
Relationship Between Dietary Nutrient Intake and Autophagy—Related Genes in Obese Humans: A Narrative Review


 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Methods
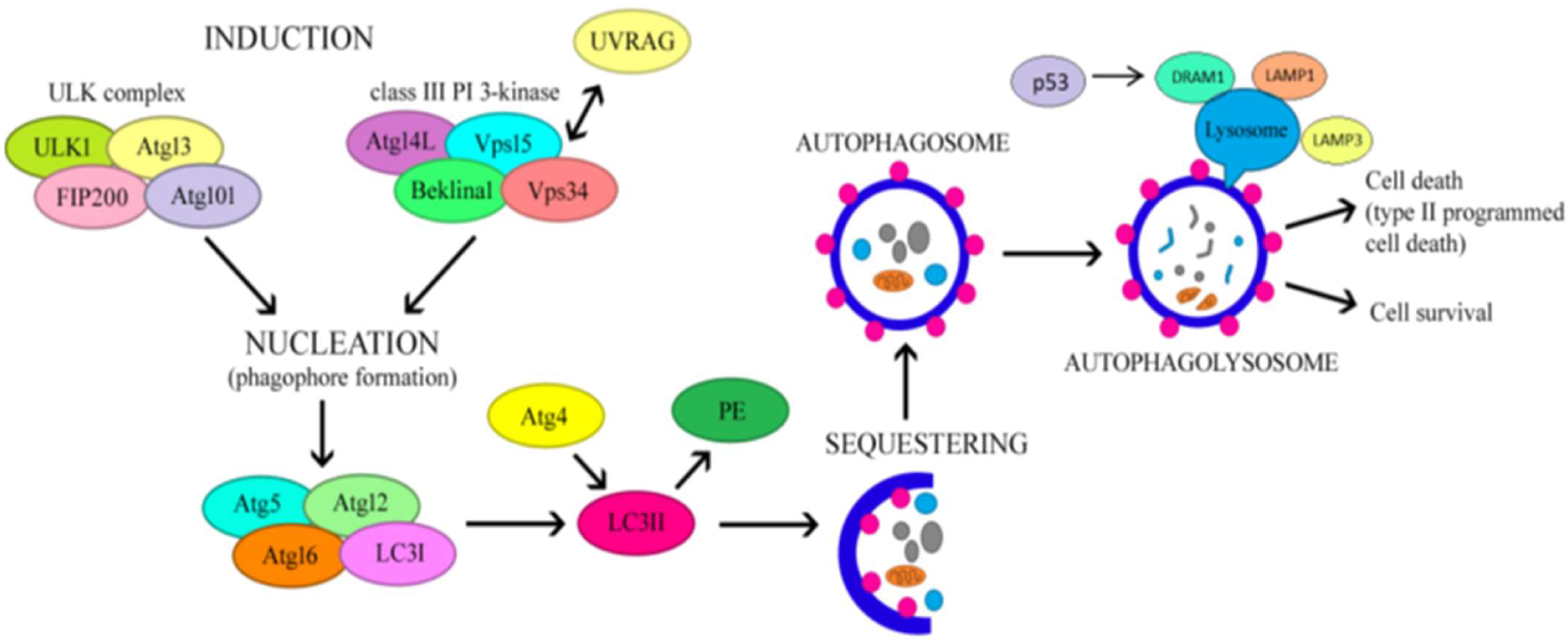
3. The Regulatory Mechanism of Autophagy
4. Autophagy and Obesity Links
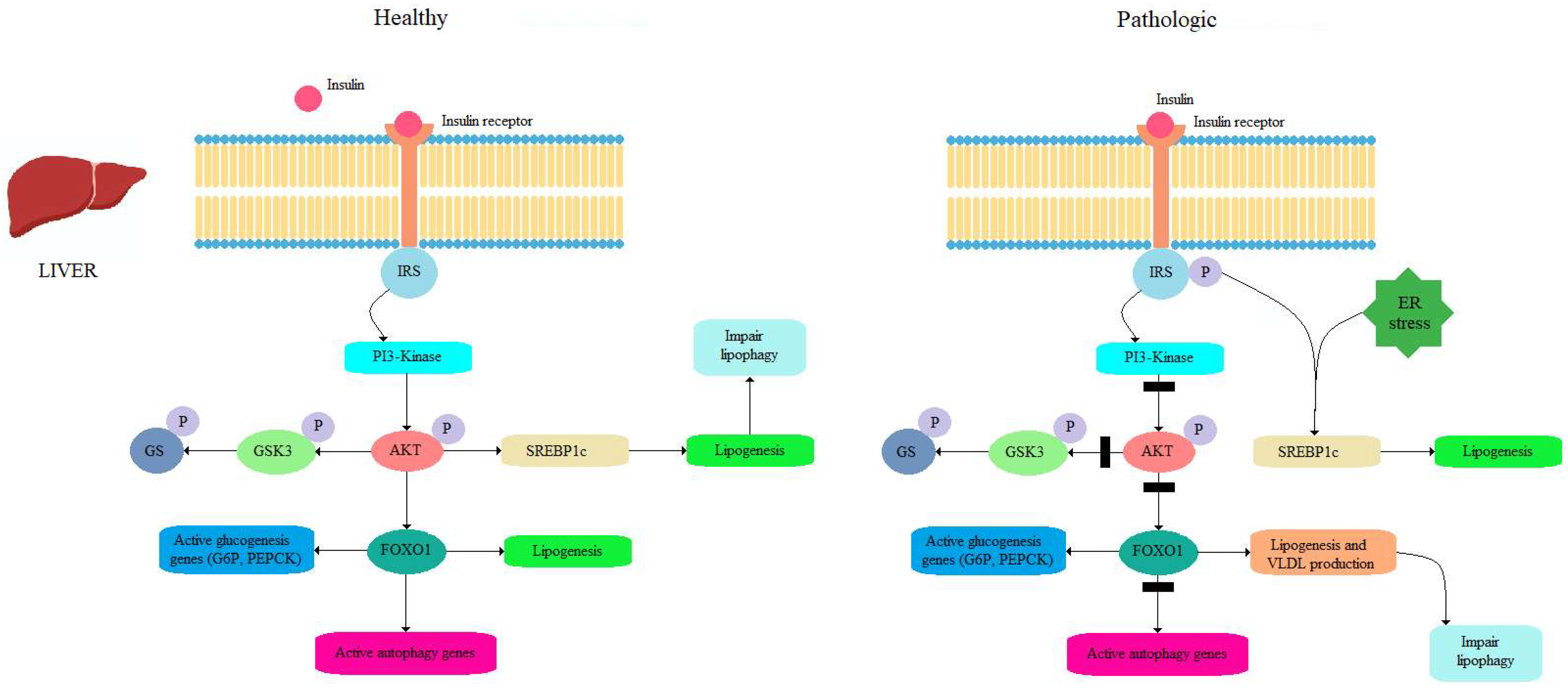
4.1. Insulin as a Potent Inhibitor of Autophagy
4.2. Role of Autophagy in Fatty Liver Modulation
5. Autophagy Related Genes in Obese Humans as a Therapeutic Target
Therapeutic Modulation of Autophagy in Obese Humans
- (a)
- Metformin—multiple signaling pathways mediate the effect of metformin, which is related to the improvement in the induction of hepatic autophagy and the inhibition of the induction of adipose tissue autophagy [87];
- (b)
- SGLT2 inhibitors—the effects of SGLT2 inhibitors promote the activation of AMPK, SIRT1, SIRT3, and SIRT6, and PGC-1α, decrease the activation of mTOR in diverse tissues under stress, and increase biomarkers of autophagy flux (LAMP-1, Beclin-1) [88];
- (c)
- DPP4 (Dipeptidyl-Peptidase 4) inhibitors promote the autophagy process to decrease triglycerides (TGs), low-density lipoproteins (LDLs) and high-density lipoproteins (HDLs), as well as nitro-oxidative stress, via a mechanism dependent on autophagy modulation [89];
- (d)
- GLP-1 receptor agonists (GLP-1 RA) play a crucial role in regulating autophagic flux, as GLP-1R knockdown suppresses the autophagy induction and mTOR inhibition induced by GLP-1 peptide treatment. Additionally, GLP-1R expression was found to be decreased in certain conditions, such as high-fat diet-induced liver steatosis [90];
- (e)
- GLP/GIP receptor agonist—this novel antidiabetic, anti-obesity drug displayed a protective cardiac effect, preventing cell death, fibrosis and hypertrophy with a potential positive impact on cardiac remodeling. It also plays a crucial role in the autophagic process via the AMPK/mTOR pathway [91].
6. Obesity Inhibits or Activates Autophagy?
7. Nutritional Interventions for Autophagy Modulations in Overweight/Obese Humans
7.1. Calorie Restriction; Intermittent Fasting
7.2. Mediterranean Diet (Met Diet)
7.3. Dietary Polyphenols
7.4. Dietary Fatty Acids
7.5. Diet Modifications
7.6. Protein Intake

{kind=link}
{kind=link}
{kind=link}
| Dietary Strategies | Model of Obesity | Parameter Studies | Effect on Autophagy | References |
|---|---|---|---|---|
| Calorie restriction or Intermittent fasting | Skeletal muscle of body fat-matched endurance athletes; skeletal muscle of obese women; obese humans (subcutaneous, white adipose tissue) | Decreased mTOR signaling through reducing insulin and IGF-1 levels and increased the AMP/ATP ratio, which leads to the activation of AMPK as well as several other products involved in the stimulation of this process (ATG 5, ATG6, ATG7, ATG8, LC3-II, Beclin1, p62, SIRT1, LAMP2, ULK1 and ATG101) | Enhanced | [96,97,98,99,100,101,102,103,104] |
| Calorie restriction 25% for 7 weeks | Peripheral blood mononuclear cells (PBMNCs) of overweight male | Activated AMPK and SIRT1 | Enhanced | [105] |
| Mediterranean diet (MD) vs. Mediterranean diet with almonds (MDSA) | Obese humans (subcutaneous, white adipose tissue) | Elevated levels of autophagy-related proteins ATG7 and ATG12 were observed in the VAT of the MDSA and ATG5 showed a non-significant trend | Enhanced | [125] |
| Epigallocatechin-3-gallate + resveratrol (280 mg + 80 mg/d) vs. placebo—12 weeks | Obese humans (subcutaneous, white adipose tissue) | Activated gene expressions of ATP6V1A, ATP6V1H, CD68, HSL/LIPE, LAMP2, PI4K2A, UCP2, GAPDH | Enhanced | [128] |
| Resveratrol 150 mg once daily for 30 days | Obese men (skeletal muscle) | Activated AMPK, increased SIRT1 and PGC-1α protein levels | Enhanced | [132] |
| Resveratrol 150 mg once daily for 30 days | Obese men (skeletal muscle) | Activated TFEB (transcriptional factor EB) expression; inhibited mTOR activity | Enhanced | [133] |
| Resveratrol (500 mg/d) vs. Calorie restriction (1000 kcal/d) | Overweight humans (blood) | Resveratrol and caloric restriction significantly increased serum concentrations of SIRT1 proteins | Enhanced | [136] |
| 4 diets followed a period of 12 weeks: a diet high in saturated fatty acids (HSFA), a diet rich in monounsaturated fatty acids (HMUFA), and two low-fat, high-complex-carbohydrate diets, one of which was supplemented with long-chain n-3 polyunsaturated fatty acids (LFHCC n-3) while the other received a placebo (LFHCC) | Obese humans (subcutaneous); white adipose tissue | HMUFA diet significantly increased expression of BECN1 and ATG7. LFHCC and LFHCC n-3 diets led to an increase in the expression of the apoptosis-related gene CASP3. Additionally, the LFHCC n-3 diet showed a tendency to increase the expressions of other autophagy markers, such as LC3, LAMP2, and ULK1. | Enhanced | [145] |
| Low-fat, high-carbohydrate diet (LF) vs. moderate-fat, low-carbohydrate diet (MF) for 10 weeks | Obese humans (subcutaneous); white adipose tissue | Expressions of FABP4, SIRT3, NR3C1, GABARAPL2, and FNTA genes were 15–65% higher in the MF than the LF | Enhanced in MF diet vs. LF | [146,147] |
| Hypocaloric diet (1500–1600 kcal/day) and low protein LP (10%) vs. hypocaloric (1500–1600 kcal/day) and high protein (30E%) for 3 weeks prior to bariatric surgery | Liver sample collected during surgery | Significantly elevated autophagy flux and FGF21 levels in the livers of patients in the LP diet versus HP | Enhanced in LP diet vs. HP | [148] |
8. Limitations
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Obesity Federation. World Obesity Atlas 2024; World Obesity Federation: London, UK, 2024; Available online: https://data.worldobesity.org/publications/ (accessed on 5 September 2024).
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 29 June 2024).
- Purnell, J. Definitions, Classification, and Epidemiology of Obesity. In Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2023; Available online: https://www.ncbi.nlm.nih.gov/books/NBK279167/ (accessed on 5 September 2024).
- Available online: https://www.worldobesity.org/what-we-do/our-policy-priorities/obesity-as-a-disease (accessed on 28 August 2024).
- Li, X.; Qi, L. Gene-Environment Interactions on Body Fat Distribution. Int. J. Mol. Sci. 2019, 20, 3690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- World Health Organization. Noncommunicable Diseases. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases#%7Etext=Noncommunicable.20diseases.20(NCDs).20kill.2041-.20and.20middle-income (accessed on 5 September 2024).
- Abarca-Gómez, L.; Abdeen, Z.A.; Abdul, Z.; Hamid, Z.A.; Abu-Rmeileh, N.; Acosta-Cazares, B.; Acuin, C.; Adams, R.; Aekplakorn, W.; Afsana, K.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, I.; Després, J.P. Metabolic syndrome: Past, present and future. Nutrients 2020, 12, 3501. [Google Scholar] [CrossRef]
- Fathi Dizaji, B. The investigations of genetic determinants of the metabolic syndrome. Diabetes Metab. Syndr. 2018, 12, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.C.; Méndez-Sánchez, N. Screening for MAFLD: Who, when and how? Ther. Adv. Endocrinol. Metab. 2023, 14, 20420188221145650. [Google Scholar] [CrossRef]
- Jakubek, P.; Pakula, B.; Rossmeisl, M.; Pinton, P.; Rimessi, A.; Wieckowski, M.R. Autophagy alterations in obesity, type 2 diabetes, and metabolic dysfunction-associated steatotic liver disease: The evidence from human studies. Intern. Emerg. Med. 2024, 19, 1473–1491. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Hassanpour, M.; Cheraghi, O.; Rahbarghazi, R.; Nouri, M. Autophagy stimulation delayed biological aging and decreased cardiac differentiation in rabbit mesenchymal stem cells. J. Cardiovasc. Thorac. Res. 2021, 13, 234–240. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, Q.; Mariman, E.C.; Blaak, E.E.; Jocken, J.W. Pharmacological agents targeting autophagy and their effects on lipolysis in human adipocytes. Mol. Cell Endocrinol. 2022, 544, 111555. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- García-Barrado, M.J.; Iglesias-Osma, M.C.; Pérez-García, E.; Carrero, S.; Blanco, E.J.; Carretero-Hernández, M.; Carretero, J. Role of Flavonoids in The Interactions among Obesity, Inflammation, and Autophagy. Pharmaceuticals 2020, 13, 342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cai, J.; Pires, K.M.; Ferhat, M.; Chaurasia, B.; Buffolo, M.A.; Smalling, R.; Sargsyan, A.; Atkinson, D.L.; Summers, S.A.; Graham, T.E.; et al. Autophagy Ablation in Adipocytes Induces Insulin Resistance and Reveals Roles for Lipid Peroxide and Nrf2 Signaling in Adipose-Liver Crosstalk. Cell Rep. 2018, 25, 1708–1717.e5. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Haczeyni, F.; Bell-Anderson, K.S.; Farrell, G.C. Causes and mechanisms of adipocyte enlargement and adipose expansion. Obes. Rev. 2018, 19, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Feve, B. Adipogenesis: Cellular and molecular aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 483–499. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Zmarzły, N.; Grabarek, B.; Mazurek, U.; Muc-Wierzgoń, M. Genes involved in the regulation of different types of autophagy and their participation in cancer pathogenesis. Oncotarget 2018, 9, 34413–34428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Budi, Y.P.; Li, Y.H.; Huang, C.; Wang, M.E.; Lin, Y.C.; Jong, D.S.; Chiu, C.H.; Jiang, Y.F. The role of autophagy in high-fat diet-induced insulin resistance of adipose tissues in mice. PeerJ 2022, 10, e13867. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lopez-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Clària, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2015, 112, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Yasuda-Yamahara, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Haneda, M.; Ugi, S.; et al. Lamp-2 deficiency prevents high-fat diet-induced obese diabetes via enhancing energy expenditure. Biochem. Biophys. Res. Commun. 2015, 465, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm 2019, 16, 8. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar]
- Terasawa, K.; Tomabechi, Y.; Ikeda, M.; Ehara, H.; Kukimoto-Niino, M.; Wakiyama, M.; Podyma-Inoue, K.A.; Rajapakshe, A.R.; Watabe, T.; Shirouzu, M.; et al. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem. Biophys. Res. Commun. 2016, 479, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Li, S.; Zhang, M.; Qin, X. Caveolin-1 in autophagy: A potential therapeutic target in atherosclerosis. Clin. Chim. Acta. 2021, 513, 25–33. [Google Scholar] [CrossRef]
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, G.; Xu, M.; Cui, Y.; He, W.; Zeng, H.; Zeng, T.; Cheng, R.; Li, X. Caspase-1 Deficiency Modulates Adipogenesis through Atg7-Mediated Autophagy: An Inflammatory-Independent Mechanism. Biomolecules 2024, 14, 501. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Z.; Huang, C.; Xu, C.; Xie, L.; Liang, J.J.; Liu, L.; Pang, C.P.; Ng, T.K.; Zhang, M. Caveolin-1 regulates human trabecular meshwork cell adhesion, endocytosis, and autophagy. J. Cell Biochem. 2019, 120, 13382–13391. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yang, X.; Jia, X.; Rong, Y.; Chen, L.; Zeng, T.; Deng, X.; Li, W.; Wu, G.; Wang, L.; et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis. Autophagy 2020, 16, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Sekar, M.; Thirumurugan, K. Autophagic Regulation of Adipogenesis Through TP53INP2: Insights from In Silico and In Vitro Analysis. Mol. Biotechnol. 2024, 66, 1188–1205. [Google Scholar] [CrossRef] [PubMed]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity Find the latest version: Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef]
- Soussi, H.; Clément, K.; Dugail, I. Adipose tissue autophagy status in obesity: Expression and flux—Two faces of the picture. Autophagy 2016, 12, 588–589. [Google Scholar] [CrossRef]
- Zhang, C.; He, Y.; Okutsu, M.; Ong, L.C.; Jin, Y.; Zheng, L.; Chow, P.; Yu, S.; Zhang, M.; Yan, Z. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARγ2 degradation. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E530–E539. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol 2018, 9, 473. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vegiopoulos, A.; Rohm, M.; Herzig, S. Adipose tissue: Between the extremes. EMBO J. 2017, 36, 1999–2017. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Y.; Gao, L.; Yang, Z.; Lin, J.; Ren, S.; Li, F.; Chen, J.; Wang, Z.; Dong, Z.; et al. PPAR-γ integrates obesity and adipocyte clock through epigenetic regulation of Bmal1. Theranostics 2022, 12, 1589–1606. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fujimori, K. Prostaglandin D2 and F2α as Regulators of Adipogenesis and Obesity. Biol. Pharmacol. 2022, 45, 8. [Google Scholar]
- Armani, A.; Berry, A.; Cirulli, F.; Caprio, M. Molecular mechanisms underlying metabolic syndrome: The expanding role of the adipocyte. FASEB J. 2017, 31, 4240–4255. [Google Scholar] [CrossRef] [PubMed]
- Ferhat, M.; Funai, K.; Boudina, S. Autophagy in Adipose Tissue Physiology and Pathophysiology. Antioxid. Redox Signal. 2019, 31, 487–501. [Google Scholar] [CrossRef]
- Pietrocola, F.; Bravo-San Pedro, J.M. Targeting Autophagy to Counteract Obesity-Associated Oxidative Stress. Antioxidants 2021, 10, 102. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef]
- Frendo-Cumbo, S.; Tokarz, V.L.; Bilan, P.J.; Brumell, J.H.; Klip, A. Communication Between Autophagy and Insulin Action: At the Crux of Insulin Action-Insulin Resistance? Front. Cell Dev. Biol. 2021, 9, 708431. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Häuselmann, S.P.; Dalmas, E.; Meier, D.T.; Thienel, C.; Traub, S.; Schulze, F.; Steiger, L.; Dror, E.; Martin, P.; et al. β Cell-Specific Deletion of the IL-1 Receptor Antagonist Impairs β Cell Proliferation and Insulin Secretion. Cell Rep. 2018, 22, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta. 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Zhang, Z.; Orwig, A.; Chen, S.; Ding, W.X.; Xu, Y.; Kunz, R.C.; Lind, N.R.L.; Stamler, J.S.; Yang, L.S. S-Nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes 2018, 67, 193–207. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Bárcena, C.; Martínez-García, G.G.; Tamargo-Gómez, I.; Suárez, M.F.; Pietrocola, F.; Castoldi, F.; Esteban, L.; Sierra-Filardi, E.; Boya, P.; et al. Autophagy couteracts weight gain, lipotoxicity and pancreatic β-cell death upon hypercaloric pro-diabetic regimens. Cell Death Dis. 2017, 8, e2970. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Jang, I.; Park, H.; Reilly, S.; Saltiel, A.R.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Meng, Q.; Cai, D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J. Biol. Chem. 2011, 286, 32324–32332. [Google Scholar] [CrossRef]
- Öst, A.; Svensson, K.; Ruishalme, I.; Brännmark, C.; Franck, N.; Krook, H.; Sandström, P.; Kjolhede, P.; Strålfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef]
- Parmar, U.M.; Jalgaonkar, M.P.; Kulkarni, Y.A.; Oza, M.J. Autophagy-nutrient sensing pathways in diabetic complications. Pharmacol. Res. 2022, 184, 106408. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Shen, C.; Li, J.; Sun, C.; Song, Z. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936–952. [Google Scholar] [CrossRef]
- de Luxán-Delgado, B.; Potes, Y.; Rubio-González, A.; Caballero, B.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Rodrigues Moreira Guimarães, M.; Vega-Naredo, I.; Boga, J.A.; et al. Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J. Pineal Res. 2016, 61, 108–123. [Google Scholar] [CrossRef]
- Moulis, M.; Vindis, C. Autophagy in metabolic age-related human diseases. Cells 2018, 7, 149. [Google Scholar] [CrossRef]
- Jansen, H.J.; vanEssen, P.; Koenen, T.; Joosten, L.A.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 8, 758311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Magdy, M.; Yousef, O.A.E.; Ibrahim, W.; Gharib, D.M. Circulating MicroRNA-30a, Beclin1 and Their Association with Different Variables in Females with Metabolically Healthy/Unhealthy Obesity. Diabetes Metab. Syndr. Obes. 2023, 16, 3065–3074. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maixner, N.; Bechor, S.; Vershinin, Z.; Pecht, T.; Goldstein, N.; Haim, Y.; Rudich, A. Transcriptional Dysregulation of Adipose Tissue Autophagy in Obesity. Physiology 2016, 31, 270–282. [Google Scholar] [CrossRef]
- Maixner, N.; Pecht, T.; Haim, Y.; Chalifa-Caspi, V.; Goldstein, N.; Tarnovscki, T.; Liberty, I.F.; Kirshtein, B.; Golan, R.; Berner, O.; et al. A TRAIL-TL1A Paracrine Network Involving Adipocytes, Macrophages, and Lymphocytes Induces Adipose Tissue Dysfunction Downstream of E2F1 in Human Obesity. Diabetes 2020, 69, 2310–2323. [Google Scholar] [CrossRef]
- Haim, Y.; Blüuher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Tinahones, A.; El Bekay, R.; Malagón, M.M.; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites 2020, 10, 179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, Q.; Mariman, E.C.M.; Roumans, N.J.T.; Vink, R.G.; Goossens, G.H.; Blaak, E.E.; Jocken, J.W.E. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte 2018, 7, 12–19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nuñez, C.E.; Rodrigues, V.S.; Gomes, F.S.; Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Critical Reanalysis of the Mechanisms Underlying the Cardiorenal Benefits of SGLT2 Inhibitors and Reaffirmation of the Nutrient Deprivation Signaling/Autophagy Hypothesis. Circulation 2022, 146, 1383–1405. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ashrafizadeh, M.; Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Effects of newly introduced antidiabetic drugs on autophagy. Diabetes Metab. Syndr. 2019, 13, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F. GLP-1-based therapies to boost autophagy in cardiometabolic patients: From experimental evidence to clinical trials. Vasc. Pharmacol. 2019, 115, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Taktaz, F.; Scisciola, L.; Fontanella, R.A.; Pesapane, A.; Ghosh, P.; Franzese, M.; Tortorella, G.; Puocci, A.; Sommella, E.; Signoriello, G.; et al. Evidence that tirzepatide protects against diabetes-related cardiac damages. Cardiovasc. Diabetol. 2024, 23, 112. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- American Dietetic Association. International Dietetics and Nutrition Terminology (IDNT) Reference Manual: Standardized Language for the Nutrition Care Proces, 3rd ed.; American Dietetic Association: Chicago, IL, USA, 2011. [Google Scholar]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Alrushud, A.S.; Rushton, A.B.; Kanavaki, A.M.; Greig, C.A. Effect of physical activity and dietary restriction interventions on weight loss and the musculoskeletal function of overweight and obese older adults with knee osteoarthritis: A systematic review and mixed method data synthesis. BMJ Open 2017, 7, e014537. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. Autophagy-dependent beneficial effects of exercise. Cold Spring Harb. Perspect. Med. 2017, 7, a029777. [Google Scholar] [CrossRef]
- Chung, K.W.; Chung, H.Y. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shabkhizan, R.; Haiaty, S.; Moslehian, M.S.; Bazmani, A.; Sadeghsoltani, F.; Saghaei Bagheri, H.; Rahbarghazi, R.; Sakhinia, E. The Beneficial and Adverse Effects of Autophagic Response to Caloric Restriction and Fasting. Adv. Nutr. 2023, 14, 1211–1225. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, L.; Xiong, H.; Chen, L.; Dai, X.; Yan, X.; Wu, Y.; Yang, M.; Shan, M.; Li, T.; Yao, J.; et al. Deacetylation of ATG4B promotes autophagy initiation under starvation. Sci. Adv. 2022, 8, eabo0412. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Liu, B.; Bensalem, J.; Sargeant, T.J.; Page, A.J.; Wittert, G.A.; Hutchison, A.T.; Heilbronn, L.K. Intermittent fasting activates markers of autophagy in mouse liver, but not muscle from mouse or humans. Nutrition 2022, 101, 111662. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Kocot, A.M.; Wróblewska, B. Nutritional strategies for autophagy activation and health consequences of autophagy impairment. Nutrition 2022, 103–104, 111686. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Song, D.K.; Kim, Y.W. Beneficial effects of intermittent fasting: A narrative review. J. Yeungnam Med. Sci. 2023, 40, 4–11. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Tsuda, S.; Kanasaki, K.; Koya, D. Calorie restriction in overweight males ameliorates obesity-related metabolic alterations and cellular adaptations through anti-aging effects, possibly including AMPK and SIRT1 activation. Biochim. Biophys. Acta. 2013, 1830, 4820–4827. [Google Scholar] [CrossRef]
- Hanningan, A.; Gorski, S. Macroautophagy. Autophagy 2009, 5, 140–151. [Google Scholar] [CrossRef]
- Mariani, S.; di Giorgio, M.R.; Martini, P.; Persichetti, A.; Barbaro, G.; Basciani, S.; Contini, S.; Poggiogalle, E.; Sarnicola, A.; Genco, A.; et al. Inverse Association of Circulating SIRT1 and Adiposity: A Study on Underweight, Normal Weight, and Obese Patients. Front. Endocrinol. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baeken, M.W. Sirtuins and their influence on autophagy. J. Cell Biochem. 2023, 125, e30377. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saini, S.K.; Singh, A.; Saini, M.; Gonzalez-Freire, M.; Leeuwenburgh, C.; Anton, S.D. Time-Restricted Eating Regimen Differentially Affects Circulatory miRNA Expression in Older Overweight Adults. Nutrients 2022, 14, 1843. [Google Scholar] [CrossRef]
- Ammendola, S.; Scotto d’Abusco, A. Nutraceuticals and the Network of Obesity Modulators. Nutrients 2022, 14, 5099. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, C.; Ji, L.; Hu, J.; Zhao, Y.; Johnston, L.J.; Zhang, X.; Ma, X. Functional Amino Acids and Autophagy: Diverse Signal Transduction and Application. Int. J. Mol. Sci. 2021, 22, 11427. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ortega, M.A.; Fraile-Martinez, O.; de Leon-Oliva, D.; Boaru, D.L.; Lopez-Gonzalez, L.; García-Montero, C.; Alvarez-Mon, M.A.; Guijarro, L.G.; Torres-Carranza, D.; Saez, M.A.; et al. Autophagy in Its (Proper) Context: Molecular Basis, Biological Relevance, Pharmacological Modulation, and Lifestyle Medicine. Int. J. Biol. Sci. 2024, 20, 2532–2554. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vafaeipour, Z.; Razavi, B.M.; Hosseinzadeh, H. Effects of turmeric (Curcuma longa) and its constituent (curcumin) on the metabolic syndrome: An updated review. J. Integr. Med. 2022, 20, 193–203. [Google Scholar] [CrossRef]
- Sbai, O.; Torrisi, F.; Fabrizio, F.P.; Rabbeni, G.; Perrone, L. Effect of the Mediterranean Diet (MeDi) on the Progression of Retinal Disease: A Narrative Review. Nutrients 2024, 16, 3169. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef] [PubMed]
- García-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.M.; Pekarek, L.; Castellanos, A.J.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional components in western diet versus mediterranean diet at the gut microbiota-immune system interplay. implications for health and disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Franco, G.A.; Interdonato, L.; Cordaro, M.; Cuzzocrea, S.; Di Paola, R. Bioactive Compounds of the Mediterranean Diet as Nutritional Support to Fight Neurodegenerative Disease. Int. J. Mol. Sci. 2023, 24, 7318. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Muscogiuri, G.; Verde, L.; Sulu, C.; Katsiki, N.; Hassapidou, M.; Frias-Toral, E.; Cucalón, G.; Pazderska, A.; Yumuk, V.D.; Colao, A.; et al. Mediterranean Diet and Obesity-related Disorders: What is the Evidence? Curr. Obes. Rep. 2022, 11, 287–304. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sokal, A.; Stocerz, K.; Olczyk, P.; Kadela-Tomanek, M. Therapeutic potential of flavonoids used in traditional Chinese medicine—A comparative study of galangin, kaempferol, chrysin and fisetin. AAMS 2024, 78, 49–60. [Google Scholar] [CrossRef]
- Domonese, N.; Di Bella, G.; Cusumano, C.; Parisi, A.; Tagliaferri, F.; Ciriminna, S.; Barbagallo, M. Mediterranean diet in the management and prevention of obesity. Exp. Gerontol. 2023, 174, 112121. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Conles, Ó.; Olbeyra, R.; Moizé, V.; Ibarzabal, A.; Giró, O.; Viaplana, J.; Jiménez, A.; Vidal, J.; de Hollanda, A. Positive Effects of a Mediterranean Diet Supplemented with Almonds on Female Adipose Tissue Biology in Severe Obesity. Nutrients 2022, 14, 2617. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brimson, J.M.; Prasanth, M.I.; Malar, D.S.; Thitilertdecha, P.; Kabra, A.; Tencomnao, T.; Prasansuklab, A. Plant Polyphenols for Aging Health: Implication from Their Autophagy Modulating Properties in Age-Associated Diseases. Pharmaceuticals 2021, 14, 982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.; Quan, W.; Zeng, M.; Wang, Z.; Chen, Q.; Chen, J.; Christian, M.; He, Z. Regulation of autophagy by plant-based polyphenols: A critical review of current advances in glucolipid metabolic diseases and food industry applications. Food Front. 2023, 4, 1039–1067. [Google Scholar] [CrossRef]
- Most, J.; Warnke, I.; Boekschoten, M.V.; Jocken, J.W.E.; de Groot, P.; Friedel, A.; Bendik, I.; Goossens, G.H.; Blaak, E.E. The effects of polyphenol supplementation on adipose tissue morphology and gene expression in overweight and obese humans. Adipocyte 2018, 7, 190–196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, L.X.; Li, C.X.; Kakar, M.U.; Khan, M.S.; Wu, P.F.; Amir, R.M.; Dai, D.F.; Naveed, M.; Li, Q.Y.; Saeed, M.; et al. Resveratrol (RV): A pharmacological review and call for further research. Biomed. Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Widlund, A.L.; Baur, J.A.; Vang, O. mTOR: More targets of resveratrol? Expert Rev. Mol. Med. 2013, 15, e10. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- Konings, E.; Timmers, S.; Boekschoten, M.V.; Goossens, G.H.; Jocken, J.W.; Afman, L.A.; Müller, M.; Schrauwen, P.; Mariman, E.C.; Blaak, E.E. The effects of 30 days resveratrol supplementation on adipose tissue morphology and gene expression patterns in obese men. Int. J. Obes. 2014, 38, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Moskot, M.; Montefusco, S.; Jakóbkiewicz-Banecka, J.; Mozolewski, P.; Węgrzyn, A.; Di Bernardo, D.; Węgrzyn, G.; Medina, D.L.; Ballabio, A.; Gabig-Cimińska, M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 2014, 289, 17054–17069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mansur, A.P.; Roggerio, A.; Goes, M.F.S.; Avakian, S.D.; Leal, D.P.; Maranhão, R.C.; Strunz, C.M.C. Serum concentrations and gene expression of sirtuin 1 in healthy and slightly overweight subjects after caloric restriction or resveratrol supplementation: A randomized trial. Int. J. Cardiol. 2017, 227, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Aloo, S.O.; Ofosu, F.K.; Kim, N.H.; Kilonzi, S.M.; Oh, D.H. Insights on Dietary Polyphenols as Agents against Metabolic Disorders: Obesity as a Target Disease. Antioxidants 2023, 12, 416. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tian, Y.; Song, W.; Li, D.; Cai, L.; Zhao, Y. Resveratrol as A Natural Regulator of Autophagy for Prevention and Treatment of Cancer. Onco Targets Ther. 2019, 12, 8601–8609. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Phillips, C.M.; Kesse-Guyot, E.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. High dietary saturated fat intake accentuates obesity risk associated with the fat mass and obesity-associated gene in adults. J. Nutr. 2012, 142, 824–831. [Google Scholar] [CrossRef]
- Liu, W.; Zhu, M.; Gong, M.; Zheng, W.; Zeng, X.; Zheng, Q.; Li, X.; Fu, F.; Chen, Y.; Cheng, J.; et al. Comparison of the Effects of Monounsaturated Fatty Acids and Polyunsaturated Fatty Acids on Liver Lipid Disorders in Obese Mice. Nutrients 2023, 15, 3200. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ntuli, T.; Wang, J.; Nie, D. Modulation autophagy by Fatty acids. In Cell Death—Autophagy, Apoptosis and Necrosis; IntechOpen: London, UK, 2015; ISBN 978-953-51-4211-9. [Google Scholar]
- O’Rourke, E.J.; Kuballa, P.; Xavier, R.; Ruvkun, G. ω-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 2013, 27, 429–440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ciesielska, K.; Gajewska, M. Fatty Acids as Potent Modulators of Autophagy Activity in White Adipose Tissue. Biomolecules 2023, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, Y.; Wu, M.; Li, X.; Mai, K.; Ai, Q. ω-6 Polyunsaturated fatty acids (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop via TOR-dependent and TOR-independent signaling pathways. Cell Death Dis. 2020, 11, 607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Camargo, A.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.; Gomez-Delgado, F.; Delgado-Lista, J.; García-Carpintero, S.; Marín, C.; Almadén, Y.; Yubero-Serrano, E.M.; López-Moreno, J.; et al. Dietary fat may modulate adipose tissue homeostasis through the processes of autophagy and apoptosis. Eur. J. Nutr. 2017, 56, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Viguerie, N.; Vidal, H.; Arner, P.; Holst, C.; Verdich, C.; Avizou, S.; Astrup, A.; Saris, W.H.; Macdonald, I.A.; Klimcakova, E.; et al. Nutrient-Gene Interactions in Human Obesity—Implications for Dietary Guideline (NUGENOB) project. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia 2005, 48, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Capel, F.; Viguerie, N.; Vega, N.; Dejean, S.; Arner, P.; Klimcakova, E.; Martinez, J.A.; Saris, W.H.; Holst, C.; Taylor, M.; et al. Contribution of energy restriction and macronutrient composition to changes in adipose tissue gene expression during dietary weight-loss programs in obese women. J. Clin. Endocrinol. Metab. 2008, 93, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Markova, M.; Seebeck, N.; Loft, A.; Hornemann, S.; Gantert, T.; Kabisch, S.; Herz, K.; Loske, J.; Ost, M.; et al. High-protein diet more effectively reduces hepatic fat than low-protein diet despite lower autophagy and FGF21 levels. Liver Int. 2020, 40, 2982–2997. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bednarczyk, M.; Dąbrowska-Szeja, N.; Łętowski, D.; Dzięgielewska-Gęsiak, S.; Waniczek, D.; Muc-Wierzgoń, M. Relationship Between Dietary Nutrient Intake and Autophagy—Related Genes in Obese Humans: A Narrative Review. Nutrients 2024, 16, 4003. https://doi.org/10.3390/nu16234003
Bednarczyk M, Dąbrowska-Szeja N, Łętowski D, Dzięgielewska-Gęsiak S, Waniczek D, Muc-Wierzgoń M. Relationship Between Dietary Nutrient Intake and Autophagy—Related Genes in Obese Humans: A Narrative Review. Nutrients. 2024; 16(23):4003. https://doi.org/10.3390/nu16234003
Chicago/Turabian StyleBednarczyk, Martyna, Nicola Dąbrowska-Szeja, Dariusz Łętowski, Sylwia Dzięgielewska-Gęsiak, Dariusz Waniczek, and Małgorzata Muc-Wierzgoń. 2024. "Relationship Between Dietary Nutrient Intake and Autophagy—Related Genes in Obese Humans: A Narrative Review" Nutrients 16, no. 23: 4003. https://doi.org/10.3390/nu16234003
APA StyleBednarczyk, M., Dąbrowska-Szeja, N., Łętowski, D., Dzięgielewska-Gęsiak, S., Waniczek, D., & Muc-Wierzgoń, M. (2024). Relationship Between Dietary Nutrient Intake and Autophagy—Related Genes in Obese Humans: A Narrative Review. Nutrients, 16(23), 4003. https://doi.org/10.3390/nu16234003