
CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases


Abstract
:1. Introduction: Canonical and Non-Canonical Activities of All-Trans Retinoic Acid (atRA)
2. CRABP1-Signalsomes
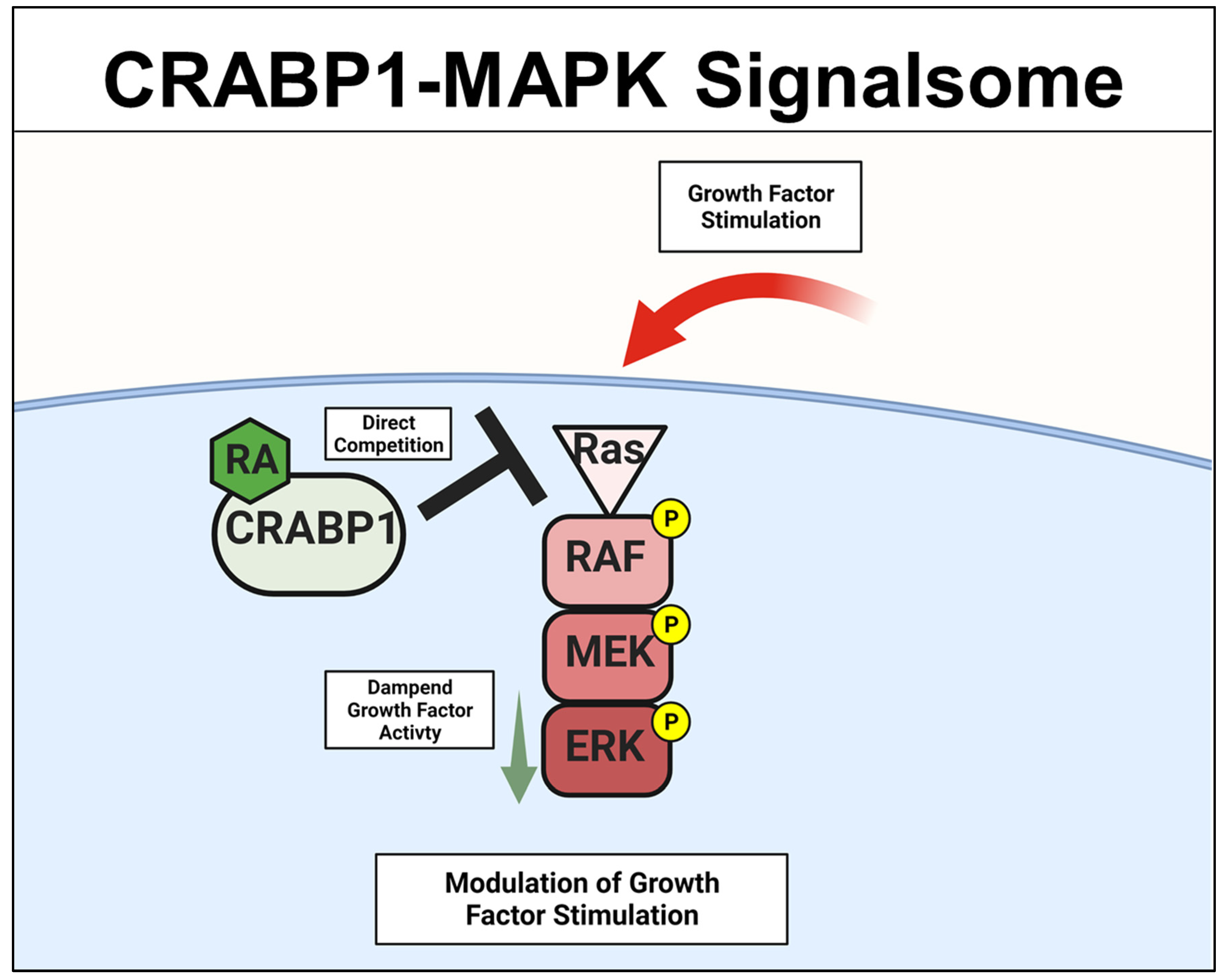
2.1. CRABP1-MAPK (RAF-MEK-ERK) Signalsome in Stem Cells, Cancers
2.2. Crabp1-MAPK Signalsome in Metabolism and Immunity
2.3. CRABP1-CaMKII Signalsome in Cardiomyocytes and Motor Neurons (MNs)
2.3.1. CRABP1-CaMKII Signalsome in Cardiomyocytes
2.3.2. Crabp1-CaMKII Signalsome in MNs
3. Crabp1 in Two Common Human Diseases: Cancer and Neurodegeneration
3.1. CRABP1 in Cancers
3.2. CRABP1 in Neurodegeneration
3.3. CRABP1 in Rare Human Diseases
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanumihardjo, S.A.; Russell, R.M.; Stephensen, C.B.; Gannon, B.M.; Craft, N.E.; Haskell, M.J.; Lietz, G.; Schulze, K.; Raiten, D.J. Biomarkers of nutrition for development (BOND)-vitamin A review. J. Nutr. 2016, 146, 1816S–1848S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G. Retinoic acid as cause of cell proliferation or cell growth inhibition depending on activation of one of two different nuclear receptors. Nutr. Rev. 2008, 66, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, N. Between death and survival: Retinoic acid in regulation of apoptosis. Annu. Rev. Nutr. 2010, 30, 201–217. [Google Scholar] [CrossRef]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.-S.D.T.; Mannino, S.; Milunsky, A. Teratogenicity of High Vitamin A Intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Shenefelt, R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: Relation to dose and stage at treatment. Teratology 5:103-18. 1972. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin a deficiency. Effects of restoration of vitamin a at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef]
- Duong, V.; Rochette-Egly, C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Topletz, A.R.; Thatcher, J.E.; Zelter, A.; Lutz, J.D.; Tay, S.; Nelson, W.L.; Isoherranen, N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 2012, 83, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topletz, A.R.; Tripathy, S.; Foti, R.S.; Shimshoni, J.A.; Nelson, W.L.; Isoherranen, N. Induction of CYP26A1 by metabolites of retinoic acid: Evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Mol. Pharmacol. 2015, 87, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L. Functions of intracellular retinoid binding-proteins. Subcell. Biochem. 2016, 81, 21–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol Ther 2017, 173, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevison, F.; Jing, J.; Tripathy, S.; Isoherranen, N. Role of Retinoic Acid-Metabolizing Cytochrome P450s, CYP26, in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 373–412. [Google Scholar]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, A.; Petrescu, A.D.; Xiong, Y.; Noys, N. Nuclear translocation of cellular retinoic acid-binding protein II is regulated by retinoic acid-controlled SUMOylation. J. Biol. Chem. 2011, 286, 42749–42757. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Ho, P.-C.; Huq, M.M.; Ha, S.G.; Park, S.W.; Khan, A.A.; Tsai, N.-P.; Wei, L.-N. Retinoic acid-stimulated sequential phosphorylation, PML recruitment, and SUMOylation of nuclear receptor TR2 to suppress Oct4 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 11424–11429. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-S.S.; Huang, W.-H.H.; Park, S.W.; Persaud, S.D.; Hung, C.-H.H.; Ho, P.-C.C.; Wei, L.-N.N. Promyelocytic leukemia protein in retinoic acid-induced chromatin remodeling of Oct4 gene promoter. Stem Cells 2011, 29, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persaud, S.D.; Lin, Y.-W.; Wu, C.-Y.; Kagechika, H.; Wei, L.-N. Cellular retinoic acid binding protein I mediates rapid non-canonical activation of ERK1/2 by all-trans retinoic acid. Cell. Signal. 2013, 25, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Persaud, S.D.; Wei, L.N. Retinoic Acid Induces Ubiquitination-Resistant RIP140/LSD1 Complex to Fine-Tune Pax6 Gene in Neuronal Differentiation. Stem Cells 2016, 34, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persaud, S.D.; Park, S.W.; Ishigami-Yuasa, M.; Koyano-Nakagawa, N.; Kagechika, H.; Wei, L.N. All trans-retinoic acid analogs promote cancer cell apoptosis through non-genomic Crabp1 mediating ERK1/2 phosphorylation. Sci. Rep. 2016, 6, 22396. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Persaud, S.D.; Nhieu, J.; Wei, L.N. Cellular Retinoic Acid-Binding Protein 1 Modulates Stem Cell Proliferation to Affect Learning and Memory in Male Mice. Endocrinology 2017, 158, 3004–3014. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Persaud, S.D.; Ogokeh, S.; Meyers, T.A.; Townsend, D.W.; Wei, L.N. CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J. Endocrinol. 2018, 236, 151–165. [Google Scholar] [CrossRef]
- Park, S.W.; Nhieu, J.; Lin, Y.W.; Wei, L.N. All-trans retinoic acid attenuates isoproterenol-induced cardiac dysfunction through Crabp1 to dampen CaMKII activation. Eur. J. Pharmacol. 2019, 858, 172485. [Google Scholar] [CrossRef]
- Lin, Y.W.; Park, S.W.; Lin, Y.L.; Burton, F.H.; Wei, L.N. Cellular retinoic acid binding protein 1 protects mice from high-fat diet-induced obesity by decreasing adipocyte hypertrophy. Int. J. Obes. 2020, 44, 466–474. [Google Scholar] [CrossRef]
- Wook Park, S.; Nhieu, J.; Persaud, S.D.; Miller, M.C.; Xia, Y.; Lin, Y.W.; Lin, Y.L.; Kagechika, H.; Mayo, K.H.; Wei, L.N. A new regulatory mechanism for Raf kinase activation, retinoic acid-bound Crabp1. Sci. Rep. 2019, 9, 10929. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.Y.L.W.; Nhieu, J.; Zhang, X.; Wei, L.N. Sonic hedgehog-gli1 signaling and cellular retinoic acid binding protein 1 gene regulation in motor neuron differentiation and diseases. Int. J. Mol. Sci. 2020, 21, 4125. [Google Scholar] [CrossRef]
- Lin, Y.W.; Nhieu, J.; Wei, C.W.; Lin, Y.L.; Kagechika, H.; Wei, L.N. Regulation of exosome secretion by cellular retinoic acid binding protein 1 contributes to systemic anti-inflammation. Cell Commun. Signal. 2021, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Wei, C.W.; Lerdall, T.A.; Nhieu, J.; Wei, L.N. Crabp1 modulates hpa axis homeostasis and anxiety-like behaviors by altering fkbp5 expression. Int. J. Mol. Sci. 2021, 22, 12240. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.Y.-W.; Nhieu, J.; Liu, P.-Y.; Le, G.; Lee, D.J.; Wei, C.-W.; Lin, Y.-L.Y.-W.; Oh, S.-H.; Lowe, D.; Wei, L.-N. CRABP1-CaMKII-Agrn regulates the maintenance of neuromuscular junction in spinal motor neuron. Cell Death Differ. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ong, D.E.; Chytil, F. Cellular retinoic acid-binding protein from rat testis. Purification and characterization. J. Biol. Chem. 1978, 253, 4551–4554. [Google Scholar] [CrossRef]
- Fiorella, P.D.; Giguère, V.; Napoli, J.L. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli: Characterization and comparison to cellular retinoic acid-binding protein (type I). J. Biol. Chem. 1993, 268, 21545–21552. [Google Scholar] [CrossRef]
- Norris, A.W.; Cheng, L.; Giguère, V.; Rosenberger, M.; Li, E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta 1994, 1209, 10–18. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Yan, H. Structure-function relationships of cellular retinoic acid-binding proteins: Quantitative analysis of the ligand binding properties of the wild-type proteins and site-directed mutants. J. Biol. Chem. 1997, 272, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.N.; Chang, L.; Lee, C.H. Studies of over-expressing cellular retinoic acid binding protein-I in cultured cells and transgenic mice. Transgenics 1997, 2, 201–209. [Google Scholar]
- Wei, L.N.; Chang, L.; Hu, X. Studies of the type I cellular retinoic acid-binding protein mutants and their biological activities. Mol. Cell. Biochem. 1999, 200, 69–76. [Google Scholar] [CrossRef]
- Nelson, C.H.; Peng, C.C.; Lutz, J.D.; Yeung, C.K.; Zelter, A.; Isoherranen, N. Direct protein–protein interactions and substrate channeling between cellular retinoic acid binding proteins and CYP26B1. FEBS Lett. 2016, 590, 2527–2535. [Google Scholar] [CrossRef] [Green Version]
- Eller, M.S.; Oleksiak, M.F.; McQuaid, T.J.; McAfee, S.G.; Gilchrest, B.A. The molecular cloning and expression of two CRABP cDNAs from human skin. Exp. Cell Res. 1992, 198, 328–336. [Google Scholar] [CrossRef]
- Tang, Z.; Li, Y.; Wan, P.; Li, X.; Zhao, S.; Liu, B.; Fan, B.; Zhu, M.; Yu, M.; Li, K. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 2007, 8, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.M.; Curley, R.W. Affinity purification of retinoic acid-binding proteins using immobilized 4-(2-Hydroxyethoxy)retinoic acid. Protein Expr. Purif. 1990, 1, 63–69. [Google Scholar] [CrossRef]
- Stoner, C.M.; Gudas, L.J. Mouse Cellular Retinoic Acid Binding Protein: Cloning, Complementary DNA Sequence, and Messenger RNA Expression during the Retinoic Acid-induced Differentiation of F9 Wild Type and RA-3-10 Mutant Teratocarcinoma Cells. Cancer Res. 1989, 49, 1497–1504. [Google Scholar] [PubMed]
- Nilsson, M.H.L.; Spurr, N.K.; Saksena, P.; Busch, C.; Nordlinder, H.; Peterson, P.A.; Rask, L.; Sundelin, J. Isolation and characterization of a cDNA clone corresponding to bovine cellular retinoic-acid-binding protein and chromosomal localization of the corresponding human gene. Eur. J. Biochem. 1988, 173, 45–51. [Google Scholar] [CrossRef]
- Wei, L.-N. Non-canonical activity of retinoic acid in epigenetic control of embryonic stem cell. Transcription 2013, 4, 158–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.N. Cellular retinoic acid binding proteins: Genomic and non-genomic functions and their regulation. Subcell. Biochem. 2016, 81, 163–178. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T.; LIU, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Guidez, F.; Parks, S.; Wong, H.; Jovanovic, J.V.; Mays, A.; Gilkes, A.F.; Mills, K.I.; Guillemin, M.-C.C.; Hobbs, R.M.; Pandolfi, P.P.; et al. RARalpha-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 18694–18699. [Google Scholar] [CrossRef] [Green Version]
- Pfoertner, S.; Goelden, U.; Hansen, W.; Toepfer, T.; Geffers, R.; Ukena, S.N.; von Knobloch, R.; Hofmann, R.; Buer, J.; Schrader, A.J. Cellular retinoic acid binding protein I: Expression and functional influence in renal cell carcinoma. Tumour Biol. 2005, 26, 313–323. [Google Scholar] [CrossRef]
- Tanaka, K.; Imoto, I.; Inoue, J.; Kozaki, K.; Tsuda, H.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Iwai, T.; Kawano, T.; et al. Frequent methylation-associated silencing of a candidate tumor-suppressor, CRABP1, in esophageal squamous-cell carcinoma. Oncogene 2007, 26, 6456–6468. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Ueda, Y.; Matsuzaki, S.; Miyatake, T.; Yoshino, K.; Fujita, M.; Nomura, T.; Enomoto, T.; Kimura, T. CRABP1-reduced expression is associated with poorer prognosis in serous and clear cell ovarian adenocarcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Lothe, R.A.; Ahlquist, T.; Silins, I.; Tropé, C.G.; Micci, F.; Nesland, J.M.; Suo, Z.; Lind, G.E. DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1, and CRABP1 as novel targets. Mol. Cancer 2007, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawthorn, L.; Stein, L.; Varma, R.; Wiseman, S.; Loree, T.; Tan, D.F. TIMP1 and SERPIN-A overexpression and TFF3 and CRABP1 underexpression as biomarkers for papillary thyroid carcinoma. Head Neck 2004, 26, 1069–1083. [Google Scholar] [CrossRef]
- Celestino, R.; Nome, T.; Pestana, A.; Hoff, A.M.; Gonçalves, A.P.; Pereira, L.; Cavadas, B.; Eloy, C.; Bjøro, T.; Sobrinho-Simões, M.; et al. CRABP1, C1QL1 and LCN2 are biomarkers of differentiated thyroid carcinoma, and predict extrathyroidal extension. BMC Cancer 2018, 18, 68. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; de la Chapelle, A.; Pellegata, N.S. Hypermethylation, but not LOH, is associated with the low expression of MT1G and CRABP1 in papillary thyroid carcinoma. Int. J. Cancer 2003, 104, 735–744. [Google Scholar] [CrossRef]
- Lind, G.E.; Kleivi, K.; Meling, G.I.; Teixeira, M.R.; Thiis-Evensen, E.; Rognum, T.O.; Lothe, R.A. ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Cell. Oncol. 2006, 28, 259–272. [Google Scholar] [CrossRef]
- Won, J.Y.; Nam, E.C.; Yoo, S.J.; Kwon, H.J.; Um, S.J.; Han, H.S.; Kim, S.H.; Byun, Y.; Kim, S.Y. The effect of cellular retinoic acid binding protein-I expression on the CYP26-mediated catabolism of all-trans retinoic acid and cell proliferation in head and neck squamous cell carcinoma. Metabolism 2004, 53, 1007–1012. [Google Scholar] [CrossRef]
- Kainov, Y.; Favorskaya, I.; Delektorskaya, V.; Chemeris, G.; Komelkov, A.; Zhuravskaya, A.; Trukhanova, L.; Zueva, E.; Tavitian, B.; Dyakova, N.; et al. CRABP1 provides high malignancy of transformed mesenchymal cells and contributes to the pathogenesis of mesenchymal and neuroendocrine tumors. Cell Cycle 2014, 13, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Choi, N.; Park, J.; Lee, J.S.; Yoe, J.; Park, G.Y.; Kim, E.; Jeon, H.; Cho, Y.M.; Roh, T.Y.; Lee, Y. miR-93/miR-106b/miR-375-CIC-CRABP1: A novel regulatory axis in prostate cancer progression. Oncotarget 2015, 6, 23533–23547. [Google Scholar] [CrossRef]
- Liu, R.Z.; Garcia, E.; Glubrecht, D.D.; Poon, H.Y.; Mackey, J.R.; Godbout, R. CRABP1 is associated with a poor prognosis in breast cancer: Adding to the complexity of breast cancer cell response to retinoic acid. Mol. Cancer 2015, 14, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, I.; Wei, L.N. All-trans retinoic acid as a versatile cytosolic signal modulator mediated by CRABP1. Int. J. Mol. Sci. 2019, 20, 3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; Noy, N. All-trans-Retinoic Acid Represses Obesity and Insulin Resistance by Activating both Peroxisome Proliferation-Activated Receptor β/δ and Retinoic Acid Receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyakumar, S.M.; Vajreswari, A.; Sesikeran, B.; Giridharan, N.V. Vitamin A supplementation induces adipose tissue loss through apoptosis in lean but not in obese rats of the WNIN/Ob strain. J. Mol. Endocrinol. 2005, 35, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamei, Y.; Kawada, T.; Mizukami, J.; Sugimoto, E. The prevention of adipose differentiation of 3T3-L1 cells caused by retinoic acid is elicited through retinoic acid receptor alpha. Life Sci. 1994, 55, PL307–PL312. [Google Scholar] [CrossRef]
- Murray, T.; Russell, T.R. Inhibition of adipose conversion in 3T3-L2 cells by retinoic acid. J. Supramol. Cell. Biochem. 1980, 14, 255–266. [Google Scholar] [CrossRef]
- Lee, B.; Ho, P.-C.; Wei, L.-N. Nuclear Receptor-Interacting Protein 1 (NRIP1). In Encyclopedia of Signaling Molecules; Springer: Cham, Switzerland, 2018; pp. 3606–3616. [Google Scholar]
- Erickson, J.R. Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 2014, 5 APR, 59. [Google Scholar] [CrossRef] [Green Version]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Karandur, D.; Kuriyan, J. Structural insights into the regulation of Ca2+/calmodulin-dependent protein kinase II (Camkii). Cold Spring Harb. Perspect. Biol. 2020, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; Chao, L.H.; Schulman, H.; Kuriyan, J. Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Curr. Opin. Struct. Biol. 2013, 23, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P. CaMKII: The molecular villain that aggravates cardiovascular disease. Exp. Ther. Med. 2017, 13, 815–820. [Google Scholar] [CrossRef] [Green Version]
- Ashpole, N.M.; Hudmon, A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell. Neurosci. 2011, 46, 720–730. [Google Scholar] [CrossRef]
- Garg, M.; Khanna, D. Exploration of pharmacological interventions to prevent isoproterenol-induced myocardial infarction in experimental models. Ther. Adv. Cardiovasc. Dis. 2014, 8, 155–169. [Google Scholar] [CrossRef]
- Nichtova, Z.; Novotova, M.; Kralova, E.; Stankovicova, T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen. Physiol. Biophys. 2012, 31, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Martinez-Pena, Y. Valenzuela, I.; Mouslim, C.; Akaaboune, M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J. Neurosci. 2010, 30, 12455–12465. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.H.; Popova, E.; Thomas, U.; Griffith, L.C.; Budnik, V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 1999, 98, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, J.M.; Hodge, J.J.L. CASK regulates CaMKII autophosphorylation in neuronal growth, calcium signaling, and learning. Front. Mol. Neurosci. 2013, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, E.R. Role of Ca 2+/calmodulin-dependent kinases in skeletal muscle plasticity. J. Appl. Physiol. 2005, 99, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.J.; Kiens, B.; Richter, E.A. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J. Physiol. 2006, 574, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64. [Google Scholar] [CrossRef]
- Glass, D.J.; Bowen, D.C.; Stitt, T.N.; Radziejewski, C.; Bruno, J.A.; Ryan, T.E.; Gies, D.R.; Shah, S.; Mattsson, K.; Burden, S.J.; et al. Agrin acts via a MuSK receptor complex. Cell 1996, 85, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 236–251. [Google Scholar] [CrossRef]
- Rizzo, F.; Nizzardo, M.; Vashisht, S.; Molteni, E.; Melzi, V.; Taiana, M.; Salani, S.; Santonicola, P.; Di Schiavi, E.; Bucchia, M.; et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain 2019, 142, 276–294. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kino, Y. Expression profiles of RNA-Seq-based grey matter-specific genes versus white matter-specific genes in grey matter lesions of multiple sclerosis. Clin. Exp. Neuroimmunol. 2015, 6, 289–298. [Google Scholar] [CrossRef]
- Scholtissek, B.; Zahn, S.; Maier, J.; Klaeschen, S.; Braegelmann, C.; Hoelzel, M.; Bieber, T.; Barchet, W.; Wenzel, J. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbari, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gonzalez, J.; Cueto, I.; Franks, A.G.; Krueger, J.G. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J. Investig. Dermatol. 2014, 134, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regazzetti, C.; Joly, F.; Marty, C.; Rivier, M.; Mehul, B.; Reiniche, P.; Mounier, C.; Rival, Y.; Piwnica, D.; Cavalié, M.; et al. Transcriptional analysis of vitiligo skin reveals the alteration of WNT pathway: A promising target for repigmenting vitiligo patients. J. Investig. Dermatol. 2015, 135, 3105–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, I.; Silva, P.; da Mata, S.; Magro, F.; Carneiro, F.; Peixoto, A.; Silva, M.; Sousa, H.T.; Roseira, J.; Parra, J.; et al. Methylation patterns in dysplasia in inflammatory bowel disease patients. Scand. J. Gastroenterol. 2020, 55, 646–655. [Google Scholar] [CrossRef]
- Kim, S.K.; Yoo, J.I.; Cho, B.K.; Hong, S.J.; Kim, Y.K.; Moon, J.A.; Kim, J.H.; Chung, Y.N.; Wang, K.C. Elevation of CRABP-I in the Cerebrospinal Fluid of Patients with Moyamoya Disease. Stroke 2003, 34, 2835–2841. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.; Sullivan, K.A.; Pande, M.; Hong, Y.; Sima, A.A.F.; Jagadish, H.V.; Kretzler, M.; Feldman, E.L. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain 2011, 134, 3222–3235. [Google Scholar] [CrossRef]
- Carr, A.; Samaras, K.; Chisholm, D.J.; Cooper, D.A. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 1998, 351, 1881–1883. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 269. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, P.; Lindsey, B.W.; Leopold, C.; Rafuse, V.F. Easy and Rapid Differentiation of Embryonic Stem Cells into Functional Motoneurons Using Sonic Hedgehog-Producing Cells. Stem Cells 2007, 25, 1697–1706. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya Disease and Moyamoya Syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreeland, A.C.; Yu, S.; Levi, L.; de Barros Rossetto, D.; Noy, N. Transcript Stabilization by the RNA-Binding Protein HuR Is Regulated by Cellular Retinoic Acid-Binding Protein 2. Mol. Cell. Biol. 2014, 34, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Vreeland, A.C.; Noy, N. RNA-binding protein HuR regulates nuclear import of protein. J. Cell Sci. 2016, 129, 4025–4033. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.S.; Noy, N. Direct Channeling of Retinoic Acid between Cellular Retinoic Acid-Binding Protein II and Retinoic Acid Receptor Sensitizes Mammary Carcinoma Cells to Retinoic Acid-Induced Growth Arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfanos, C.E.; Ehlert, R.; Gollnick, H. The Retinoids: A Review of their Clinical Pharmacology and Therapeutic Use. Drugs 1987, 34, 459–503. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun. 2020, 11, 4265. [Google Scholar] [CrossRef]
- Bremner, J.D.; Shearer, K.D.; McCaffery, P.J. Retinoic acid and affective disorders: The evidence for an association. J. Clin. Psychiatry 2012, 73, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Wood, E. The Retinoids, Volumes I and II. Biochem. Educ. 1985, 13, 147. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Hattan, D.G.; Jenkins, M.Y.; McDonald, J.T.; Sundaresan, P.R.; Wilkening, V.L. Evaluation of vitamin A toxicity. Am. J. Clin. Nutr. 1990, 52, 183–202. [Google Scholar] [CrossRef]
- Bendich, A.; Langseth, L. Safety of vitamin A. Am. J. Clin. Nutr. 1989, 49, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Yob, E.H.; Pochi, P.E. Side Effects and Long-term Toxicity of Synthetic Retinoids. Arch. Dermatol. 1987, 123, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Thielitz, A.; Gollnick, H. Topical retinoids in acne vulgaris: Update on efficacy and safety. Am. J. Clin. Dermatol. 2008, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-B.B.; Zhang, J.; Wang, Z.-Y.Y.; Chen, S.-J.J.; Chen, Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 959–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, C.W.E.; Cornish, K.A.S.; Whiting, A.; Pohl, E. Structure-functional relationship of cellular retinoic acid-binding proteins i and II interacting with natural and synthetic ligands. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 164–175. [Google Scholar] [CrossRef]
- Zheng, C.; Zhu, Y.; Liu, Q.; Luo, T.; Xu, W. Maprotiline Suppresses Cholesterol Biosynthesis and Hepatocellular Carcinoma Progression Through Direct Targeting of CRABP1. Front. Pharmacol. 2021, 12, 1243. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | CRABP1 Status | Reference |
|---|---|---|
| Breast Cancer | Over-Expression | [61] |
| Prostate Cancer | Over-Expression | [60] |
| Mesenchymal & Neuroendocrine Tumors | Over-Expression | [59] |
| Head and Neck Squamous Cell Carcinoma (HNSCC) | Over-Expression | [58] |
| Colorectal Cancer | Silenced (Promoter Hypermethylation) | [57] |
| Thyroid Cancer | Silenced (Promoter Hypermethylation) | [56] |
| Reduced Expression | [54] | |
| Ovarian Cancer | Silenced (Promoter Hypermethylation) | [53] |
| Reduced Expression | [52] | |
| Esophageal Squamous-Cell Carcinoma (ESCC) | Silenced (Promoter Hypermethylation) | [51] |
| Renal Cell Carcinoma | Reduced Expression | [50] |
| Acute myeloid leukemia (AML) | Silenced (Promoter Hypermethylation) | [49] |
| Neurodegenerative Diseases | CRABP1 Status | Reference |
| Amyotrophic Lateral Sclerosis (ALS) | Reduced Expression | [88] |
| Spinal Muscular Atrophy (SMA) | Reduced Expression | [89] |
| Late-Stage Age-Related Macular Degeneration (AMD) | Reduced Expression | [90] |
| Immune Disorders | CRABP1 Status | Reference |
| Multiple Sclerosis | Reduced Expression | [91] |
| Cutaneous Lupus Erythematosus (CLE) | Reduced Expression | [92] # E-MTAB-5542 |
| Psoriasis | Reduced Expression | [93] # E-GEOD-52471 |
| Vitiligo | Reduced Expression | [94] # E-GEOD-65127 |
| Inflammatory Bowel Disease (IBD) | Silenced (Promoter Hypermethylation) | [95] |
| Other Diseases | CRABP1 Status | Reference |
| Moyamoya Disease (MMD) | Increased Protein Level | [96] |
| Diabetic Neuropathy | Increased Expression | [97] |
| HIV Therapy-Associated Lipodystrophy and Metabolic Syndrome | Inhibited Function | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nhieu, J.; Lin, Y.-L.; Wei, L.-N. CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases. Nutrients 2022, 14, 1528. https://doi.org/10.3390/nu14071528
Nhieu J, Lin Y-L, Wei L-N. CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases. Nutrients. 2022; 14(7):1528. https://doi.org/10.3390/nu14071528
Chicago/Turabian StyleNhieu, Jennifer, Yu-Lung Lin, and Li-Na Wei. 2022. "CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases" Nutrients 14, no. 7: 1528. https://doi.org/10.3390/nu14071528
APA StyleNhieu, J., Lin, Y.-L., & Wei, L.-N. (2022). CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases. Nutrients, 14(7), 1528. https://doi.org/10.3390/nu14071528