
High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. HDL Structure
3. HDL’s Distinct Cargo: Proteins, Hormones, Vitamins, and RNAs
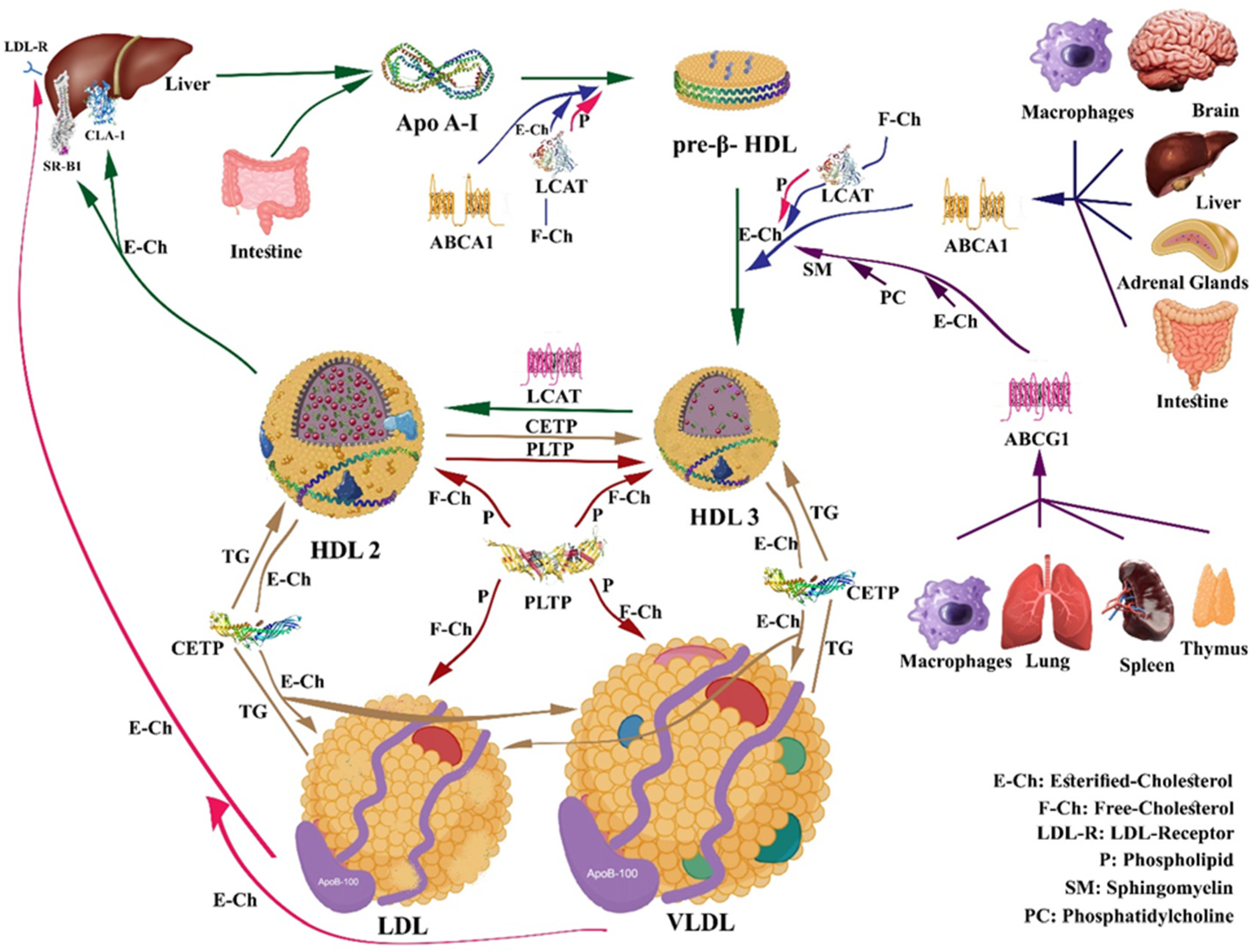
4. HDL Metabolism and Reverse Cholesterol Transport (RCT)
5. HDL Interaction with Major Proteins and Receptors Involved in Cholesterol Efflux
5.1. HDL and ABCA1 Transporter
5.2. HDL and ABCG1 Transporter
5.3. HDL and SR-BI Receptor
5.4. HDL and ectoF1-ATPase Receptor
5.5. HDL and LCAT
5.6. HDL and CETP
5.7. HDL and PLTP
5.8. HDL and Paraoxonase 1 (PON1)
6. Principal HDL Functions Related to CVDs
6.1. Antioxidant/Pro-Oxidant Properties of HDL
6.2. Anti-Inflammatory/Pro-Inflammatory and Immunomodulatory Effects of HDL
7. Aging, HDL, and CVD
8. Effect of Healthy Diet on HDL
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [PubMed]
- Sagaro, G.G.; Battineni, G. Self-Reported Modifiable Risk Factors of Cardiovascular Disease among Seafarers: A Cross-Sectional Study of Prevalence and Clustering. J. Pers. Med. 2021, 11, 512. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.; Abbott, R.D.; Castelli, W.P. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arterioscler. Off. J. Am. Heart Assoc. Inc. 1988, 8, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Barzi, F.; Lam, T.; Czernichow, S.; Fang, X.; Welborn, T.; Shaw, J.; Ueshima, H.; Zimmet, P.; Jee, S. Asia Pacific Cohort Studies Collaboration and the Obesity in Asia Collaboration: Isolated low levels of high-density lipoprotein cholesterol are associated with an increased risk of coronary heart disease: An individual participant data meta-analysis of 23 studies in the Asia-Pacific region. Circulation 2011, 124, 2056. [Google Scholar]
- Kaplan, H.; Thompson, R.C.; Trumble, B.C.; Wann, L.S.; Allam, A.H.; Beheim, B.; Frohlich, B.; Sutherland, M.L.; Sutherland, J.D.; Stieglitz, J.; et al. Coronary atherosclerosis in indigenous South American Tsimane: A cross-sectional cohort study. Lancet 2017, 389, 1730–1739. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R. Genetic variation in the ABCA1 gene, HDL cholesterol, and risk of ischemic heart disease in the general population. Atherosclerosis 2010, 208, 305–316. [Google Scholar] [CrossRef]
- Liu, C.; Dhindsa, D.; Almuwaqqat, Z.; Ko, Y.A.; Mehta, A.; Alkhoder, A.A.; Alras, Z.; Desai, S.R.; Patel, K.J.; Hooda, A.; et al. Association Between High-Density Lipoprotein Cholesterol Levels and Adverse Cardiovascular Outcomes in High-risk Populations. JAMA Cardiol. 2022, 7, 672–680. [Google Scholar] [CrossRef]
- Cabou, C.; Honorato, P.; Briceño, L.; Ghezali, L.; Duparc, T.; León, M.; Combes, G.; Frayssinhes, L.; Fournel, A.; Abot, A.; et al. Pharmacological inhibition of the F1-ATPase/P2Y1 pathway suppresses the effect of apolipoprotein A1 on endothelial nitric oxide synthesis and vasorelaxation. Acta Physiol. 2019, 226, e13268. [Google Scholar] [CrossRef] [PubMed]
- Kunnen, S.; Van Eck, M. Lecithin: Cholesterol acyltransferase: Old friend or foe in atherosclerosis? J. Lipid Res. 2012, 53, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Ford, H.Z.; Byrne, H.M.; Myerscough, M.R. A lipid-structured model for macrophage populations in atherosclerotic plaques. J. Theor. Biol. 2019, 479, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, L.; Jiang, R.; Zheng, L.; Liu, D. Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J. Atheroscler. Thromb. 2014, 21, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dhindsa, D.; Almuwaqqat, Z.; Sun, Y.V.; Quyyumi, A.A. Very High High-Density Lipoprotein Cholesterol Levels and Cardiovascular Mortality. Am. J. Cardiol. 2022, 167, 43–53. [Google Scholar] [CrossRef]
- Fogacci, F.; Borghi, C.; Cicero, A.F.G. New evidences on the association between high-density lipoprotein cholesterol and cardiovascular risk: A never ending research story. Eur. J. Prev. Cardiol. 2022, 29, 842–843. [Google Scholar] [CrossRef]
- Turner, S.; Voogt, J.; Davidson, M.; Glass, A.; Killion, S.; Decaris, J.; Mohammed, H.; Minehira, K.; Boban, D.; Murphy, E. Measurement of reverse cholesterol transport pathways in humans: In vivo rates of free cholesterol efflux, esterification, and excretion. J. Am. Heart Assoc. 2012, 1, e001826. [Google Scholar] [CrossRef]
- Soran, H.; Schofield, J.D.; Durrington, P.N. Antioxidant properties of HDL. Front. Pharmacol. 2015, 6, 222. [Google Scholar] [CrossRef]
- Ansell, B.J.; Navab, M.; Watson, K.E.; Fonarow, G.C.; Fogelman, A.M. Anti-inflammatory properties of HDL. Rev. Endocr. Metab. Disord. 2004, 5, 351–358. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Moreno, P.; Nabel, E.G.; Hachinski, V.; Fuster, V. Cellular senescence, vascular disease, and aging: Part 2 of a 2-part review: Clinical vascular disease in the elderly. Circulation 2011, 123, 1900–1910. [Google Scholar] [CrossRef]
- Luengo-Fernandez, R.; Walli-Attaei, M.; Gray, A. Economic burden of cardiovascular diseases in the European Union: A population-based cost study. Eur. Heart J. 2023, 44, 4752–4767. [Google Scholar] [CrossRef]
- US Census Bureau. International Database. Table 094. Midyear Population, by Age and Sex; US Census Bureau: Washington, DC, USA, 2004.
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef]
- Curtis, A.B.; Karki, R.; Hattoum, A.; Sharma, U.C. Arrhythmias in Patients ≥80 Years of Age: Pathophysiology, Management, and Outcomes. J. Am. Coll. Cardiol. 2018, 71, 2041–2057. [Google Scholar] [CrossRef] [PubMed]
- Berrougui, H.; Isabelle, M.; Cloutier, M.; Grenier, G.; Khalil, A. Age-related impairment of HDL-mediated cholesterol efflux. J. Lipid Res. 2007, 48, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, G.; Pagel, P.; Pfahlert, V.; Genser, B.; Scharnagl, H.; Kleber, M.E.; Delgado, G.; Ohrui, H.; Ritsch, A.; Grammer, T.B. High-density lipoprotein subclasses, coronary artery disease, and cardiovascular mortality. Clin. Chem. 2017, 63, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Cupples, L.A.; Demissie, S.; Horvath, K.V.; Cox, C.E.; Batista, M.C.; Schaefer, E.J. High-density lipoprotein subpopulation profile and coronary heart disease prevalence in male participants of the Framingham Offspring Study. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Collins, D.; Cupples, L.A.; Demissie, S.; Horvath, K.V.; Bloomfield, H.E.; Robins, S.J.; Schaefer, E.J. Value of high-density lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events in the Veterans Affairs HDL Intervention Trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Ritsch, A.; Scharnagl, H.; Maerz, W. HDL cholesterol efflux capacity and cardiovascular events. N. Engl. J. Med. 2015, 372, 1870–1871. [Google Scholar]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol efflux capacity, high-density lipoprotein particle number, and incident cardiovascular events: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Kontush, A. HDL particle number and size as predictors of cardiovascular disease. Front. Pharmacol. 2015, 6, 218. [Google Scholar] [CrossRef]
- Rohrer, L.; Hersberger, M.; von Eckardstein, A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr. Opin. Lipidol. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome1. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef]
- Tselepis, A.D.; Chapman, M.J. Inflammation, bioactive lipids and atherosclerosis: Potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Atheroscler. Suppl. 2002, 3, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, H.; Vincent, V.; Sen, A.; Singh, A.; Roy, A. Changing perspectives on HDL: From simple quantity measurements to functional quality assessment. J. Lipids 2021, 2021. [Google Scholar] [CrossRef]
- Okada, T.; Ohama, T.; Takafuji, K.; Kanno, K.; Matsuda, H.; Sairyo, M.; Zhu, Y.; Saga, A.; Kobayashi, T.; Masuda, D. Shotgun proteomic analysis reveals proteome alterations in HDL of patients with cholesteryl ester transfer protein deficiency. J. Clin. Lipidol. 2019, 13, 317–325. [Google Scholar] [CrossRef]
- Rader, D.J. Molecular regulation of HDL metabolism and function: Implications for novel therapies. J. Clin. Investig. 2006, 116, 3090–3100. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.-R.; Remaley, A. Tangier disease: Still more questions than answers. Cell. Mol. Life Sci. CMLS 2005, 62, 2150–2160. [Google Scholar] [CrossRef]
- Saleheen, D.; Natarajan, P.; Armean, I.M.; Zhao, W.; Rasheed, A.; Khetarpal, S.A.; Won, H.-H.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Samocha, K.E. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017, 544, 235–239. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Hofland, J., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- AbdelHafez, M.A. Protective and therapeutic potentials of HDL and ApoA1 in COVID-19 elderly and chronic illness patients. Bull. Natl. Res. Cent. 2022, 46, 222. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Goux, A.; Badiou, S.; Patrier, L.; Canaud, B.; Maudelonde, T.; Cristol, J.-P.; Solassol, J. HDL proteome in hemodialysis patients: A quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE 2012, 7, e34107. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Cahnmann, H.J.; Rader, D.; Kindt, M.; Facchiano, A.; Robbins, J. Thyroid hormone binding to isolated human apolipoproteins A-II, CI, C-II, and C-III: Homology in thyroxine binding sites. Thyroid 1994, 4, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.E.; Harrison, E.H. Mechanisms of selective delivery of xanthophylls to retinal pigment epithelial cells by human lipoproteins. J. Lipid Res. 2016, 57, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.H.; Navab, M.; Dwyer, K.M.; Hassan, K.; Sun, P.; Shircore, A.; Hama-Levy, S.; Hough, G.; Wang, X.; Drake, T. Oxygenated carotenoid lutein and progression of early atherosclerosis: The Los Angeles atherosclerosis study. Circulation 2001, 103, 2922–2927. [Google Scholar] [CrossRef]
- Alwaili, K.; Bailey, D.; Awan, Z.; Bailey, S.D.; Ruel, I.; Hafiane, A.; Krimbou, L.; Laboissiere, S.; Genest, J. The HDL proteome in acute coronary syndromes shifts to an inflammatory profile. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 405–415. [Google Scholar] [CrossRef]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, J.; Xu, N.; Han, G.; Geng, Q.; Song, J.; Li, S.; Zhao, J.; Chen, H. Signature of circulating microRNAs as potential biomarkers in vulnerable coronary artery disease. PLoS ONE 2013, 8, e80738. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Röxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Remaley, A.T. HDL and cholesterol: Life after the divorce? J. Lipid Res. 2014, 55, 4–12. [Google Scholar] [CrossRef]
- Ono, K. Functions of microRNA-33a/b and microRNA therapeutics. J. Cardiol. 2016, 67, 28–33. [Google Scholar] [CrossRef]
- Hyötyläinen, T.; Mattila, I.; Wiedmer, S.K.; Koivuniemi, A.; Taskinen, M.-R.; Yki-Järvinen, H.; Orešič, M. Metabolomic analysis of polar metabolites in lipoprotein fractions identifies lipoprotein-specific metabolic profiles and their association with insulin resistance. Mol. Biosyst. 2012, 8, 2559–2565. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef]
- Tall, A. Metabolic and genetic control of HDL cholesterol levels. J. Intern. Med. 1992, 231, 661–668. [Google Scholar] [CrossRef]
- Kuusi, T.; Saarinen, P.; Nikkilä, E.A. Evidence for the role of hepatic endothelial lipase in the metabolism of plasma high density lipoprotein2 in man. Atherosclerosis 1980, 36, 589–593. [Google Scholar] [CrossRef]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Calvo, D.; Gómez-Coronado, D.; Lasunción, M.A.; Vega, M.A. CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arter. Thromb. Vasc. Biol. 1997, 17, 2341–2349. [Google Scholar] [CrossRef]
- Singh, I.M.; Shishehbor, M.H.; Ansell, B.J. High-density lipoprotein as a therapeutic target: A systematic review. JAMA 2007, 298, 786–798. [Google Scholar] [CrossRef]
- Jones, P.H.; Davidson, M.H.; Stein, E.A.; Bays, H.E.; McKenney, J.M.; Miller, E.; Cain, V.A.; Blasetto, J.W. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am. J. Cardiol. 2003, 92, 152–160. [Google Scholar] [CrossRef]
- Kameda, T.; Horiuchi, Y.; Shimano, S.; Yano, K.; Lai, S.J.; Ichimura, N.; Tohda, S.; Kurihara, Y.; Tozuka, M.; Ohkawa, R. Effect of myeloperoxidase oxidation and N-homocysteinylation of high-density lipoprotein on endothelial repair function. Biol. Chem. 2022, 403, 265–277. [Google Scholar] [CrossRef]
- Kovanen, P.T. Mast cells: Multipotent local effector cells in atherothrombosis. Immunol. Rev. 2007, 217, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R.; Cai, L.; Ziemba, K.S.; Yu, J.; Kindy, M.S.; van der Westhuyzen, D.R.; de Beer, F.C. The fate of HDL particles in vivo after SR-BI-mediated selective lipid uptake. J. Lipid Res. 2002, 43, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Singaraja, R.R.; Stahmer, B.; Brundert, M.; Merkel, M.; Heeren, J.; Bissada, N.; Kang, M.; Timmins, J.M.; Ramakrishnan, R.; Parks, J.S. Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Kee, P.; Rye, K.-A.; Taylor, J.L.; Barrett, P.H.R.; Barter, P.J. Metabolism of apoA-I as lipid-free protein or as component of discoidal and spherical reconstituted HDLs: Studies in wild-type and hepatic lipase transgenic rabbits. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Brundert, M.; Heeren, J.; Greten, H.; Rinninger, F. Hepatic lipase mediates an increase in selective uptake of HDL-associated cholesteryl esters by cells in culture independent from SR-BI. J. Lipid Res. 2003, 44, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Maugeais, C.; Tietge, U.J.; Broedl, U.C.; Marchadier, D.; Cain, W.; McCoy, M.G.; Lund-Katz, S.; Glick, J.M.; Rader, D.J. Dose-dependent acceleration of high-density lipoprotein catabolism by endothelial lipase. Circulation 2003, 108, 2121–2126. [Google Scholar] [CrossRef] [PubMed]
- Woollett, L.A.; Spady, D.K. Kinetic parameters for high density lipoprotein apoprotein AI and cholesteryl ester transport in the hamster. J. Clin. Investig. 1997, 99, 1704–1713. [Google Scholar] [CrossRef][Green Version]
- Kozyraki, R.; Fyfe, J.; Kristiansen, M.; Gerdes, C.; Jacobsen, C.; Cui, S.; Christensen, E.I.; Aminoff, M.; De La Chapelle, A.; Krahe, R. The intrinsic factor–vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein AI receptor facilitating endocytosis of high-density lipoprotein. Nat. Med. 1999, 5, 656–661. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Nielsen, L.B. The role of the kidney in lipid metabolism. Curr. Opin. Lipidol. 2005, 16, 301–306. [Google Scholar] [CrossRef]
- Zannis, V.I.; Fotakis, P.; Koukos, G.; Kardassis, D.; Ehnholm, C.; Jauhiainen, M.; Chroni, A. HDL Biogenesis, Remodeling, and Catabolism. In High Density Lipoproteins: From Biological Understanding to Clinical Exploitation; von Eckardstein, A., Kardassis, D., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 53–111. [Google Scholar] [CrossRef]
- Koukos, G.; Chroni, A.; Duka, A.; Kardassis, D.; Zannis, V.I. LCAT can rescue the abnormal phenotype produced by the natural ApoA-I mutations (Leu141Arg) Pisa and (Leu159Arg) FIN. Biochemistry 2007, 46, 10713–10721. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.M.; Lee, J.-Y.; Boudyguina, E.; Kluckman, K.D.; Brunham, L.R.; Mulya, A.; Gebre, A.K.; Coutinho, J.M.; Colvin, P.L.; Smith, T.L. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Investig. 2005, 115, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.C.; VandenBroek, J.M.; Cooper, P.S. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid Res. 2004, 45, 1594–1607. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Demosky, S.J., Jr.; Stonik, J.A.; Combs, C.; Remaley, A.T.; Duverger, N.; Santamarina-Fojo, S.; Brewer, H.B., Jr. The ABCA1 transporter functions on the basolateral surface of hepatocytes. Biochem. Biophys. Res. Commun. 2002, 297, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Remaley, A.T.; Stonik, J.A.; Demosky, S.J.; Neufeld, E.B.; Bocharov, A.V.; Vishnyakova, T.G.; Eggerman, T.L.; Patterson, A.P.; Duverger, N.J.; Santamarina-Fojo, S. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 2001, 280, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Daniil, G.; Phedonos, A.A.; Holleboom, A.G.; Motazacker, M.M.; Argyri, L.; Kuivenhoven, J.A.; Chroni, A. Characterization of antioxidant/anti-inflammatory properties and apoA-I-containing subpopulations of HDL from family subjects with monogenic low HDL disorders. Clin. Chim. Acta 2011, 412, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Van Eck, M.; Singaraja, R.R.; Ye, D.; Hildebrand, R.B.; James, E.R.; Hayden, M.R.; Van Berkel, T.J. Macrophage ATP-binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 929–934. [Google Scholar] [CrossRef]
- Rohrer, L.; Ohnsorg, P.M.; Lehner, M.; Landolt, F.; Rinninger, F.; von Eckardstein, A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ. Res. 2009, 104, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Ma, L.; Denis, M.; Karwatsky, J.; Li, Z.; Jiang, X.C.; Zha, X. ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J. Lipid Res. 2009, 50, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; McGillicuddy, F.C.; Hinkle, C.C.; O’Neill, S.; Glick, J.M.; Rothblat, G.H.; Reilly, M.P. Adipocyte modulation of high-density lipoprotein cholesterol. Circulation 2010, 121, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Stefulj, J.; Panzenboeck, U.; Becker, T.; Hirschmugl, B.; Schweinzer, C.; Lang, I.; Marsche, G.; Sadjak, A.; Lang, U.; Desoye, G.; et al. Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1. Circ. Res. 2009, 104, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ranalletta, M.; Matsuura, F.; Peng, F.; Tall, A.R. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.M.; Oram, J.F. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 2005, 280, 30150–30157. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Kling, J.; Pagler, T.; Li, H.; Hubbard, B.; Fisher, T.; Sparrow, C.P.; Taggart, A.K.; Tall, A.R. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1430–1438. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Ma, J.; Ling, W.; Xia, M. Adenosine monophosphate activated protein kinase regulates ABCG1-mediated oxysterol efflux from endothelial cells and protects against hypercholesterolemia-induced endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1354–1362. [Google Scholar] [CrossRef]
- Hardy, L.M.; Frisdal, E.; Le Goff, W. Critical Role of the Human ATP-Binding Cassette G1 Transporter in Cardiometabolic Diseases. Int. J. Mol. Sci. 2017, 18, 1892. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldán, Á.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A.; Trigatti, B.L.; Penman, M.; Rayburn, H.; Herz, J.; Krieger, M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 12610–12615. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.A.; Yesilaltay, A.; Pal, R.; Daniels, K.; Barker, C.; Quinones, V.; Rigotti, A.; Krieger, M.; Kocher, O. Normal hepatic cell surface localization of the high density lipoprotein receptor, scavenger receptor class B, type I, depends on all four PDZ domains of PDZK1. J. Biol. Chem. 2009, 284, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Yesilaltay, A.; Daniels, K.; Pal, R.; Krieger, M.; Kocher, O. Loss of PDZK1 causes coronary artery occlusion and myocardial infarction in Paigen diet-fed apolipoprotein E deficient mice. PLoS ONE 2009, 4, e8103. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Wilson, M.; Kelly, T.; Su, W.; Dressman, J.; Kincer, J.; Matveev, S.V.; Guo, L.; Guerin, T.; Li, X.A.; et al. HDL-associated estradiol stimulates endothelial NO synthase and vasodilation in an SR-BI-dependent manner. J. Clin. Investig. 2003, 111, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, M.; Korporaal, S.J.; Franssen, R.; Meurs, I.; Out, R.; Hovingh, G.K.; Hoekstra, M.; Sierts, J.A.; Dallinga-Thie, G.M.; Motazacker, M.M. Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 2011, 364, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Uda, S.; Spolitu, S.; Angius, F.; Collu, M.; Accossu, S.; Banni, S.; Murru, E.; Sanna, F.; Batetta, B. Role of HDL in cholesteryl ester metabolism of lipopolysaccharide-activated P388D1 macrophages. J. Lipid Res. 2013, 54, 3158–3169. [Google Scholar] [CrossRef]
- Manna, P.R.; Chandrala, S.P.; Jo, Y.; Stocco, D.M. cAMP-independent signaling regulates steroidogenesis in mouse Leydig cells in the absence of StAR phosphorylation. J. Mol. Endocrinol. 2006, 37, 81–95. [Google Scholar] [CrossRef]
- Rao, R.M.; Jo, Y.; Leers-Sucheta, S.; Bose, H.S.; Miller, W.L.; Azhar, S.; Stocco, D.M. Differential regulation of steroid hormone biosynthesis in R2C and MA-10 Leydig tumor cells: Role of SR-B1-mediated selective cholesteryl ester transport. Biol. Reprod. 2003, 68, 114–121. [Google Scholar] [CrossRef]
- Mardones, P.; Quiñones, V.; Amigo, L.; Moreno, M.; Miquel, J.F.; Schwarz, M.; Miettinen, H.E.; Trigatti, B.; Krieger, M.; VanPatten, S. Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I-deficient mice. J. Lipid Res. 2001, 42, 170–180. [Google Scholar] [CrossRef]
- Holm, T.M.; Braun, A.; Trigatti, B.L.; Brugnara, C.; Sakamoto, M.; Krieger, M.; Andrews, N.C. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood J. Am. Soc. Hematol. 2002, 99, 1817–1824. [Google Scholar]
- Schwartz, C.C.; Halloran, L.G.; Vlahcevic, Z.R.; Gregory, D.H.; Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 1978, 200, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.O.; Najib, S.; Perret, B.; Cabou, C.; Lichtenstein, L. Ecto-F1-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis 2015, 238, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Jonas, A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1529, 245–256. [Google Scholar] [CrossRef]
- Simon, J.B.; Boyer, J.L. Production of lecithin: Cholesterol acyltransferase by the isolated perfused rat liver. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1970, 218, 549–551. [Google Scholar] [CrossRef]
- Subbaiah, P.; Albers, J.; Chen, C.H.; Bagdade, J.D. Low density lipoprotein-activated lysolecithin acylation by human plasma lecithin-cholesterol acyltransferase. Identity of lysolecithin acyltransferase and lecithin-cholesterol acyltransferase. J. Biol. Chem. 1980, 255, 9275–9280. [Google Scholar] [CrossRef]
- Kuivenhoven, J.; Pritchard, H.; Hill, J.; Frohlich, J.; Assmann, G.; Kastelein, J. The molecular pathology of lecithin: Cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 1997, 38, 191–205. [Google Scholar] [CrossRef]
- McIntyre, N. Familial LCAT deficiency and fish-eye disease. J. Inherit. Metab. Dis. 1988, 11, 45–56. [Google Scholar] [CrossRef]
- Calabresi, L.; Francheschini, G. Genetic LCAT deficiency: Molecular diagnosis, plasma lipids, and atherosclerosis. In High Density Lipoproteins, Dyslipidemia, and Coronary Heart Disease; Springer: New York, NY, USA, 2010; pp. 89–93. [Google Scholar]
- Hovingh, G.K.; Hutten, B.A.; Holleboom, A.G.; Petersen, W.; Rol, P.; Stalenhoef, A.; Zwinderman, A.H.; de Groot, E.; Kastelein Md, J.J.; Kuivenhoven, J.A. Compromised LCAT function is associated with increased atherosclerosis. Circulation 2005, 112, 879–884. [Google Scholar] [CrossRef]
- Ikewaki, K.; Nishiwaki, M.; Sakamoto, T.; Ishikawa, T.; Fairwell, T.; Zech, L.; Nagano, M.; Nakamura, H.; Brewer, H.; Rader, D. Increased catabolic rate of low density lipoproteins in humans with cholesteryl ester transfer protein deficiency. J. Clin. Investig. 1995, 96, 1573–1581. [Google Scholar] [CrossRef]
- Agellon, L.; Walsh, A.; Hayek, T.; Moulin, P.; Jiang, X.C.; Shelanski, S.A.; Breslow, J.; Tall, A.R. Reduced high density lipoprotein cholesterol in human cholesteryl ester transfer protein transgenic mice. J. Biol. Chem. 1991, 266, 10796–10801. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Group, H.T.R.C. Effects of anacetrapib in patients with atherosclerotic vascular disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar]
- Kastelein, J.J.P.; Hsieh, A.; Dicklin, M.R.; Ditmarsch, M.; Davidson, M.H. Obicetrapib: Reversing the Tide of CETP Inhibitor Disappointments. Curr. Atheroscler. Rep. 2024, 26, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, J. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill Education: New York, NY, USA, 2001; pp. 2789–2816. [Google Scholar]
- Wolfrum, C.; Poy, M.N.; Stoffel, M. Apolipoprotein M is required for preβ-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 2005, 11, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, G.J.; Barter, P.J. Transfers of esterified cholesterol and triglyceride between high density and very low density lipoproteins: In vitro studies of rabbits and humans. Metabolism 1980, 29, 546–550. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Emerging Evidence that ApoC-III Inhibitors Provide Novel Options to Reduce the Residual CVD. Curr. Atheroscler. Rep. 2019, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Walter, M. Interrelationships among HDL metabolism, aging, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.; Beamer, L.J.; Tall, A.R. The implications of the structure of the bactericidal/permeability-increasing protein on the lipid-transfer function of the cholesteryl ester transfer protein. Curr. Opin. Struct. Biol. 1998, 8, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-C.; Bruce, C. Regulation of murine plasma phospholipid transfer protein activity and mRNA levels by lipopolysaccharide and high cholesterol diet (∗). J. Biol. Chem. 1995, 270, 17133–17138. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-C.; Bruce, C.; Mar, J.; Lin, M.; Ji, Y.; Francone, O.L.; Tall, A.R. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J. Clin. Investig. 1999, 103, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Forester, L.R.; Bongiovanni, G.L. Facilitation of phosphatidylcholine transfer into high density lipoproteins by an apolipoprotein in the density 1.20-1.26 g/ml fraction of plasma. J. Lipid Res. 1983, 24, 277–289. [Google Scholar] [CrossRef]
- Oka, T.; Kujiraoka, T.; Ito, M.; Egashira, T.; Takahashi, S.; Nanjee, M.N.; Miller, N.E.; Metso, J.; Olkkonen, V.M.; Ehnholm, C.; et al. Distribution of phospholipid transfer protein in human plasma: Presence of two forms of phospholipid transfer protein, one catalytically active and the other inactive. J. Lipid Res. 2000, 41, 1651–1657. [Google Scholar] [CrossRef]
- Jiang, X.C. Phospholipid transfer protein: Its impact on lipoprotein homeostasis and atherosclerosis. J. Lipid Res. 2018, 59, 764–771. [Google Scholar] [CrossRef]
- Desrumaux, C.; Risold, P.-Y.; Schroeder, H.; Deckert, V.; Masson, D.; Athias, A.; Laplanche, H.; Le Guern, N.; Blache, D.; Jiang, X.-C. Phospholipid transfer protein (PLTP) deficiency reduces brain vitamin E content and increases anxiety in mice. FASEB J. 2005, 19, 296–297. [Google Scholar] [CrossRef]
- Jauhiainen, M.; Metso, J.; Pahlman, R.; Blomqvist, S.; Van Tol, A.; Ehnholm, C. Human plasma phospholipid transfer protein causes high density lipoprotein conversion. J. Biol. Chem. 1993, 268, 4032–4036. [Google Scholar] [CrossRef]
- Rye, K.-A.; Jauhiainen, M.; Barter, P.J.; Ehnholm, C. Triglyceride-enrichment of high density lipoproteins enhances their remodelling by phospholipid transfer protein. J. Lipid Res. 1998, 39, 613–622. [Google Scholar] [CrossRef]
- van Haperen, R.; van Tol, A.; Vermeulen, P.; Jauhiainen, M.; van Gent, T.; van den Berg, P.; Ehnholm, S.; Grosveld, F.; van der Kamp, A.; de Crom, R. Human plasma phospholipid transfer protein increases the antiatherogenic potential of high density lipoproteins in transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Yazdanyar, A.; Quan, W.; Jin, W.; Jiang, X.-C. Liver-Specific Phospholipid Transfer Protein Deficiency Reduces High-Density Lipoprotein and Non–High-Density Lipoprotein Production in Mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2058–2064. [Google Scholar] [CrossRef]
- Jiang, H.; Yazdanyar, A.; Lou, B.; Chen, Y.; Zhao, X.; Li, R.; Hoang Bui, H.; Kuo, M.-S.; Navab, M.; Qin, S. Adipocyte phospholipid transfer protein and lipoprotein metabolism. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Aviram, M. Paraoxonases role in the prevention of cardiovascular diseases. Biofactors 2009, 35, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Teiber, J.F.; Kramer, G.L.; de Lemos, J.A.; Drazner, M.H.; Haley, R.W. serum paraoxonase 1 (PON1) activity is associated with indices of hypertensive heart disease and cardiac remodeling in the Dallas heart study population. Circulation 2015, 132, A17217. [Google Scholar] [CrossRef]
- Ikhlef, S.; Berrougui, H.; Kamtchueng Simo, O.; Zerif, E.; Khalil, A. Human paraoxonase 1 overexpression in mice stimulates HDL cholesterol efflux and reverse cholesterol transport. PLoS ONE 2017, 12, e0173385. [Google Scholar] [CrossRef]
- Mackness, M.I.; Arrol, S.; Durrington, P.N. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991, 286, 152–154. [Google Scholar] [CrossRef]
- Costa, L.G.; Cole, T.B.; Jarvik, G.P.; Furlong, C.E. Functional genomics of the paraoxonase (PON1) polymorphisms: Effects on pesticide sensitivity, cardiovascular disease, and drug metabolism. Annu. Rev. Med. 2003, 54, 371–392. [Google Scholar] [CrossRef]
- Draganov, D.; La Du, B.N. Pharmacogenetics of paraoxonases: A brief review. Naunyn-Schmiedebergs Arch. Pharmacol. 2004, 369, 78–88. [Google Scholar] [CrossRef]
- Draganov, D.I.; Stetson, P.L.; Watson, C.E.; Billecke, S.S.; La Du, B.N. Rabbit serum paraoxonase 3 (PON3) is a high density lipoprotein-associated lactonase and protects low density lipoprotein against oxidation. J. Biol. Chem. 2000, 275, 33435–33442. [Google Scholar] [CrossRef]
- Wallace, A.; Sutherland, W.; Mann, J.; Williams, S. The effect of meals rich in thermally stressed olive and safflower oils on postprandial serum paraoxonase activity in patients with diabetes. Eur. J. Clin. Nutr. 2001, 55, 951–958. [Google Scholar] [CrossRef]
- Calabresi, L.; Villa, B.; Canavesi, M.; Sirtori, C.R.; James, R.W.; Bernini, F.; Franceschini, G. An ω-3 Polyunsaturated Fatty Acid Concentrate Increases Plasma High-Density Lipoprotein 2 Cholesterol and Paraoxonase Levels in Patients with Familial Combined Hyperlipidemia. Metab. Clin. Exp. 2004, 53, 153–158. [Google Scholar] [CrossRef]
- Bhattacharyya, T.; Nicholls, S.J.; Topol, E.J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D.M.; Ellis, S.G. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Mackness, M.; Mackness, B. Human paraoxonase-1 (PON1): Gene structure and expression, promiscuous activities and multiple physiological roles. Gene 2015, 567, 12–21. [Google Scholar] [CrossRef]
- Jaouad, L.; Milochevitch, C.; Khalil, A. PON1 paraoxonase activity is reduced during HDL oxidation and is an indicator of HDL antioxidant capacity. Free Radic. Res. 2003, 37, 77–83. [Google Scholar] [CrossRef]
- Lopez, P.; Rodriguez-Carrio, J.; Martinez-Zapico, A.; Perez-Alvarez, A.I.; Lopez-Mejias, R.; Benavente, L.; Mozo, L.; Caminal-Montero, L.; Gonzalez-Gay, M.A.; Suarez, A. Serum levels of anti-PON1 and anti-HDL antibodies as potential biomarkers of premature atherosclerosis in systemic lupus erythematosus. Thromb. Haemost. 2017, 117, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Rasmiena, A.A.; Barlow, C.K.; Ng, T.W.; Tull, D.; Meikle, P.J. High density lipoprotein efficiently accepts surface but not internal oxidised lipids from oxidised low density lipoprotein. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 69–77. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.; Williams, A.; Schulze, I. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577.e1567. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Antiatherogenic function of HDL particle subpopulations: Focus on antioxidative activities. Curr. Opin. Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Xepapadaki, E.; Zvintzou, E.; Kalogeropoulou, C.; Filou, S.; Kypreos, K.E. Τhe antioxidant function of HDL in atherosclerosis. Angiology 2020, 71, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Koller, L.; Richter, B.; Winter, M.-P.; Sulzgruber, P.; Potolidis, C.; Liebhart, F.; Moertl, D.; Berger, R.; Goliasch, G.; Lang, I. Clusterin/apolipoprotein J is independently associated with survival in patients with chronic heart failure. J. Clin. Lipidol. 2017, 11, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Namiri-Kalantari, R.; Gao, F.; Chattopadhyay, A.; Wheeler, A.A.; Navab, K.D.; Farias-Eisner, R.; Reddy, S.T. The dual nature of HDL: Anti-Inflammatory and pro-Inflammatory. Biofactors 2015, 41, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Anderson, J.L.; Gruppen, E.G.; Lei, Y.; Bakker, S.J.; Dullaart, R.P.; Tietge, U.J. High-density lipoprotein anti-inflammatory capacity and incident cardiovascular events. Circulation 2021, 143, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Säemann, M.D.; Poglitsch, M.; Kopecky, C.; Haidinger, M.; Hörl, W.H.; Weichhart, T. The versatility of HDL: A crucial anti-inflammatory regulator. Eur. J. Clin. Investig. 2010, 40, 1131–1143. [Google Scholar] [CrossRef]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014, 15, 152–160. [Google Scholar] [CrossRef]
- Imachi, H.; Murao, K.; Cao, W.; Ohyama, T.; Sato, M.; Sasaguri, Y.; Ishida, T.; Takahara, J. Expression of HDL receptor, CLA-1 in human smooth-muscle cells and effect of interferon-γ on its regulation. Horm. Metab. Res. 2001, 33, 389–393. [Google Scholar] [CrossRef]
- Hao, W.; Friedman, A. The LDL-HDL profile determines the risk of atherosclerosis: A mathematical model. PLoS ONE 2014, 9, e90497. [Google Scholar] [CrossRef]
- Okura, H.; Yamashita, S.; Ohama, T.; Saga, A.; Yamamoto-Kakuta, A.; Hamada, Y.; Sougawa, N.; Ohyama, R.; Sawa, Y.; Matsuyama, A. HDL/apolipoprotein AI binds to macrophage-derived progranulin and suppresses its conversion into proinflammatory granulins. J. Atheroscler. Thromb. 2010, 17, 568–577. [Google Scholar] [CrossRef]
- Yu, B.l.; Wang, S.h.; Peng, D.q.; Zhao, S.p. HDL and immunomodulation: An emerging role of HDL against atherosclerosis. Immunol. Cell Biol. 2010, 88, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Drew, B.G.; Nakhla, S.; Duffy, S.J.; Murphy, A.J.; Barter, P.J.; Rye, K.A.; Chin-Dusting, J.; Hoang, A.; Sviridov, D.; et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2009, 53, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Petropoulou, P.-I.; Berbée, J.F.; Theodoropoulos, V.; Hatziri, A.; Stamou, P.; Karavia, E.A.; Spyridonidis, A.; Karagiannides, I.; Kypreos, K.E. Lack of LCAT reduces the LPS-neutralizing capacity of HDL and enhances LPS-induced inflammation in mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Lucero, D.; Islam, P.; Freeman, L.A.; Jin, X.; Pryor, M.; Tang, J.; Kruth, H.S.; Remaley, A.T. Interleukin 10 promotes macrophage uptake of HDL and LDL by stimulating fluid-phase endocytosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158537. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, H.; Ding, S.F. Sphingosine-1-phosphate promotes the proliferation and attenuates apoptosis of Endothelial progenitor cells via S1PR1/S1PR3/PI3K/Akt pathway. Cell Biol. Int. 2018, 42, 1492–1502. [Google Scholar] [CrossRef]
- Matsuo, M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef]
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc. Res. 2012, 95, 165–172. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Vinagre, C.G.; Freitas, F.R.; de Mesquita, C.H.; Vinagre, J.C.; Mariani, A.C.; Kalil-Filho, R.; Maranhão, R.C. Removal of Chylomicron Remnants from the Bloodstream is Delayed in Aged Subjects. Aging Dis. 2018, 9, 748–754. [Google Scholar] [CrossRef]
- Seres, I.; Paragh, G.; Deschene, E.; Fulop, T.; Khalil, A. Study of factors influencing the decreased HDL associated PON1 activity with aging. Exp. Gerontol. 2004, 39, 59–66. [Google Scholar] [CrossRef]
- Reaven, P.D.; Napoli, C.; Merat, S.; Witztumc, J.L. Lipoprotein modification and atherosclerosis in aging. Exp. Gerontol. 1999, 34, 527–537. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Khalil, A.; Jay-Gerin, J.-P.; Fülöp, T., Jr. Age-related increased susceptibility of high-density lipoproteins (HDL) to in vitro oxidation induced by γ-radiolysis of water. FEBS Lett. 1998, 435, 153–158. [Google Scholar] [CrossRef]
- Khalil, A.; Wagner, J.R.; Lacombe, G.; Dangoisse, V.; Fülöp, T., Jr. Increased susceptibility of low-density lipoprotein (LDL) to oxidation by γ-radiolysis with age. FEBS Lett. 1996, 392, 45–48. [Google Scholar] [CrossRef]
- Ericsson, S.; Eriksson, M.; Vitols, S.; Einarsson, K.; Berglund, L.; Angelin, B. Influence of age on the metabolism of plasma low density lipoproteins in healthy males. J. Clin. Investig. 1991, 87, 591–596. [Google Scholar] [CrossRef]
- Cherki, M.; Berrougui, H.; Isabelle, M.; Cloutier, M.; Koumbadinga, G.A.; Khalil, A. Effect of PON1 polymorphism on HDL antioxidant potential is blunted with aging. Exp. Gerontol. 2007, 42, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Nalewajska, M.; Gurazda, K.; Styczyńska-Kowalska, E. The Role of MicroRNAs in Selected Forms of Glomerulonephritis. Int. J. Mol. Sci. 2019, 20, 5050. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.L.; Murillo, A.G. Postmenopausal Women Have Higher HDL and Decreased Incidence of Low HDL than Premenopausal Women with Metabolic Syndrome. Healthcare 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Park, H.J.; Kim, S.J.; Kim, J.R. Decrease in HDL-C is Associated with Age and Household Income in Adults from the Korean National Health and Nutrition Examination Survey 2017: Correlation Analysis of Low HDL-C and Poverty. Int. J. Environ. Res. Public Health 2019, 16, 3329. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 2013, 1831, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.A.; Kast-Woelbern, H.R.; Anisfeld, A.M.; Edwards, P.A. Identification of PLTP as an LXR target gene and apoE as an FXR target gene reveals overlapping targets for the two nuclear receptors. J. Lipid Res. 2002, 43, 2037–2041. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharmacol. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Wolf, P.; Curcic, S.; Birner-Gruenberger, R.; Weger, W.; Inzinger, M.; El-Gamal, D.; Wadsack, C.; Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol efflux capacity [S]. J. Lipid Res. 2012, 53, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Föger, B.; Santamarina-Fojo, S.; Shamburek, R.D.; Parrot, C.L.; Talley, G.D.; Brewer, H.B. Plasma phospholipid transfer protein: Adenovirus-mediated overexpression in mice leads to decreased plasma high density lipoprotein (HDL) and enhanced hepatic uptake of phospholipids and cholesteryl esters from HDL. J. Biol. Chem. 1997, 272, 27393–27400. [Google Scholar] [CrossRef] [PubMed]
- van Haperen, R.; Samyn, H.; van Gent, T.; Zonneveld, A.J.; Moerland, M.; Grosveld, F.; Jansen, H.; Dallinga-Thie, G.M.; van Tol, A.; de Crom, R. Novel roles of hepatic lipase and phospholipid transfer protein in VLDL as well as HDL metabolism. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 1031–1036. [Google Scholar] [CrossRef]
- Barzilai, N.; Atzmon, G.; Schechter, C.; Schaefer, E.J.; Cupples, A.L.; Lipton, R.; Cheng, S.; Shuldiner, A.R. Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA 2003, 290, 2030–2040. [Google Scholar] [CrossRef]
- Luc, G.; Bard, J.; Lussier-Cacan, S.; Bouthillier, D.; Parra, H.; Fruchart, J.; Davignon, J. High-density lipoprotein particles in octogenarians. Metabolism 1991, 40, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Mc Auley, M.T.; Mooney, K.M. Lipid metabolism and hormonal interactions: Impact on cardiovascular disease and healthy aging. Expert Rev. Endocrinol. Metab. 2014, 9, 357–367. [Google Scholar] [CrossRef]
- Sanllorente, A.; Lassale, C. Modification of High-Density Lipoprotein Functions by Diet and Other Lifestyle Changes: A Systematic Review of Randomized Controlled Trials. J. Clin. Med. 2021, 10, 5897. [Google Scholar] [CrossRef]
- Burillo, E.; Mateo-Gallego, R.; Cenarro, A.; Fiddyment, S.; Bea, A.M.; Jorge, I.; Vázquez, J.; Civeira, F. Beneficial effects of omega-3 fatty acids in the proteome of high-density lipoprotein proteome. Lipids Health Dis. 2012, 11, 116. [Google Scholar] [CrossRef]
- Blesso, C.N.; Andersen, C.J.; Barona, J.; Volek, J.S.; Fernandez, M.L. Whole egg consumption improves lipoprotein profiles and insulin sensitivity to a greater extent than yolk-free egg substitute in individuals with metabolic syndrome. Metabolism 2013, 62, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Mutungi, G.; Waters, D.; Ratliff, J.; Puglisi, M.; Clark, R.M.; Volek, J.S.; Fernandez, M.L. Eggs distinctly modulate plasma carotenoid and lipoprotein subclasses in adult men following a carbohydrate-restricted diet. J. Nutr. Biochem. 2010, 21, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, Á.; Castañer, O.; Tresserra-Rimbau, A.; Pintó, X.; Fitó, M.; Casas, R.; Martínez-González, M.; Corella, D.; Salas-Salvadó, J.; Lapetra, J.; et al. Mediterranean Diet and Atherothrombosis Biomarkers: A Randomized Controlled Trial. Mol. Nutr. Food Res. 2020, 64, e2000350. [Google Scholar] [CrossRef] [PubMed]
- Farràs, M.; Fernández-Castillejo, S.; Rubió, L.; Arranz, S.; Catalán, Ú.; Subirana, I.; Romero, M.P.; Castañer, O.; Pedret, A.; Blanchart, G.; et al. Phenol-enriched olive oils improve HDL antioxidant content in hypercholesterolemic subjects. A randomized, double-blind, cross-over, controlled trial. J. Nutr. Biochem. 2018, 51, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Rantala, M.; Silaste, M.L.; Tuominen, A.; Kaikkonen, J.; Salonen, J.T.; Alfthan, G.; Aro, A.; Kesäniemi, Y.A. Dietary modifications and gene polymorphisms alter serum paraoxonase activity in healthy women. J. Nutr. 2002, 132, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Freese, R.; Alfthan, G.; Jauhiainen, M.; Basu, S.; Erlund, I.; Salminen, I.; Aro, A.; Mutanen, M. High intakes of vegetables, berries, and apples combined with a high intake of linoleic or oleic acid only slightly affect markers of lipid peroxidation and lipoprotein metabolism in healthy subjects. Am. J. Clin. Nutr. 2002, 76, 950–960. [Google Scholar] [CrossRef]
- Gardner, C.D.; Landry, M.J. Effect of a ketogenic diet versus Mediterranean diet on glycated hemoglobin in individuals with prediabetes and type 2 diabetes mellitus: The interventional Keto-Med randomized crossover trial. Am. J. Clin. Nutr. 2022, 116, 640–652. [Google Scholar] [CrossRef]
- Saslow, L.R.; Daubenmier, J.J. Twelve-month outcomes of a randomized trial of a moderate-carbohydrate versus very low-carbohydrate diet in overweight adults with type 2 diabetes mellitus or prediabetes. Nutr. Diabetes 2017, 7, 304. [Google Scholar] [CrossRef]
- Vidić, V.; Ilić, V.; Toskić, L.; Janković, N.; Ugarković, D. Effects of calorie restricted low carbohydrate high fat ketogenic vs. non-ketogenic diet on strength, body-composition, hormonal and lipid profile in trained middle-aged men. Clin. Nutr. 2021, 40, 1495–1502. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morvaridzadeh, M.; Zoubdane, N.; Heshmati, J.; Alami, M.; Berrougui, H.; Khalil, A. High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects? Nutrients 2024, 16, 653. https://doi.org/10.3390/nu16050653
Morvaridzadeh M, Zoubdane N, Heshmati J, Alami M, Berrougui H, Khalil A. High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects? Nutrients. 2024; 16(5):653. https://doi.org/10.3390/nu16050653
Chicago/Turabian StyleMorvaridzadeh, Mojgan, Nada Zoubdane, Javad Heshmati, Mehdi Alami, Hicham Berrougui, and Abdelouahed Khalil. 2024. "High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects?" Nutrients 16, no. 5: 653. https://doi.org/10.3390/nu16050653
APA StyleMorvaridzadeh, M., Zoubdane, N., Heshmati, J., Alami, M., Berrougui, H., & Khalil, A. (2024). High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects? Nutrients, 16(5), 653. https://doi.org/10.3390/nu16050653