
Retinoids in the Pathogenesis and Treatment of Liver Diseases


Abstract

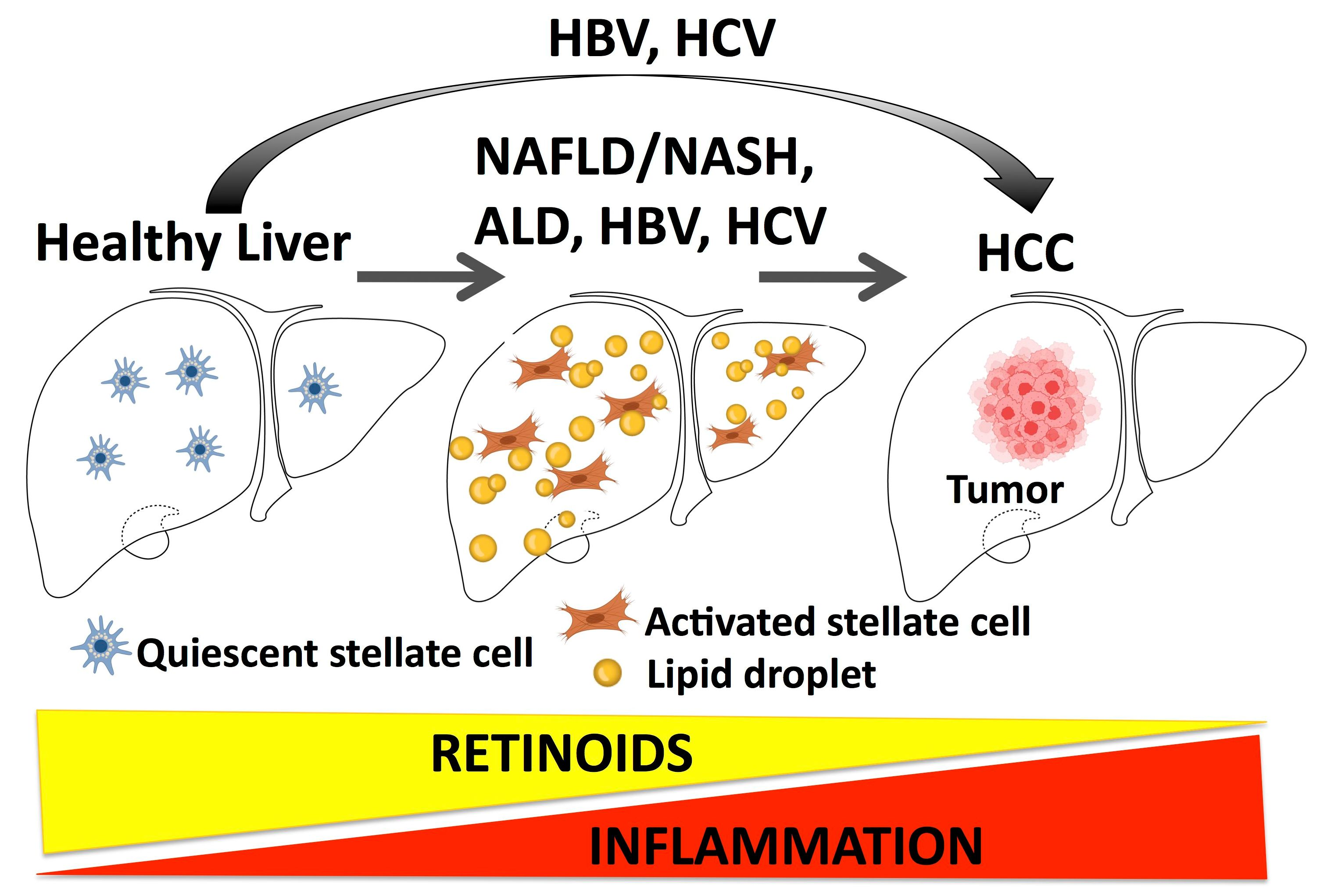{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Retinoids, Vitamin A, and Retinoic Acid Receptors
1.1. Actions of the Retinoic Acid Receptors and Retinoic Acid (RA) in Transcriptional Activation
1.2. The Actions of RARs in Sequence-Specific Translational Control
1.3. Endogenous Ligands of the RARs
2. Effects of Vitamin A Deficiency in the Liver
3. RARs Are Required to Prevent Liver Disease (Steatohepatitis) and Hepatocellular Carcinoma in a Mouse Model
4. Non-Alcohol-Associated Fatty Liver Disease
4.1. NAFLD Is Associated with Reductions in Hepatic Retinoids—A Possible Inverse Relationship between Intrahepatic Triglyceride Levels and Retinoids
4.1.1. De Novo Lipogenesis
4.1.2. Fatty Acid Oxidation
4.1.3. Free Fatty Acid Influx to the Liver and Export of Lipids from the Liver
4.2. Targeting Hepatic Stellate Cell (HSC) Activation and Fibrogenesis
4.3. Targeting Inflammation in NAFLD
4.4. Targeting the Effects of the Gut Microbiome on NAFLD Progression
5. The Roles of Retinoids in the Pathogenesis and Treatment of Alcohol-Associated Liver Disease (ALD)
5.1. Chronic Alcohol Abuse Is Associated with Depletion of Liver Retinoids
5.2. Mechanisms of Hepatic Retinoid Depletion in ALD
5.3. Effects of Retinoids on ALD
6. Retinoids in Liver Cancer
6.1. Association of Hepatitis B and C Viruses with Abnormal RARβ Function
6.2. Aberrant Regulation of Retinoid Metabolism and RARβ in Hepatocellular Carcinoma
6.3. Therapeutic Potential of Retinoids in HCC
7. General Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2010, 226, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e19. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, M.; Meyer, K.F.; Yin, J.; Duester, G. Discovery of genes required for body axis and limb formation by global identification of retinoic acid-regulated epigenetic marks. PLoS Biol. 2020, 18, e3000719. [Google Scholar] [CrossRef] [PubMed]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418.e7. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Huang, J.; Weinstein, S.J.; Yu, K.; Männistö, S.; Albanes, D. Association between serum retinol and overall and cause-specific mortality in a 30-year prospective cohort study. Nat. Commun. 2021, 12, 6418. [Google Scholar] [CrossRef]
- Tang, X.-H.; Gudas, L.J. Retinoids, Retinoic Acid Receptors, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 345–364. [Google Scholar] [CrossRef]
- Gudas, L.J. Synthetic Retinoids beyond Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 155–175. [Google Scholar] [CrossRef]
- Blaner, W.S.; Li, Y.; Brun, P.-J.; Yuen, J.J.; Lee, S.-A.; Clugston, R.D. Vitamin A Absorption, Storage and Mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar] [CrossRef]
- Belyaeva, O.V.; Adams, M.K.; Popov, K.M.; Kedishvili, N.Y. Generation of Retinaldehyde for Retinoic Acid Biosynthesis. Biomolecules 2019, 10, 5. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.-Å.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Langston, A.W.; Gudas, L.J. Identification of a retinoic acid responsive enhancer 3′ of the murine homeobox gene Hox-1.6. Mech. Dev. 1992, 38, 217–227. [Google Scholar] [CrossRef]
- Langston, A.W.; Thompson, J.R.; Gudas, L.J. Retinoic Acid-responsive Enhancers Located 3′ of the Hox A and Hox B Homeobox Gene Clusters. J. Biol. Chem. 1997, 272, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Chen, S.W.; Langston, A.W.; Gudas, L.J. A conserved retinoic acid responsive element in the murine Hoxb-1 gene is required for expression in the developing gut. Development 1998, 125, 3235–3246. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Gavalas, A.; Marshall, H.; Ariza-McNaughton, L.; Rijli, F.M.; Chambon, P.; Krumlauf, R. Genetic interactions between Hoxa1 and Hoxb1 reveal new roles in regulation of early hindbrain patterning. Development 1998, 125, 1025–1036. [Google Scholar] [CrossRef]
- Marshall, H.; Studer, M.; Pöpperl, H.; Aparicio, S.; Kuroiwa, A.; Brenner, S.; Krumlauf, R. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature 1994, 370, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Vasios, G.W.; Gold, J.D.; Petkovich, M.; Chambon, P.; Gudas, L.J. A retinoic acid-responsive element is present in the 5′ flanking region of the laminin B1 gene. Proc. Natl. Acad. Sci. USA 1989, 86, 9099–9103. [Google Scholar] [CrossRef] [PubMed]
- Vasios, G.; Mader, S.; Gold, J.D.; Leid, M.; Lutz, Y.; Gaub, M.P.; Chambon, P.; Gudas, L. The late retinoic acid induction of laminin B1 gene transcription involves RAR binding to the responsive element. EMBO J. 1991, 10, 1149–1158. [Google Scholar] [CrossRef]
- De The, H.; Vivanco, M.; Tiollais, P.; Stunnenberg, H.; Dejean, A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 1990, 343, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, M.L.; Kraus, W.L. Transcriptional activation by nuclear receptors. Essays Biochem. 2004, 40, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Burris, T.P. Small Molecule Modulation of Nuclear Receptor Conformational Dynamics: Implications for Function and Drug Discovery. Mol. Pharmacol. 2013, 83, 1–8. [Google Scholar] [CrossRef]
- Hudson, W.H.; Youn, C.; Ortlund, E.A. The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 2012, 20, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Ganti, K.P.; Chambon, P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc. Natl. Acad. Sci. USA 2015, 113, E635–E643. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Penco, S.; Ostrowski, J.; Balaguer, P.; Pons, M.; E Starrett, J.; Reczek, P.; Chambon, P.; Gronemeyer, H. RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J. 1995, 14, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Fanjul, A.; Dawson, M.I.; Hobbs, P.D.; Jong, L.; Cameron, J.F.; Harlev, E.; Graupner, G.; Lu, X.-P.; Pfahl, M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994, 372, 107–111. [Google Scholar] [CrossRef]
- Huq, M.D.; Tsai, N.P.; Khan, S.A.; Wei, L.N. Lysine trimethylation of retinoic acid receptor-alpha: A novel means to regulate receptor function. Mol. Cell Proteom. 2007, 6, 677–688. [Google Scholar] [CrossRef]
- Giannì, M.; Bauer, A.; Garattini, E.; Chambon, P.; Rochette-Egly, C. Phosphorylation by p38MAPK and recruitment of SUG-1 are required for RA-induced RARγ degradation and transactivation. EMBO J. 2002, 21, 3760–3769. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A Rapid, Extensive, and Transient Transcriptional Response to Estrogen Signaling in Breast Cancer Cells. Cell 2011, 145, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lee, J.H.; Zhang, Z.; De La Rosa, R.; Bi, M.; Tan, Y.; Liao, Y.; Hong, J.; Du, B.; Wu, Y.; et al. Enhancer RNAs Mediate Estrogen-Induced Decommissioning of Selective Enhancers by Recruiting ERα and Its Cofactor. Cell Rep. 2020, 31, 107803. [Google Scholar] [CrossRef]
- Nouspikel, T. DNA Repair in Mammalian Cells. Cell. Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER Factors Are Recruited to Active Promoters and Facilitate Chromatin Modification for Transcription in the Absence of Exogenous Genotoxic Attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef]
- Poon, M.M.; Chen, L. Retinoic acid-gated sequence-specific translational control by RARα. Proc. Natl. Acad. Sci. USA 2008, 105, 20303–20308. [Google Scholar] [CrossRef]
- Hsu, Y.-T.; Li, J.; Wu, D.; Südhof, T.C.; Chen, L. Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning. Proc. Natl. Acad. Sci. USA 2019, 116, 7113–7122. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lau, A.; Arendt, K.; Chen, L. FMRP Interacts with RARα in Synaptic Retinoic Acid Signaling and Homeostatic Synaptic Plasticity. Int. J. Mol. Sci. 2021, 22, 6579. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, W.W.M.; Folkers, G.E.; de Jonge, W.J.; Verdegem, P.J.E.; de Laat, S.W.; Lugtenburg, J.; Hendriks, H.F.J.; van der Saag, P.T.; Durston, A.J. Metabolism to a response pathway selective retinoid ligand during axial pattern formation. Proc. Natl. Acad. Sci. USA 1998, 95, 15424–15429. [Google Scholar] [CrossRef] [PubMed]
- Idres, N.; Marill, J.; Flexor, M.A.; Chabot, G.G. Activation of Retinoic Acid Receptor-dependent Transcription by All-trans-retinoic Acid Metabolites and Isomers. J. Biol. Chem. 2002, 277, 31491–31498. [Google Scholar] [CrossRef] [PubMed]
- Topletz, A.R.; Tripathy, S.; Foti, R.S.; Shimshoni, J.A.; Nelson, W.L.; Isoherranen, N. Induction of CYP26A1 by Metabolites of Retinoic Acid: Evidence That CYP26A1 Is an Important Enzyme in the Elimination of Active Retinoids. Mol. Pharmacol. 2015, 87, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Gudas, L.J. An Analysis of Retinoic Acid-induced Gene Expression and Metabolism in AB1 Embryonic Stem Cells. J. Biol. Chem. 1996, 271, 14971–14980. [Google Scholar] [CrossRef]
- Schönberger, K.; Obier, N.; Romero-Mulero, M.C.; Cauchy, P.; Mess, J.; Pavlovich, P.V.; Zhang, Y.W.; Mitterer, M.; Rettkowski, J.; Lalioti, M.-E.; et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 2021, 29, 131–148.e10. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, G.J.; Gudas, L.J. Early retinoic acid-induced F9 teratocarcinoma stem cell gene ERA-1: Alternate splicing creates transcripts for a homeobox-containing protein and one lacking the homeobox. Mol. Cell. Biol. 1988, 8, 3906–3917. [Google Scholar] [CrossRef] [PubMed]
- Langton, S.; Gudas, L.J. CYP26A1 knockout embryonic stem cells exhibit reduced differentiation and growth arrest in response to retinoic acid. Dev. Biol. 2008, 315, 331–354. [Google Scholar] [CrossRef][Green Version]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Hogarth, C.; Snyder, J.M.; Palau, L.; Topping, T.; Huang, W.; Czuba, L.; LaFrance, J.; Ghiaur, G.; Isoherranen, N. The retinoic acid hydroxylase Cyp26a1 has minor effects on postnatal vitamin A homeostasis, but is required for exogenous atRA clearance. J. Biol. Chem. 2019, 294, 11166–11179. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.M.; Zhong, G.; Hogarth, C.; Huang, W.; Topping, T.; LaFrance, J.; Palau, L.; Czuba, L.C.; Griswold, M.; Ghiaur, G.; et al. Knockout of Cyp26a1 and Cyp26b1 during postnatal life causes reduced lifespan, dermatitis, splenomegaly, and systemic inflammation in mice. FASEB J. 2020, 34, 15788–15804. [Google Scholar] [CrossRef] [PubMed]
- Baybutt, R.C.; Hu, L.; Molteni, A. Vitamin A deficiency injures lung and liver parenchyma and impairs function of rat type II pneumocytes. J. Nutr. 2000, 130, 1159–1165. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Singh, V.N.; Ramachandran, C.K. Decreased Hepatic Gluconeogenesis in Vitamin A-Deficient Rats. Exp. Biol. Med. 1981, 167, 248–253. [Google Scholar] [CrossRef]
- Seifert, W.F.; Bosma, A.; Brouwer, A.; Hendriks, H.F.; Roholl, P.J.; van Leeuwen, R.E.; van Thiel-De Ruiter, G.C.F.; Ingrid Seifert-Bock, I.; Knook, D.L. Vitamin A deficiency potentiates carbon tetrachloride-induced liver fibrosis in rats. Hepatology 1994, 19, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Barshack, I.; Onaca, N.; Goldberg, I.; Berkovich, Z.; Melzer, E.; Jonas, A.; Reifen, R. Vitamin A deficiency associated with enhanced proliferation of bile duct epithelial cells in the rat. Isr. Med. Assoc. J. IMAJ 2010, 12, 82–86. [Google Scholar] [PubMed]
- Evarts, R.P.; Hu, Z.; Omori, N.; Omori, M.; Marsden, E.R.; Thorgeirsson, S.S. Effect of vitamin A deficiency on the integrity of hepatocytes after partial hepatectomy. Am. J. Pathol. 1995, 147, 699–706. [Google Scholar] [PubMed]
- Shmarakov, I.; Jiang, H.; Yang, K.; Goldberg, I.J.; Blaner, W.S. Hepatic retinoid stores are required for normal liver regeneration. J. Lipid Res. 2013, 54, 893–908. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.-Y.; Boyer, J.L. The role of the retinoid receptor, RAR/RXR heterodimer, in liver physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166085. [Google Scholar] [CrossRef]
- Saitou, M.; Narumiya, S.; Kakizuka, A. Alteration of a single amino acid residue in retinoic acid receptor causes dominant-negative phenotype. J. Biol. Chem. 1994, 269, 19101–19107. [Google Scholar] [CrossRef]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor ? dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic Acid Treatment Enhances Lipid Oxidation and Inhibits Lipid Biosynthesis Capacities in the Liver of Mice. Cell. Physiol. Biochem. 2010, 25, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Petrov, P.; Bonet, M.L.; Ribot, J.; Palou, A. Induction of carnitine palmitoyl transferase 1 and fatty acid oxidation by retinoic acid in HepG2 cells. Int. J. Biochem. Cell Biol. 2012, 44, 2019–2027. [Google Scholar] [CrossRef]
- Tanaka, N.; Kimura, T.; Fujimori, N.; Nagaya, T.; Komatsu, M.; Tanaka, E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J. Gastroenterol. 2019, 25, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. What’s new in NAFLD pathogenesis, biomarkers and treatment? Nat. Rev. Gastroenterol. Hepatol. 2019, 17, 70–71. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, M.D.; Sanyal, A.J. Burden of Disease due to Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. N. Am. 2020, 49, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA J. Am. Med. Assoc. 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Arase, Y.; Tsuji, H.; Fujihara, K.; Saito, K.; Hsieh, S.D.; Tanaka, S.; Kodama, S.; Hara, S.; Sone, H. Metabolically Healthy Obesity, Presence or Absence of Fatty Liver, and Risk of Type 2 Diabetes in Japanese Individuals: Toranomon Hospital Health Management Center Study 20 (TOPICS 20). J. Clin. Endocrinol. Metab. 2014, 99, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Dudek, M.; Knolle, P. Non-alcoholic fatty liver disease: The interplay between metabolism, microbes and immunity. Nat. Metab. 2021, 3, 1596–1607. [Google Scholar] [CrossRef]
- Trasino, S.E.; Tang, X.-H.; Jessurun, J.; Gudas, L.J. Obesity Leads to Tissue, but not Serum Vitamin A Deficiency. Sci. Rep. 2015, 5, 15893. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 10, 29. [Google Scholar] [CrossRef]
- Saeed, A.; Bartuzi, P.; Heegsma, J.; Dekker, D.; Kloosterhuis, N.; de Bruin, A.; Jonker, J.W.; van de Sluis, B.; Faber, K.N. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 309–325.e3. [Google Scholar] [CrossRef] [PubMed]
- Bissig-Choisat, B.; Alves-Bezerra, M.; Zorman, B.; Ochsner, S.A.; Barzi, M.; Legras, X.; Yang, D.; Borowiak, M.; Dean, A.M.; York, R.B.; et al. A human liver chimeric mouse model for non-alcoholic fatty liver disease. JHEP Rep. 2021, 3, 100281. [Google Scholar] [CrossRef]
- Zhong, G.; Kirkwood, J.; Won, K.-J.; Tjota, N.; Jeong, H.-Y.; Isoherranen, N. Characterization of Vitamin A Metabolome in Human Livers with and without Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2019, 370, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Chaves, G.V.; Pereira, S.E.; Saboya, C.J.; Spitz, D.; Rodrigues, C.S.; Ramalho, A. Association between Liver Vitamin A Reserves and Severity of Nonalcoholic Fatty Liver Disease in the Class III Obese Following Bariatric Surgery. Obes. Surg. 2014, 24, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Czuba, L.C.; Wu, X.; Huang, W.; Hollingshead, N.; Roberto, J.B.; Kenerson, H.L.; Yeung, R.S.; Crispe, I.N.; Isoherranen, N. Altered vitamin a metabolism in human liver slices corresponds to fibrogenesis. Clin. Transl. Sci. 2021, 14, 976–989. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in health and disease: A role for hepatic stellate cells in affecting retinoid levels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158674. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Leibundgut, M.; Boehringer, D.; Ban, N. Structure and function of eukaryotic fatty acid synthases. Q. Rev. Biophys. 2010, 43, 373–422. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Kothapalli, K.S.; Brenna, J.T. Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 103–110. [Google Scholar] [CrossRef]
- Kihara, A. Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem. 2012, 152, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 2012, 1821, 177–189. [Google Scholar] [CrossRef]
- Geng, C.; Xu, H.; Zhang, Y.; Gao, Y.; Yinliang, Z.; Liu, X.; Gao, M.; Wang, X.; Liu, X.; Fang, F.; et al. Retinoic acid ameliorates high-fat diet-induced liver steatosis through sirt1. Sci. China Life Sci. 2017, 60, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kim, C.-K.; Axe, D.; Cook, A.; Lee, M.; Li, T.; Smallwood, N.; Chiang, J.Y.; Hardwick, J.P.; Moore, D.D.; et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology 2013, 59, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Vuckovic, M.G.; Smullin, C.P.; Kim, M.; Lo, C.P.-S.; Devericks, E.; Yoo, H.S.; Tintcheva, M.; Deng, Y.; Napoli, J.L. Modest Decreases in Endogenous All-trans-Retinoic Acid Produced by a Mouse Rdh10 Heterozygote Provoke Major Abnormalities in Adipogenesis and Lipid Metabolism. Diabetes 2018, 67, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Ikeda, Y.; Ebata, Y.; Kojima, C.; Katsuma, R.; Tsuruyama, T.; Sakabe, T.; Shomori, K.; Komeda, N.; Oshiro, S.; et al. Retinoids ameliorate insulin resistance in a leptin-dependent manner in mice. Hepatology 2012, 56, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Trasino, S.E.; Tang, X.-H.; Jessurun, J.; Gudas, L.J. Retinoic acid receptor β2 agonists restore glycaemic control in diabetes and reduce steatosis. Diabetes Obes. Metab. 2015, 18, 142–151. [Google Scholar] [CrossRef]
- Tang, X.-H.; Melis, M.; Lu, C.; Rappa, A.; Zhang, T.; Jessurun, J.; Gross, S.S.; Gudas, L.J. A retinoic acid receptor β2 agonist attenuates transcriptome and metabolome changes underlying nonalcohol-associated fatty liver disease. J. Biol. Chem. 2021, 297, 101331. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Cree-Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic Acid Receptor β Stimulates Hepatic Induction of Fibroblast Growth Factor 21 to Promote Fatty Acid Oxidation and Control Whole-body Energy Homeostasis in Mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef]
- Trasino, S.E.; Tang, X.-H.; Jessurun, J.; Gudas, L.J. A retinoic acid receptor β2 agonist reduces hepatic stellate cell activation in nonalcoholic fatty liver disease. Klin. Wochenschr. 2016, 94, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Tang, X.-H.; Trasino, S.E.; Patel, V.M.; Stummer, D.J.; Jessurun, J.; Gudas, L.J. Effects of AM80 compared to AC261066 in a high fat diet mouse model of liver disease. PLoS ONE 2019, 14, e0211071. [Google Scholar] [CrossRef] [PubMed]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 2016, 1, e90954. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Jeong, W.-I. Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 2012, 27, 75–79. [Google Scholar] [CrossRef]
- Murakami, K.-I.; Kaji, T.; Shimono, R.; Hayashida, Y.; Matsufuji, H.; Tsuyama, S.; Maezono, R.; Kosai, K.-I.; Takamatsu, H. Therapeutic effects of vitamin A on experimental cholestatic rats with hepatic fibrosis. Pediatr. Surg. Int. 2011, 27, 863–870. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Thomas, S.; Ramachandran, A.; Pulimood, A.B.; Balasubramanian, K.A. Retinoid metabolism during development of liver cirrhosis. Arch. Biochem. Biophys. 2005, 443, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Okuno, M.; Moriwaki, H.; Imai, S.; Muto, Y.; Kawada, N.; Suzuki, Y.; Kojima, S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology 1997, 26, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.-I.; Park, O.; Suh, Y.-G.; Byun, J.-S.; Park, S.-Y.; Choi, E.; Kim, J.-K.; Ko, H.; Wang, H.; Miller, A.M.; et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, K.; Verbuyst, P.; Quartier, E.; Schuit, F.; Rombouts, K.; Chandraratna, R.A.; Schuppan, D.; Geerts, A. Differential modulation of rat hepatic stellate phenotype by natural and synthetic retinoids. Hepatology 2004, 39, 97–108. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, F.; Wroblewski, K.; O’Byrne, S.M.; Jiang, H.; Clerkin, K.; Benhammou, J.; Blaner, W.S.; Beaven, S.W. Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology 2015, 62, 615–626. [Google Scholar] [CrossRef]
- Wang, L.; Tankersley, L.R.; Tang, M.; Potter, J.J.; Mezey, E. Regulation of the murine α2(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys. 2002, 401, 262–270. [Google Scholar] [CrossRef]
- Radaeva, S.; Wang, L.; Radaev, S.; Jeong, W.-I.; Park, O.; Gao, B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Liver Physiol. 2007, 293, G809–G816. [Google Scholar] [CrossRef]
- Jeong, W.-I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand-dependent Manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Motomura, K.; Sakai, H.; Isobe, H.; Nawata, H. Effects of retinoids on the production of tumour necrosis factor-alpha and nitric oxide by lipopolysaccharide-stimulated rat Kupffer cells in vitro: Evidence for participation of retinoid X receptor signalling pathway. Cell Biochem. Funct. 1997, 15, 95–101. [Google Scholar] [CrossRef]
- Na, S.-Y.; Kang, B.Y.; Chung, S.W.; Han, S.-J.; Ma, X.; Trinchieri, G.; Im, S.-Y.; Lee, J.W.; Kim, T.S. Retinoids Inhibit Interleukin-12 Production in Macrophages through Physical Associations of Retinoid X Receptor and NFkappaB. J. Biol. Chem. 1999, 274, 7674–7680. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Lehmann, J.; Hoffmann, B.; Hermann, T.; Tzukerman, M.; Pfahl, M. Antagonism between retinoic acid receptors. Mol. Cell. Biol. 1991, 11, 4097–4103. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, S.; Schnabl, B.; Knight, R.; Loomba, R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2020, 33, 21–32. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Cha, H.-R.; Chang, S.-Y.; Chang, J.-H.; Kim, J.-O.; Yang, J.-Y.; Kim, C.-H.; Kweon, M.-N. Downregulation of Th17 Cells in the Small Intestine by Disruption of Gut Flora in the Absence of Retinoic Acid. J. Immunol. 2010, 184, 6799–6806. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Cui, T.; Yang, T.; Liu, L.; Li, T.; Chen, J. Retinoic Acid Facilitates Toll-Like Receptor 4 Expression to Improve Intestinal Barrier Function through Retinoic Acid Receptor Beta. Cell. Physiol. Biochem. 2017, 42, 1390–1406. [Google Scholar] [CrossRef]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Dunn, W.; Shah, V.H. Pathogenesis of Alcoholic Liver Disease. Clin. Liver Dis. 2016, 20, 445–456. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef]
- Clugston, R.D.; Blaner, W.S. The Adverse Effects of Alcohol on Vitamin A Metabolism. Nutrients 2012, 4, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Pappa, A.; Estey, T. Role of Human Aldehyde Dehydrogenases in Endobiotic and Xenobiotic Metabolism. Drug Metab. Rev. 2004, 36, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Zolfaghari, R. Cytochrome P450s in the Regulation of Cellular Retinoic Acid Metabolism. Annu. Rev. Nutr. 2011, 31, 65–87. [Google Scholar] [CrossRef]
- Smith, J.C.; Brown, E.; White, S.; Finkelstein, J. Letter: Plasma vitamin A and zinc concentration in patients with alcoholic cirrhosis. Lancet 1975, 305, 1251–1252. [Google Scholar] [CrossRef]
- McClain, C.J.; Van Thiel, D.H.; Parker, S.; Badzin, L.K.; Gilbert, H. Alterations in Zinc, Vitamin A, and Retinol-Binding Protein in Chronic Alcoholics: A Possible Mechanism for Night Blindness and Hypogonadism. Alcohol. Clin. Exp. Res. 1979, 3, 135–141. [Google Scholar] [CrossRef]
- Leo, M.A.; Lieber, C.S. Hepatic Vitamin a Depletion in Alcoholic Liver Injury. N. Engl. J. Med. 1982, 307, 597–601. [Google Scholar] [CrossRef]
- Leo, M.A.; Sato, M.; Lieber, C.S. Effect of hepatic vitamin A depletion on the liver in humans and rats. Gastroenterology 1983, 84, 562–572. [Google Scholar] [CrossRef]
- Majumdar, S.K.; Shaw, G.K.; Thomson, A.D. Vitamin A utilization status in chronic alcoholic patients. Int. J. Vitam. Nutr. Res. 1983, 53, 273–279. [Google Scholar] [CrossRef]
- Lieber, C.S. Relationships between Nutrition, Alcohol Use, and Liver Disease. Alcohol Res. Health 2003, 27, 220–231. [Google Scholar]
- Kim, Y.-K.; Zuccaro, M.V.; Zhang, C.; Sarkar, D.K.; Quadro, L. Alcohol exposure in utero perturbs retinoid homeostasis in adult rats. Hepatobiliary Surg. Nutr. 2015, 4, 268–277. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: A potential mechanism of ethanol toxicity. FASEB J. 2010, 24, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Hautekeete, M.L.; Dodeman, I.; Azais-Braesco, V.; Van den Berg, K.; Seynaeve, C.; Geerts, A. Hepatic stellate cells and liver retinoid content in alcoholic liver disease in humans. Alcohol. Clin. Exp. Res. 1998, 22, 494–500. [Google Scholar] [CrossRef]
- Bjørneboe, G.A.; Johnsen, J.; Bjørneboe, A.; Mørland, J.; Drevon, C.A. Effect of heavy alcohol consumption on serum concentrations of fat-soluble vitamins and selenium. Alcohol. Alcohol. Suppl. 1987, 1, 533–537. [Google Scholar] [PubMed]
- Bjørneboe, G.E.; Johnsen, J.; Bjørneboe, A.; Rousseau, B.; Pedersen, J.I.; Norum, K.R.; Mørland, J.; Drevon, C.A. Effect of alcohol consumption on serum concentration of 25-hydroxyvitamin D3, retinol, and retinol-binding protein. Am. J. Clin. Nutr. 1986, 44, 678–682. [Google Scholar] [CrossRef]
- French, S.W. The mallory body: Structure, composition, and pathogenesis. Hepatology 1981, 1, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Celli, R.; Zhang, X. Pathology of Alcoholic Liver Disease. J. Clin. Transl. Hepatol. 2014, 2, 103–109. [Google Scholar] [CrossRef]
- Clugston, R.D.; Huang, L.-S.; Blaner, W.S. Chronic alcohol consumption has a biphasic effect on hepatic retinoid loss. FASEB J. 2015, 29, 3654–3667. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.-Y.; Dapito, D.H.; Pradere, J.-P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.P.; Ross, A.C.; Stephensen, C.B.; Bohn, T.; Tanumihardjo, S.A. Metabolic Effects of Inflammation on Vitamin A and Carotenoids in Humans and Animal Models. Adv. Nutr. Int. Rev. J. 2017, 8, 197–212. [Google Scholar] [CrossRef]
- Sato, M.; Lieber, C.S. Hepatic Vitamin a Depletion after Chronic Ethanol Consumption in Baboons and Rats. J. Nutr. 1981, 111, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Russell, R.M.; Seitz, H.K.; Wang, X.-D. Ethanol enhances retinoic acid metabolism into polar metabolites in rat liver via induction of cytochrome P4502E1. Gastroenterology 2001, 120, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Microsomal Ethanol-Oxidizing System (MEOS): The First 30 Years (1968–1998)—A Review. Alcohol. Clin. Exp. Res. 1999, 23, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chung, J.; Seitz, H.K.; Russell, R.M.; Wang, X.-D. Chlormethiazole treatment prevents reduced hepatic vitamin A levels in ethanol-fed rats. Alcohol. Clin. Exp. Res. 2002, 26, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Ferdouse, A.; Agrawal, R.R.; Gao, M.A.; Jiang, H.; Blaner, W.S.; Clugston, R.D. Alcohol induced hepatic retinoid depletion is associated with the induction of multiple retinoid catabolizing cytochrome P450 enzymes. PLoS ONE 2022, 17, e0261675. [Google Scholar] [CrossRef]
- Melis, M.; Tang, X.; Attarwala, N.; Chen, Q.; Prishker, C.; Qin, L.; Gross, S.S.; Gudas, L.J.; Trasino, S.E. A retinoic acid receptor β2 agonist protects against alcohol liver disease and modulates hepatic expression of canonical retinoid metabolism genes. BioFactors 2021. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Effects of ethanol on physiological retinoic acid levels. IUBMB Life 2011, 63, 701–706. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, P.; Koul, O.; Smith, D.; Napoli, J.L.; Chen, N.; Ullman, M.D. Ethanol increases retinoic acid production in cerebellar astrocytes and in cerebellum. Dev. Brain Res. 2004, 153, 233–241. [Google Scholar] [CrossRef]
- Kane, M.A.; Napoli, J.L. Quantification of Endogenous Retinoids. Myelin 2010, 652, 1–54. [Google Scholar] [CrossRef]
- Jones, J.W.; Pierzchalski, K.; Yu, J.; Kane, M.A. Use of Fast HPLC Multiple Reaction Monitoring Cubed for Endogenous Retinoic Acid Quantification in Complex Matrices. Anal. Chem. 2015, 87, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Quantitative Profiling of Endogenous Retinoic Acid In Vivo and In Vitro by Tandem Mass Spectrometry. Anal. Chem. 2008, 80, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Dollé, P.; Niederreither, K. The Retinoids: Biology, Biochemistry, and Disease, 1st ed.; Wiley Blackwell: Hoboken, NJ, USA, 2015; 585p. [Google Scholar]
- Serio, R.N.; Laursen, K.B.; Urvalek, A.M.; Gross, S.S.; Gudas, L.J. Ethanol promotes differentiation of embryonic stem cells through retinoic acid receptor-gamma. J. Biol. Chem. 2019, 294, 5536–5548. [Google Scholar] [CrossRef] [PubMed]
- Motomura, K.; Ohata, M.; Satre, M.; Tsukamoto, H. Destabilization of TNF-α mRNA by retinoic acid in hepatic macrophages: Implications for alcoholic liver disease. Am. J. Physiol. Metab. 2001, 281, E420–E429. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Liu, C.; Smith, D.E.; Seitz, H.K.; Russell, R.M.; Wang, X.-D. Restoration of retinoic acid concentration supresses ethanol-enhanced c-Jun expression and hepatocyte proliferation in rat liver. Carcinogenesis 2001, 22, 1213–1219. [Google Scholar] [CrossRef]
- Chung, J.; Chavez, P.R.; Russell, R.M.; Wang, X.-D. Retinoic acid inhibits hepatic Jun N-terminal kinase-dependent signaling pathway in ethanol-fed rats. Oncogene 2002, 21, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Dan, Z.; Fu, Y.; Tang, W.; Lin, J. Low-dose ATRA supplementation abolishes PRM formation in rat liver and ameliorates ethanol-induced liver injury. J. Huazhong Univ. Sci. Technol. 2006, 26, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.A.; Arai, M.; Sato, M.; Lieber, C.S. Hepatotoxicity of vitamin A and ethanol in the rat. Gastroenterology 1982, 82, 194–205. [Google Scholar] [CrossRef]
- Leo, M.A.; Lieber, C.S. Hepatic Fibrosis after Long-Term Administration of Ethanol and Moderate Vitamin A Supplementation in the Rat. Hepatology 2007, 3, 1–11. [Google Scholar] [CrossRef]
- Ahmed, S.; Leo, M.A.; Lieber, C.S. Interactions between alcohol and β-carotene in patients with alcoholic liver disease. Am. J. Clin. Nutr. 1994, 60, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Marucci, L.; Benedetti, A.; Mancini, R.; Jezequel, A.-M.; Orlandi, F. Chronic ethanol feeding increases apoptosis and cell proliferation in rat liver. J. Hepatol. 1994, 20, 508–513. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Cifelli, C.J.; Zolfaghari, R.; Li, N.-Q. Multiple cytochromeP-450 genes are concomitantly regulated by vitamin A under steady-state conditions and by retinoic acid during hepatic first-pass metabolism. Physiol. Genom. 2011, 43, 57–67. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Gong, L.; Fang, Y.; Zhan, Q.; Liu, H.-X.; Lu, Y.; Guo, G.L.; Lehman-McKeeman, L.; Fang, J.; Wan, Y.-J.Y. The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling. BMC Genom. 2013, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dan, Z.; Wang, H.; Lin, J. Effect of ATRA on contents of liver retinoids, oxidative stress and hepatic injury in rat model of extrahepatic cholestasis. J. Huazhong Univ. Sci. Technol. 2007, 27, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Potter, J.J.; Rennie-Tankersley, L.; Novitskiy, G.; Sipes, J.; Mezey, E. Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice. Biochim. Biophys. Acta 2007, 1772, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Akechi, Y.; Ikeda, R.; Nishio, R.; Sakabe, T.; Terabayashi, K.; Matsumi, Y.; Ashla, A.A.; Hoshikawa, Y.; Kurimasa, A.; et al. Suppressive Effects of Retinoids on Iron-Induced Oxidative Stress in the Liver. Gastroenterology 2009, 136, 341–350.e8. [Google Scholar] [CrossRef]
- Kartasheva-Ebertz, D.M.; Pol, S.; Lagaye, S. Retinoic Acid: A New Old Friend of IL-17A in the Immune Pathogeny of Liver Fibrosis. Front. Immunol. 2021, 12, 2295. [Google Scholar] [CrossRef]
- Muindi, J.; Frankel, S.R.; Miller, W.H.; Jr Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P., Jr. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: Implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood 1992, 79, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Thomas, G.; Gourmel, B.; Agadir, A.; Castaigne, S.; Dreux, C.; Degos, L.; Chomienne, C. Pharmacokinetics of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Leukemia 1991, 5, 1054–1058. [Google Scholar] [PubMed]
- Diesinger, T.; Buko, V.; Lautwein, A.; Dvorsky, R.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Buckert, D.; et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS ONE 2020, 15, e0235990. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.W.; Piu, F.; Gauthier, N.K.; Eeg, A.; Currier, E.; Sherbukhin, V.; Brann, M.R.; Hacksell, U.; Olsson, R. Discovery of a Potent, Orally Available, and Isoform-Selective Retinoic Acid β2 Receptor Agonist. J. Med. Chem. 2005, 48, 7517–7519. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; Reynolds, C.P.; Cho, H.; Kang, M.H. Clinical development of fenretinide as an antineoplastic drug: Pharmacology perspectives. Exp. Biol. Med. 2017, 242, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-H.; Melis, M.; Mai, K.; Gudas, L.J.; Trasino, S.E. Fenretinide Improves Intestinal Barrier Function and Mitigates Alcohol Liver Disease. Front. Pharmacol. 2021, 12, 630557. [Google Scholar] [CrossRef] [PubMed]
- Sabichi, A.L.; Xu, H.; Fischer, S.; Zou, C.; Yang, X.; Steele, V.E.; Kelloff, G.J.; Lotan, R.; Clifford, J.L. Retinoid receptor-dependent and independent biological activities of novel fenretinide analogues and metabolites. Clin. Cancer Res. 2003, 9, 4606–4613. [Google Scholar]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Njei, B.; Rotman, Y.; Ditah, I.; Lim, J.K. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015, 61, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Peres, W.A.F.; Chaves, G.V.; Gonçalves, J.C.S.; Ramalho, A.; Coelho, H.S.M. Vitamin A deficiency in patients with hepatitis C virus-related chronic liver disease. Br. J. Nutr. 2011, 106, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Datfar, T.; Doulberis, M.; Papaefthymiou, A.; Hines, I.N.; Manzini, G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens 2021, 10, 1366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Xu, C.-F.; Yu, C.-H.; Chen, W.-X.; Li, Y.-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Irshad, M.; Gupta, P.; Irshad, K. Immunopathogenesis of Liver Injury during Hepatitis C Virus Infection. Viral Immunol. 2019, 32, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bitetto, D.; Bortolotti, N.; Falleti, E.; Vescovo, S.; Fabris, C.; Fattovich, G.; Cussigh, A.; Cmet, S.; Fornasiere, E.; Ceriani, E.; et al. Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology 2012, 57, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Read, S.A.; Shackel, N.A.; Hebbard, L.; George, J.; Ahlenstiel, G. The Role of Micronutrients in the Infection and Subsequent Response to Hepatitis C Virus. Cells 2019, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, S.; Fukuda, R.; Ishimura, N.; Rumi, M.A.K.; Kazumori, H.; Uchida, Y.; Kadowaki, Y.; Ishihara, S.; Kinoshita, Y. 9-cis Retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor. J. Lab. Clin. Med. 2003, 141, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Ikeda, M.; Abe, K.-I.; Dansako, H.; Ohkoshi, S.; Aoyagi, Y.; Kato, N. Comprehensive Analysis of the Effects of Ordinary Nutrients on Hepatitis C Virus RNA Replication in Cell Culture. Antimicrob. Agents Chemother. 2007, 51, 2016–2027. [Google Scholar] [CrossRef]
- Bang, B.-R.; Li, M.; Tsai, K.-N.; Aoyagi, H.; Lee, S.-A.; Machida, K.; Aizaki, H.; Jung, J.U.; Ou, J.-H.J.; Saito, T. Regulation of Hepatitis C Virus Infection by Cellular Retinoic Acid Binding Proteins through the Modulation of Lipid Droplet Abundance. J. Virol. 2019, 93, e02302-18. [Google Scholar] [CrossRef]
- Böcher, W.O.; Wallasch, C.; Höhler, T.; Galle, P.R. All-trans retinoic acid for treatment of chronic hepatitis C. Liver Int. 2008, 28, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Dejean, A.; Bougueleret, L.; Grzeschik, K.-H.; Tiollais, P. Hepatitis B virus DNA integration in a sequence homologous to verb-A and steroid receptor genes in a hepatocellular carcinoma. Nature 1986, 322, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Brand, N.; Petkovich, M.; Krust, A.; Chambon, P.; De Thé, H.; Marchio, A.; Tiollais, P.; Dejean, A. Identification of a second human retinoic acid receptor. Nature 1988, 332, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; de Thé, H.; Tiollais, P.; Samarut, J.; Dejean, A. A hepatitis B virus pre-S-retinoic acid receptor beta chimera transforms erythrocytic progenitor cells in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.K.; Park, S.-H.; Jang, K.L. Hepatitis B virus X protein overcomes the growth-inhibitory potential of retinoic acid by downregulating retinoic acid receptor-β2 expression via DNA methylation. J. Gen. Virol. 2009, 91 Pt 2, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Takayama, T.; Murakami, K.; Saiki, I.; Makuuchi, M. Overexpression of retinoic acid receptor alpha in hepatocellular carcinoma. Clin. Cancer Res. 2003, 9 Pt 1, 3679–3683. [Google Scholar]
- Cortes, E.; Lachowski, D.; Rice, A.; Chronopoulos, A.; Robinson, B.; Thorpe, S.; Lee, D.A.; Possamai, L.A.; Wang, H.; Pinato, D.J.; et al. Retinoic Acid Receptor-β Is Downregulated in Hepatocellular Carcinoma and Cirrhosis and Its Expression Inhibits Myosin-Driven Activation and Durotaxis in Hepatic Stellate Cells. Hepatology 2019, 69, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Simbrunner, B.; Semmler, G.; Stadlmann, A.; Scheiner, B.; Schwabl, P.; Paternostro, R.; Bucsics, T.; Bauer, D.; Eigenbauer, E.; Pinter, M.; et al. Vitamin A levels reflect disease severity and portal hypertension in patients with cirrhosis. Hepatol. Int. 2020, 14, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Lee, S.-A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. 2012, 1821, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.E.; Hennings, L.; Ronis, M.J.J. Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis. Adv. Exp. Med. Biol. 2015, 815, 185–195. [Google Scholar] [CrossRef]
- Mercer, K.E.; Hennings, L.; Sharma, N.; Lai, K.; Cleves, M.A.; Wynne, R.A.; Badger, T.M.; Ronis, M.J. Alcohol Consumption Promotes Diethylnitrosamine-Induced Hepatocarcinogenesis in Male Mice through Activation of the Wnt/β-Catenin Signaling Pathway. Cancer Prev. Res. 2014, 7, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Sever, C.E.; Locker, J. Expression of retinoic acid α and β receptor genes in liver and hepatocellular carcinoma. Mol. Carcinog. 1991, 4, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Shackel, N.A.; Warner, F.J. Identification of resident hepatic stem cell populations. Hepatology 2007, 46, 2042–2044. [Google Scholar] [CrossRef] [PubMed]
- Tzartzeva, K.; Obi, J.; Rich, N.E.; Parikh, N.D.; Marrero, J.A.; Yopp, A.; Waljee, A.K.; Singal, A.G. Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients with Cirrhosis: A Meta-analysis. Gastroenterology 2018, 154, 1706–1718.e1. [Google Scholar] [CrossRef]
- Hu, X.; Chen, R.; Wei, Q.; Xu, X. The Landscape Of Alpha Fetoprotein In Hepatocellular Carcinoma: Where Are We? Int. J. Biol. Sci. 2022, 18, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J.; Wang, J.; Yan, Y.; Zhang, C. Alpha-fetoprotein accelerates the progression of hepatocellular carcinoma by promoting Bcl-2 gene expression through an RA-RAR signalling pathway. J. Cell. Mol. Med. 2020, 24, 13804–13812. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; Li, C.; Guo, L.; Liu, H.; Zhou, S.; Liu, X.; Chen, Z.; Shi, S.; Wei, J.; et al. Cytoplasmic alpha-fetoprotein functions as a co-repressor in RA-RAR signaling to promote the growth of human hepatoma Bel 7402 cells. Cancer Lett. 2009, 285, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Clotet-Freixas, S.; Baciu, C.; Pasini, E.; Hammad, A.; Ivanics, T.; Reid, S.; Azhie, A.; Angeli, M.; Ghanekar, A.; et al. Combined proteomic/transcriptomic signature of recurrence post-liver transplantation for hepatocellular carcinoma beyond Milan. Clin. Proteom. 2021, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Gräff, A.; Ertelt, V.; Allgaier, H.; Koelble, K.; Olschewski, M.; Nitschke, R.; Bochaton-Piallat, M.; Gabbiani, G.; Blum, H.E. Cellular retinol-binding protein-1 in hepatocellular carcinoma correlates with β-catenin, Ki-67 index, and patient survival. Hepatology 2003, 38, 470–480. [Google Scholar] [CrossRef]
- Nakanishi, M.; Tomaru, Y.; Miura, H.; Hayashizaki, Y.; Suzuki, M. Identification of transcriptional regulatory cascades in retinoic acid-induced growth arrest of HepG2 cells. Nucleic Acids Res. 2008, 36, 3443–3454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liao, X.-H.; Zhang, A.L.; Zheng, M.; Li, M.-Q.; Chen, C.P.; Xu, H.; Chu, Q.-S.; Yang, D.; Lu, W.; Tsai, T.-F.; et al. Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways. Sci. Rep. 2017, 7, 43639. [Google Scholar] [CrossRef]
- Yang, H.; Bushue, N.; Bu, P.; Wan, Y.-J.Y. Induction and intracellular localization of Nur77 dictate fenretinide-induced apoptosis of human liver cancer cells. Biochem. Pharmacol. 2010, 79, 948–954. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, D.; Shao, D.; Liu, H.; Zhou, Q.; Gui, S.; Wei, W.; Wang, Y. Fenretinide inhibits the proliferation and migration of human liver cancer HepG2 cells by downregulating the activation of myosin light chain kinase through the p38-MAPK signaling pathway. Oncol. Rep. 2018, 40, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Wan, Y.-J.Y. Fenretinide-induced apoptosis of Huh-7 hepatocellular carcinoma is retinoic acid receptor beta dependent. BMC Cancer 2007, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhan, Q.; Wan, Y.-J.Y. Enrichment of Nur77 mediated by retinoic acid receptor β leads to apoptosis of human hepatocellular carcinoma cells induced by fenretinide and histone deacetylase inhibitors. Hepatology 2010, 53, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Qin, W.; Zhang, Y.; Luo, Y.; Niu, B.; An, Q.; Yang, B.; Shi, K.; Yu, Z.; Chen, J.; et al. Sulfarotene, a synthetic retinoid, overcomes stemness and sorafenib resistance of hepatocellular carcinoma via suppressing SOS2-RAS pathway. J. Exp. Clin. Cancer Res. 2021, 40, 280. [Google Scholar] [CrossRef]
- Muto, Y.; Moriwaki, H.; Omori, M. In vitro binding affinity of novel synthetic polyprenoids (polyprenoic acids) to cellular retinoid-binding proteins. Gann Gan 1981, 72, 974–977. [Google Scholar] [PubMed]
- Muto, Y.; Moriwaki, H.; Ninomiya, M.; Adachi, S.; Saito, A.; Takasaki, K.T.; Tanaka, T.; Tsurumi, K.; Okuno, M.; Tomita, E.; et al. Prevention of Second Primary Tumors by an Acyclic Retinoid, Polyprenoic Acid, in Patients with Hepatocellular Carcinoma. N. Engl. J. Med. 1996, 334, 1561–1568. [Google Scholar] [CrossRef]
- Woo, H.Y.; Yoo, S.Y.; Heo, J. Peretinoin, an Acyclic Retinoid, for the Secondary Prevention of Hepatocellular Carcinoma. Molecules 2021, 26, 295. [Google Scholar] [CrossRef]
- Okada, H.; Takabatake, R.; Honda, M.; Takegoshi, K.; Yamashita, T.; Nakamura, M.; Shirasaki, T.; Sakai, Y.; Shimakami, T.; Nagata, N.; et al. Peretinoin, an acyclic retinoid, suppresses steatohepatitis and tumorigenesis by activating autophagy in mice fed an atherogenic high-fat diet. Oncotarget 2017, 8, 39978–39993. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Kagawa, M.; Okuno, M.; Ishibashi, N.; Hashimoto, M.; Yamamoto, M.; Suzuki, R.; Kohno, H.; Matsushima-Nishiwaki, R.; Takano, Y.; et al. Prevention of Rat Hepatocarcinogenesis by Acyclic Retinoid Is Accompanied by Reduction in Emergence of Both TGF-α-Expressing Oval-Like Cells and Activated Hepatic Stellate Cells. Nutr. Cancer 2005, 51, 197–206. [Google Scholar] [CrossRef]
- Honda, M.; Yamashita, T.; Yamashita, T.; Arai, K.; Sakai, Y.; Sakai, A.; Nakamura, M.; Mizukoshi, E.; Kaneko, S. Peretinoin, an acyclic retinoid, improves the hepatic gene signature of chronic hepatitis C following curative therapy of hepatocellular carcinoma. BMC Cancer 2013, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Honda, M.; Campbell, J.S.; Sakai, Y.; Yamashita, T.; Takebuchi, Y.; Hada, K.; Shirasaki, T.; Takabatake, R.; Nakamura, M.; et al. Acyclic Retinoid Targets Platelet-Derived Growth Factor Signaling in the Prevention of Hepatic Fibrosis and Hepatocellular Carcinoma Development. Cancer Res. 2012, 72, 4459–4471. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, F.; Zhang, L.; Zhou, Q.; Gui, S.; Wang, Y. A novel all-trans retinoic acid derivative 4-amino-2-trifluoromethyl-phenyl retinate inhibits the proliferation of human hepatocellular carcinoma HepG2 cells by inducing G0/G1 cell cycle arrest and apoptosis via upregulation of p53 and ASPP1 and downregulation of iASPP. Oncol. Rep. 2016, 36, 333–341. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melis, M.; Tang, X.-H.; Trasino, S.E.; Gudas, L.J. Retinoids in the Pathogenesis and Treatment of Liver Diseases. Nutrients 2022, 14, 1456. https://doi.org/10.3390/nu14071456
Melis M, Tang X-H, Trasino SE, Gudas LJ. Retinoids in the Pathogenesis and Treatment of Liver Diseases. Nutrients. 2022; 14(7):1456. https://doi.org/10.3390/nu14071456
Chicago/Turabian StyleMelis, Marta, Xiao-Han Tang, Steven E. Trasino, and Lorraine J. Gudas. 2022. "Retinoids in the Pathogenesis and Treatment of Liver Diseases" Nutrients 14, no. 7: 1456. https://doi.org/10.3390/nu14071456
APA StyleMelis, M., Tang, X.-H., Trasino, S. E., & Gudas, L. J. (2022). Retinoids in the Pathogenesis and Treatment of Liver Diseases. Nutrients, 14(7), 1456. https://doi.org/10.3390/nu14071456