone Body Exposure. Biomedicines 2022, 10, 1665. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar] [CrossRef]
- He, F.; Ye, L.; Miao, P.; Zhou, J.; Ding, Y.; Wang, S. Long-term ketogenic diet therapy improves mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS): A case report. CNS Neurosci. Ther. 2023, 29, 2717–2720. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. PCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review. Pediatr. Neurol. 2020, 105, 3–9. [Google Scholar] [CrossRef]
- Trivisano, M.; Specchio, N. The role of PCDH19 in refractory status epilepticus. Epilepsy Behav. 2019, 101, 106539. [Google Scholar] [CrossRef]
- Talwar, D.; Hammer, M.F. SCN8A Epilepsy, Developmental Encephalopathy, and Related Disorders. Pediatr. Neurol. 2021, 122, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, Y.; Zhang, J.; Lu, Y.; Men, X.; Wang, X. Ketogenic Diet in Infants with Early-Onset Epileptic Encephalopathy and SCN2A Mutation. Yonsei Med. J. 2021, 62, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yang, D.; Kim, S.H.; Kim, B.; Kim, H.D.; Lee, J.S.; Choi, J.R.; Lee, S.T.; Kang, H.C. The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy. Epileptic Disord. 2020, 22, 563–570. [Google Scholar] [CrossRef]
- Su, D.J.; Lu, J.F.; Lin, L.J.; Liang, J.S.; Hung, K.L. SCN2A mutation in an infant presenting with migrating focal seizures and infantile spasm responsive to a ketogenic diet. Brain Dev. 2018, 40, 724–727. [Google Scholar] [CrossRef]
- Appavu, B.; Vanatta, L.; Condie, J.; Kerrigan, J.F.; Jarrar, R. Ketogenic diet treatment for pediatric super-refractory status epilepticus. Seizure 2016, 41, 62–65. [Google Scholar] [CrossRef]
- Manville, R.W.; Papanikolaou, M.; Abbott, G.W. M-Channel Activation Contributes to the Anticonvulsant Action of the Ketone Body beta-Hydroxybutyrate. J. Pharmacol. Exp. Ther. 2020, 372, 148–156. [Google Scholar] [CrossRef]
- Strehlow, V.; Myers, K.A.; Morgan, A.T.; Scheffer, I.E.; Lemke, J.R. GRIN2A-Related Disorders. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lagae, L. Rational treatment options with AEDs and ketogenic diet in Landau-Kleffner syndrome: Still waiting after all these years. Epilepsia 2009, 50 (Suppl. S7), 59–62. [Google Scholar] [CrossRef]
- Duque Lasio, M.L.; Lehman, A.N.; Ahmad, A.; Bedoyan, J.K. Pyruvate Carboxylase Deficiency. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- García-Cazorla, A.; Rabier, D.; Touati, G.; Chadefaux-Vekemans, B.; Marsac, C.; de Lonlay, P.; Saudubray, J.M. Pyruvate carboxylase deficiency: Metabolic characteristics and new neurological aspects. Ann. Neurol. 2006, 59, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Liu, G.S.; Yi, B. Primary carnitine deficiency in two sisters with intractable epilepsy and reversible metabolic cardiomyopathy: Two case reports. Exp. Ther. Med. 2020, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Zweers, H.E.E.; Kroesen, S.H.; Beerlink, G.; Buit, E.; Gerrits, K.; Dorhout, A.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B.; Timmers, S.; et al. Ketogenic diet in adult patients with mitochondrial myopathy. Mol. Genet. Metab. 2024, 143, 108610. [Google Scholar] [CrossRef] [PubMed]
- Dynka, D.; Kowalcze, K.; Paziewska, A. The Role of Ketogenic Diet in the Treatment of Neurological Diseases. Nutrients 2022, 14, 5003. [Google Scholar] [CrossRef]
- Mason, E.; Hindmarch, C.C.T.; Dunham-Snary, K.J. Medium-chain Acyl-COA dehydrogenase deficiency: Pathogenesis, diagnosis, and treatment. Endocrinol. Diabetes Metab. 2023, 6, e385. [Google Scholar] [CrossRef]
- Van Calcar, S.C.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.U.; Pendyal, S.; Wallace, L.S.; Hansen, J.G.; Stembridge, A.; et al. Nutrition management guideline for very-long chain acyl-CoA dehydrogenase deficiency (VLCAD): An evidence- and consensus-based approach. Mol. Genet. Metab. 2020, 131, 23–37. [Google Scholar] [CrossRef]
- Rücklová, K.; Hrubá, E.; Pavlíková, M.; Hanák, P.; Farolfi, M.; Chrastina, P.; Vlášková, H.; Kousal, B.; Smolka, V.; Foltenová, H.; et al. Impact of Newborn Screening and Early Dietary Management on Clinical Outcome of Patients with Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Medium Chain Acyl-CoA Dehydrogenase Deficiency-A Retrospective Nationwide Study. Nutrients 2021, 13, 2925. [Google Scholar] [CrossRef]
- Pedersen, Z.O.; Holm-Yildiz, S.; Dysgaard, T. Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety. Int. J. Mol. Sci. 2022, 23, 10658. [Google Scholar] [CrossRef]
- Wesol-Kucharska, D.; Greczan, M.; Kaczor, M.; Ehmke Vel Emczynska-Seliga, E.; Hajdacka, M.; Czekuc-Kryskiewicz, E.; Piekutowska-Abramczuk, D.; Halat-Wolska, P.; Ciara, E.; Jaworski, M.; et al. Efficacy and Safety of Ketogenic Diet Treatment in Pediatric Patients with Mitochondrial Disease. Nutrients 2024, 16, 812. [Google Scholar] [CrossRef]
- Newmaster, K.; Zhu, Z.; Bolt, E.; Chang, R.J.; Day, C.; Mhanna, A.; Paudel, S.; Farooq, O.; Swaminathan, A.; Acharya, P.; et al. A Review of the Multi-Systemic Complications of a Ketogenic Diet in Children and Infants with Epilepsy. Children 2022, 9, 1372. [Google Scholar] [CrossRef]
- Lin, A.; Turner, Z.; Doerrer, S.C.; Stanfield, A.; Kossoff, E.H. Complications During Ketogenic Diet Initiation: Prevalence, Treatment, and Influence on Seizure Outcomes. Pediatr. Neurol. 2017, 68, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Na, J.H.; Kim, H.D.; Lee, Y.M. Effective and safe diet therapies for Lennox-Gastaut syndrome with mitochondrial dysfunction. Ther. Adv. Neurol. Disord. 2020, 13, 1756286419897813. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Nochi, Z.; Olsen, R.K.J.; Gregersen, N. Short-chain acyl-CoA dehydrogenase deficiency: From gene to cell pathology and possible disease mechanisms. J. Inherit. Metab. Dis. 2017, 40, 641–655. [Google Scholar] [CrossRef]
- Stinton, C.; Fraser, H.; Geppert, J.; Johnson, R.; Connock, M.; Johnson, S.; Clarke, A.; Taylor-Phillips, S. Newborn Screening for Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase and Mitochondrial Trifunctional Protein Deficiencies Using Acylcarnitines Measurement in Dried Blood Spots—A Systematic Review of Test Accuracy. Front. Pediatr. 2021, 9, 606194. [Google Scholar] [CrossRef]
- Falsaperla, R.; Sortino, V.; Striano, P.; Kluger, G.; Ramantani, G.; Ruggieri, M.; Network for Therapy in Rare, E. Is ketogenic diet a ’precision medicine’? Recent developments and future challenges. Eur. J. Paediatr. Neurol. 2024, 48, 13–16. [Google Scholar] [CrossRef]
- Qiao, Y.N.; Li, L.; Hu, S.H.; Yang, Y.X.; Ma, Z.Z.; Huang, L.; An, Y.P.; Yuan, Y.Y.; Lin, Y.; Xu, W.; et al. Ketogenic diet-produced beta-hydroxybutyric acid accumulates brain GABA and increases GABA/glutamate ratio to inhibit epilepsy. Cell Discov. 2024, 10, 17. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. beta-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Dussmann, H.; Watters, O.; Connolly, N.M.C.; Prehn, J.H.M. Ketone body beta-hydroxybutyrate (BHB) preserves mitochondrial bioenergetics. Sci. Rep. 2023, 13, 19664. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Leu, C.; Balestrini, S.; Mudge, J.M.; Steward, C.A.; Frankish, A.; Leung, M.A.; Mackay, M.; Scheffer, I.; Williams, R.; et al. Genome-wide association study: Exploring the genetic basis for responsiveness to ketogenic dietary therapies for drug-resistant epilepsy. Epilepsia 2018, 59, 1557–1566. [Google Scholar] [CrossRef]
- Jiang, Z.; Yin, X.; Wang, M.; Chen, T.; Wang, Y.; Gao, Z.; Wang, Z. Effects of Ketogenic Diet on Neuroinflammation in Neurodegenerative Diseases. Aging Dis. 2022, 13, 1146–1165. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Category | Gene | Associated Disorder | Impact on cKD Response | Mechanistic Considerations | References |
|---|---|---|---|---|---|
| Positive Response | SLC2A1 | Glucose Transporter Type 1 Deficiency Syndrome (GLUT1DS) | Strongly Positive | Ketones provide an alternative energy source due to defective glucose transport | [39,40] |
| PDHA1 | Pyruvate Dehydrogenase Complex Deficiency (PDCD) | Strongly Positive | cKD bypasses defective pyruvate metabolism by providing ketones as an energy source | [34,35] | |
| SCN1A | Dravet Syndrome | Positive | Ketones stabilize neuronal hyperexcitability in sodium channel dysfunction | [43,45] | |
| KCNQ2/KCNQ3 | KCNQ2/3-related Developmental and Epileptic Encephalopathy (DEE) | Positive | β-hydroxybutyrate enhances M-current, stabilizing neuronal excitability | [72] | |
| TSC1/TSC2 | Tuberous Sclerosis Complex (TSC) | Positive | cKD reduces mTORC1 overactivation and oxidative stress | [51] | |
| CDKL5 | CDKL5 Deficiency Disorder (CDD) | Variable to Positive | cKD may stabilize neuronal excitability but response varies | [53,54] | |
| STXBP1 | STXBP1-related Epilepsy | Variable to Positive | Potential benefits via synaptic stabilization and mitochondrial support | [56] | |
| SCN8A | SCN8A-related Epileptic Encephalopathy | Variable to Positive | Some cases respond well, possibly through mitochondrial modulation | [67] | |
| GRIN2A | Landau–Kleffner Syndrome (LKS) | Variable to Positive | Some reports indicate benefits, but more studies are needed | [74] | |
| MT-TL1 | MELAS Syndrome | Positive with Caution | cKD may improve mitochondrial function but requires careful monitoring | [63,64] | |
| Contraindications/Cautious Use | PC | Pyruvate Carboxylase Deficiency | Contraindicated | cKD exacerbates metabolic acidosis due to impaired anaplerotic metabolism | [75,76] |
| SLC22A5 | Primary Carnitine Deficiency | Contraindicated | Fat metabolism dysfunction prevents ketone utilization | [8,77] | |
| mtDNA deletions | Mitochondrial DNA Deletion Syndromes (e.g., Kearns-Sayre) | Contraindicated | cKD may exacerbate metabolic decompensation, but LGIT may be safer | [63,78,79] | |
| ACADM | Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCADD) | Contraindicated | cKD leads to hypoglycemia and energy deficits | [80] | |
| ACADVL | Very Long-chain Acyl-CoA Dehydrogenase Deficiency (VLCADD) | Contraindicated | Fatty acid oxidation defects cause metabolic instability | [81] | |
| HADHA/HADHB | Long-chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency (LCHADD) | Contraindicated | Long-chain fatty acid metabolism impaired, but MCTD may be cautiously considered | [82] | |
| POLG | POLG-related Mitochondrial Disorders | Highly Cautious Use | Risk of liver toxicity, metabolic crises, and worsening mitochondrial function | [63,83] |
| Complication Category | Complication | Proposed Mechanism | Genetic Associations | Implications for cKD Use | References |
|---|---|---|---|---|---|
| Metabolic | Metabolic acidosis | Impaired buffering capacity, increased ketone production, accumulation of lactate | PC, SLC22A5 | Absolute contraindication due to risk of severe acidosis | [8,75] |
| Hypoglycemia | Enhanced insulin sensitivity, reduced glycogen stores | No strong genetic predisposition identified | Close glucose monitoring required, particularly in young children | [8,86] | |
| Mitochondrial Dysfunction | Worsening of energy metabolism | Inability to utilize fatty acids, increased lactate production | mtDNA deletions, PDHA1 | Relative contraindication; LGIT may be safer due to partial glucose allowance | [63,85] |
| Gastrointestinal | Constipation, nausea, vomiting | Altered gut motility due to high fat intake, gut microbiota shifts | No specific monogenic link, but gut microbiota composition may influence tolerance | Dietary fiber, hydration, and probiotics may improve symptoms | [23,26,31,32,85] |
| Pancreatitis | Hyperlipidemia-induced pancreatic stress, impaired lipid metabolism | Susceptibility in LPL (Lipoprotein lipase) deficiency, though rare | Avoid in individuals with a history of pancreatitis or hypertriglyceridemia | [26,85] | |
| Cardiovascular | Prolonged QT, cardiomyopathy | Electrolyte imbalances, selenium deficiency, altered lipid metabolism | SLC22A5 (Primary Carnitine Deficiency) may contribute to cardiac dysfunction | Requires regular ECG and selenium/carnitine monitoring | [77,85] |
| Renal | Nephrolithiasis | Increased urinary calcium excretion, hypocitraturia, reduced urinary citrate | No strong genetic predisposition identified | Potassium citrate supplementation recommended for prevention | [17,23,25,85] |
| Liver Dysfunction | Hepatic steatosis, elevated liver enzymes | Altered lipid metabolism, increased hepatic fat accumulation | Some mitochondrial disorders (POLG-related hepatopathy) may predispose to liver dysfunction | Regular liver function test monitoring required | [63,85] |
| Hematologic | Neutropenia, platelet dysfunction | Increased ketone metabolism affecting bone marrow function | No strong genetic predisposition identified | Routine blood count monitoring advised in long-term KD use | [23,85] |
| Neurological | Exacerbation of seizures | Inability to metabolize ketones effectively, paradoxical seizure worsening | POLG mutations can lead to progressive myoclonic epilepsy | Extreme caution required, as worsening seizures may occur | [79,85] |
| Fatty Acid Oxidation Disorders | Energy failure, metabolic crisis | Impaired β-oxidation, dependence on glucose metabolism | ACADM, ACADVL, HADHA, HADHB, ACADS | Absolute contraindication for cKD; MCTD may be cautiously considered in LCHAD patients under strict metabolic monitoring | [80,82,85] |
| Nutritional Deficiencies | Selenium, carnitine, vitamin D deficiency | Decreased intake due to dietary restrictions, increased nutrient loss | No single-gene association, but SLC22A5 (Carnitine Transporter Deficiency) may increase carnitine loss | Routine supplementation of selenium, carnitine, and vitamin D required | [17,85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, J.-H.; Lee, H.; Lee, Y.-M. Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy. Nutrients 2025, 17, 979. https://doi.org/10.3390/nu17060979
Na J-H, Lee H, Lee Y-M. Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy. Nutrients. 2025; 17(6):979. https://doi.org/10.3390/nu17060979
Chicago/Turabian StyleNa, Ji-Hoon, Hyunjoo Lee, and Young-Mock Lee. 2025. "Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy" Nutrients 17, no. 6: 979. https://doi.org/10.3390/nu17060979
APA StyleNa, J.-H., Lee, H., & Lee, Y.-M. (2025). Clinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy. Nutrients, 17(6), 979. https://doi.org/10.3390/nu17060979