
Maternal and Placental DNA Methylation Changes Associated with the Pathogenesis of Gestational Diabetes Mellitus

Abstract
1. Introduction
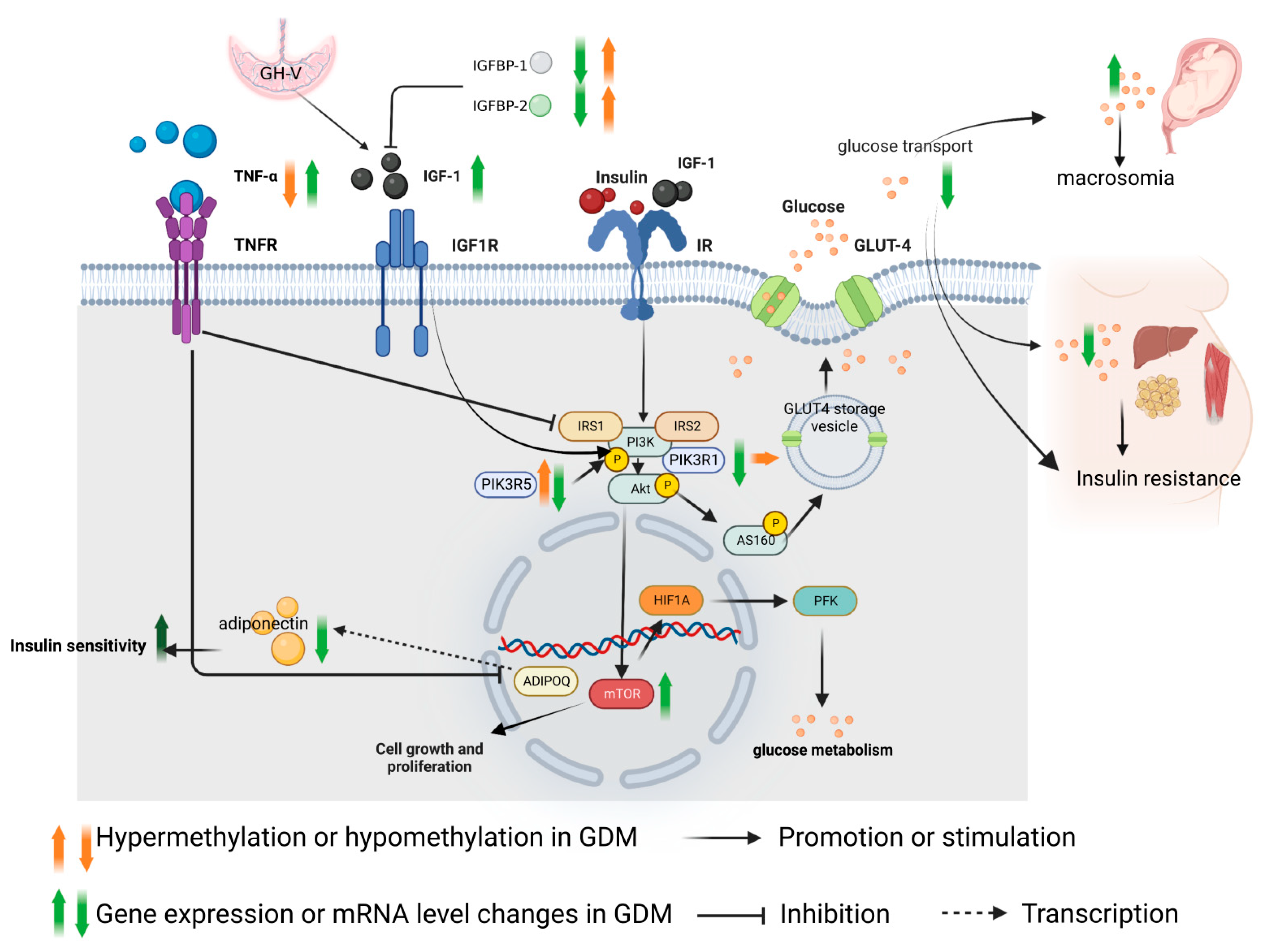
2. Role of IGF-1 Axis in the Pathogenesis of GDM and Methylation Changes
2.1. Regulation Mechanism of IGF-1
2.2. Role of IGF-1 in the Pathogenesis of GDM
3. Adipokine
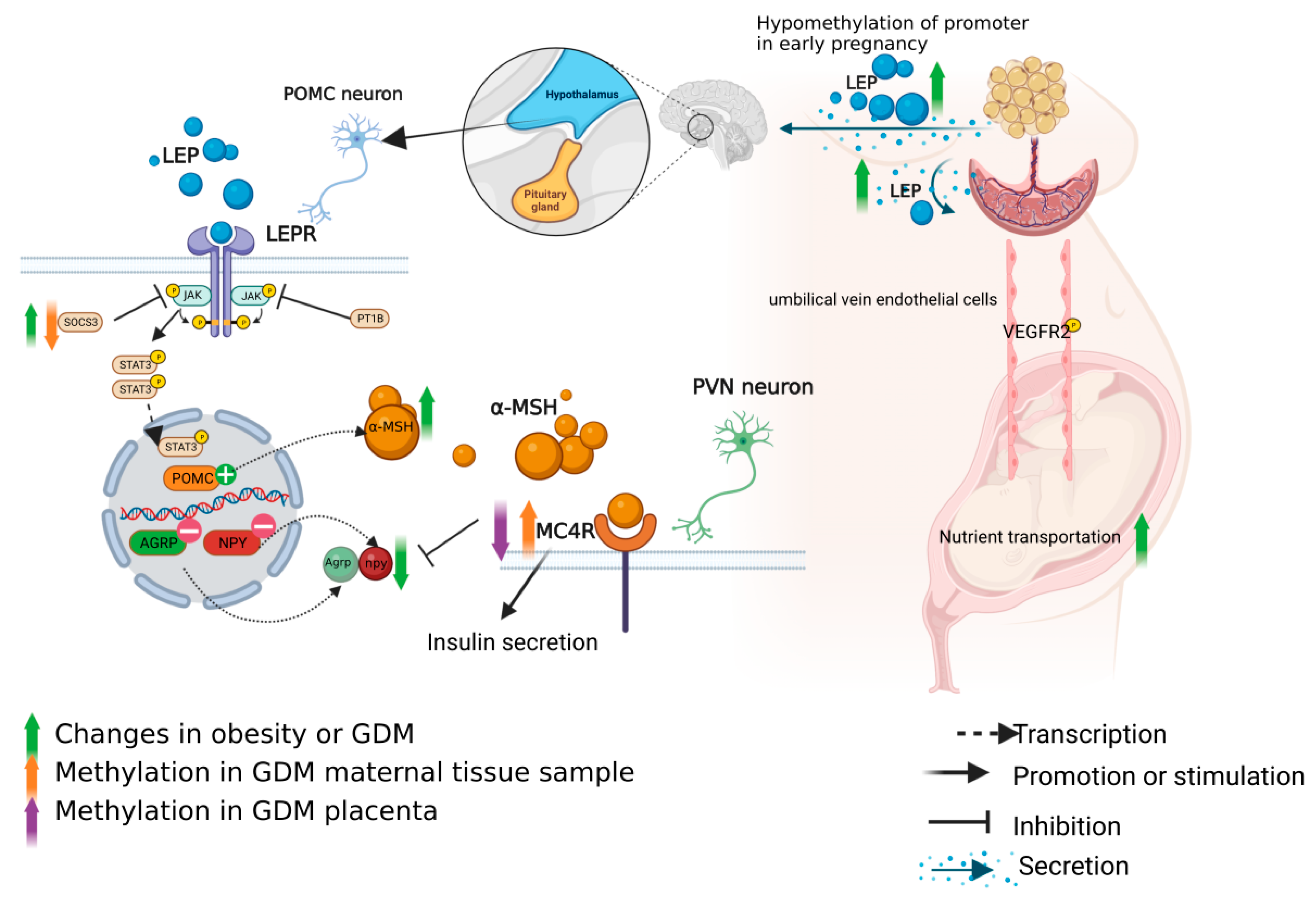
3.1. Role of Leptin Axis in the Pathogenesis of GDM and Methylation Changes
3.1.1. Leptin in the Pathogenesis of GDM
3.1.2. Regulation of Leptin Axis
3.1.3. Methylation Changes of the Leptin Axis in GDM Patients
3.2. Function of Adiponectin in GDM and Methylation Changes
3.3. Role of Chemerin in GDM and Methylation Changes
4. GDM Insulin Resistance Related Mechanisms and Methylation Changes
4.1. Molecular Mechanism of GDM Insulin Resistance
4.2. Methylation Changes of Insulin Resistance in GDM
5. Changes and Roles of Cytokines in GDM and Their Methylation Changes
5.1. TNF-α
5.2. IL-10
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association. Gestational diabetes mellitus. Diabetes Care 2004, 27 (Suppl. 1), S88–S90. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.F.; Has, P.; Tarabulsi, G.; Lee, J.; Satin, A. Early postpartum glucose testing in women with gestational diabetes mellitus. Am. J. Perinatol. 2016, 33, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef]
- Luo, Z.C.; Nuyt, A.M.; Delvin, E.; Audibert, F.; Girard, I.; Shatenstein, B.; Cloutier, A.; Cousineau, J.; Djemli, A.; Deal, C.; et al. Maternal and fetal IGF-I and IGF-II levels, fetal growth, and gestational diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1720–1728. [Google Scholar] [CrossRef]
- Grissa, O.; Yessoufou, A.; Mrisak, I.; Hichami, A.; Amoussou-Guenou, D.; Grissa, A.; Djrolo, F.; Moutairou, K.; Miled, A.; Khairi, H.; et al. Growth factor concentrations and their placental mRNA expression are modulated in gestational diabetes mellitus: Possible interactions with macrosomia. BMC Pregnancy Childbirth 2010, 10, 7. [Google Scholar] [CrossRef]
- Halle, M.; Berg, A.; Northoff, H.; Keul, J. Importance of TNF-alpha and leptin in obesity and insulin resistance: A hypothesis on the impact of physical exercise. Exerc. Immunol. Rev. 1998, 4, 77–94. [Google Scholar]
- Lee, M.H.; Jeon, Y.J.; Lee, S.M.; Park, M.H.; Jung, S.C.; Kim, Y.J. Placental gene expression is related to glucose metabolism and fetal cord blood levels of insulin and insulin-like growth factors in intrauterine growth restriction. Early Hum. Dev. 2010, 86, 45–50. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Zeck, W.; Russell, A. Placental growth hormone, fetal growth and the IGF axis in normal and diabetic pregnancy. Curr. Diabetes Rev. 2009, 5, 185–189. [Google Scholar] [CrossRef]
- Langer, O.; Yogev, Y.; Most, O.; Xenakis, E.M. Gestational diabetes: The consequences of not treating. Am. J. Obstet. Gynecol. 2005, 192, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Kumar, S.; Creatsas, G.; Mastorakos, G. The effects of adipose tissue and adipocytokines in human pregnancy. Ann. N. Y. Acad. Sci. 2010, 1205, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Martín-Estal, I.; Castilla-Cortázar, I.; Castorena-Torres, F. The placenta as a target for alcohol during pregnancy: The close relation with IGFs signaling pathway. Rev. Physiol. Biochem. Pharmacol. 2021, 180, 119–153. [Google Scholar] [CrossRef] [PubMed]
- Kavran, J.M.; McCabe, J.M.; Byrne, P.O.; Connacher, M.K.; Wang, Z.; Ramek, A.; Sarabipour, S.; Shan, Y.; Shaw, D.E.; Hristova, K.; et al. How IGF-1 activates its receptor. eLife 2014, 3, e03772. [Google Scholar] [CrossRef]
- Moltke, I.; Grarup, N.; Jørgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T.; et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef]
- Karlsson, H.K.; Zierath, J.R.; Kane, S.; Krook, A.; Lienhard, G.E.; Wallberg-Henriksson, H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 2005, 54, 1692–1697. [Google Scholar] [CrossRef]
- Hwa, V.; Oh, Y.; Rosenfeld, R.G. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar] [CrossRef]
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [CrossRef]
- Shang, M.; Wen, Z. Increased placental IGF-1/mTOR activity in macrosomia born to women with gestational diabetes. Diabetes Res. Clin. Pract. 2018, 146, 211–219. [Google Scholar] [CrossRef]
- Yoon, M.S. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed]
- Steyn, A.; Crowther, N.J.; Norris, S.A.; Rabionet, R.; Estivill, X.; Ramsay, M. Epigenetic modification of the pentose phosphate pathway and the IGF-axis in women with gestational diabetes mellitus. Epigenomics 2019, 11, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef]
- Šimják, P.; Cinkajzlová, A.; Anderlová, K.; Pařízek, A.; Mráz, M.; Kršek, M.; Haluzík, M. The role of obesity and adipose tissue dysfunction in gestational diabetes mellitus. J. Endocrinol. 2018, 238, R63–R77. [Google Scholar] [CrossRef] [PubMed]
- De Luccia, T.P.B.; Pendeloski, K.P.T.; Ono, E.; Mattar, R.; Pares, D.B.S.; Yazaki Sun, S.; Daher, S. Unveiling the pathophysiology of gestational diabetes: Studies on local and peripheral immune cells. Scand. J. Immunol. 2020, 91, e12860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Kralisch, S.; Bluher, M.; Paschke, R.; Stumvoll, M.; Fasshauer, M. Adipokines and adipocyte targets in the future management of obesity and the metabolic syndrome. Mini Rev. Med. Chem. 2007, 7, 39–45. [Google Scholar] [CrossRef]
- Lisboa, P.C.; Oliveira, E.; Fagundes, A.T.; Santos-Silva, A.P.; Conceição, E.P.; Passos, M.C.; Moura, E.G. Postnatal low protein diet programs leptin signaling in the hypothalamic-pituitary-thyroid axis and pituitary TSH response to leptin in adult male rats. Horm. Metab. Res. Horm. Stoffwechselforsch. Horm. Metab. 2012, 44, 114–122. [Google Scholar] [CrossRef]
- D’Souza, A.M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The pathophysiology of gestational diabetes mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Powe, C.E.; Huston Presley, L.P.; Locascio, J.J.; Catalano, P.M. Augmented insulin secretory response in early pregnancy. Diabetologia 2019, 62, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Koestler, D.C.; Padbury, J.F.; Marsit, C.J. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol. Cell. Endocrinol. 2013, 381, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Tessier, D.R.; Ferraro, Z.M.; Gruslin, A. Role of leptin in pregnancy: Consequences of maternal obesity. Placenta 2013, 34, 205–211. [Google Scholar] [CrossRef]
- Kinalski, M.; Sledziewski, A.; Kowalska, I.; Telejko, B.; Kuźmicki, M.; Kretowski, A.; Majkowicz-Młynarczyk, A.; Kinalska, I. Postpartum maternal plasma leptin levels and their relationship to gestational diabetes mellitus. Med. Wieku Rozw. 2004, 8, 703–710. [Google Scholar]
- Pérez-Pérez, A.; Maymó, J.L.; Gambino, Y.P.; Guadix, P.; Dueñas, J.L.; Varone, C.L.; Sánchez-Margalet, V. Activated translation signaling in placenta from pregnant women with gestational diabetes mellitus: Possible role of leptin. Horm. Metab. Res. = Horm. Stoffwechselforsch. = Horm. Metab. 2013, 45, 436–442. [Google Scholar] [CrossRef]
- Lewandowski, K.C.; Biesiada, L.; Grzesiak, M.; Sakowicz, A. C-Peptide and leptin system in dichorionic, small and appropriate for gestational age twins-possible link to metabolic programming? Nutr. Diabetes 2020, 10, 29. [Google Scholar] [CrossRef]
- Garonna, E.; Botham, K.M.; Birdsey, G.M.; Randi, A.M.; Gonzalez-Perez, R.R.; Wheeler-Jones, C.P. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS ONE 2011, 6, e18823. [Google Scholar] [CrossRef]
- Catalano, P.M.; Presley, L.; Minium, J.; Hauguel-de Mouzon, S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef]
- van der Klaauw, A.A. Neuropeptides in Obesity and Metabolic Disease. Clin. Chem. 2018, 64, 173–182. [Google Scholar] [CrossRef]
- Maejima, Y.; Sakuma, K.; Santoso, P.; Gantulga, D.; Katsurada, K.; Ueta, Y.; Hiraoka, Y.; Nishimori, K.; Tanaka, S.; Shimomura, K.; et al. Oxytocinergic circuit from paraventricular and supraoptic nuclei to arcuate POMC neurons in hypothalamus. FEBS Lett. 2014, 588, 4404–4412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hernandez-Sanchez, D.; Herzog, H. Regulation of feeding-related behaviors by arcuate neuropeptide y neurons. Endocrinology 2019, 160, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef]
- Krishna, R.; Gumbiner, B.; Stevens, C.; Musser, B.; Mallick, M.; Suryawanshi, S.; Maganti, L.; Zhu, H.; Han, T.H.; Scherer, L.; et al. Potent and selective agonism of the melanocortin receptor 4 with MK-0493 does not induce weight loss in obese human subjects: Energy intake predicts lack of weight loss efficacy. Clin. Pharmacol. Ther. 2009, 86, 659–666. [Google Scholar] [CrossRef]
- Mansour, M.; White, D.; Wernette, C.; Dennis, J.; Tao, Y.X.; Collins, R.; Parker, L.; Morrison, E. Pancreatic neuronal melanocortin-4 receptor modulates serum insulin levels independent of leptin receptor. Endocrine 2010, 37, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Wang, Y.; Shi, C.; Zhang, X.; Gong, H.; Dong, Y. Islet MC4R Regulates PC1/3 to Improve insulin secretion in T2DM mice via the cAMP and β-arrestin-1 pathways. Appl. Biochem. Biotechnol. 2022, 194, 6164–6178. [Google Scholar] [CrossRef]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Li, Z.; Padbury, J.F.; Marsit, C.J. Maternal obesity and gestational diabetes are associated with placental leptin DNA methylation. Am. J. Obstet. Gynecol. 2014, 211, 654.e1–654.e9. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Porreca, A.; D’Ardes, M.; Di Nicola, M.; Di Tizio, L.; Liberati, M.; Stuppia, L.; Vitacolonna, E. The obesogenic environment: Epigenetic modifications in placental melanocortin 4 receptor gene connected to gestational diabetes and smoking. Front. Nutr. 2022, 9, 879526. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, R.C.; Ott, R.; Ziska, T.; Schellong, K.; Melchior, K.; Henrich, W.; Plagemann, A. Visceral adipose tissue inflammatory factors (TNF-Alpha, SOCS3) in gestational diabetes (GDM): Epigenetics as a clue in GDM pathophysiology. Int. J. Mol. Sci. 2020, 21, 479. [Google Scholar] [CrossRef] [PubMed]
- Mandò, C.; Abati, S.; Anelli, G.M.; Favero, C.; Serati, A.; Dioni, L.; Zambon, M.; Albetti, B.; Bollati, V.; Cetin, I. Epigenetic profiling in the saliva of obese pregnant women. Nutrients 2022, 14, 2122. [Google Scholar] [CrossRef]
- Brochu-Gaudreau, K.; Rehfeldt, C.; Blouin, R.; Bordignon, V.; Murphy, B.D.; Palin, M.F. Adiponectin action from head to toe. Endocrine 2010, 37, 11–32. [Google Scholar] [CrossRef]
- Bao, W.; Baecker, A.; Song, Y.; Kiely, M.; Liu, S.; Zhang, C. Adipokine levels during the first or early second trimester of pregnancy and subsequent risk of gestational diabetes mellitus: A systematic review. Metab. Clin. Exp. 2015, 64, 756–764. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M.; Stumvoll, M. Adipokines in gestational diabetes. Lancet Diabetes Endocrinol. 2014, 2, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.W.; Vatish, M.; Randeva, H.S. Secretion of adiponectin by human placenta: Differential modulation of adiponectin and its receptors by cytokines. Diabetologia 2006, 49, 1292–1302. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.; Naimo, G.D.; Ricchio, E.; Panno, M.L.; Andò, S. Cross-talk between adiponectin and IGF-IR in breast cancer. Front. Oncol. 2015, 5, 157. [Google Scholar] [CrossRef] [PubMed]
- Ott, R.; Stupin, J.H.; Melchior, K.; Schellong, K.; Ziska, T.; Dudenhausen, J.W.; Henrich, W.; Rancourt, R.C.; Plagemann, A. Alterations of adiponectin gene expression and DNA methylation in adipose tissues and blood cells are associated with gestational diabetes and neonatal outcome. Clin. Epigenet. 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Houshmand-Oeregaard, A.; Hansen, N.S.; Hjort, L.; Kelstrup, L.; Broholm, C.; Mathiesen, E.R.; Clausen, T.D.; Damm, P.; Vaag, A. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin. Epigenet. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Estienne, A.; Bongrani, A.; Reverchon, M.; Ramé, C.; Ducluzeau, P.H.; Froment, P.; Dupont, J. Involvement of novel adipokines, chemerin, visfatin, resistin and apelin in reproductive functions in normal and pathological conditions in humans and animal models. Int. J. Mol. Sci. 2019, 20, 4431. [Google Scholar] [CrossRef]
- Tsiotra, P.C.; Halvatsiotis, P.; Patsouras, K.; Maratou, E.; Salamalekis, G.; Raptis, S.A.; Dimitriadis, G.; Boutati, E. Circulating adipokines and mRNA expression in adipose tissue and the placenta in women with gestational diabetes mellitus. Peptides 2018, 101, 157–166. [Google Scholar] [CrossRef]
- Fatima, S.S.; Rehman, R.; Muhammad, J.S.; Martins, R.; Mohammed, N.; Khan, U. Association of chemerin gene promoter methylation in maternal blood and breast milk during gestational diabetes. J. Dev. Orig. Health Dis. 2022, 13, 108–114. [Google Scholar] [CrossRef]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Hancock, J.A.; Golichowski, A.M. Multiple defects in the adipocyte glucose transport system cause cellular insulin resistance in gestational diabetes. Heterogeneity in the number and a novel abnormality in subcellular localization of GLUT4 glucose transporters. Diabetes 1993, 42, 1773–1785. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Klemm, D.J. Adipose tissue insulin sensitivity and macrophage recruitment: Does PI3K pick the pathway? Adipocyte 2013, 2, 135–142. [Google Scholar] [CrossRef]
- Metz, H.E.; Houghton, A.M. Insulin receptor substrate regulation of phosphoinositide 3-kinase. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule phosphatidylinositol 3-kinase inhibitors prescribed for the treatment of malignancies. Pharmacol. Res. 2021, 168, 105579. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, R.C.; Ott, R.; Schellong, K.; Melchior, K.; Ziska, T.; Henrich, W.; Plagemann, A. Visceral adipose tissue alteration of PI3KR1 expression is associated with gestational diabetes but not promoter DNA methylation. Adipocyte 2019, 8, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Farrell, W.E.; Haworth, K.E.; Emes, R.D.; Kitchen, M.O.; Glossop, J.R.; Hanna, F.W.; Fryer, A.A. Maternal genome-wide DNA methylation profiling in gestational diabetes shows distinctive disease-associated changes relative to matched healthy pregnancies. Epigenetics 2018, 13, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Mollet, I.G.; Malm, H.A.; Wendt, A.; Orho-Melander, M.; Eliasson, L. Integrator of stress responses calmodulin binding transcription activator 1 (Camta1) regulates miR-212/miR-132 expression and insulin secretion. J. Biol. Chem. 2016, 291, 18440–18452. [Google Scholar] [CrossRef]
- Dias, S.; Adam, S.; Rheeder, P.; Louw, J.; Pheiffer, C. Altered genome-wide DNA methylation in peripheral blood of South African women with gestational diabetes mellitus. Int. J. Mol. Sci. 2019, 20, 5828. [Google Scholar] [CrossRef]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef]
- Dong, Y.; Chauhan, M.; Betancourt, A.; Belfort, M.; Yallampalli, C. Adipose tissue inflammation and adrenomedullin overexpression contribute to lipid dysregulation in diabetic pregnancies. J. Clin. Endocrinol. Metab. 2018, 103, 3810–3818. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Oliva, K.; Georgiou, H.M.; Permezel, J.M.; Rice, G.E. Glucose-induced release of tumour necrosis factor-alpha from human placental and adipose tissues in gestational diabetes mellitus. Diabet. Med. J. Br. Diabet. Assoc. 2001, 18, 921–927. [Google Scholar] [CrossRef]
- Peraldi, P.; Spiegelman, B. TNF-alpha and insulin resistance: Summary and future prospects. Mol. Cell. Biochem. 1998, 182, 169–175. [Google Scholar] [CrossRef]
- Laffer, B.; Bauer, D.; Wasmuth, S.; Busch, M.; Jalilvand, T.V.; Thanos, S.; Meyer Zu Hörste, G.; Loser, K.; Langmann, T.; Heiligenhaus, A.; et al. Loss of IL-10 promotes differentiation of microglia to a M1 phenotype. Front. Cell. Neurosci. 2019, 13, 430. [Google Scholar] [CrossRef] [PubMed]
- Charles, B.A.; Doumatey, A.; Huang, H.; Zhou, J.; Chen, G.; Shriner, D.; Adeyemo, A.; Rotimi, C.N. The roles of IL-6, IL-10, and IL-1RA in obesity and insulin resistance in African-Americans. J. Clin. Endocrinol. Metab. 2011, 96, E2018–E2022. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Liu, B.; Li, Q.; Wang, Z.; Fan, S.; Wang, H.; Wang, L. Functional defects of regulatory T Cell through interleukin 10 mediated mechanism in the induction of gestational diabetes mellitus. DNA Cell Biol. 2018, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, C.N.; Li, H.Y.; Hsu, K.H.; Wang, S.H.; Lin, S.Y. Association of interleukin-10 methylation levels with gestational diabetes in a Taiwanese population. Front. Genet. 2018, 9, 222. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name | Maternal Obesity | GDM |
|---|---|---|---|
| Cytokines | TNF-α | ↑ or → | ↑ |
| IL-6 | ↑ | ↑ | |
| Adipokines | Leptin | ↑ | ↑ |
| Adiponectin | ↓ | ↓ | |
| Immune cells | M1 macrophage | ↑ | ↑ or → |
| NK cells | ↑ | cytotoxic and cytokine-secreting cells ↑ | |
| Th1 and Tc lymphocytes | ↑ | ↑ | |
| Tregs | ↓ | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Dong, S.; Wu, L.; Bai, Y.; Bi, X.; Li, Y.; Shu, C. Maternal and Placental DNA Methylation Changes Associated with the Pathogenesis of Gestational Diabetes Mellitus. Nutrients 2023, 15, 70. https://doi.org/10.3390/nu15010070
Xu P, Dong S, Wu L, Bai Y, Bi X, Li Y, Shu C. Maternal and Placental DNA Methylation Changes Associated with the Pathogenesis of Gestational Diabetes Mellitus. Nutrients. 2023; 15(1):70. https://doi.org/10.3390/nu15010070
Chicago/Turabian StyleXu, Peng, Shuai Dong, Linlin Wu, Yule Bai, Xueqing Bi, Yaping Li, and Chang Shu. 2023. "Maternal and Placental DNA Methylation Changes Associated with the Pathogenesis of Gestational Diabetes Mellitus" Nutrients 15, no. 1: 70. https://doi.org/10.3390/nu15010070
APA StyleXu, P., Dong, S., Wu, L., Bai, Y., Bi, X., Li, Y., & Shu, C. (2023). Maternal and Placental DNA Methylation Changes Associated with the Pathogenesis of Gestational Diabetes Mellitus. Nutrients, 15(1), 70. https://doi.org/10.3390/nu15010070