
Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms



Abstract
1. Introduction
2. Epidemiological Studies
2.1. Ecological Studies
2.2. Observational Studies Based on Residential UVB Doses
2.3. Observational Studies Based on Serum 25(OH)D Concentrations


2.4. RCTs of Vitamin D and Cancer Risk
- Baseline 25(OH)D concentrations should be measured and used as a criterion for inclusion in the study;
- The vitamin D dose should be large enough to increase 25(OH)D concentration to the point at which it would have an observable effect on health outcomes;
- Achieved 25(OH)D concentrations should be measured;
- Conutrient status must be optimized to ensure that vitamin D is the only nutrient-related limiting factor in the response.
3. Perspectives on Epidemiological Studies
3.1. Ecological Studies
3.2. Observational Studies
3.3. Historical Overview
4. Mechanisms Introduction
4.1. Inhibition of Tumor Cell Proliferation
4.2. Sensitization to Apoptosis, Combined Action with Chemotherapy and Radiotherapy
4.3. Regulation of Autophagy
4.4. Induction of Cell Differentiation, Inhibition of Epithelial-to-Mesenchymal Transition
4.5. Antagonism of Wnt/β-Catenin Signaling Pathway
4.6. Inhibition of Angiogenesis
4.7. Inhibition of Cancer Cell Migration, Invasion and Metastasis
4.8. Stromal Effects: Cancer-Associated Fibroblasts
4.9. Effects on Cancer Stem Cells
4.10. Effects on the Immune System
4.11. Animal Models
4.12. Systemic Effects: Detoxification and Microbiome
4.12.1. Detoxification
4.12.2. Microbiome
4.13. Discussion of Mechanistic Studies
5. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Peller, S. Carcinogenesis as a means of reducing cancer mortlity. Lancet 1936, 228, 552–556. [Google Scholar] [CrossRef]
- Peller, S.; Stephenson, C.S. Skin ittittion and cancer in the United States Navy. Am. J. Med. Sci. 1937, 194, 326–333. [Google Scholar] [CrossRef]
- Apperly, F.L. The Relation of Solar Radiation to Cancer Mortality in North America. Cancer Res. 1941, 1, 191–195. [Google Scholar]
- Ainsleigh, H.G. Beneficial effects of sun exposure on cancer mortality. Prev. Med. 1993, 22, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.J.; McKay, F.W.; Hoover, R.; Blot, W.J.; Fraumeni, J.F., Jr. Atlas of Cancer Mortality for U.S. Counties: 1950–1969; U.S. Department of Health, Education, and Welfare: Washington, DC, USA, 1975. [Google Scholar]
- Garland, C.F.; Garland, F.C. Do sunlight and vitamin D reduce the likelihood of colon cancer? Int. J. Epidemiol. 1980, 9, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.; Shekelle, R.B.; Barrett-Connor, E.; Criqui, M.H.; Rossof, A.H.; Paul, O. Dietary vitamin D and calcium and risk of colorectal cancer: A 19-year prospective study in men. Lancet 1985, 1, 307–309. [Google Scholar] [CrossRef]
- Garland, C.F.; Comstock, G.W.; Garland, F.C.; Helsing, K.J.; Shaw, E.K.; Gorham, E.D. Serum 25-hydroxyvitamin D and colon cancer: Eight-year prospective study. Lancet 1989, 2, 1176–1178. [Google Scholar] [CrossRef]
- Garland, F.C.; Garland, C.F.; Gorham, E.D.; Young, J.F. Geographic variation in breast cancer mortality in the United States: A hypothesis involving exposure to solar radiation. Prev. Med. 1990, 19, 614–622. [Google Scholar] [CrossRef]
- Lefkowitz, E.S.; Garland, C.F. Sunlight, vitamin D, and ovarian cancer mortality rates in US women. Int. J. Epidemiol. 1994, 23, 1133–1136. [Google Scholar] [CrossRef]
- Garland, C. The Summer of 1974, Or...How I Found My Life’s Mission. Available online: https://www.grassrootshealth.net/?s=The+Summer+of+1974%2C+Or...How+I+Found+My+Life%27s+Mission (accessed on 23 February 2022).
- Devesa, S.S.; Grauman, D.J.; Blot, W.J.; Pennello, G.A.; Hoover, R.N.; Fraumeni, J.F., Jr. Atlas of Cancer Mortality in the United States, 1950–1994; National Institutes of Health; National Cancer Institue: Bethesda, MD, USA, 1999. [Google Scholar]
- Grant, W.B. An estimate of premature cancer mortality in the U.S. due to inadequate doses of solar ultraviolet-B radiation. Cancer 2002, 94, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Garland, C.F. The association of solar ultraviolet B (UVB) with reducing risk of cancer: Multifactorial ecologic analysis of geographic variation in age-adjusted cancer mortality rates. Anticancer Res. 2006, 26, 2687–2699. [Google Scholar] [PubMed]
- Grant, W.B. Lower vitamin-D production from solar ultraviolet-B irradiance may explain some differences in cancer survival rates. J. Natl. Med. Assoc. 2006, 98, 357–364. [Google Scholar] [PubMed]
- Ames, B.N.; Grant, W.B.; Willett, W.C. Does the High Prevalence of Vitamin D Deficiency in African Americans Contribute to Health Disparities? Nutrients 2021, 13, 499. [Google Scholar] [CrossRef] [PubMed]
- Moukayed, M.; Grant, W.B. Molecular link between vitamin D and cancer prevention. Nutrients 2013, 5, 3993–4021. [Google Scholar] [CrossRef]
- Chen, W.; Clements, M.; Rahman, B.; Zhang, S.; Qiao, Y.; Armstrong, B.K. Relationship between cancer mortality/incidence and ambient ultraviolet B irradiance in China. Cancer Causes Control 2010, 21, 1701–1709. [Google Scholar] [CrossRef]
- Fioletov, V.E.; McArthur, L.J.; Mathews, T.W.; Marrett, L. Estimated ultraviolet exposure levels for a sufficient vitamin D status in North America. J. Photochem. Photobiol. B 2010, 100, 57–66. [Google Scholar] [CrossRef]
- Herman, J.R.; Krotkov, N.; Celarier, E.; Larko, D.; Lebow, G. Distribution of UV radiation at the Earth’s surface from TOMSmeasured UV-backscattered radiances. J. Geophys. Res. 1999, 104, 12059–12076. [Google Scholar] [CrossRef]
- Mizoue, T. Ecological study of solar radiation and cancer mortality in Japan. Health Phys. 2004, 87, 532–538. [Google Scholar] [CrossRef]
- Boscoe, F.P.; Schymura, M.J. Solar ultraviolet-B exposure and cancer incidence and mortality in the United States, 1993–2002. BMC Cancer 2006, 6, 264. [Google Scholar] [CrossRef]
- Fukuda, Y.; Nakaya, T.; Nakao, H.; Yahata, Y.; Imai, H. Multilevel analysis of solar radiation and cancer mortality using ecological data in Japan. Biosci. Trends 2008, 2, 235–240. [Google Scholar]
- Borisenkov, M.F. Latitude of residence and position in time zone are predictors of cancer incidence, cancer mortality, and life expectancy at birth. Chronobiol. Int. 2011, 28, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. Role of solar UVB irradiance and smoking in cancer as inferred from cancer incidence rates by occupation in Nordic countries. Dermatoendocrinology 2012, 4, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Pukkala, E.; Martinsen, J.I.; Lynge, E.; Gunnarsdottir, H.K.; Sparen, P.; Tryggvadottir, L.; Weiderpass, E.; Kjaerheim, K. Occupation and cancer—Follow-up of 15 million people in five Nordic countries. Acta Oncol. 2009, 48, 646–790. [Google Scholar] [CrossRef]
- Grant, W.B. A meta-analysis of second cancers after a diagnosis of nonmelanoma skin cancer: Additional evidence that solar ultraviolet-B irradiance reduces the risk of internal cancers. J. Steroid Biochem. Mol. Biol. 2007, 103, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kenborg, L.; Jorgensen, A.D.; Budtz-Jorgensen, E.; Knudsen, L.E.; Hansen, J. Occupational exposure to the sun and risk of skin and lip cancer among male wage earners in Denmark: A population-based case-control study. Cancer Causes Control 2010, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.W.; Wheeler, D.C.; Park, Y.; Cahoon, E.K.; Hollenbeck, A.R.; Freedman, D.M.; Abnet, C.C. Prospective study of ultraviolet radiation exposure and risk of cancer in the United States. Int. J. Cancer 2012, 131, E1015–E1023. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, F.; van Geffen, J.; van Weele, M.; Zgaga, L. Annual Ambient UVB at Wavelengths that Induce Vitamin D Synthesis is Associated with Reduced Esophageal and Gastric Cancer Risk: A Nested Case-Control Study. Photochem. Photobiol. 2018, 94, 797–806. [Google Scholar] [CrossRef]
- Sempos, C.T.; Durazo-Arvizu, R.A.; Binkley, N.; Jones, J.; Merkel, J.M.; Carter, G.D. Developing vitamin D dietary guidelines and the lack of 25-hydroxyvitamin D assay standardization: The ever-present past. J. Steroid Biochem. Mol. Biol. 2016, 164, 115–119. [Google Scholar] [CrossRef]
- Ginde, A.A.; Liu, M.C.; Camargo, C.A., Jr. Demographic differences and trends of vitamin D insufficiency in the US population, 1988–2004. Arch. Intern. Med. 2009, 169, 626–632. [Google Scholar] [CrossRef]
- Crowe, F.L.; Steur, M.; Allen, N.E.; Appleby, P.N.; Travis, R.C.; Key, T.J. Plasma concentrations of 25-hydroxyvitamin D in meat eaters, fish eaters, vegetarians and vegans: Results from the EPIC-Oxford study. Public Health Nutr. 2011, 14, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Cashman, K.D.; O’Sullivan, S.M.; Galvin, K.; Ryan, M. Contribution of Vitamin D2 and D3 and Their Respective 25-Hydroxy Metabolites to the Total Vitamin D Content of Beef and Lamb. Curr. Dev. Nutr. 2020, 4, nzaa112. [Google Scholar] [CrossRef] [PubMed]
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D status and ill health: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 76–89. [Google Scholar] [CrossRef]
- Autier, P.; Mullie, P.; Macacu, A.; Dragomir, M.; Boniol, M.; Coppens, K.; Pizot, C.; Boniol, M. Effect of vitamin D supplementation on non-skeletal disorders: A systematic review of meta-analyses and randomised trials. Lancet Diabetes Endocrinol. 2017, 5, 986–1004. [Google Scholar] [CrossRef]
- Smolders, J.; van den Ouweland, J.; Geven, C.; Pickkers, P.; Kox, M. Letter to the Editor: Vitamin D deficiency in COVID-19: Mixing up cause and consequence. Metabolism 2021, 115, 154434. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. C-reactive protein levels in the early stage of COVID-19. Med. Mal. Infect. 2020, 50, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Allin, K.H.; Bojesen, S.E.; Nordestgaard, B.G. Baseline C-reactive protein is associated with incident cancer and survival in patients with cancer. J. Clin. Oncol. 2009, 27, 2217–2224. [Google Scholar] [CrossRef]
- Grant, W.B. Effect of interval between serum draw and follow-up period on relative risk of cancer incidence with respect to 25-hydroxyvitamin D level: Implications for meta-analyses and setting vitamin D guidelines. Dermatoendocrinology 2011, 3, 199–204. [Google Scholar] [CrossRef]
- Grant, W.B. 25-hydroxyvitamin D and breast cancer, colorectal cancer, and colorectal adenomas: Case-control versus nested case-control studies. Anticancer Res. 2015, 35, 1153–1160. [Google Scholar]
- Grant, W.B. Effect of follow-up time on the relation between prediagnostic serum 25-hydroxyvitamin D and all-cause mortality rate. Dermatoendocrinology 2012, 4, 198–202. [Google Scholar] [CrossRef]
- Wu, K.; Feskanich, D.; Fuchs, C.S.; Willett, W.C.; Hollis, B.W.; Giovannucci, E.L. A nested case control study of plasma 25-hydroxyvitamin D concentrations and risk of colorectal cancer. J. Natl. Cancer Inst. 2007, 99, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Feskanich, D.; Ma, J.; Fuchs, C.S.; Kirkner, G.J.; Hankinson, S.E.; Hollis, B.W.; Giovannucci, E.L. Plasma vitamin D metabolites and risk of colorectal cancer in women. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1502–1508. [Google Scholar]
- Gorham, E.D.; Garland, C.F.; Garland, F.C.; Grant, W.B.; Mohr, S.B.; Lipkin, M.; Newmark, H.L.; Giovannucci, E.; Wei, M.; Holick, M.F. Optimal vitamin D status for colorectal cancer prevention: A quantitative meta analysis. Am. J. Prev. Med. 2007, 32, 210–216. [Google Scholar] [CrossRef] [PubMed]
- McCullough, M.L.; Zoltick, E.S.; Weinstein, S.J.; Fedirko, V.; Wang, M.; Cook, N.R.; Eliassen, A.H.; Zeleniuch-Jacquotte, A.; Agnoli, C.; Albanes, D.; et al. Circulating Vitamin D and Colorectal Cancer Risk: An International Pooling Project of 17 Cohorts. J. Natl. Cancer Inst. 2019, 111, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.J.; Yu, K.; Horst, R.L.; Ashby, J.; Virtamo, J.; Albanes, D. Serum 25-hydroxyvitamin D and risks of colon and rectal cancer in Finnish men. Am. J. Epidemiol. 2011, 173, 499–508. [Google Scholar] [CrossRef]
- Lee, J.E.; Li, H.; Chan, A.T.; Hollis, B.W.; Lee, I.M.; Stampfer, M.J.; Wu, K.; Giovannucci, E.; Ma, J. Circulating levels of vitamin D and colon and rectal cancer: The Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev. Res. 2011, 4, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Kakourou, A.; Koutsioumpa, C.; Lopez, D.S.; Hoffman-Bolton, J.; Bradwin, G.; Rifai, N.; Helzlsouer, K.J.; Platz, E.A.; Tsilidis, K.K. Interleukin-6 and risk of colorectal cancer: Results from the CLUE II cohort and a meta-analysis of prospective studies. Cancer Causes Control 2015, 26, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Wu, K.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E.L. Plasma 25-hydroxyvitamin D and risk of colorectal cancer after adjusting for inflammatory markers. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Langseth, H.; Gislefoss, R.E.; Martinsen, J.I.; Dillner, J.; Ursin, G. Cohort Profile: The Janus Serum Bank Cohort in Norway. Int. J. Epidemiol. 2017, 46, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Jenab, M.; Bueno-de-Mesquita, H.B.; Ferrari, P.; van Duijnhoven, F.J.; Norat, T.; Pischon, T.; Jansen, E.H.; Slimani, N.; Byrnes, G.; Rinaldi, S.; et al. Association between pre-diagnostic circulating vitamin D concentration and risk of colorectal cancer in European populations:a nested case-control study. BMJ 2010, 340, b5500. [Google Scholar] [CrossRef]
- Woolcott, C.G.; Wilkens, L.R.; Nomura, A.M.; Horst, R.L.; Goodman, M.T.; Murphy, S.P.; Henderson, B.E.; Kolonel, L.N.; Le Marchand, L. Plasma 25-hydroxyvitamin D levels and the risk of colorectal cancer: The multiethnic cohort study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 130–134. [Google Scholar] [CrossRef]
- McCullough, M.L.; Robertson, A.S.; Rodriguez, C.; Jacobs, E.J.; Chao, A.; Carolyn, J.; Calle, E.E.; Willett, W.C.; Thun, M.J. Calcium, vitamin D, dairy products, and risk of colorectal cancer in the Cancer Prevention Study II Nutrition Cohort (United States). Cancer Causes Control 2003, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Iwasaki, M.; Sasazuki, S.; Inoue, M.; Tsugane, S.; Japan Public Health Center-Based Prospective Study, G. Plasma vitamin D and risk of colorectal cancer: The Japan Public Health Center-Based Prospective Study. Br. J. Cancer 2007, 97, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Goodman, G.E.; Thornquist, M.D.; Barnett, M.J.; Beresford, S.A.; LaCroix, A.Z.; Zheng, Y.; Neuhouser, M.L. Estimated intake of vitamin D and its interaction with vitamin A on lung cancer risk among smokers. Int. J. Cancer 2014, 135, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.J.; Purdue, M.P.; Smith-Warner, S.A.; Mondul, A.M.; Black, A.; Ahn, J.; Huang, W.Y.; Horst, R.L.; Kopp, W.; Rager, H.; et al. Serum 25-hydroxyvitamin D, vitamin D binding protein and risk of colorectal cancer in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Int. J. Cancer 2015, 136, E654–E664. [Google Scholar] [CrossRef]
- Tangrea, J.; Helzlsouer, K.; Pietinen, P.; Taylor, P.; Hollis, B.; Virtamo, J.; Albanes, D. Serum levels of vitamin D metabolites and the subsequent risk of colon and rectal cancer in Finnish men. Cancer Causes Control 1997, 8, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Sperati, F.; Razavi, P.; Agnoli, C.; Sieri, S.; Berrino, F.; Krogh, V.; Abbagnato, C.; Grioni, S.; Blandino, G.; et al. Endogenous sex steroids in premenopausal women and risk of breast cancer: The ORDET cohort. Breast Cancer Res. 2013, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, A.J.; Jones, M.E.; Schoemaker, M.J.; Hemming, J.; Thomas, D.; Williamson, J.; Ashworth, A. The Breakthrough Generations Study: Design of a long-term UK cohort study to investigate breast cancer aetiology. Br. J. Cancer 2011, 105, 911–917. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Manson, J.E.; Millen, A.; Pettinger, M.; Margolis, K.; Jacobs, E.T.; Shikany, J.M.; Vitolins, M.; Adams-Campbell, L.; Liu, S.; et al. The influence of health and lifestyle characteristics on the relation of serum 25-hydroxyvitamin D with risk of colorectal and breast cancer in postmenopausal women. Am. J. Epidemiol. 2012, 175, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Chandler, P.D.; Buring, J.E.; Manson, J.E.; Giovannucci, E.L.; Moorthy, M.V.; Zhang, S.; Lee, I.M.; Lin, J.H. Circulating Vitamin D Levels and Risk of Colorectal Cancer in Women. Cancer Prev. Res. 2015, 8, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Scarmo, S.; Afanasyeva, Y.; Lenner, P.; Koenig, K.L.; Horst, R.L.; Clendenen, T.V.; Arslan, A.A.; Chen, Y.; Hallmans, G.; Lundin, E.; et al. Circulating levels of 25-hydroxyvitamin D and risk of breast cancer: A nested case-control study. Breast Cancer Res. 2013, 15, R15. [Google Scholar] [CrossRef]
- Oh, E.Y.; Ansell, C.; Nawaz, H.; Yang, C.H.; Wood, P.A.; Hrushesky, W.J. Global breast cancer seasonality. Breast Cancer Res. Treat. 2010, 123, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Linseisen, J.; Slanger, T.; Kropp, S.; Mutschelknauss, E.J.; Flesch-Janys, D.; Chang-Claude, J. Serum 25-hydroxyvitamin D and risk of post-menopausal breast cancer--results of a large case-control study. Carcinogenesis 2008, 29, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Chang-Claude, J.; Linseisen, J. Plasma 25-hydroxyvitamin D and premenopausal breast cancer risk in a German case-control study. Int. J. Cancer 2009, 124, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Deng, Y.; Liu, K.; Zhou, L.; Li, N.; Zheng, Y.; Hao, Q.; Yang, S.; Wu, Y.; Zhai, Z.; et al. Vitamin D intake, blood vitamin D levels, and the risk of breast cancer: A dose-response meta-analysis of observational studies. Aging 2019, 11, 12708–12732. [Google Scholar] [CrossRef]
- McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Baggerly, L.L.; Garland, C.F.; Gorham, E.D.; Hollis, B.W.; Trump, D.L.; Lappe, J.M. Breast cancer risk markedly lower with serum 25-hydroxyvitamin D concentrations >/=60 vs >20 ng/mL (150 vs 50 nmol/L): Pooled analysis of two randomized trials and a prospective cohort. PLoS ONE 2018, 13, e0199265. [Google Scholar] [CrossRef]
- Han, J.; Guo, X.; Yu, X.; Liu, S.; Cui, X.; Zhang, B.; Liang, H. 25-Hydroxyvitamin D and Total Cancer Incidence and Mortality: A Meta-Analysis of Prospective Cohort Studies. Nutrients 2019, 11, 2295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, C.; Pan, W.; Gao, M.; He, W.; Mao, R.; Lin, T.; Huang, J. Comparative efficacy of vitamin D status in reducing the risk of bladder cancer: A systematic review and network meta-analysis. Nutrition 2016, 32, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alonso, P.; Boughanem, H.; Canudas, S.; Becerra-Tomas, N.; Fernandez de la Puente, M.; Babio, N.; Macias-Gonzalez, M.; Salas-Salvado, J. Circulating vitamin D levels and colorectal cancer risk: A meta-analysis and systematic review of case-control and prospective cohort studies. Crit. Rev. Food Sci. Nutr. 2021, 61, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Gorham, E.D. Dose-response of serum 25-hydroxyvitamin D in association with risk of colorectal cancer: A meta-analysis. J. Steroid Biochem. Mol. Biol. 2017, 168, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhu, G.; Xu, Y.; Zheng, S.; Tang, B.; Huang, H.; Wu, I.X.Y.; Huang, D.; Liu, Y.; Zhang, X. Association between Vitamin D Exposure and Head and Neck Cancer: A Systematic Review with Meta-Analysis. Front. Immunol. 2021, 12, 627226. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.F.; Zhao, T.; Han, J.M.; Li, S.; Li, D. Vitamin D and liver cancer risk: A meta-analysis of prospective studies. Asia Pac. J. Clin. Nutr. 2020, 29, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, X.; Li, X.; Gaman, M.A.; Kord-Varkaneh, H.; Rahmani, J.; Salehi-Sahlabadi, A.; Day, A.S.; Xu, Y. Serum Vitamin D Levels and Risk of Liver Cancer: A Systematic Review and Dose-Response Meta-Analysis of Cohort Studies. Nutr. Cancer 2021, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dong, Y.; Lu, C.; Wang, Y.; Peng, L.; Jiang, M.; Tang, Y.; Zhao, Q. Meta-analysis of the correlation between vitamin D and lung cancer risk and outcomes. Oncotarget 2017, 8, 81040–81051. [Google Scholar] [CrossRef]
- Feng, J.; Shan, L.; Du, L.; Wang, B.; Li, H.; Wang, W.; Wang, T.; Dong, H.; Yue, X.; Xu, Z.; et al. Clinical improvement following vitamin D3 supplementation in Autism Spectrum Disorder. Nutr. Neurosci. 2017, 20, 284–290. [Google Scholar] [CrossRef]
- Wei, H.; Jing, H.; Wei, Q.; Wei, G.; Heng, Z. Associations of the risk of lung cancer with serum 25-hydroxyvitamin D level and dietary vitamin D intake: A dose-response PRISMA meta-analysis. Medicine 2018, 97, e12282. [Google Scholar] [CrossRef]
- Xu, J.; Chen, K.; Zhao, F.; Huang, D.; Zhang, H.; Fu, Z.; Xu, J.; Wu, Y.; Lin, H.; Zhou, Y.; et al. Association between vitamin D/calcium intake and 25-hydroxyvitamin D and risk of ovarian cancer: A dose-response relationship meta-analysis. Eur. J. Clin. Nutr. 2021, 75, 417–429. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, X.Z.; Chen, W.J.; Wu, J.; Chen, Y.; Wu, C.C.; Wang, Z.N. Plasma 25-hydroxyvitamin D levels, vitamin D intake, and pancreatic cancer risk or mortality: A meta-analysis. Oncotarget 2017, 8, 64395–64406. [Google Scholar] [CrossRef]
- Gao, J.; Wei, W.; Wang, G.; Zhou, H.; Fu, Y.; Liu, N. Circulating vitamin D concentration and risk of prostate cancer: A dose-response meta-analysis of prospective studies. Ther. Clin. Risk Manag. 2018, 14, 95–104. [Google Scholar] [CrossRef]
- Wu, J.; Yang, N.; Youan, M. Dietary and circulating vitamin D and risk of renal cell carcinoma: A meta-analysis of observational studies. Int. Braz. J. Urol. 2021, 47, 733–744. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Zhang, Z.; Zhou, X.; Yao, J.; Zhang, R.; Liao, L.; Dong, J. Vitamin D deficiency as a risk factor for thyroid cancer: A meta-analysis of case-control studies. Nutrition 2019, 57, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Canc.cer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Lee, D.H.; Greenwood, D.C.; Manson, J.E.; Giovannucci, E. Vitamin D supplementation and total cancer incidence and mortality: A meta-analysis of randomized controlled trials. Ann. Oncol. 2019, 30, 733–743. [Google Scholar] [CrossRef]
- Zhang, X.; Niu, W. Meta-analysis of randomized controlled trials on vitamin D supplement and cancer incidence and mortality. Biosci. Rep. 2019, 39, BSR20190396. [Google Scholar] [CrossRef] [PubMed]
- Ekmekcioglu, C.; Haluza, D.; Kundi, M. 25-Hydroxyvitamin D Status and Risk for Colorectal Cancer and Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Epidemiological Studies. Int. J. Environ. Res. Public Health 2017, 14, 20127. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.C.; Zhang, Z.L.; Wan, Z.; Wang, L.; Weber, P.; Eggersdorfer, M.; Qin, L.Q.; Zhang, W. Circulating 25-hydroxyvitamin D and risk of lung cancer: A dose-response meta-analysis. Cancer Causes Control 2015, 26, 1719–1728. [Google Scholar] [CrossRef]
- Song, Z.Y.; Yao, Q.; Zhuo, Z.; Ma, Z.; Chen, G. Circulating vitamin D level and mortality in prostate cancer patients: A dose-response meta-analysis. Endocr. Connect. 2018, 7, R294–R303. [Google Scholar] [CrossRef]
- Pilz, S.; Trummer, C.; Theiler-Schwetz, V.; Grubler, M.R.; Verheyen, N.D.; Odler, B.; Karras, S.N.; Zittermann, A.; Marz, W. Critical Appraisal of Large Vitamin D Randomized Controlled Trials. Nutrients 2022, 14, 303. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef]
- Trivedi, D.P.; Doll, R.; Khaw, K.T. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: Randomised double blind controlled trial. BMJ 2003, 326, 469. [Google Scholar] [CrossRef]
- Wactawski-Wende, J.; Kotchen, J.M.; Anderson, G.L.; Assaf, A.R.; Brunner, R.L.; O’Sullivan, M.J.; Margolis, K.L.; Ockene, J.K.; Phillips, L.; Pottern, L.; et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N. Engl. J. Med. 2006, 354, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Lappe, J.M.; Travers-Gustafson, D.; Davies, K.M.; Recker, R.R.; Heaney, R.P. Vitamin D and calcium supplementation reduces cancer risk: Results of a randomized trial. Am. J. Clin. Nutr. 2007, 85, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Stuart, A.L.; Williamson, E.J.; Simpson, J.A.; Kotowicz, M.A.; Young, D.; Nicholson, G.C. Annual high-dose oral vitamin D and falls and fractures in older women: A randomized controlled trial. JAMA 2010, 303, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Avenell, A.; MacLennan, G.S.; Jenkinson, D.J.; McPherson, G.C.; McDonald, A.M.; Pant, P.R.; Grant, A.M.; Campbell, M.K.; Anderson, F.H.; Cooper, C.; et al. Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J. Clin. Endocrinol. Metab. 2012, 97, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Lappe, J.; Garland, C.; Gorham, E. Vitamin D Supplementation and Cancer Risk. JAMA 2017, 318, 299–300. [Google Scholar] [CrossRef]
- Scragg, R.; Khaw, K.T.; Toop, L.; Sluyter, J.; Lawes, C.M.M.; Waayer, D.; Giovannucci, E.; Camargo, C.A., Jr. Monthly High-Dose Vitamin D Supplementation and Cancer Risk: A Post Hoc Analysis of the Vitamin D Assessment Randomized Clinical Trial. JAMA Oncol. 2018, 4, e182178. [Google Scholar] [CrossRef]
- Neale, R.E.; Baxter, C.; Romero, B.D.; McLeod, D.S.A.; English, D.R.; Armstrong, B.K.; Ebeling, P.R.; Hartel, G.; Kimlin, M.G.; O’Connell, R.; et al. The D-Health Trial: A randomised controlled trial of the effect of vitamin D on mortality. Lancet Diabetes Endocrinol. 2022, 10, 120–128. [Google Scholar] [CrossRef]
- Heaney, R.P. Guidelines for optimizing design and analysis of clinical studies of nutrient effects. Nutr. Rev. 2014, 72, 48–54. [Google Scholar] [CrossRef]
- Grant, W.B.; Boucher, B.J.; Bhattoa, H.P.; Lahore, H. Why vitamin D clinical trials should be based on 25-hydroxyvitamin D concentrations. J. Steroid Biochem. Mol. Biol. 2018, 177, 266–269. [Google Scholar] [CrossRef]
- Hrushesky, W.J.; Sothern, R.B.; Rietveld, W.J.; Du Quiton, J.; Boon, M.E. Season, sun, sex, and cervical cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1940–1947. [Google Scholar] [CrossRef][Green Version]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Merrill, S.J.; Subramanian, M.; Godar, D.E. Worldwide cutaneous malignant melanoma incidences analyzed by sex, age, and skin type over time (1955–2007): Is HPV infection of androgenic hair follicular melanocytes a risk factor for developing melanoma exclusively in people of European-ancestry? Dermatoendocrinology 2016, 8, e1215391. [Google Scholar] [CrossRef] [PubMed]
- Loomis, D.; Huang, W.; Chen, G. The International Agency for Research on Cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: Focus on China. Chin. J. Cancer 2014, 33, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Grant, W.B. How strong is the evidence that solar ultraviolet B and vitamin D reduce the risk of cancer?: An examination using Hill’s criteria for causality. Dermatoendocrinology 2009, 1, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.B.; Gorham, E.D.; Alcaraz, J.E.; Kane, C.I.; Macera, C.A.; Parsons, J.K.; Wingard, D.L.; Garland, C.F. Does the evidence for an inverse relationship between serum vitamin D status and breast cancer risk satisfy the Hill criteria? Dermatoendocrinology 2012, 4, 152–157. [Google Scholar] [CrossRef]
- Frieden, T.R. Evidence for Health Decision Making—Beyond Randomized, Controlled Trials. N. Engl. J. Med. 2017, 377, 465–475. [Google Scholar] [CrossRef]
- Giovannucci, E.; Liu, Y.; Rimm, E.B.; Hollis, B.W.; Fuchs, C.S.; Stampfer, M.J.; Willett, W.C. Prospective study of predictors of vitamin D status and cancer incidence and mortality in men. J. Natl. Cancer Inst. 2006, 98, 451–459. [Google Scholar] [CrossRef]
- Colston, K.; Colston, M.J.; Feldman, D. 1,25-dihydroxyvitamin D3 and malignant melanoma: The presence of receptors and inhibition of cell growth in culture. Endocrinology 1981, 108, 1083–1086. [Google Scholar] [CrossRef]
- Abe, E.; Miyaura, C.; Sakagami, H.; Takeda, M.; Konno, K.; Yamazaki, T.; Yoshiki, S.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1alpha,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Mayorga, G.; Larriba, M.J.; Crespo, P.; Muñoz, A. Mechanisms of action of vitamin D in colon cancer. J. Steroid Biochem. Mol. Biol. 2019, 185, 1–6. [Google Scholar] [CrossRef]
- Wu, X.; Hu, W.; Lu, L.; Zhao, Y.; Zhou, Y.; Xiao, Z.; Zhang, L.; Zhang, H.; Li, X.; Li, W.; et al. Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm. Sinica B 2019, 9, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Antoszczak, M.; Kojs, Z.; Bednarek, W.; Markowska, J.; Huczynski, A. Role of vitamin D3 in selected malignant neoplasms. Nutrition 2020, 79, 110964. [Google Scholar] [CrossRef]
- Carlberg, C.; Velleuer, E. Vitamin D and the risk for cancer: A molecular analysis. Biochem. Pharmacol. 2022, 196, 114735. [Google Scholar] [CrossRef] [PubMed]
- Vanhevel, J.; Verlinden, L.; Doms, S.; Wildiers, H.; Verstuyf, A. The role of vitamin D in breast cancer risk and progression. Endocr. Relat. Cancer 2022, 29, R33–R55. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Chen, J.-Y.; Hung, W.-C. Vitamin D3 receptor/Sp1 complex is required for the induction of p27KIP1 expression by vitamin D3. Oncogene 2004, 23, 4856–4861. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, E.S.; Burnstein, K.L. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J. Biol. Chem. 2003, 278, 46862–46868. [Google Scholar] [CrossRef]
- Li, P.; Li, C.; Zhao, X.; Zhang, X.; Nicosia, S.V.; Bai, W. p27Kip1 stabilization and G1 arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. J. Biol. Chem. 2004, 279, 25260–25267. [Google Scholar] [CrossRef]
- Washington, M.N.; Kim, J.S.; Weigel, N.L. 1alpha,25-dihydroxyvitamin D3 inhibits C4-2 prostate cancer cell growth via a retinoblastoma protein (Rb)-independent G1 arrest. Prostate 2011, 71, 98–110. [Google Scholar] [CrossRef]
- Toropainen, S.; Väisänen, S.; Heikkinen, S.; Carlberg, C. The down-regulation of the human MYC gene by the nuclear hormone 1alpha,25-dihydroxyvitamin D3 is associated with cycling of corepressors and histone deacetylases. J. Mol. Biol. 2010, 400, 284–294. [Google Scholar] [CrossRef]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; García de Herreros, A.; Lafarga, M.; et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of b-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez-Morán, P.; Larriba, M.J.; Pálmer, H.G.; Valero, R.A.; Barbáchano, A.; Duñach, M.; García de Herreros, A.; Villalobos, C.; Berciano, M.T.; Lafarga, M.; et al. RhoA-ROCK and p38MAPK-MSK1 mediate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J. Cell Biol. 2008, 183, 697–710. [Google Scholar] [CrossRef]
- Álvarez-Díaz, S.; Valle, N.; García, J.M.; Peña, C.; Freije, J.M.; Quesada, V.; Astudillo, A.; Bonilla, F.; López-Otín, C.; Muñoz, A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J. Clin. Investig. 2009, 119, 2343–2358. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Tabar, R.; Nguyen-Yamamoto, L.; Tavera-Mendoza, L.E.; Quail, T.; Dimitrov, V.; An, B.S.; Glass, L.; Goltzman, D.; White, J.H. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc. Natl. Acad. Sci. USA 2012, 109, 18827–18832. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, S.; Guo, B. Vitamin D Suppresses Ovarian Cancer Growth and Invasion by Targeting Long Non-Coding RNA CCAT2. Int. J. Mol. Sci. 2020, 21, 72334. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Tabar, R.; Memari, B.; Wong, H.; Dimitrov, V.; Rochel, N.; White, J.H. The Tumor Suppressor FBW7 and the Vitamin D Receptor Are Mutual Cofactors in Protein Turnover and Transcriptional Regulation. Mol. Cancer Res. MCR 2019, 17, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Pálmer, H.G.; Sánchez-Carbayo, M.; Ordóñez-Morán, P.; Larriba, M.J.; Cordón-Cardó, C.; Muñoz, A. Genetic signatures of differentiation induced by 1a,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003, 63, 7799–7806. [Google Scholar] [PubMed]
- Zhu, Y.; Chen, P.; Gao, Y.; Ta, N.; Zhang, Y.; Cai, J.; Zhao, Y.; Liu, S.; Zheng, J. MEG3 Activated by Vitamin D Inhibits Colorectal Cancer Cells Proliferation and Migration via Regulating Clusterin. EBioMedicine 2018, 30, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, Z.; Cai, J.; Pan, C.; Lin, S.; Zhang, Y.; Chen, Y.; Leng, M.; He, C.; Zhou, P.; et al. VDR Signaling via the Enzyme NAT2 Inhibits Colorectal Cancer Progression. Front. Pharmacol. 2021, 12, 727704. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Y.; Jiang, H.; Xiao, Z.; Wu, X.; Zhang, H.; Zhao, Y.; Du, F.; Chen, Y.; Wu, Z.; et al. Vitamin D suppressed gastric cancer cell growth through downregulating CD44 expression in vitro and in vivo. Nutrition 2021, 91, 111413. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Weider, R.; Christakos, S. CCAAT enhancer-binding protein alpha is a molecular target of 1,25-dihydroxyvitamin D3 in MCF-7 breast cancer cells. J. Biol. Chem. 2009, 284, 3086–3095. [Google Scholar] [CrossRef]
- Boyle, B.J.; Zhao, X.Y.; Cohen, P.; Feldman, D. Insulin-like growth factor binding protein-3 mediates 1 alpha,25-dihydroxyvitamin d(3) growth inhibition in the LNCaP prostate cancer cell line through p21/WAF1. J. Urol. 2001, 165, 1319–1324. [Google Scholar] [CrossRef]
- Chang, S.; Gao, L.; Yang, Y.; Tong, D.; Guo, B.; Liu, L.; Li, Z.; Song, T.; Huang, C. miR-145 mediates the antiproliferative and gene regulatory effects of vitamin D3 by directly targeting E2F3 in gastric cancer cells. Oncotarget 2015, 6, 7675–7685. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wang, K.; Zheng, R.; Derwahl, M. 1,25 dihydroxyvitamin D3 inhibits the proliferation of thyroid cancer stem-like cells via cell cycle arrest. Endocr. Res. 2016, 41, 71–80. [Google Scholar] [CrossRef]
- Kulling, P.M.; Olson, K.C.; Olson, T.L.; Feith, D.J.; Loughran, T.P., Jr. Vitamin D in hematological disorders and malignancies. Eur. J. Haematol. 2017, 98, 187–197. [Google Scholar] [CrossRef]
- Tong, W.-M.; Kállay, E.; Hofer, H.; Hulla, W.; Manhardt, T.; Peterlik, M.; Cross, H.S. Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. Eur. J. Cancer 1998, 34, 2119–2125. [Google Scholar] [CrossRef]
- Tong, W.-M.; Hofer, H.; Ellinger, A.; Peterlik, M.; Cross, H.S. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: Relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol. Res. 1999, 11, 77–84. [Google Scholar] [PubMed]
- Andl, C.D.; Rustgi, A.K. No one-way street: Cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: A mechanism to regulate RTK activity. Cancer Biol. Ther. 2005, 4, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Barbáchano, A.; Ordóñez-Morán, P.; García, J.M.; Sánchez, A.; Pereira, F.; Larriba, M.J.; Martínez, N.; Hernández, J.; Landolfi, S.; Bonilla, F.; et al. SPROUTY-2 and E-cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene 2010, 29, 4800–4813. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, U.; Mustafi, R.; Sadiq, F.; Almoghrabi, A.; Mustafi, D.; Kreisheh, M.; Sundaramurthy, S.; Liu, W.; Konda, V.J.; Pekow, J.; et al. The renin-angiotensin system mediates EGF receptor-vitamin d receptor cross-talk in colitis-associated colon cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5848–5859. [Google Scholar] [CrossRef]
- Oh, Y.S.; Kim, E.J.; Schaffer, B.S.; Kang, Y.H.; Binderup, L.; MacDonald, R.G.; Park, J.H.Y. Synthetic low-calcaemic vitamin D3 analogues inhibit secretion of insulin-like growth factor II and stimulate production of insulin-like growth factor-binding protein-6 in conjunction with growth suppression of HT-29 colon cancer cells. Mol. Cell. Endocrinol. 2001, 183, 141–149. [Google Scholar] [CrossRef]
- Leng, S.L.; Leeding, K.S.; Whitehead, R.H.; Bach, L.A. Insulin-like growth factor (IGF)-binding protein-6 inhibits IGF-II-induced but not basal proliferation and adhesion of LIM 1215 colon cancer cells. Mol. Cell. Endocrinol. 2001, 174, 121–127. [Google Scholar] [CrossRef]
- Rosli, S.N.; Shintani, T.; Toratani, S.; Usui, E.; Okamoto, T. 1alpha,25(OH)(2)D(3) inhibits FGF-2 release from oral squamous cell carcinoma cells through down-regulation of HBp17/FGFBP-1. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 802–806. [Google Scholar] [CrossRef]
- Higaki, M.; Shintani, T.; Hamada, A.; Rosli, S.N.Z.; Okamoto, T. Eldecalcitol (ED-71)-induced exosomal miR-6887-5p suppresses squamous cell carcinoma cell growth by targeting heparin-binding protein 17/fibroblast growth factor-binding protein-1 (HBp17/FGFBP-1). In Vitro Cell. Dev. Biol. Anim. 2020, 56, 222–233. [Google Scholar] [CrossRef]
- Nazarova, N.; Golovko, O.; Blauer, M.; Tuohimaa, P. Calcitriol inhibits growth response to Platelet-Derived Growth Factor-BB in human prostate cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 189–196. [Google Scholar] [CrossRef]
- Wu, F.S.; Zheng, S.S.; Wu, L.J.; Teng, L.S.; Ma, Z.M.; Zhao, W.H.; Wu, W. Calcitriol inhibits the growth of MHCC97 heptocellular cell lines by down-modulating c-met and ERK expressions. Liver Int. 2007, 27, 700–707. [Google Scholar] [CrossRef]
- Inaba, M.; Koyama, H.; Hino, M.; Okuno, S.; Terada, M.; Nishizawa, Y.; Nishino, T.; Morii, H. Regulation of release of hepatocyte growth factor from human promyelocytic leukemia cells, HL-60, by 1,25-dihydroxyvitamin D3, 12-O-tetradecanoylphorbol 13-acetate, and dibutyryl cyclic adenosine monophosphate. Blood 1993, 82, 53–59. [Google Scholar] [CrossRef]
- Larriba, M.J.; González-Sancho, J.M.; Bonilla, F.; Muñoz, A. Interaction of vitamin D with membrane-based signaling pathways. Front. Physiol. 2014, 5, 60. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Swami, S.; Feldman, D. Vitamin D and breast cancer: Inhibition of estrogen synthesis and signaling. J. Steroid Biochem. Mol. Biol. 2010, 121, 343–348. [Google Scholar] [CrossRef]
- Zheng, W.; Cao, L.; Ouyang, L.; Zhang, Q.; Duan, B.; Zhou, W.; Chen, S.; Peng, W.; Xie, Y.; Fan, Q.; et al. Anticancer activity of 1,25-(OH)2D3 against human breast cancer cell lines by targeting Ras/MEK/ERK pathway. OncoTargets Ther. 2019, 12, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Seoane, S.; García-Caballero, T.; Gallego, R.; Macia, M.; González, L.O.; Vizoso, F.; Pérez-Fernández, R. Deregulation of the Pit-1 transcription factor in human breast cancer cells promotes tumor growth and metastasis. J. Clin. Investig. 2010, 120, 4289–4302. [Google Scholar] [CrossRef]
- Perez-Fernandez, R.; Seoane, S.; Garcia-Caballero, T.; Segura, C.; Macia, M. Vitamin D, Pit-1, GH, and PRL: Possible roles in breast cancer development. Curr. Med. Chem. 2007, 14, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Díaz, S.; Valle, N.; Ferrer-Mayorga, G.; Lombardía, L.; Herrera, M.; Domínguez, O.; Segura, M.F.; Bonilla, F.; Hernando, E.; Muñoz, A. MicroRNA-22 is induced by vitamin D and contributes to its antiproliferative, antimigratory and gene regulatory effects in colon cancer cells. Hum. Mol. Genet. 2012, 21, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zou, H.; Mo, J.; Jin, C.; Jiang, H.; Yu, C.; Jiang, Z.; Yang, Y.; He, B.; Wang, K. Micro1278 Leads to Tumor Growth Arrest, Enhanced Sensitivity to Oxaliplatin and Vitamin D and Inhibits Metastasis via KIF5B, CYP24A1, and BTG2, Respectively. Front. Oncol. 2021, 11, 637878. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, J.; Venkateswarlu, S.; Ko, T.; Brattain, M.G. Autocrine TGFbeta signaling mediates vitamin D3 analog-induced growth inhibition in breast cells. J. Cell. Physiol. 2001, 188, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Davis, B.H.; Sitrin, M.D.; Brasitus, T.A.; Bissonnette, M. Transforming growth factor-b 1 signaling contributes to Caco-2 cell growth inhibition induced by 1,25(OH)2D3. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G864–G874. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Honda, A.; Kurokawa, M. Impact of vitamin D level at diagnosis and transplantation on the prognosis of hematological malignancy: A meta-analysis. Blood Adv. 2021, 6, 1499–1511. [Google Scholar] [CrossRef]
- Gerousi, M.; Psomopoulos, F.; Kotta, K.; Tsagiopoulou, M.; Stavroyianni, N.; Anagnostopoulos, A.; Anastasiadis, A.; Gkanidou, M.; Kotsianidis, I.; Ntoufa, S.; et al. The Calcitriol/Vitamin D Receptor System Regulates Key Immune Signaling Pathways in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 285. [Google Scholar] [CrossRef]
- Olson, K.C.; Kulling, P.M.; Olson, T.L.; Tan, S.F.; Rainbow, R.J.; Feith, D.J.; Loughran, T.P., Jr. Vitamin D decreases STAT phosphorylation and inflammatory cytokine output in T-LGL leukemia. Cancer Biol. Ther. 2017, 18, 290–303. [Google Scholar] [CrossRef]
- McGlorthan, L.; Paucarmayta, A.; Casablanca, Y.; Maxwell, G.L.; Syed, V. Progesterone induces apoptosis by activation of caspase-8 and calcitriol via activation of caspase-9 pathways in ovarian and endometrial cancer cells in vitro. Apoptosis Int. J. Program. Cell Death 2021, 26, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Bao, J.; Li, P.; Nicosia, S.V.; Bai, W. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down-regulation of telomerase. J. Biol. Chem. 2004, 279, 53213–53221. [Google Scholar] [CrossRef] [PubMed]
- Kasiappan, R.; Shen, Z.; Tse, A.K.; Jinwal, U.; Tang, J.; Lungchukiet, P.; Sun, Y.; Kruk, P.; Nicosia, S.V.; Zhang, X.; et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J. Biol. Chem. 2012, 287, 41297–41309. [Google Scholar] [CrossRef]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Abu El Maaty, M.A.; Wölfl, S. Effects of 1,25(OH)(2)D(3) on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents. Nutrients 2017, 9, 87. [Google Scholar] [CrossRef]
- Kaler, P.; Galea, V.; Augenlicht, L.; Klampfer, L. Tumor associated macrophages protect colon cancer cells from TRAIL-induced apoptosis through IL-1beta-dependent stabilization of Snail in tumor cells. PLoS ONE 2010, 5, e11700. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, R.; Du, L.; Zhao, Z.; McMillan, E.; Kosti, A.; Yang, C.R.; Suraokar, M.; Wistuba, I.I.; Gazdar, A.F.; Minna, J.D.; et al. Genetic mutation of p53 and suppression of the miR-17 approximately 92 cluster are synthetic lethal in non-small cell lung cancer due to upregulation of vitamin D Signaling. Cancer Res. 2015, 75, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K. Vitamin D in autophagy signaling for health and diseases: Insights on potential mechanisms and future perspectives. J. Nutr. Biochem. 2022, 99, 108841. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Suares, A.; Tapia, C.; Gonzalez-Pardo, V. VDR agonists down regulate PI3K/Akt/mTOR axis and trigger autophagy in Kaposi’s sarcoma cells. Heliyon 2019, 5, e02367. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lian, H.; Zhao, Y.; Kauss, M.A.; Spindel, S. Vitamin D3 induces autophagy of human myeloid leukemia cells. J. Biol. Chem. 2008, 283, 25596–25605. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Tang, Y.; Zhong, M.; Wu, W. Antitumor effects and mechanisms of 1,25(OH)2D3 in the Pfeiffer diffuse large B lymphoma cell line. Mol. Med. Rep. 2019, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, F.; Li, J.; Wan, J.; Zhang, L.; Li, H.; Chen, A.; Chen, J.; Cai, T.; He, X.; et al. DNA damage-inducible transcript 4 is an innate guardian for human squamous cell carcinoma and an molecular vector for anti-carcinoma effect of 1,25(OH)2 D3. Exp. Dermatol. 2019, 28, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, J.; Chen, K.; Wang, J.; Dong, Q.; Xie, J.; Yuan, Y. Vitamin D promotes autophagy in AML cells by inhibiting miR-17-5p-induced Beclin-1 overexpression. Mol. Cell. Biochem. 2021, 476, 3951–3962. [Google Scholar] [CrossRef] [PubMed]
- Demasters, G.; Di, X.; Newsham, I.; Shiu, R.; Gewirtz, D.A. Potentiation of radiation sensitivity in breast tumor cells by the vitamin D3 analogue, EB 1089, through promotion of autophagy and interference with proliferative recovery. Mol. Cancer Ther. 2006, 5, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D 3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Goehe, R.W.; Di, X.; Hicks, M.A., 2nd; Torti, S.V.; Torti, F.M.; Harada, H.; Gewirtz, D.A. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy 2014, 10, 2346–2361. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.H.; Kang, S.H.; Choi, D.R.; Gil, M.N.; Yu, K.S.; Jeong, J.H.; Lee, N.S.; Lee, J.H.; Jeong, Y.G.; Kim, D.K.; et al. Autophagy enhancement contributes to the synergistic effect of vitamin D in temozolomide-based glioblastoma chemotherapy. Exp. Ther. Med. 2016, 11, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; Garcia de Herreros, A.; Muñoz, A. Vitamin D and the Epithelial to Mesenchymal Transition. Stem Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Barral, A.; Bustamante-Madrid, P.; Ferrer-Mayorga, G.; Barbáchano, A.; Larriba, M.J.; Muñoz, A. Vitamin D Effects on Cell Differentiation and Stemness in Cancer. Cancers 2020, 12, 2413. [Google Scholar] [CrossRef] [PubMed]
- Pendás-Franco, N.; González-Sancho, J.M.; Suarez, Y.; Aguilera, O.; Steinmeyer, A.; Gamallo, C.; Berciano, M.T.; Lafarga, M.; Muñoz, A. Vitamin D regulates the phenotype of human breast cancer cells. Differ. Res. Biol. Divers. 2007, 75, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J. Vitamin D and Breast Cancer: Mechanistic Update. J. Bone Miner. Res. Plus 2021, 5, e10582. [Google Scholar] [CrossRef] [PubMed]
- Kouchi, Z.; Fujiwara, Y.; Yamaguchi, H.; Nakamura, Y.; Fukami, K. Phosphatidylinositol 5-phosphate 4-kinase type II beta is required for vitamin D receptor-dependent E-cadherin expression in SW480 cells. Biochem. Biophys. Res. Commun. 2011, 408, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; Carvalho, J.; Duraes, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. 2012, 32, 249–257. [Google Scholar] [PubMed]
- Upadhyay, S.K.; Verone, A.; Shoemaker, S.; Qin, M.; Liu, S.; Campbell, M.; Hershberger, P.A. 1,25-Dihydroxyvitamin D3 (1,25(OH)2D3) Signaling Capacity and the Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer (NSCLC): Implications for Use of 1,25(OH)2D3 in NSCLC Treatment. Cancers 2013, 5, 1504–1521. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Chen, S.C.; Yeh, C.N.; Pang, J.H.; Shen, S.C.; Hsu, J.T.; Liu, Y.Y.; Chen, L.W.; Kuo, S.F.; Takano, M.; et al. MART-10, a less calcemic vitamin D analog, is more potent than 1alpha,25-dihydroxyvitamin D3 in inhibiting the metastatic potential of MCF-7 breast cancer cells in vitro. J. Steroid Biochem. Mol. Biol. 2014, 139, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Yeh, C.N.; Hsu, J.T.; Jan, Y.Y.; Chen, L.W.; Kuo, S.F.; Takano, M.; Kittaka, A.; Chen, T.C.; Chen, W.T.; et al. The vitamin D analog, MART-10, represses metastasis potential via downregulation of epithelial-mesenchymal transition in pancreatic cancer cells. Cancer Lett. 2014, 354, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Findlay, V.J.; Moretz, R.E.; Wang, C.; Vaena, S.G.; Bandurraga, S.G.; Ashenafi, M.; Marshall, D.T.; Watson, D.K.; Camp, E.R. Slug expression inhibits calcitriol-mediated sensitivity to radiation in colorectal cancer. Mol. Carcinog. 2014, 53, E130–E139. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.F.; Gao, S.H.; Wang, P.; Zhang, H.M.; Liu, L.Z.; Ye, M.X.; Zhou, G.M.; Zhang, Z.L.; Li, B.Y. 1alpha,25(OH)(2)D(3) Suppresses the Migration of Ovarian Cancer SKOV-3 Cells through the Inhibition of Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2016, 17, 1285. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Barbáchano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Muñoz, A.; Larriba, M.J. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665. [Google Scholar] [CrossRef] [PubMed]
- Barbáchano, A.; Fernández-Barral, A.; Pereira, F.; Segura, M.F.; Ordóñez-Morán, P.; Carrillo-de Santa Pau, E.; González-Sancho, J.M.; Hanniford, D.; Martinez, N.; Costales-Carrera, A.; et al. SPROUTY-2 represses the epithelial phenotype of colon carcinoma cells via upregulation of ZEB1 mediated by ETS1 and miR-200/miR-150. Oncogene 2016, 35, 2991–3003. [Google Scholar] [CrossRef] [PubMed]
- Koeffler, H.P.; Amatruda, T.; Ikekawa, N.; Kobayashi, Y.; DeLuca, H.F. Induction of macrophage differentiation of human normal and leukemic myeloid stem cells by 1,25-dihydroxyvitamin D3 and its fluorinated analogues. Cancer Res. 1984, 44, 5624–5628. [Google Scholar] [PubMed]
- Tanaka, H.; Abe, E.; Miyaura, C.; Shiina, Y.; Suda, T. 1 alpha,25-dihydroxyvitamin D3 induces differentiation of human promyelocytic leukemia cells (HL-60) into monocyte-macrophages, but not into granulocytes. Biochem. Biophys. Res. Commun. 1983, 117, 86–92. [Google Scholar] [CrossRef]
- Abe, J.; Moriya, Y.; Saito, M.; Sugawara, Y.; Suda, T.; Nishii, Y. Modulation of cell growth, differentiation, and production of interleukin-3 by 1 alpha,25-dihydroxyvitamin D3 in the murine myelomonocytic leukemia cell line WEHI-3. Cancer Res. 1986, 46, 6316–6321. [Google Scholar]
- Gocek, E.; Studzinski, G.P. Vitamin D and differentiation in cancer. Crit. Rev. Clin. Lab. Sci. 2009, 46, 190–209. [Google Scholar] [CrossRef]
- Hmama, Z.; Nandan, D.; Sly, L.; Knutson, K.L.; Herrera-Velit, P.; Reiner, N.E. 1alpha,25-dihydroxyvitamin D(3)-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J. Exp. Med. 1999, 190, 1583–1594. [Google Scholar] [CrossRef]
- Ji, Y.; Studzinski, G.P. Retinoblastoma protein and CCAAT/enhancer-binding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res. 2004, 64, 370–377. [Google Scholar] [CrossRef]
- Marchwicka, A.; Marcinkowska, E. Regulation of Expression of CEBP Genes by Variably Expressed Vitamin D Receptor and Retinoic Acid Receptor alpha in Human Acute Myeloid Leukemia Cell Lines. Int. J. Mol. Sci. 2018, 19, 1918. [Google Scholar] [CrossRef]
- Song, J.H.; Park, E.; Kim, M.S.; Cho, K.M.; Park, S.H.; Lee, A.; Song, J.; Kim, H.J.; Koh, J.T.; Kim, T.S. l-Asparaginase-mediated downregulation of c-Myc promotes 1,25(OH)2 D3-induced myeloid differentiation in acute myeloid leukemia cells. Int. J. Cancer 2017, 140, 2364–2374. [Google Scholar] [CrossRef]
- Sabatier, M.; Boet, E.; Zaghdoudi, S.; Guiraud, N.; Hucteau, A.; Polley, N.; Cognet, G.; Saland, E.; Lauture, L.; Farge, T.; et al. Activation of Vitamin D Receptor Pathway Enhances Differentiating Capacity in Acute Myeloid Leukemia with Isocitrate Dehydrogenase Mutations. Cancers 2021, 13, 5243. [Google Scholar] [CrossRef]
- Hickish, T.; Cunningham, D.; Colston, K.; Millar, B.C.; Sandle, J.; Mackay, A.G.; Soukop, M.; Sloane, J. The effect of 1,25-dihydroxyvitamin D3 on lymphoma cell lines and expression of vitamin D receptor in lymphoma. Br. J. Cancer 1993, 68, 668–672. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [PubMed]
- The_Cancer_Genome_Atlas_Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- González-Sancho, J.M.; Larriba, M.J.; Muñoz, A. Wnt and Vitamin D at the Crossroads in Solid Cancer. Cancers 2020, 12, 3434. [Google Scholar] [CrossRef]
- Aguilera, O.; Peña, C.; García, J.M.; Larriba, M.J.; Ordóñez-Morán, P.; Navarro, D.; Barbáchano, A.; López de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007, 28, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Beildeck, M.E.; Islam, M.; Shah, S.; Welsh, J.; Byers, S.W. Control of TCF-4 expression by VDR and vitamin D in the mouse mammary gland and colorectal cancer cell lines. PLoS ONE 2009, 4, e7872. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Zhang, Y.G.; Wu, S.; Lu, R.; Lin, Z.; Zheng, Y.; Chen, H.; Cs-Szabo, G.; Sun, J. Vitamin D receptor is a novel transcriptional regulator for Axin1. J. Steroid Biochem. Mol. Biol. 2017, 165, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Nguyen, P.; Kershaw, K.M.; Lay, A.R.; Ostertag-Hill, C.A.; Sherman, M.H.; Downes, M.; Liddle, C.; Evans, R.M.; Dawson, D.W. Calcipotriol Targets LRP6 to Inhibit Wnt Signaling in Pancreatic Cancer. Mol. Cancer Res. MCR 2015, 13, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Katz, L.H.; Munoz, N.M.; Gu, S.; Shin, J.H.; Jogunoori, W.S.; Lee, M.H.; Belkin, M.D.; Kim, S.B.; White, J.C.; et al. Vitamin D Deficiency Promotes Liver Tumor Growth in Transforming Growth Factor-beta/Smad3-Deficient Mice Through Wnt and Toll-like Receptor 7 Pathway Modulation. Sci. Rep. 2016, 6, 30217. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Z.H.; Fu, L.; Song, J.; Xie, D.D.; Yu, D.X.; Xu, D.X.; Sun, G.P. Calcitriol inhibits migration and invasion of renal cell carcinoma cells by suppressing Smad2/3-, STAT3- and beta-catenin-mediated epithelial-mesenchymal transition. Cancer Sci. 2020, 111, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, N.I.; Pálmer, H.G.; García, M.; González-Martín, A.; del Rio, M.; Barettino, D.; Volpert, O.; Muñoz, A.; Jiménez, B. 1a,25-Dihydroxyvitamin D3 regulates the expression of Id1 and Id2 genes and the angiogenic phenotype of human colon carcinoma cells. Oncogene 2005, 24, 6533–6544. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1alpha,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. Cancer Ther. 2007, 6, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Pendás-Franco, N.; García, J.M.; Peña, C.; Valle, N.; Pálmer, H.G.; Heinaniemi, M.; Carlberg, C.; Jiménez, B.; Bonilla, F.; Muñoz, A.; et al. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene 2008, 27, 4467–4477. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, W.H.; Suh, D.H.; Kim, K.; No, J.H.; Kim, Y.B. Calcitriol Combined with Platinum-based Chemotherapy Suppresses Growth and Expression of Vascular Endothelial Growth Factor of SKOV-3 Ovarian Cancer Cells. Anticancer Res. 2021, 41, 2945–2952. [Google Scholar] [CrossRef]
- Piotrowska, A.; Beserra, F.P.; Wierzbicka, J.M.; Nowak, J.I.; Zmijewski, M.A. Vitamin D Enhances Anticancer Properties of Cediranib, a VEGFR Inhibitor, by Modulation of VEGFR2 Expression in Melanoma Cells. Front. Oncol. 2021, 11, 763895. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.Y.; Yao, J.; Lee, Y.F. 1alpha, 25-dihydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis 2006, 27, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- García-Quiroz, J.; Rivas-Suárez, M.; Garcia-Becerra, R.; Barrera, D.; Martínez-Reza, I.; Ordaz-Rosado, D.; Santos-Martinez, N.; Villanueva, O.; Santos-Cuevas, C.L.; Avila, E.; et al. Calcitriol reduces thrombospondin-1 and increases vascular endothelial growth factor in breast cancer cells: Implications for tumor angiogenesis. J. Steroid Biochem. Mol. Biol. 2014, 144, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Mantell, D.J.; Owens, P.E.; Bundred, N.J.; Mawer, E.B.; Canfield, A.E. 1 alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ. Res. 2000, 87, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.J.; Johnson, C.S.; Modzelewski, R.A.; Trump, D.L. Antiproliferative effects of 1alpha,25-dihydroxyvitamin D(3) and vitamin D analogs on tumor-derived endothelial cells. Endocrinology 2002, 143, 2508–2514. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Wong, M.K.; Flynn, G.; Yu, W.D.; Johnson, C.S.; Trump, D.L. Differential antiproliferative effects of calcitriol on tumor-derived and matrigel-derived endothelial cells. Cancer Res. 2006, 66, 8565–8573. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.; Chung, I.; Yu, W.D.; Romano, M.; Modzelewski, R.A.; Johnson, C.S.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) selectively inhibits proliferation of freshly isolated tumor-derived endothelial cells and induces apoptosis. Oncology 2006, 70, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.; Feldman, D. 1,25-Dihydroxyvitamin D3 decreases human prostate cancer cell adhesion and migration. Mol. Cell. Endocrinol. 2000, 164, 133–143. [Google Scholar] [CrossRef]
- Tokar, E.J.; Webber, M.M. Cholecalciferol (vitamin D3) inhibits growth and invasion by up-regulating nuclear receptors and 25-hydroxylase (CYP27A1) in human prostate cancer cells. Clin. Exp. Metastasis 2005, 22, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, J.; Zuo, S.; Ma, J.; Zhang, J.; Chen, G.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. 1,25(OH)2D3 attenuates TGF-beta1/beta2-induced increased migration and invasion via inhibiting epithelial-mesenchymal transition in colon cancer cells. Biochem. Biophys. Res. Commun. 2015, 468, 130–135. [Google Scholar] [CrossRef]
- Hsu, J.W.; Yasmin-Karim, S.; King, M.R.; Wojciechowski, J.C.; Mickelsen, D.; Blair, M.L.; Ting, H.J.; Ma, W.L.; Lee, Y.F. Suppression of prostate cancer cell rolling and adhesion to endothelium by 1alpha,25-dihydroxyvitamin D3. Am. J. Pathol. 2011, 178, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luco, A.L.; Camirand, A.; St-Arnaud, R.; Kremer, R. Vitamin D Regulates CXCL12/CXCR4 and Epithelial-to-Mesenchymal Transition in a Model of Breast Cancer Metastasis to Lung. Endocrinology 2021, 162, bqab049. [Google Scholar] [CrossRef]
- González-Sancho, J.M.; Alvarez-Dolado, M.; Muñoz, A. 1,25-Dihydroxyvitamin D3 inhibits tenascin-C expression in mammary epithelial cells. FEBS Lett. 1998, 426, 225–228. [Google Scholar] [CrossRef]
- Koli, K.; Keski-Oja, J. 1alpha,25-dihydroxyvitamin D3 and its analogues down-regulate cell invasion-associated proteases in cultured malignant cells. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2000, 11, 221–229. [Google Scholar]
- Bao, B.Y.; Yeh, S.D.; Lee, Y.F. 1alpha,25-dihydroxyvitamin D3 inhibits prostate cancer cell invasion via modulation of selective proteases. Carcinogenesis 2006, 27, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wilmanski, T.; Barnard, A.; Parikh, M.R.; Kirshner, J.; Buhman, K.; Burgess, J.; Teegarden, D. 1alpha,25-Dihydroxyvitamin D Inhibits the Metastatic Capability of MCF10CA1a and MDA-MB-231 Cells in an In Vitro Model of Breast to Bone Metastasis. Nutr. Cancer 2016, 68, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Vanoirbeek, E.; Eelen, G.; Verlinden, L.; Carmeliet, G.; Mathieu, C.; Bouillon, R.; O’Connor, R.; Xiao, G.; Verstuyf, A. PDLIM2 expression is driven by vitamin D and is involved in the pro-adhesion, and anti-migration and -invasion activity of vitamin D. Oncogene 2014, 33, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, C.J.; Grebenc, D.; Balinth, S.; Welsh, J.E. Vitamin D regulation of HAS2, hyaluronan synthesis and metabolism in triple negative breast cancer cells. J. Steroid Biochem. Mol. Biol. 2020, 201, 105688. [Google Scholar] [CrossRef]
- Ma, Y.; Luo, W.; Bunch, B.L.; Pratt, R.N.; Trump, D.L.; Johnson, C.S. 1,25D3 differentially suppresses bladder cancer cell migration and invasion through the induction of miR-101-3p. Oncotarget 2017, 8, 60080–60093. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.H.; Chiang, E.I.; Syu, J.N.; Chao, C.Y.; Lin, H.Y.; Lin, C.C.; Yang, M.D.; Tsai, S.Y.; Tang, F.Y. Treatment of 13-cis retinoic acid and 1,25-dihydroxyvitamin D3 inhibits TNF-alpha-mediated expression of MMP-9 protein and cell invasion through the suppression of JNK pathway and microRNA 221 in human pancreatic adenocarcinoma cancer cells. PLoS ONE 2021, 16, e0247550. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Pure, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Ben Tov, A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.; Hernández, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Wang, B.; Zheng, Y.; Jiang, X.; Pan, Z.; Ren, J. Vitamin D prevents the intestinal fibrosis via induction of vitamin D receptor and inhibition of transforming growth factor-beta1/Smad3 pathway. Dig. Dis. Sci. 2015, 60, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.T.; Brentani, H.; Roela, R.A.; Katayama, M.L.; Lima, L.; Rolim, C.F.; Milani, C.; Folgueira, M.A.; Brentani, M.M. Differences in transcriptional effects of 1alpha,25 dihydroxyvitamin D3 on fibroblasts associated to breast carcinomas and from paired normal breast tissues. J. Steroid Biochem. Mol. Biol. 2013, 133, 12–24. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Niell, N.; Larriba, M.J.; Ferrer-Mayorga, G.; Sánchez-Pérez, I.; Cantero, R.; Real, F.X.; Del Peso, L.; Muñoz, A.; González-Sancho, J.M. The human PKP2/plakophilin-2 gene is induced by Wnt/beta-catenin in normal and colon cancer-associated fibroblasts. Int. J. Cancer 2018, 142, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Mayorga, G.; Niell, N.; Cantero, R.; González-Sancho, J.M.; Del Peso, L.; Muñoz, A.; Larriba, M.J. Vitamin D and Wnt3A have additive and partially overlapping modulatory effects on gene expression and phenotype in human colon fibroblasts. Sci. Rep. 2019, 9, 8085. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Li, L.; Wang, G.; Deng, X.; Li, Z.; Kong, X. VDR signaling inhibits cancer-associated-fibroblasts’ release of exosomal miR-10a-5p and limits their supportive effects on pancreatic cancer cells. Gut 2019, 68, 950–951. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernandez Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef]
- Fujii, M.; Sato, T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat. Mater. 2021, 20, 156–169. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Barbachano, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Prieto, I.; Rodriguez-Salas, N.; Larriba, M.J.; Muñoz, A. Organoids and Colorectal Cancer. Cancers 2021, 13, 2657. [Google Scholar] [CrossRef]
- Fernández-Barral, A.; Costales-Carrera, A.; Buira, S.P.; Jung, P.; Ferrer-Mayorga, G.; Larriba, M.J.; Bustamante-Madrid, P.; Dominguez, O.; Real, F.X.; Guerra-Pastrián, L.; et al. Vitamin D differentially regulates colon stem cells in patient-derived normal and tumor organoids. FEBS J. 2020, 287, 53–72. [Google Scholar] [CrossRef]
- Costales-Carrera, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Dominguez, O.; Guerra-Pastrián, L.; Cantero, R.; Del Peso, L.; Burgos, A.; Barbáchano, A.; Muñoz, A. Comparative Study of Organoids from Patient-Derived Normal and Tumor Colon and Rectal Tissue. Cancers 2020, 12, 2302. [Google Scholar] [CrossRef]
- Vaughan-Shaw, P.G.; Blackmur, J.P.; Grimes, G.; Ooi, L.Y.; Ochocka-Fox, A.M.; Dunbar, K.; von Kriegsheim, A.; Rajasekaran, V.; Timofeeva, M.; Walker, M.; et al. Vitamin D treatment induces in vitro and ex vivo transcriptomic changes indicating anti-tumor effects. FASEB J. 2022, 36, e22082. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zhu, Z.; Chen, Y.; Song, J.; Huang, Y.; Song, K.; Zhong, J.; Xu, X.; Wei, J.; Wang, C.; et al. Small-molecule activating SIRT6 elicits therapeutic effects and synergistically promotes anti-tumor activity of vitamin D3 in colorectal cancer. Theranostics 2020, 10, 5845–5864. [Google Scholar] [CrossRef] [PubMed]
- McCray, T.; Pacheco, J.V.; Loitz, C.C.; Garcia, J.; Baumann, B.; Schlicht, M.J.; Valyi-Nagy, K.; Abern, M.R.; Nonn, L. Vitamin D sufficiency enhances differentiation of patient-derived prostate epithelial organoids. iScience 2021, 24, 101974. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.L.; Minden, A.; Furmanski, P.; Bak, M.J.; Cai, L.; Wernyj, R.; Sargsyan, D.; Cheng, D.; Wu, R.; Kuo, H.D.; et al. Analysis of the Transcriptome: Regulation of Cancer Stemness in Breast Ductal Carcinoma In Situ by Vitamin D Compounds. Cancer Prev. Res. 2020, 13, 673–686. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Wahler, J.; Das Gupta, S.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Suh, N. HES1-mediated inhibition of Notch1 signaling by a Gemini vitamin D analog leads to decreased CD44(+)/CD24(-/low) tumor-initiating subpopulation in basal-like breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 111–121. [Google Scholar] [CrossRef]
- Wahler, J.; So, J.Y.; Cheng, L.C.; Maehr, H.; Uskokovic, M.; Suh, N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 148–155. [Google Scholar] [CrossRef]
- Ferronato, M.J.; Nadal Serrano, M.; Arenas Lahuerta, E.J.; Bernado Morales, C.; Paolillo, G.; Martinez-Sabadell Aliguer, A.; Santalla, H.; Mascaro, M.; Vitale, C.; Fall, Y.; et al. Vitamin D analogues exhibit antineoplastic activity in breast cancer patient-derived xenograft cells. J. Steroid Biochem. Mol. Biol. 2021, 208, 105735. [Google Scholar] [CrossRef] [PubMed]
- Ao, T.; Kikuta, J.; Ishii, M. The Effects of Vitamin D on Immune System and Inflammatory Diseases. Biomolecules 2021, 11, 1624. [Google Scholar] [CrossRef] [PubMed]
- Hanel, A.; Neme, A.; Malinen, M.; Hamalainen, E.; Malmberg, H.R.; Etheve, S.; Tuomainen, T.P.; Virtanen, J.K.; Bendik, I.; Carlberg, C. Common and personal target genes of the micronutrient vitamin D in primary immune cells from human peripheral blood. Sci. Rep. 2020, 10, 21051. [Google Scholar] [CrossRef]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Catala-Moll, F.; Ferrete-Bonastre, A.G.; Godoy-Tena, G.; Morante-Palacios, O.; Ciudad, L.; Barbera, L.; Fondelli, F.; Martínez-Cáceres, E.M.; Rodriguez-Ubreva, J.; Li, T.; et al. Vitamin D receptor, STAT3, and TET2 cooperate to establish tolerogenesis. Cell Rep. 2022, 38, 110244. [Google Scholar] [CrossRef] [PubMed]
- Korf, H.; Wenes, M.; Stijlemans, B.; Takiishi, T.; Robert, S.; Miani, M.; Eizirik, D.L.; Gysemans, C.; Mathieu, C. 1,25-Dihydroxyvitamin D3 curtails the inflammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology 2012, 217, 1292–1300. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, M.; Guo, Y.; Song, Z.; Liu, B. 1,25-Dihydroxyvitamin D(3) Promotes High Glucose-Induced M1 Macrophage Switching to M2 via the VDR-PPARgamma Signaling Pathway. BioMed Res. Int. 2015, 2015, 157834. [Google Scholar] [CrossRef]
- Von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, A.; Malki, A. Vitamin D Signaling in Inflammation and Cancer: Molecular Mechanisms and Therapeutic Implications. Molecules 2020, 25, 3219. [Google Scholar] [CrossRef]
- Dankers, W.; Colin, E.M.; van Hamburg, J.P.; Lubberts, E. Vitamin D in Autoimmunity: Molecular Mechanisms and Therapeutic Potential. Front. Immunol. 2016, 7, 697. [Google Scholar] [CrossRef] [PubMed]
- Karkeni, E.; Morin, S.O.; Bou Tayeh, B.; Goubard, A.; Josselin, E.; Castellano, R.; Fauriat, C.; Guittard, G.; Olive, D.; Nunes, J.A. Vitamin D Controls Tumor Growth and CD8+ T Cell Infiltration in Breast Cancer. Front. Immunol. 2019, 10, 1307. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C.; Burcham, G.N.; Calvert, R.D.; Elzey, B.D.; Ratliff, T.L. 1alpha, 25 Dihydroxyvitamin D (1,25(OH)2D) inhibits the T cell suppressive function of myeloid derived suppressor cells (MDSC). J. Steroid Biochem. Mol. Biol. 2020, 198, 105557. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, F.; Xing, J.C.; Zhang, F.; Xu, J.Z.; Zhang, Z.H. 1,25(OH)2 D3 inhibited Th17 cells differentiation via regulating the NF-kappaB activity and expression of IL-17. Cell Prolif. 2018, 51, e12461. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Lahav, M.; Shany, S.; Tobvin, D.; Chaimovitz, C.; Douvdevani, A. Vitamin D decreases NFkappaB activity by increasing IkappaBalpha levels. Nephrol. Dial. Transpl. 2006, 21, 889–897. [Google Scholar] [CrossRef]
- Tse, A.K.; Zhu, G.Y.; Wan, C.K.; Shen, X.L.; Yu, Z.L.; Fong, W.F. 1alpha,25-Dihydroxyvitamin D3 inhibits transcriptional potential of nuclear factor kappa B in breast cancer cells. Mol. Immunol. 2010, 47, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Feldman, D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 311–336. [Google Scholar] [CrossRef]
- Moreno, J.; Krishnan, A.V.; Swami, S.; Nonn, L.; Peehl, D.M.; Feldman, D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005, 65, 7917–7925. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.C.; Fedirko, V.; Baron, J.A.; Barry, E.L.; Flanders, W.D.; McCullough, M.L.; Yacoub, R.; Raavi, T.; Rutherford, R.E.; Seabrook, M.E.; et al. Inflammation Modulation by Vitamin D and Calcium in the Morphologically Normal Colorectal Mucosa of Patients with Colorectal Adenoma in a Clinical Trial. Cancer Prev. Res. 2021, 14, 65–76. [Google Scholar] [CrossRef]
- Bruns, H.; Buttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra247. [Google Scholar] [CrossRef] [PubMed]
- Min, D.; Lv, X.B.; Wang, X.; Zhang, B.; Meng, W.; Yu, F.; Hu, H. Downregulation of miR-302c and miR-520c by 1,25(OH)2D3 treatment enhances the susceptibility of tumour cells to natural killer cell-mediated cytotoxicity. Br. J. Cancer 2013, 109, 723–730. [Google Scholar] [CrossRef]
- Neumann, F.; Acker, F.; Schormann, C.; Pfreundschuh, M.; Bittenbring, J.T. Determination of optimum vitamin D3 levels for NK cell-mediated rituximab- and obinutuzumab-dependent cellular cytotoxicity. Cancer Immunol. Immunother. 2018, 67, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Bittenbring, J.T.; Neumann, F.; Altmann, B.; Achenbach, M.; Reichrath, J.; Ziepert, M.; Geisel, J.; Regitz, E.; Held, G.; Pfreundschuh, M. Vitamin D deficiency impairs rituximab-mediated cellular cytotoxicity and outcome of patients with diffuse large B-cell lymphoma treated with but not without rituximab. J. Clin. Oncol. 2014, 32, 3242–3248. [Google Scholar] [CrossRef] [PubMed]
- Mortara, L.; Gariboldi, M.B.; Bosi, A.; Bregni, M.; Pinotti, G.; Guasti, L.; Squizzato, A.; Noonan, D.M.; Monti, E.; Campiotti, L. Vitamin D Deficiency has a Negative Impact on Cetuximab-Mediated Cellular Cytotoxicity against Human Colon Carcinoma Cells. Target. Oncol. 2018, 13, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lipplaa, A.; Fernandes, R.; Marshall, A.; Lorigan, P.; Dunn, J.; Myers, K.A.; Barker, E.; Newton-Bishop, J.; Middleton, M.R.; Corrie, P.G. 25-hydroxyvitamin D serum levels in patients with high risk resected melanoma treated in an adjuvant bevacizumab trial. Br. J. Cancer 2018, 119, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, V.; Bouttier, M.; Boukhaled, G.; Salehi-Tabar, R.; Avramescu, R.G.; Memari, B.; Hasaj, B.; Lukacs, G.L.; Krawczyk, C.M.; White, J.H. Hormonal vitamin D up-regulates tissue-specific PD-L1 and PD-L2 surface glycoprotein expression in humans but not mice. J. Biol. Chem. 2017, 292, 20657–20668. [Google Scholar] [CrossRef] [PubMed]
- Bendix, M.; Greisen, S.; Dige, A.; Hvas, C.L.; Bak, N.; Jorgensen, S.P.; Dahlerup, J.F.; Deleuran, B.; Agnholt, J. Vitamin D increases programmed death receptor-1 expression in Crohn’s disease. Oncotarget 2017, 8, 24177–24186. [Google Scholar] [CrossRef]
- Stucci, L.S.; D’Oronzo, S.; Tucci, M.; Macerollo, A.; Ribero, S.; Spagnolo, F.; Marra, E.; Picasso, V.; Orgiano, L.; Marconcini, R.; et al. Vitamin D in melanoma: Controversies and potential role in combination with immune check-point inhibitors. Cancer Treat. Rev. 2018, 69, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kutuzova, G.D.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 regulates genes responsible for detoxification in intestine. Toxicol. Appl. Pharmacol. 2007, 218, 37–44. [Google Scholar] [CrossRef]
- Lindh, J.D.; Bjorkhem-Bergman, L.; Eliasson, E. Vitamin D and drug-metabolising enzymes. Photochem. Photobiol. Sci. 2012, 11, 1797–1801. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Echchgadda, I.; Song, C.S. Vitamin D receptor regulation of the steroid/bile acid sulfotransferase SULT2A1. Methods Enzymol. 2005, 400, 165–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schuetz, E.G.; Xu, Y.; Thummel, K.E. Interplay between vitamin D and the drug metabolizing enzyme CYP3A4. J. Steroid Biochem. Mol. Biol. 2013, 136, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef]
- Peterlik, M. Role of bile acid secretion in human colorectal cancer. Wien. Med. Wochenschr. 2008, 158, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Matsunawa, M.; Akagi, D.; Uno, S.; Endo-Umeda, K.; Yamada, S.; Ikeda, K.; Makishima, M. Vitamin D receptor activation enhances benzo[a]pyrene metabolism via CYP1A1 expression in macrophages. Drug Metab. Dispos. 2012, 40, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y. The Role of the Gut Microbiome in Colorectal Cancer. Clin. Colon Rectal Surg. 2018, 31, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, C.; Zhong, Y.N.; Zhao, F.; Hao, Z.; Xu, Y.; Lai, R.; Shen, G.; Yin, X. Effect and mechanism of vitamin D on the development of colorectal cancer based on intestinal flora disorder. J. Gastroenterol. Hepatol. 2020, 35, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L. Vitamin D and microbiota: Two sides of the same coin in the immunomodulatory aspects. Int. Immunopharmacol. 2020, 79, 106112. [Google Scholar] [CrossRef]
- Wang, J.; Thingholm, L.B.; Skieceviciene, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Ruhlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Lu, R.; Shang, M.; Zhang, Y.G.; Jiao, Y.; Xia, Y.; Garrett, S.; Bakke, D.; Bauerl, C.; Martinez, G.P.; Kim, C.H.; et al. Lactic Acid Bacteria Isolated From Korean Kimchi Activate the Vitamin D Receptor-autophagy Signaling Pathways. Inflamm. Bowel Dis. 2020, 26, 1199–1211. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Lu, R.; Wu, S.; Chatterjee, I.; Zhou, D.; Xia, Y.; Sun, J. Vitamin D Receptor Protects Against Dysbiosis and Tumorigenesis via the JAK/STAT Pathway in Intestine. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 729–746. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.M.; Murphy, L.; Zgaga, L.; Bennett, K. De novo vitamin D supplement use post-diagnosis is associated with breast cancer survival. Breast Cancer Res. Treat. 2018, 172, 179–190. [Google Scholar] [CrossRef]
- Carlberg, C.; Haq, A. The concept of the personal vitamin D response index. J. Steroid Biochem. Mol. Biol. 2018, 175, 12–17. [Google Scholar] [CrossRef]
- Dimitrov, V.; Barbier, C.; Ismailova, A.; Wang, Y.; Dmowski, K.; Salehi-Tabar, R.; Memari, B.; Groulx-Boivin, E.; White, J.H. Vitamin D-regulated Gene Expression Profiles: Species-specificity and Cell-specific Effects on Metabolism and Immunity. Endocrinology 2021, 162, bqaa218. [Google Scholar] [CrossRef]


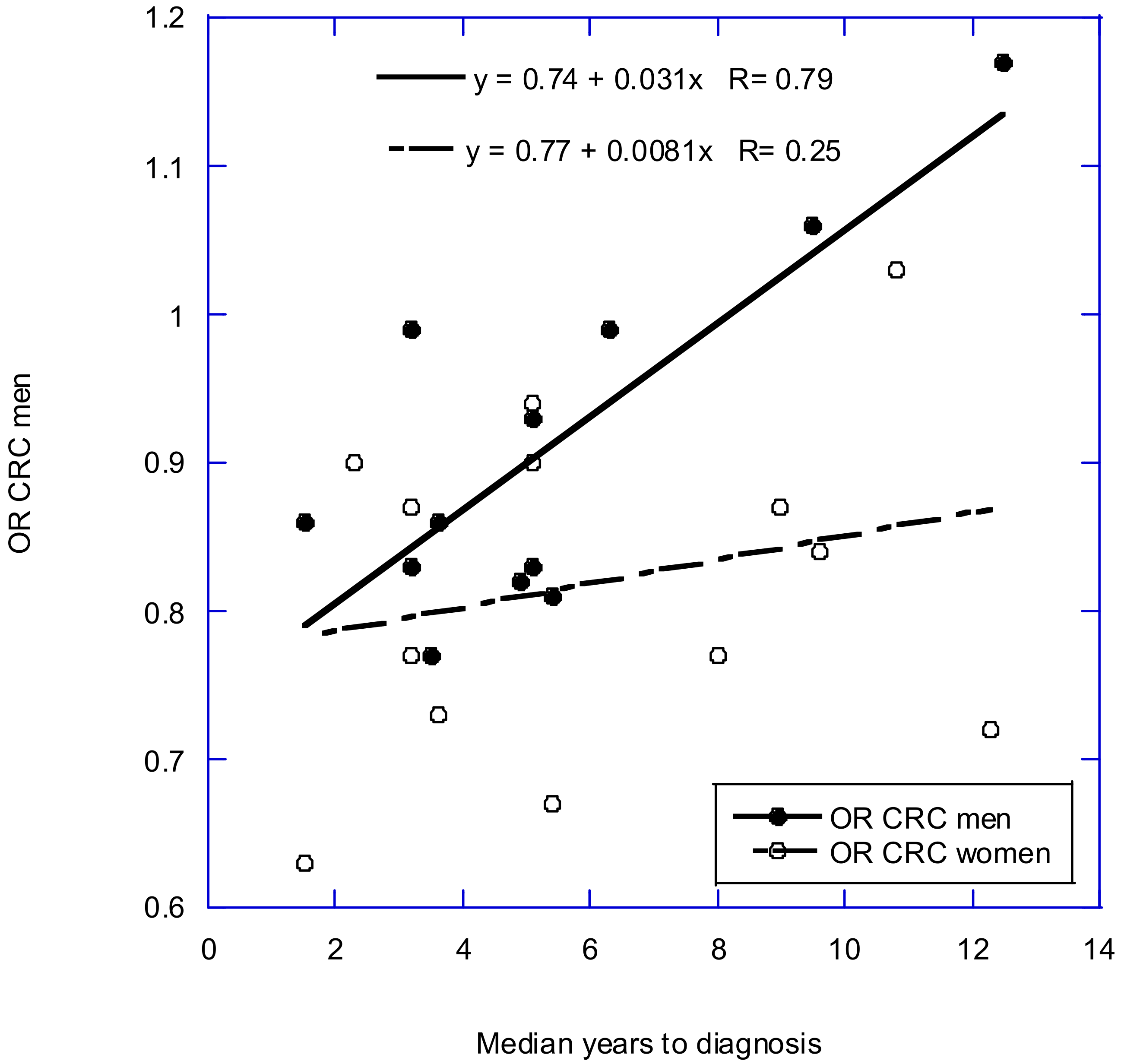{kind=link}
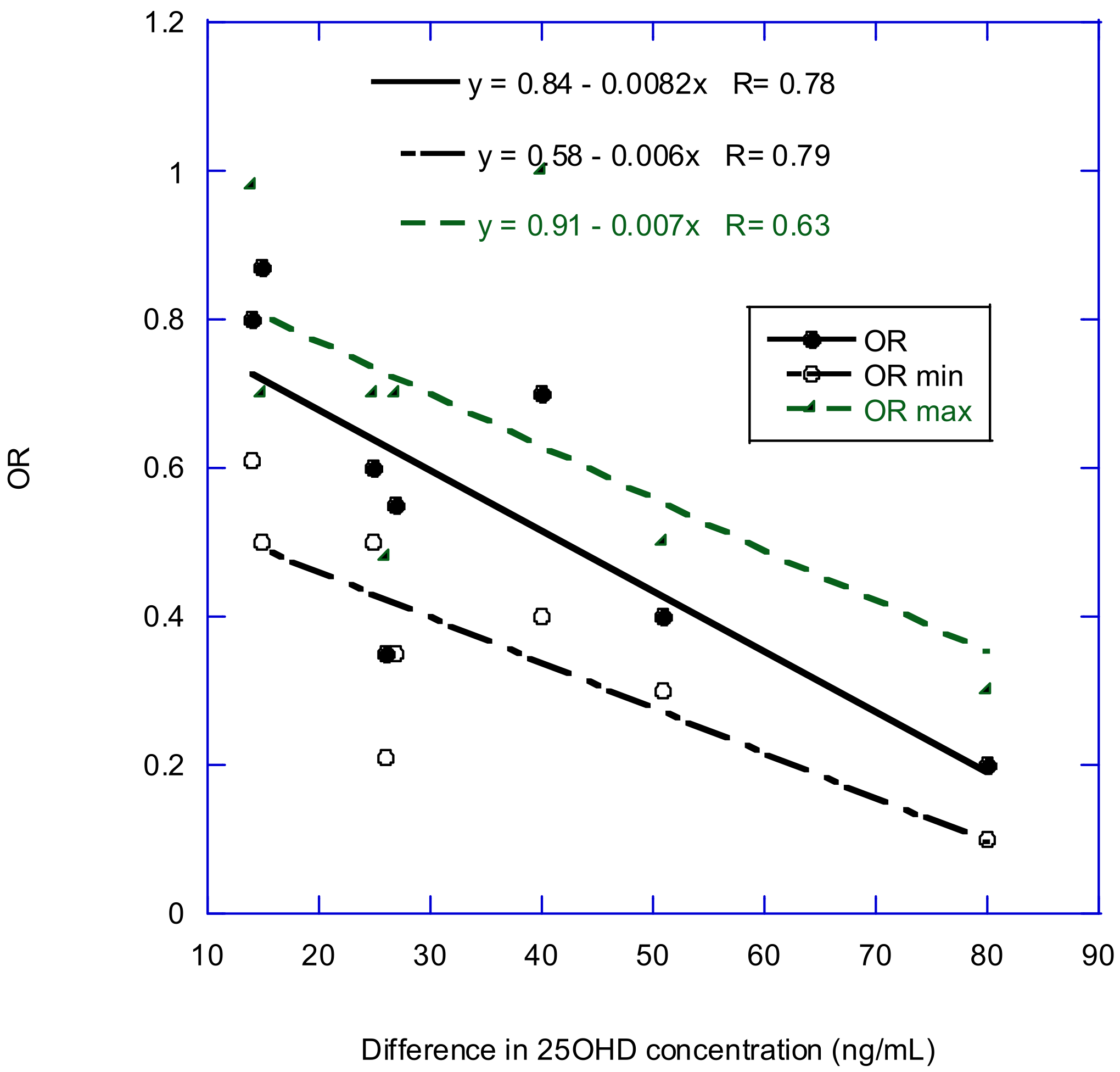{kind=link}
{kind=link}
| Country(ies) | Solar UVB Index | Latitude (°N) | Incidence or Mortality; Years of Data | No. of Cases | Confounding Factors | Ref. |
|---|---|---|---|---|---|---|
| U.S. | Surface UVB, July 1992, TOMS | 25–45 | Mortality, 1950–1994 | 9.5 million, 1970–1994 | None | [13] |
| Japan | Annual hours of solar radiation | 30–45 | Mortality, 2000 | 180,000 | Fat intake for colon, rectum, and prostate; salt intake for stomach cancer | [21] |
| U.S. (white pop.) | Surface UVB, July 1992 | 25–45 | Mortality, 1950–1994 | 9.5 million, 1970–1994 | Alcohol consumption, Hispanic heritage, lung cancer (index for smoking), poverty, urban/rural residence | [14] |
| U.S. | 300–320 nm, TOMS, north vs. south | 25–45 | Incidence, 1998–2002; mortality, 1993–2002 | Incidence, 3.4 million; mortality, 3.5 million | Age, air quality, alcohol, exercise, income, outdoor occupation, poverty, smoking, urban/rural residence | [22] |
| Japan | Global solar radiation | 30–45 | Mortality, 1998–2002 | ~900,000 | Dietary factors, smoking, socioeconomic conditions | [23] |
| China | TOMS, 305 nm | 22–50 | Incidence, 1998–2002; mortality, 1990–1992 | Urban/rural residence | [18] | |
| Russia | Latitude | 43–69 | Incidence, mortality, 2008 | incidence, ~250,000; deaths, ~140,000 | None | [24] |
| Nordic countries | Lip cancer less lung cancer incidence | 55–70 | Incidence, 1961–2005 | 2.8 million | Lung cancer | [25] |
| Incidence [29] (×1000) | Cancer | USA [22] | China [18] | Russia [24] | Nordic [25] |
|---|---|---|---|---|---|
| 219.4 | Lung | –M, FNS, –R, –U | M, FNS | ||
| 194.3 | Breast | F | –F, –R, –U | M, F | |
| 192.3 | Prostate | M | –M | MNS | |
| 147.0 | Colorectal | M, F, R | |||
| 106.1 | Colon | M, F | M, F | ||
| 71.0 | Bladder, urinary | M, F | –M, –F, –R, –U | M, F | |
| 68.7 | Melanoma | –M, –F | M + F | M | |
| 66.0 | Non-Hodgkin lymphoma | M, F | NS | ||
| 57.8 | Kidney | M, F | M + F | M, FNS | |
| 44.8 | Leukemia | M, F | MNS, FNS, R, –U | ||
| 42.5 | Pancreas | M, F | M + F | M, FNS | |
| 42.2 | Uterus, corpus | F | FNS | ||
| 40.9 | Rectum | M, F | M, FNS | ||
| 37.2 | Thyroid | MNS, F | |||
| 35.7 | Oral cavity and pharynx | –M, –F | |||
| 23.1 | Oral | M | |||
| 22.6 | Myeloma | M, F | M + F | ||
| 22.6 | Liver | –M, –F, –R, –U | M, FNS | ||
| 22.1 | Brain | M | |||
| 21.6 | Ovary | FNS | |||
| 21.1 | Stomach (gastric) | M, F | M, F, R, –U | M + F | M?, FNS |
| 16.5 | Esophagus | M | M, F, R, –U | M + F | MNS |
| 12.6 | Pharynx | –M, –F, –R, –U | –(M + F) | ||
| 12.3 | Larynx | M | |||
| 11.3 | Cervix | –F | F, R,–U | ||
| 9.8 | Gallbladder | F | M | ||
| 9.8 | Biliary, other | M, F | M + F | ||
| 8.5 | Hodgkin lymphoma | M, F | |||
| 8.4 | Testis | NS | |||
| 6.2 | Small intestine | M, F | M | ||
| 5.9 | Skin, other | –M, –F | –(M + F) | –M | |
| 5.3 | Anus, etc. | –M, –F | |||
| 3.6 | Vulva | F |
| Mortality [29] (×1000) | Cancer | Japan [23] | USA [14] | USA [22] | China [18] | Russia [24] |
|---|---|---|---|---|---|---|
| 159.4 | Lung | M, F | M, F, R, U | |||
| 69.1 | Colorectal | M | M, F, R | |||
| 49.9 | Colon | M, F | M, F | M + F | ||
| 40.6 | Breast | FNS | M, F | F | F, R | –(M + F) |
| 35.2 | Pancreas | M, F | M, FNS | M, F | M + F | |
| 27.4 | Prostate | MNS | MNS | M | M | |
| 21.9 | Leukemia | M, F | MNS, FNS | |||
| 19.5 | Non-Hodgkin lymphoma | M, F | M, F | |||
| 19.2 | Rectum | M, F | M, F | M + F | ||
| 18.2 | Liver | M | –M, –F | M, F, R | ||
| 14.6 | Ovary | F | F | F | ||
| 14.5 | Esophagus | M | M, F | M | M, F, R | M + F |
| 14.3 | Bladder, urinary | M, F | M, F | M, F, R | M + F | |
| 13.9 | Kidney | M, F | M, F | M + F | ||
| 12.9 | Brain | –M, –F | ||||
| 10.6 | Myeloma | M, F | M + F | |||
| 10.6 | Stomach (gastric) | M, FNS | M, F | M, F | M, F, U | M + F |
| 8.7 | Melanoma | –M, –F | M + F | |||
| 7.8 | Uterus, corpus | F | F | |||
| 7.6 | Oral cavity and pharynx | –M, –F | ||||
| 5.4 | Oral | MNS, FNS | ||||
| 4.1 | Cervix | F | –F | –F, –R, –U | ||
| 3.7 | Larynx | M, F? | MNS, FNS | M + F | ||
| 3.4 | Gallbladder | MNS, F | M, F | M, F | ||
| 3.4 | Biliary, other | M. F | ||||
| 2.9 | Skin, other | –M, –F | –(M + F) | |||
| 2.2 | Pharynx | –M. –F, –R, –U | ||||
| 1.6 | Thyroid | MNS, F | ||||
| 1.5 | Bone and joint | –M, –F | ||||
| 1.3 | Hodgkin lymphoma | M, F | M, F | |||
| 1.1 | Small intestine | MNS, F | ||||
| 0.9 | Vulva | F | F | |||
| 0.7 | Anus, etc. | –M, –F | M + F |
| Study | Follow-Up (Years) | RR | Ref. |
|---|---|---|---|
| Men | |||
| ATBC2 | 12.5 | 1.17 | [48] |
| PHS | 9.50 | 1.06 | [49] |
| CLUE II | 3.20 | 0.99 | [50] |
| HPFS | 6.30 | 0.99 | [51] |
| JANUS | 5.10 | 0.93 | [52] |
| EPIC | 3.60 | 0.86 | [53] |
| MEC | 1.50 | 0.86 | [54] |
| CPS-II | 3.20 | 0.83 | [55] |
| JPHC | 5.10 | 0.83 | [56] |
| CARET | 4.90 | 0.82 | [57] |
| PLCO | 5.40 | 0.81 | [58] |
| ABCT1 | 3.50 | 0.77 | [59] |
| Women | |||
| ORDET | 10.8 | 1.03 | [60] |
| JPHC | 5.10 | 0.94 | [56] |
| JANUS | 5.10 | 0.90 | [52] |
| BGS | 2.30 | 0.90 | [61] |
| CLUE-II | 9.00 | 0.87 | [50] |
| WHI | 3.20 | 0.87 | [62] |
| NHS | 9.60 | 0.84 | [51] |
| CPS-II | 3.20 | 0.77 | [55] |
| WHS | 8.00 | 0.77 | [63] |
| EPIC | 3.60 | 0.73 | [53] |
| NYUWHS | 12.3 | 0.72 | [64] |
| PLCO | 5.40 | 0.67 | [58] |
| MEC | 1.50 | 0.63 | [54] |
| Cancer Site | N Studies, Cases, Controls | Type of Study | Follow-Up (Years) | RR (95% CI), High vs. Low | Ref. |
|---|---|---|---|---|---|
| All | 8, —, — | Prospective, incidence | 5–28 | 0.86 (0.73–1.02) | [70] |
| All | 17, —, — | Prospective, mortality | 5–28 | 0.81 (0.71–0.93) | [70] |
| Bladder | 5, 1251, 1332 | CC and NCC, incidence | 0 (4), 12, 13 | 0.70 (0.56–0.88) | [71] |
| Bladder | 2, 2264, 2258 | Cohort, incidence | 14, 28 | 0.80 (0.67–0.94) | [71] |
| Breast | 44, 29,095, 53,060 | CC and NCC, incidence | 0.57 (0.48–0.66) | [68] | |
| Breast | 6, 2257, — | Cohort, incidence | 1.17 (0.92–1.48) | [68] | |
| Colorectal | 11, —, — | 1 CC, 9 NCC, 1 meta-analysis, incidence | 0–20 | 0.60 (0.53–0.68) | [72] |
| Colorectal | 6, 1252, — | Cohort, incidence | 8–20 | 0.80 (0.66–0.97) | [72] |
| Colorectal | 15, 6691, — | NCC, incidence | 0.67 (0.59–0.76) | [73] | |
| Head and neck | 5, —, — | Cohort, incidence | 7, 15 | 0.68 (0.59–0.78) | [74] |
| Liver | 8, 992, — | Cohort, incidence | 6–28 | 0.78 (0.63–0.95) | [75] |
| Liver | 6, 776, — | Cohort, incidence | (0.75), 16–22 | 0.53 (0.41–0.68) | [76] |
| Lung | 8, 1386, — | Cohort, incidence | 7–26 | 0.72 (0.61–0.85) | [77] |
| Lung | 9, —, — | 7 Cohort, 2 CC, incidence | 0.84 (0.74–0.95) | [78] | |
| Lung | 3, —, — | 1 Cohort, 2 CC, mortality | 0.76 (0.61–0.94) | [78] | |
| Lung | 12, —, — | 7 Cohort, 5 CC | 1.05 (0.95–1.16) | [79] | |
| Ovarian | 8, —, — | CC, cohort, NCC | 0.86 (0.56–1.33) | [80] | |
| Pancreatic | 5, 1068, — | 2 Cohort, 3 NCC, incidence | 6.5–21 | 1.02 (0.66–1.57) | [81] |
| Pancreatic | 5, 2003, — | Cohort, mortality | 6.5–21 | 0.81 (0.68–0.96) | [81] |
| Prostate | 19, 12,786 | 16 NCC, 3 cohort, incidence | 1.15 (1.06–1.24) | [82] | |
| Renal | 5, —, — | 4 Cohort (+1 CC, 3.5% weighting), incidence | (0), 7–22 | 0.76 (0.64–0.89) | [83] |
| Renal | 1, —, — | CC, incidence | 0 | 0.30 (0.13–0.72) | [83] |
| Thyroid | 6, 387, 457 | CC, incidence | 0 | Deficiency, 1.30 (1.00–1.69), p = 0.05 | [84] |
| Cancer Site | N Studies | Type of Study | RR (95% CI), High vs. Low Vitamin D Intake | Ref. |
|---|---|---|---|---|
| Breast | 17 | 8 CC, 9 cohorts | 0.97 (0.92–1.07), per 400 IU/d | [68] |
| Colorectal | 12 | CC | 0.75 (0.67–0.81) | [72] |
| Colorectal | 6 | Cohort | 0.89 (0.80–1.02) | [72] |
| Head and neck | 3 | 0.75 (0.58–0.97) | [74] | |
| Lung | 6 | Cohort | 0.89 (0.83–0.97) | [77] |
| Lung | 5 | Cohort | 0.85 (0.74–0.98) | [79] |
| Renal | 4 | CC | 0.80 (0.67–0.95) | [83] |
| Renal | 4 | Cohort | 0.97 (0.77–1.22) | [83] |
| Overall cancer death | 0.84 (0.74–0.95) | [87] |
| Cancer | Min 25(OH)D (ng/mL) | Max 25(OH)D (ng/mL) | OR (95% CI) | Ref. |
|---|---|---|---|---|
| All, inc | 2 | 25 | ~0.6 | [70] |
| Bladder, inc | 3 | 30 | ~0.55 (0.35–0.70) | [71] |
| Breast, inc (Song et al.) | 5 | 85 | ~0.2 (0.1–0.3) | [68] |
| Breast, inc | 15 | 70 | 0.18 (0.04–0.62) | [69] |
| Colorectal, inc | 4 | 55 | ~0.4 (0.3–0.5) | [73] |
| Colorectal, inc | 10 | 50 | ~0.7 (0.4–1.0) | [88] |
| Liver, inc | 4 | 30 | 0.35 (0.21–0.48) | [76] |
| Liver, inc | 5 | 30 | ~0.6 (0.5–0.7) | [75] |
| Lung, inc | 6 | 21 | 0.87 (0.76–0.97) | [89] |
| Lung, inc | 10 | 24 | 0.80 (0.61–0.98) | [78] |
| Lung, mort | 10 | 42 | 0.37 (0.25–0.53) | [78] |
| Prostate, inc | 0 | 60 | ~1.3 (1.1–1.8) | [82] |
| Prostate, mort | 4 | 43 | ~0.55 (0.2–1.1) | [90] |
| Location | Mean Baseline and Achieved 25(OH)D (ng/mL), Treatment Arm | Vitamin D Dose (IU) Frequency in Treatment Arm | Duration (Years) | Mean BMI (kg/m2) | Original Purpose | Reference |
|---|---|---|---|---|---|---|
| UK | 100,000/ (4 months) | 5.5 | 24 ± 3 | fracture incidence, cause of death | [93] | |
| USA | 400/day + 1 g/day Ca | 7 | 28? | colorectal cancer incidence, mortality | [94] | |
| Nebraska, USA | 29, 38 | 1100/day + 1.5 g/day Ca; 1.5 g/day Ca | 4 | 29 ± 6 | fracture incidence | [95] |
| Australia | 21, 24–48 | 500,000/year | falls and fractures | [96] | ||
| England, Scotland | 800/day; 1 g/d Ca; 800/day + 1 g/day Ca | 3 | [97] | |||
| Nebraska, USA | 33, 44 | 2000/day + 1500 mg/day Ca | 4 | 30 ± 7 | cancer | [98] |
| New Zealand | 26, -- | 100,000/mo | 3.3 ± 0.8 | 28 ± 5 | disease incidence with respect to bolus dose of vitamin D | [99] |
| USA | 30, 41 | 2000/day | 5.3 | 31 | cancer and cardiovascular disease risk | [85] |
| Australia | 31 ± 10, 46 ± 12 | 60,000/ month | 5 | 27? | mortality by disease | [100] |
| Location | Number of Participants, Cancer Cases, Deaths, Treatment Arm | Number of Participants, Cancer Cases, Deaths, Non-Vitamin D Arm | RR, Incidence (95% CI) | RR, Mortality (95% CI) | Reference |
|---|---|---|---|---|---|
| UK | 1345, 163, 63 | 1341, 147, 72 | 1.11 (0.86–1.42) | 0.86 (0.61–1.20) | [93] |
| USA | 18,176, 1634, 344 | 18,106, 1655, 382 | 0.98 (0.91–1.05) | 0.89 (0.77–1.03) | [94] |
| Nebraska, USA | 446, 13, -- | 733, 37, -- | 0.76 (0.38–1.55) | [95] | |
| Australia | 1131, 7 | 1125, 10 | 0.70 (0.27–1.82) | [96] | |
| England, Scotland | 1306, 182, 78; 1311, 189, 95 | 1343, 187, 73; 1332, 165, 83 | 1.24 (0.80–2.28) | 1.26 (0.73–3.26) | [97] |
| Nebraska, USA | 1156, 45, -- | 1147, 64, -- | 0.70 (0.47–1.02) | [98] | |
| New Zealand | 2558, 302, -- | 2550, 293, -- | 1.01 (0.81–1.25) | [99] | |
| USA | 12,927, 793, 154 | 12,946, 824, 187 | 0.96 (0.88–1.06) | 0.83 (0.67–1.02) | [85] |
| Meta-analysis for ten incidence trials and five mortality rate trials | 0.98 (0.93–1.03) | 0.87 (0.79–0.96) | [86] | ||
| Australia | 21,315, --, 221 | 10,662, --, 189 | 1.15 (0.96–1.39) | [100] |
| Year | Finding | Reference |
|---|---|---|
| 1936 | Sun exposure can cause skin cancer but reduce risk of internal cancer. | [1] |
| 1937 | US Navy personnel highly exposed to sun had high skin cancer rates but low internal cancer rates. | [2] |
| 1941 | Cancer mortality rates for whites in the U.S. found inversely related to a solar radiation index while skin cancer (melanoma) mortality rates were directly related. | [3] |
| 1980 | Annual solar radiation dose inversely correlated with colon cancer mortality rate, USA, vitamin D production suggested. | [6] |
| 1985 | Dietary vitamin D and calcium inversely correlated with colorectal cancer incidence. | [7] |
| 1989 | Serum 25(OH)D concentration inversely correlated with colon cancer incidence. | [8] |
| 1990 | Annual solar radiation dose inversely correlated with breast cancer mortality rate in the U.S. | [9] |
| 2002 | Mortality rates for thirteen types of cancer are inversely correlated with solar UVB doses in the U.S., 1970–1994. | [13] |
| 2006 | A Harvard cohort study finding that incidence of several types of cancer were inversely correlated with predicted 25(OH)D concentration. | [111] |
| 2006 | An ecological study in the U.S. finding that incidence and mortality rates for many types of cancer were inversely correlated with solar UVB doses. | [22] |
| 2007 | A meta-analysis presenting a 25(OH)D concentration-colorectal cancer incidence relationship. | [46] |
| 2007 | An RCT conducted in the U.S. finding that vitamin D supplementation significantly reduced risk of all-cancer incidence rate. | [95] |
| Mechanism | Cancer Type Model | References |
|---|---|---|
| Inhibition of cell proliferation | Breast, prostate, colon, ovarian, gastric thyroid, hepatocellular, leukemias, lymphomas | [111,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,152,153,154,155,156,158,159,161,162] |
| Induction of differentiation | Leukemia, colon, breast | [112,124,125,126,138,176,185,187,196,197,198,199,200,201,202,203,204,205] |
| EMT inhibition | Colon, ovarian, breast, pancreas | [126,142,189,190,191,192,193,194,195] |
| Sensitization of autophagy | Colon, prostate, breast, ovarian, lung | [115,116,118,163,164,165,168,169] |
| Induction of autophagy | Breast, Kaposi’s sarcoma, lymphoma, cutaneous squamous cell carcinoma, leukemia | [171,172,173,174,175,176,177,178,179,180,181] |
| Wnt/β-catenin antagonism | Colon, breast, ovarian, hepatocellular, renal, head and neck, Kaposi’s sarcoma | [124,210,211,212,213,214,215,216,217] |
| Invasion, angiogenesis, metastasis | Colon, prostate, breast, ovarian, renal, pancreas | [193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,218,219,220,221,222,223,230,231,232,233,234,235,236,237,238,239,240,241,242] |
| Cancer-associated fibroblasts | Breast, colon, pancreas, liver | [248,250,252,253,254,255,256,257] |
| Normal/cancer stem cells | Breast, colon, pancreas, liver | [261,262,263,264,265,266,267,268,269] |
| Detoxification and microbiome | Colon, perhaps other cancer types | [296,297,298,299,300,301,302,303,305,306,307,308,309] |
| Immune system regulation | Many | [272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288] |
| Combination with immunotherapy | Lymphoma, melanoma, colon, breast | [289,290,291,292,293,294,295] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, A.; Grant, W.B. Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms. Nutrients 2022, 14, 1448. https://doi.org/10.3390/nu14071448
Muñoz A, Grant WB. Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms. Nutrients. 2022; 14(7):1448. https://doi.org/10.3390/nu14071448
Chicago/Turabian StyleMuñoz, Alberto, and William B. Grant. 2022. "Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms" Nutrients 14, no. 7: 1448. https://doi.org/10.3390/nu14071448
APA StyleMuñoz, A., & Grant, W. B. (2022). Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms. Nutrients, 14(7), 1448. https://doi.org/10.3390/nu14071448