
Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Human Liver Tissue and Preparation of PCLSs
2.2. PCLS Culture and Induction of Steatosis
2.3. ATP Measurement
2.4. Triglyceride Quantitation
2.5. Total Protein Measurement
2.6. RNA Sequencing and Quantitative Real-Time PCR
2.7. Measurement of Excreted Proteins
2.8. Histology
2.9. Statistics
3. Results
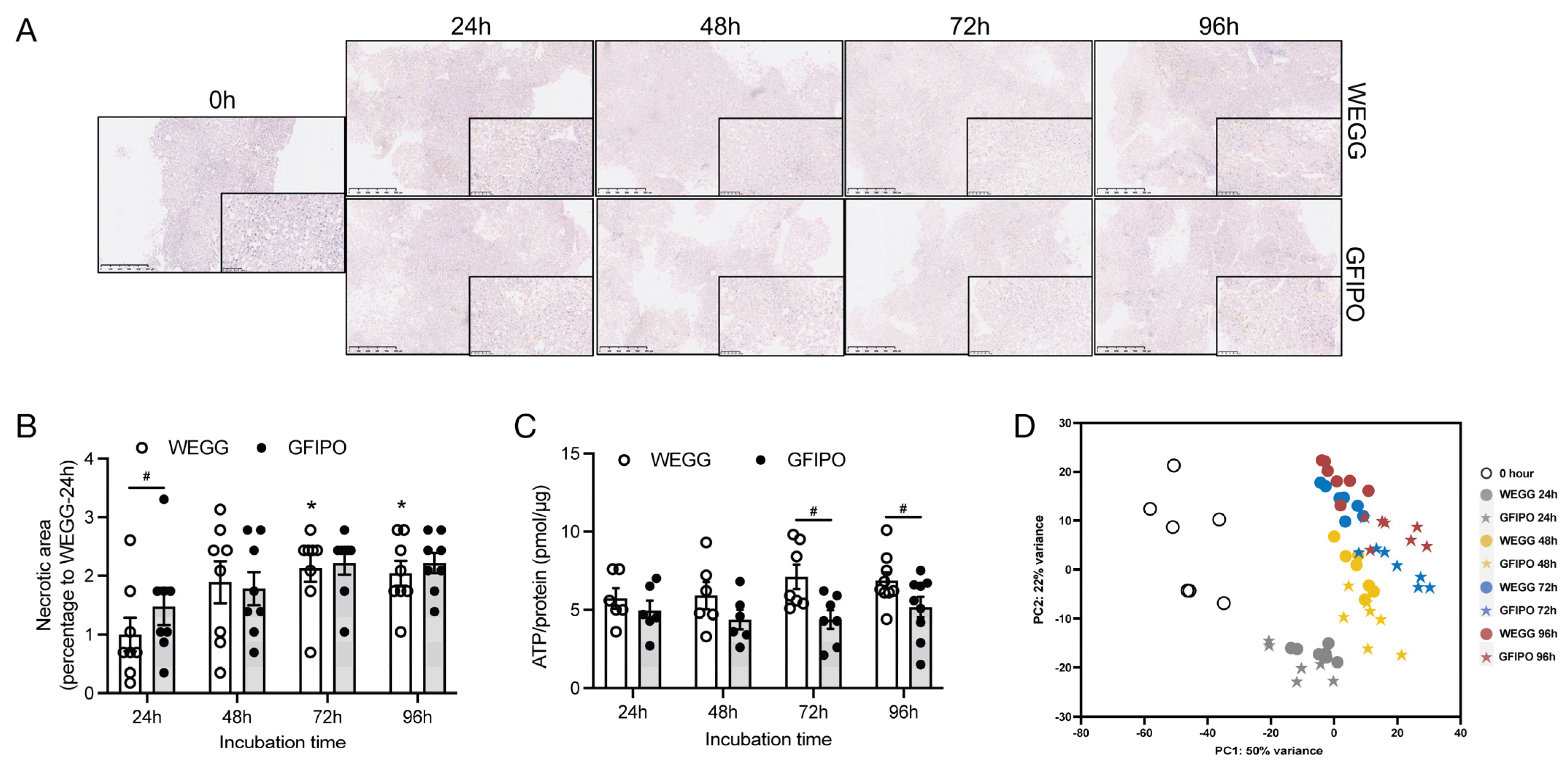
3.1. Characterization of PCLSs
3.1.1. Slices Retain Viability after Incubation
3.1.2. Principal Component Analysis (PCA)
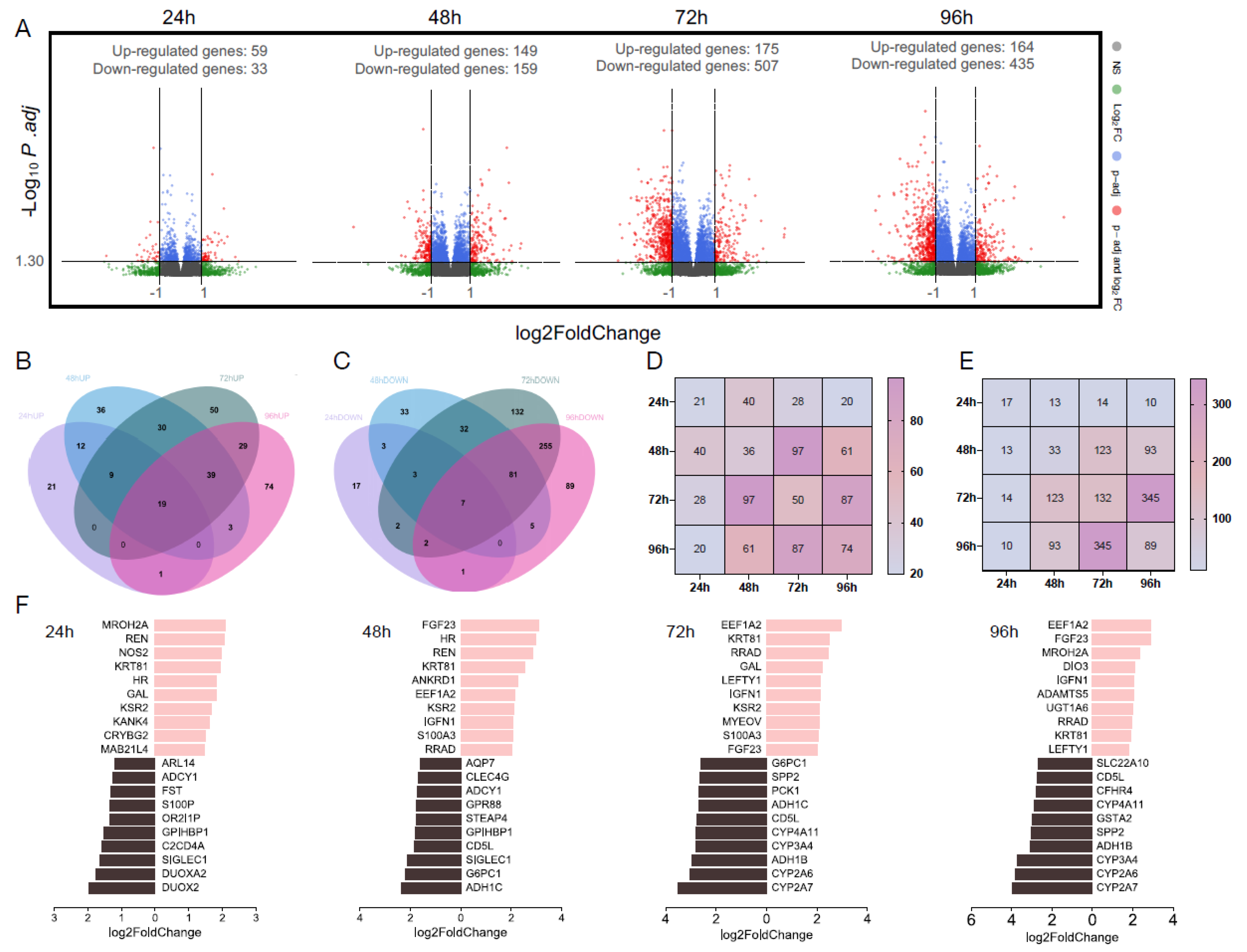
3.1.3. Total Number of Differentially Expressed Genes
3.1.4. Overlapping Differentially Expressed Genes
3.1.5. Top 10 Significantly Differentially Expressed Genes
3.2. Characterization of PCLSs
3.2.1. Genes Related to Fatty Acid Metabolism
3.2.2. Fatty Acid Metabolism Pathway
3.2.3. Phenotype of Fat Accumulation on PCLSs
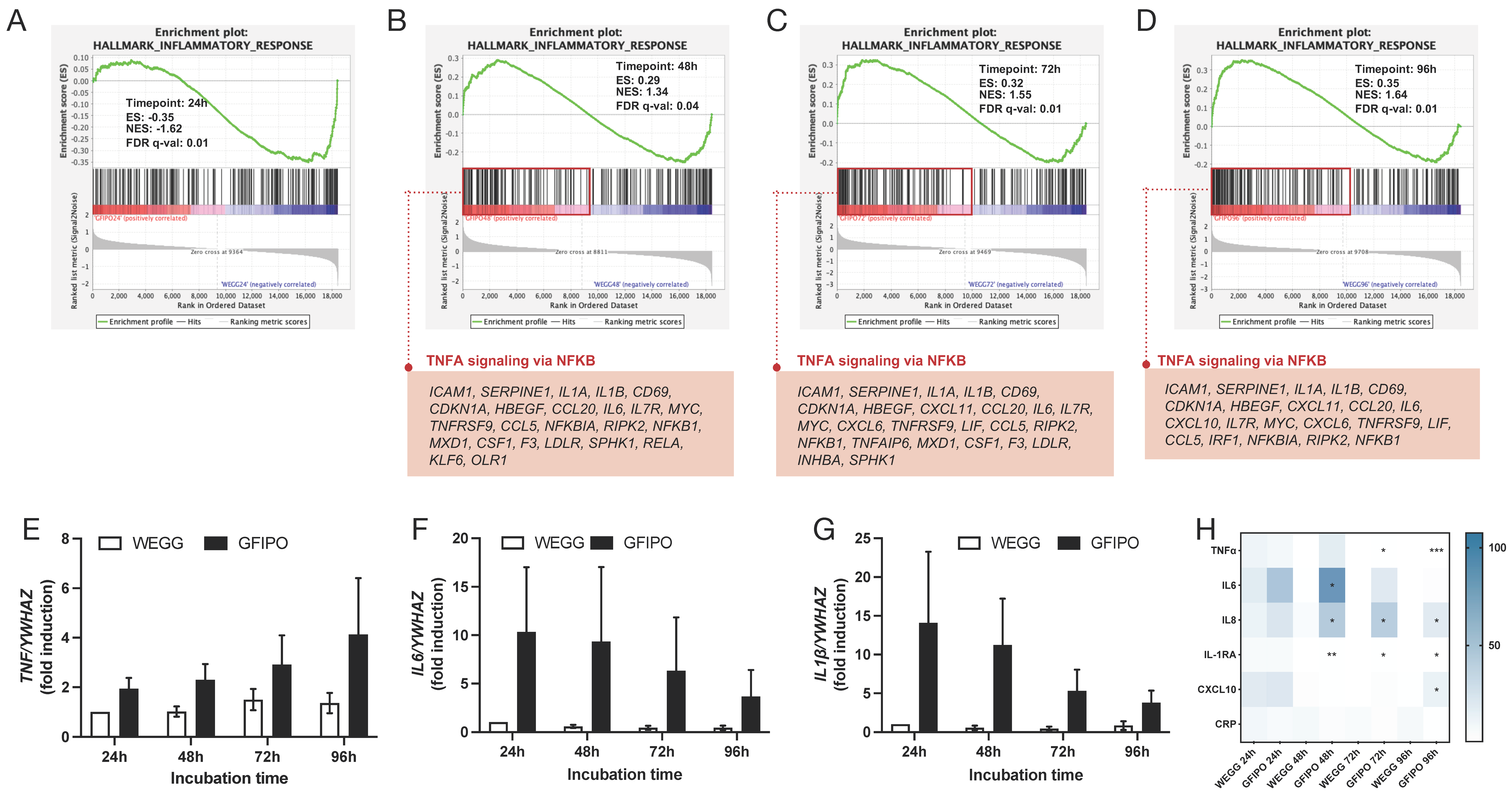
3.3. Inflammatory Response in PCLSs by GFIPO Compared to WEGG
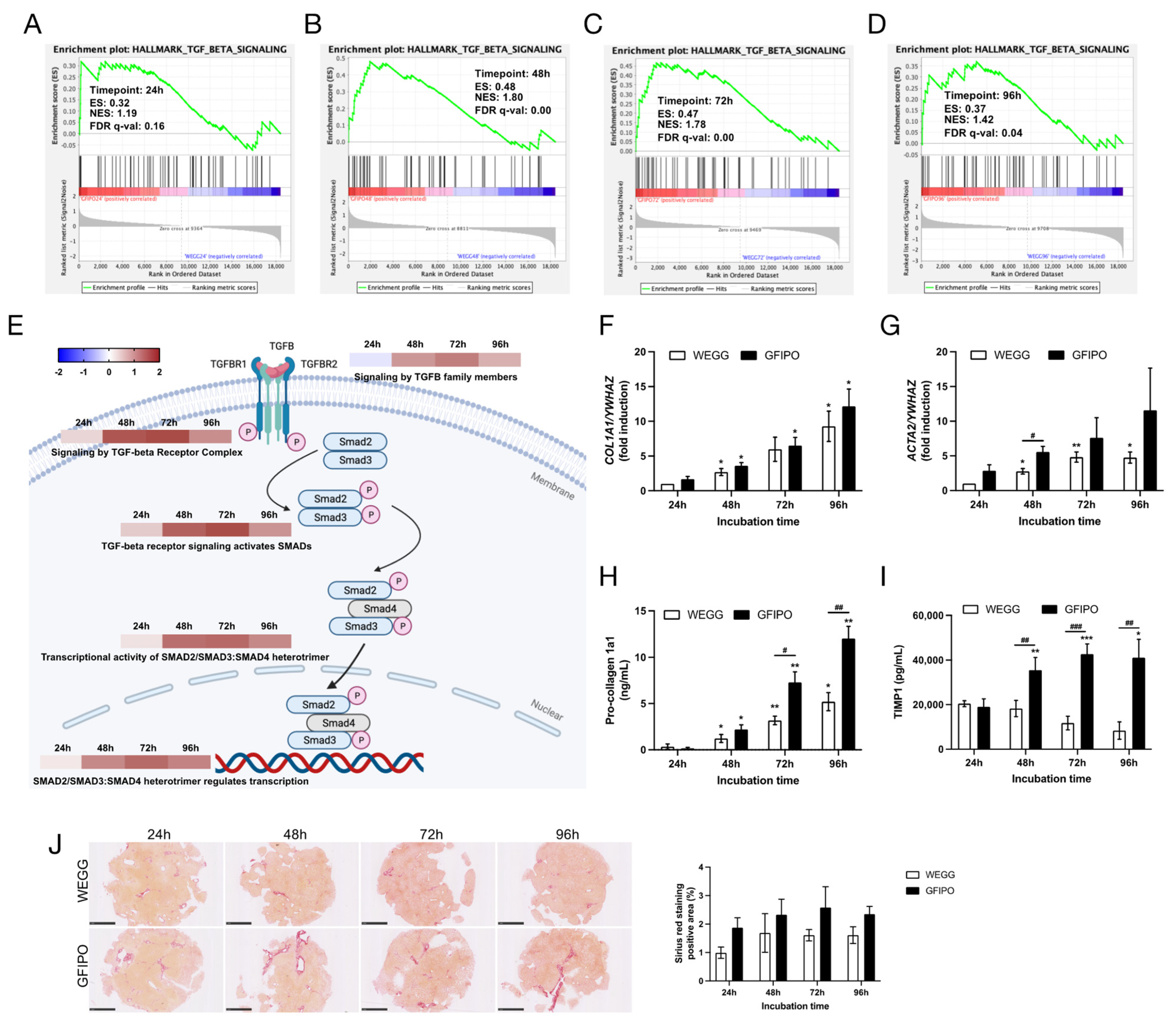
3.4. Development of Liver Fibrosis in PCLSs by GFIPO Compared to WEGG
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Non-alcoholic fatty liver disease. BMC Med. 2017, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Willebrords, J.; Pereira, I.V.A.; Maes, M.; Yanguas, S.C.; Colle, I.; Bossche, B.V.D.; Silva, T.C.D.; Oliveira, C.P.M.S.d.; Andraus, W.; Alves, V.A.; et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog. Lipid Res. 2015, 59, 106–125. [Google Scholar] [CrossRef]
- Graaf, I.A.M.d.; Groothuis, G.M.M.; Olinga, P. Precision-cut tissue slices as a tool to predict metabolism of novel drugs. Expert. Opin. Drug Met. 2007, 3, 879–898. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.H.; Rios-Morales, M.; Gerding, A.; Reijngoud, D.J.; Olinga, P.; Bakker, B.M. The Effects of Butyrate on Induced Metabolic-Associated Fatty Liver Disease in Precision-Cut Liver Slices. Nutrients 2021, 13, 4203. [Google Scholar] [CrossRef]
- Rius, B.; Duran-Guell, M.; Flores-Costa, R.; Lopez-Vicario, C.; Lopategi, A.; Alcaraz-Quiles, J.; Casulleras, M.; Lozano, J.J.; Titos, E.; Claria, J. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J. 2017, 31, 5384–5398. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.H.; Luangmonkong, T.; Oosterhuis, D.; Mutsaers, H.A.M.; Dekker, F.J.; Olinga, P. A Pathophysiological Model of Non-Alcoholic Fatty Liver Disease Using Precision-Cut Liver Slices. Nutrients 2019, 11, 507. [Google Scholar] [CrossRef]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Karim, S.; Newsome, P.N.; Lalor, P.F. Inhibition of vascular adhesion protein-1 modifies hepatic steatosis in vitro and in vivo. World J. Hepatol. 2020, 12, 931–948. [Google Scholar] [CrossRef]
- Gore, E. Integrating an Ex Vivo Model into Fibrosis Research; University of Groningen: Groningen, The Netherlands, 2019. [Google Scholar]
- Graaf, I.A.M.d.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 22 January 2024).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [PubMed]
- Oligschlaeger, Y.; Shiri-Sverdlov, R. NAFLD preclinical models: More than a handful, less of a concern? Biomedicines 2020, 8, 28. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Diehl, A.M. Towards a definite mouse model of NAFLD. J. Hepatol. 2018, 69, 272–274. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008, 34, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Walewski, J.; Anglade, D.; Berk, P. Regulation of Hepatocellular Fatty Acid Uptake in Mouse Models of Fatty Liver Disease with and without Functional Leptin Signaling: Roles of NfKB and SREBP-1C and the Effects of Spexin. Semin. Liver Dis. 2016, 36, 360–372. [Google Scholar] [CrossRef]
- Ring, A.; Le Lay, S.; Pohl, J.; Verkade, P.; Stremmel, W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2006, 1761, 416–423. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Rosa, M.D.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Cai, Y. Concurrent exercise improves insulin resistance and nonalcoholic fatty liver disease by upregulating PPAR-γ and genes involved in the beta-oxidation of fatty acids in ApoE-KO mice fed a high-fat diet. Lipids Health Dis. 2019, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.H. Metabolic-Associated Fatty Liver Disease: A Major Problem Miniaturised; University of Groningen: Groningen, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Zhou, Y.; Zhou, L.; Li, S.W.; Wang, B.M. Identification of Key Genes and Immune Infiltrate in Nonalcoholic Steatohepatitis: A Bioinformatic Analysis. Biomed. Res. Int. 2021, 2021, 7561645. [Google Scholar] [CrossRef]
- Söderberg, C.; Marmur, J.; Eckes, K.; Glaumann, H.; Sällberg, M.; Frelin, L.; Rosenberg, P.; Stål, P.E.R.; Hultcrantz, R. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011, 119, 412–420. [Google Scholar] [CrossRef]
- Mackay, C.R.; Imhof, B.A. Cell adhesion in the immune system. Immunol. Today 1993, 14, 99–102. [Google Scholar] [CrossRef]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113869. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Ahmed, H.; Umar, M.I.; Imran, S.; Javaid, F.; Syed, S.K.; Riaz, R.; Hassan, W. TGF-β1 signaling can worsen NAFLD with liver fibrosis backdrop. Exp. Mol. Pathol. 2022, 124, 104733. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Dijke, P.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in hepatic stellate cell activation and liver fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef]
- Nishida, N. Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers 2021, 13, 3666. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Larsen, F.T.; van den Heuvel, M.C.; Gier, K.; Gorter, A.R.; Oosterhuis, D.; Bijzet, J.; de Meijer, V.E.; Ravnskjaer, K.; Nagelkerke, A.; et al. Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions. Nutrients 2024, 16, 626. https://doi.org/10.3390/nu16050626
Li M, Larsen FT, van den Heuvel MC, Gier K, Gorter AR, Oosterhuis D, Bijzet J, de Meijer VE, Ravnskjaer K, Nagelkerke A, et al. Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions. Nutrients. 2024; 16(5):626. https://doi.org/10.3390/nu16050626
Chicago/Turabian StyleLi, Mei, Frederik T. Larsen, Marius C. van den Heuvel, Konstanze Gier, Alan R. Gorter, Dorenda Oosterhuis, Johan Bijzet, Vincent E. de Meijer, Kim Ravnskjaer, Anika Nagelkerke, and et al. 2024. "Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions" Nutrients 16, no. 5: 626. https://doi.org/10.3390/nu16050626
APA StyleLi, M., Larsen, F. T., van den Heuvel, M. C., Gier, K., Gorter, A. R., Oosterhuis, D., Bijzet, J., de Meijer, V. E., Ravnskjaer, K., Nagelkerke, A., & Olinga, P. (2024). Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions. Nutrients, 16(5), 626. https://doi.org/10.3390/nu16050626