
Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome?


Abstract
1. Trisomy 21: Overview
2. The Search for Therapies for DS and the Hypothesis: Can Fatty Acids Become a Therapeutic Strategy for DS?
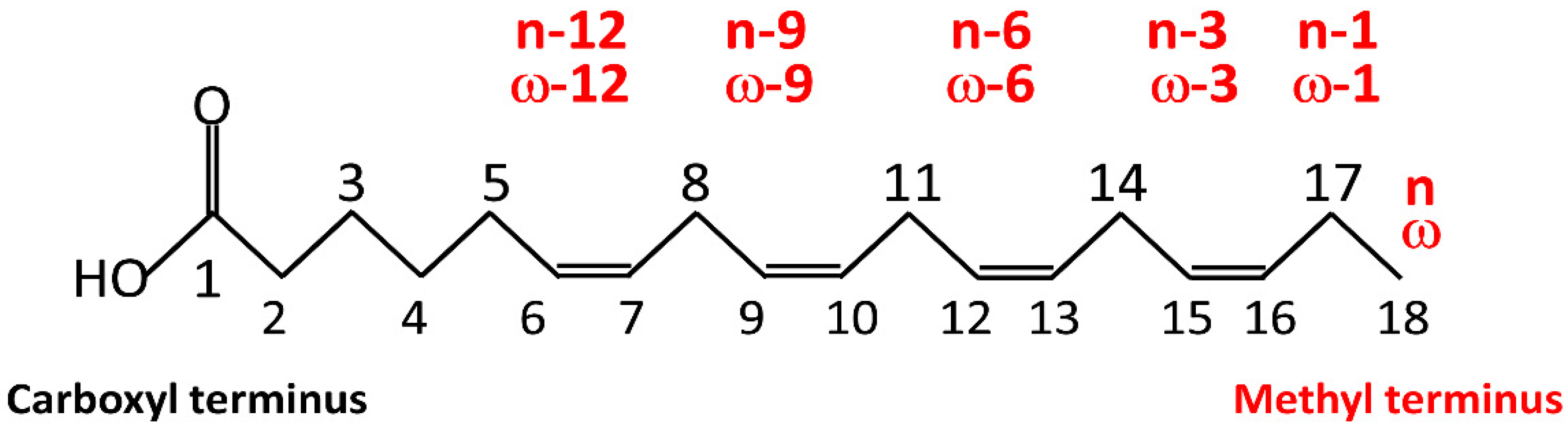
3. Overview of Fatty Acids: Chemistry and Nomenclature
4. Overview of Fatty Acids in the Brain
4.1. Major FAs in the Brain
4.2. Sources of Transport of FAs in Neuronal Cells
4.3. Functions of FAs in Neuronal Cells
4.3.1. Components of Cell Membranes
4.3.2. Production of Energy
4.3.3. Signaling
5. Fatty Acids and Neurogenesis
5.1. Short Outline of Neurogenesis in Humans and Rodents
5.2. Effects of Fatty Acids on Neurogenesis
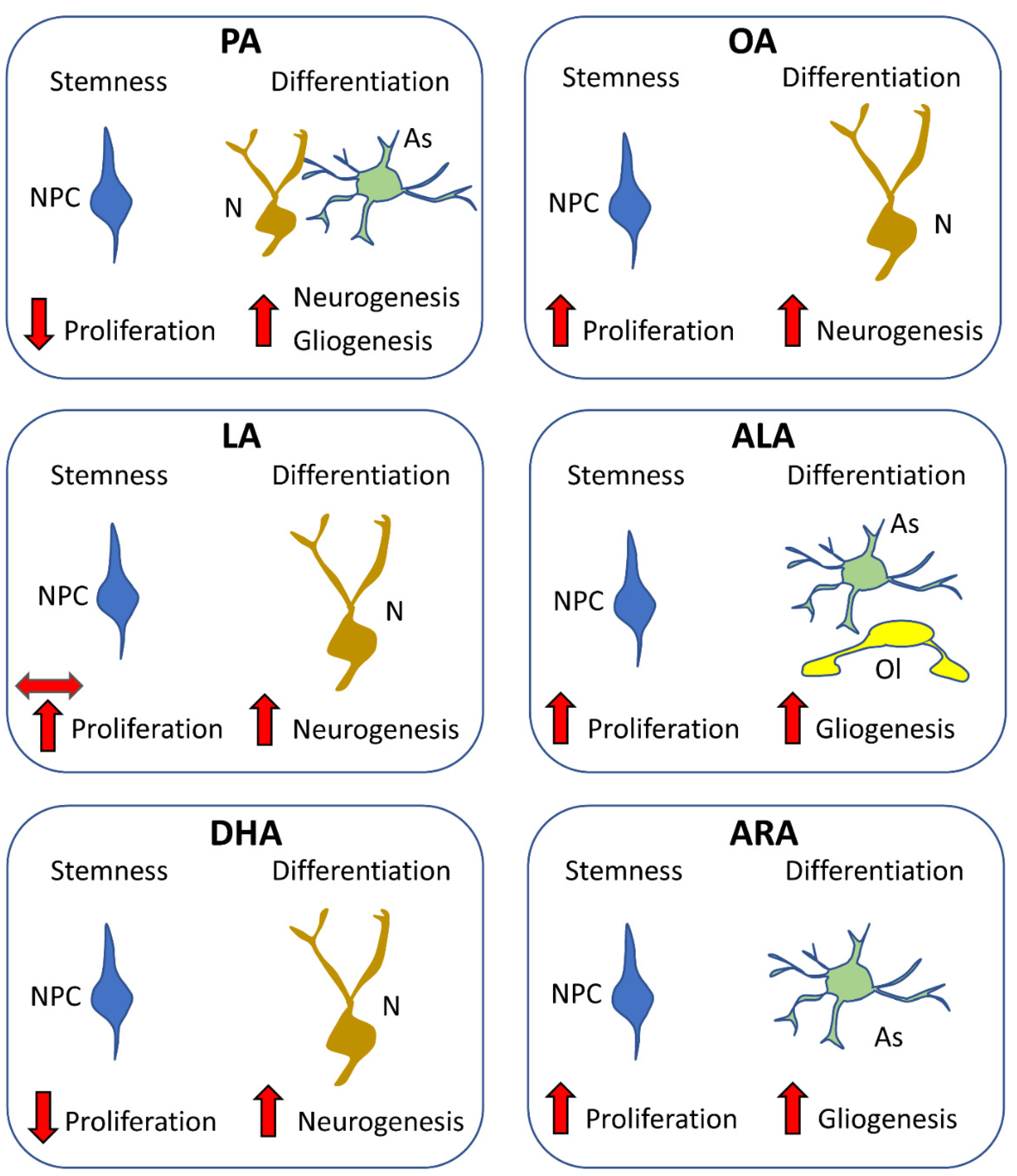
5.2.1. Overview
5.2.2. Effects of Saturated Fatty Acids
5.2.3. Effects of Unsaturated Fatty Acids: MUFAs
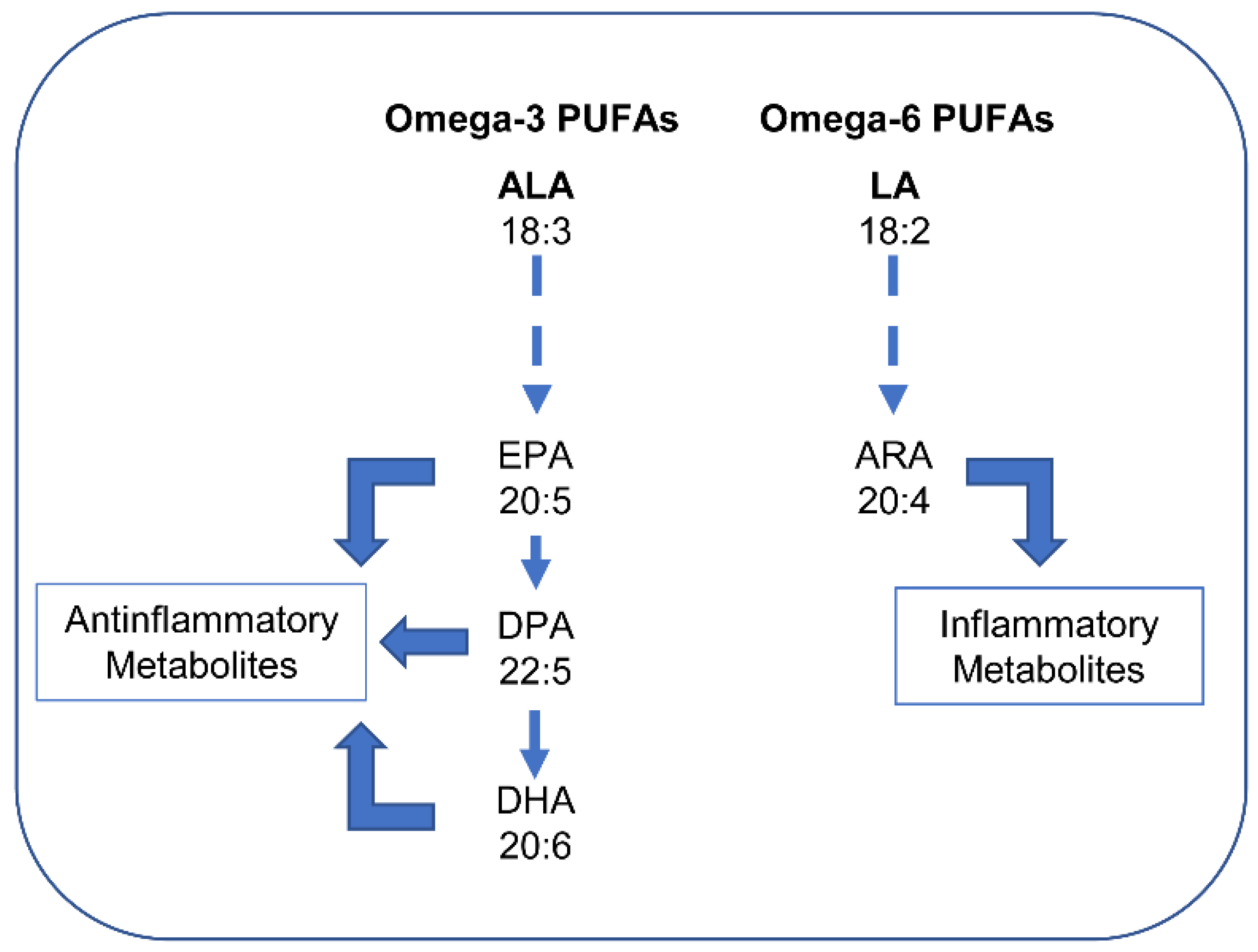
5.2.4. Effects of Unsaturated Fatty Acids: PUFAs
5.2.5. Conclusions
6. Fatty Acids and Neuron Maturation
6.1. Overview of Neuron Maturation in Humans and Rodents
6.2. Effects of FA on Neuron Maturation
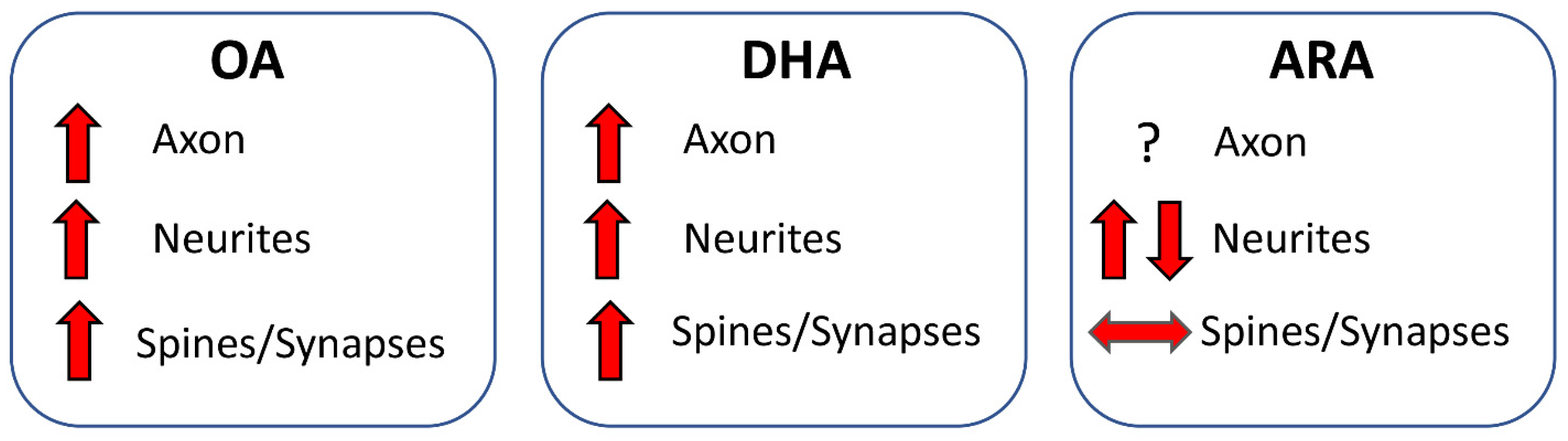
6.2.1. Axon
6.2.2. Dendrites
6.2.3. Dendritic Spines and Synaptic Proteins
6.2.4. Conclusions
7. Fatty Acids and Mitochondrial Function
8. Fatty Acids and Cognition
9. Fatty Acids and Alzheimer’s Disease-Related Dementia
9.1. Altered Brain- Lipid Profile in AD
9.2. Supplementation with FA to Prevent or Reduce AD-Related Dementia
10. Fatty Acids and Down Syndrome
10.1. Brain-Lipid-Profile Alterations in DS
10.1.1. Maternal Blood
10.1.2. Fetuses with DS
10.1.3. Adults with DS
10.2. Effects of Fatty Acids in a DS Model
10.2.1. Fetal Treatment
10.2.2. Early Postnatal Treatment
10.2.3. Adult Treatment
10.3. Effects of Fatty Acids on Individuals with DS
10.4. Early Therapies with FA in DS: A Promising Strategy
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef]
- Hughes-McCormack, L.A.; McGowan, R.; Pell, J.P.; Mackay, D.; Henderson, A.; O’Leary, L.; Cooper, S.A. Birth incidence, deaths and hospitalisations of children and young people with Down syndrome, 1990–2015: Birth cohort study. BMJ Open 2020, 10, e033770. [Google Scholar] [CrossRef]
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef]
- Zigman, W.B. Atypical aging in Down syndrome. Dev. Disabil. Res. Rev. 2013, 18, 51–67. [Google Scholar] [CrossRef]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in Down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Zigman, W.B.; Lott, I.T. Alzheimer’s disease in Down syndrome: Neurobiology and risk. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 237–246. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Bartesaghi, R. Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic. Biol. Med. 2018, 114, 15–32. [Google Scholar] [CrossRef]
- Stagni, F.; Bartesaghi, R. The Challenging Pathway of Treatment for Neurogenesis Impairment in Down Syndrome: Achievements and Perspectives. Front. Cell. Neurosci. 2022, 16, 903729. [Google Scholar] [CrossRef]
- Ponroy Bally, B.; Murai, K.K. Astrocytes in Down Syndrome Across the Lifespan. Front. Cell. Neurosci. 2021, 15, 334. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Kang, H.J.; Tyler, W.A.; Silbereis, J.C.; Cheng, F.; Zhu, Y.; Pletikos, M.; Jankovic-Rapan, L.; Cramer, N.P.; Galdzicki, Z.; et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 2016, 89, 1208–1222. [Google Scholar] [CrossRef]
- Abraham, H.; Vincze, A.; Veszpremi, B.; Kravjak, A.; Gomori, E.; Kovacs, G.G.; Seress, L. Impaired myelination of the human hippocampal formation in Down syndrome. Int. J. Dev. Neurosci. 2012, 30, 147–158. [Google Scholar] [CrossRef]
- Takashima, S.; Becker, L.E.; Armstrong, D.L.; Chan, F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down’s syndrome. A quantitative and qualitative Golgi study. Brain Res. 1981, 225, 1–21. [Google Scholar] [CrossRef]
- Takashima, S.; Iida, K.; Mito, T.; Arima, M. Dendritic and histochemical development and ageing in patients with Down’s syndrome. J. Intellect. Disabil. Res. 1994, 38, 265–273. [Google Scholar] [CrossRef]
- Becker, L.E.; Armstrong, D.L.; Chan, F. Dendritic atrophy in children with Down’s syndrome. Ann. Neurol. 1986, 20, 520–526. [Google Scholar] [CrossRef]
- Prinz, M.; Prinz, B.; Schulz, E. The growth of non-pyramidal neurons in the primary motor cortex of man: A Golgi study. Histol. Histopathol. 1997, 12, 895–900. [Google Scholar]
- Marin-Padilla, M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. J. Comp. Neurol. 1976, 167, 63–81. [Google Scholar] [CrossRef]
- Purpura, D.P. Normal and aberrant neuronal development in the cerebral cortex of human fetus and young infant. UCLA Forum. Med. Sci. 1975, 18, 141–169. [Google Scholar] [CrossRef]
- Gotti, S.; Caricati, E.; Panzica, G. Alterations of brain circuits in Down syndrome murine models. J. Chem. Neuroanat. 2011, 42, 317–326. [Google Scholar] [CrossRef]
- Bartesaghi, R.; Guidi, S.; Ciani, E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011, 22, 419–455. [Google Scholar] [CrossRef]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Esbensen, A.J. Chapter Four-Health Conditions Associated with Aging and End of Life of Adults with Down Syndrome. In International Review of Research in Mental Retardation; Urbano, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 39, pp. 107–126. [Google Scholar]
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down syndrome and Alzheimer’s disease. Free Radic Biol. Med. 2018, 114, 110–121. [Google Scholar] [CrossRef]
- Godfrey, M.; Lee, N.R. A comprehensive examination of the memory profile of youth with Down syndrome in comparison to typically developing peers. Child Neuropsychol. 2020, 26, 721–738. [Google Scholar] [CrossRef]
- Pennington, B.F.; Moon, J.; Edgin, J.; Stedron, J.; Nadel, L. The neuropsychology of Down syndrome: Evidence for hippocampal dysfunction. Child Dev. 2003, 74, 75–93. [Google Scholar] [CrossRef]
- Byrne, A.; MacDonald, J.; Buckley, S. Reading, language and memory skills: A comparative longitudinal study of children with Down syndrome and their mainstream peers. Br. J. Educ. Psychol. 2002, 72, 513–529. [Google Scholar] [CrossRef]
- Jarrold, C.; Baddeley, A.D.; Phillips, C. Long-term memory for verbal and visual information in Down syndrome and Williams syndrome: Performance on the Doors and People test. Cortex 2007, 43, 233–247. [Google Scholar] [CrossRef]
- Vicari, S. Implicit versus explicit memory function in children with Down and Williams syndrome. Downs Syndr. Res. Pract. 2001, 7, 35–40. [Google Scholar] [CrossRef]
- Grieco, J.; Pulsifer, M.; Seligsohn, K.; Skotko, B.; Schwartz, A. Down syndrome: Cognitive and behavioral functioning across the lifespan. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Tungate, A.S.; Conners, F.A. Executive function in Down syndrome: A meta-analysis. Res. Dev. Disabil. 2021, 108, 103802. [Google Scholar] [CrossRef]
- Del Hoyo Soriano, L.; Rosser, T.; Hamilton, D.; Wood, T.; Abbeduto, L.; Sherman, S. Gestational age is related to symptoms of attention-deficit/hyperactivity disorder in late-preterm to full-term children and adolescents with down syndrome. Sci. Rep. 2020, 10, 20345. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.C.; Dosen, A.; Buitelaar, J.K.; Janzing, J.G. Depression in Down syndrome: A review of the literature. Res. Dev. Disabil. 2011, 32, 1432–1440. [Google Scholar] [CrossRef]
- Marino, M.; Scala, I.; Scicolone, O.; Strisciuglio, P.; Bravaccio, C. Distribution and age of onset of psychopathological risk in a cohort of children with Down syndrome in developmental age. Ital. J. Pediatr. 2019, 45, 92. [Google Scholar] [CrossRef]
- Dykens, E.M.; Shah, B.; Davis, B.; Baker, C.; Fife, T.; Fitzpatrick, J. Psychiatric disorders in adolescents and young adults with Down syndrome and other intellectual disabilities. J. Neurodev. Disord. 2015, 7, 9. [Google Scholar] [CrossRef]
- Palumbo, M.L.; McDougle, C.J. Pharmacotherapy of Down syndrome. Expert Opin. Pharm. 2018, 19, 1875–1889. [Google Scholar] [CrossRef]
- Bartesaghi, R.; Vicari, S.; Mobley, W.C. Prenatal and Postnatal Pharmacotherapy in Down Syndrome: The Search to Prevent or Ameliorate Neurodevelopmental and Neurodegenerative Disorders. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 211–233. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Herault, Y.; Delabar, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Yu, E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Model. Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef] [PubMed]
- Muñiz Moreno, M.D.M.; Brault, V.; Birling, M.C.; Pavlovic, G.; Herault, Y. Modeling Down syndrome in animals from the early stage to the 4.0 models and next. Prog. Brain Res. 2020, 251, 91–143. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.; Scott-McKean, J.J. Prospects for improving brain function in individuals with down syndrome. CNS Drugs 2013, 27, 679–702. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesaghi, R. Timing of therapies for Down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef]
- Gardiner, K.J. Pharmacological approaches to improving cognitive function in Down syndrome: Current status and considerations. Drug Des. Devel. Ther. 2015, 9, 103–125. [Google Scholar] [CrossRef]
- Rueda, N.; Florez, J.; Dierssen, M.; Martinez-Cue, C. Translational validity and implications of pharmacotherapies in preclinical models of Down syndrome. Prog. Brain Res. 2020, 251, 245–268. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- Hart, S.J.; Visootsak, J.; Tamburri, P.; Phuong, P.; Baumer, N.; Hernandez, M.C.; Skotko, B.G.; Ochoa-Lubinoff, C.; Liogier D’Ardhuy, X.; Kishnani, P.S.; et al. Pharmacological interventions to improve cognition and adaptive functioning in Down syndrome: Strides to date. Am. J. Med. Genet. A 2017, 173, 3029–3041. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Tian, D.; Guo, Y.; Yan, Y.; Yang, H. Regulation of Neurogenesis and Neuronal Differentiation by Natural Compounds. Curr. Stem Cell Res. Ther. 2021, 34493197. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Djuricic, I.; Calder, P.C. Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty Acids on Human Health: An Update for 2021. Nutrients 2021, 13, 2421. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance-A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, S.; Arsic, A. Fatty Acids. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 623–631. [Google Scholar] [CrossRef]
- Hussain, G.; Schmitt, F.; Loeffler, J.P.; Gonzalez de Aguilar, J.L. Fatting the brain: A brief of recent research. Front. Cell Neurosci. 2013, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.I.; Kiliaan, A.J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: The influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef]
- Kang, J.X.; Wan, J.B.; He, C. Concise review: Regulation of stem cell proliferation and differentiation by essential fatty acids and their metabolites. Stem. Cells 2014, 32, 1092–1098. [Google Scholar] [CrossRef]
- Tułowiecka, N.; Kotlęga, D.; Bohatyrewicz, A.; Szczuko, M. Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment? Int. J. Mol. Sci. 2021, 22, 4207. [Google Scholar] [CrossRef]
- Ghasemi Fard, S.; Cameron-Smith, D.; Sinclair, A.J. n-3 Docosapentaenoic acid: The iceberg n-3 fatty acid. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 134–138. [Google Scholar] [CrossRef]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef]
- Patterson, E.; Cryan, J.F.; Fitzgerald, G.F.; Ross, R.P.; Dinan, T.G.; Stanton, C. Gut microbiota, the pharmabiotics they produce and host health. Proc. Nutr. Soc. 2014, 73, 477–489. [Google Scholar] [CrossRef]
- Bhat, M.I.; Kapila, R. Dietary metabolites derived from gut microbiota: Critical modulators of epigenetic changes in mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. MMBR 2017, 81, e00036-17. [Google Scholar] [CrossRef]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The Healthy Microbiome-What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Cândido, F.G.; Valente, F.X.; Grześkowiak, Ł.M.; Moreira, A.P.B.; Rocha, D.; Alfenas, R.C.G. Impact of dietary fat on gut microbiota and low-grade systemic inflammation: Mechanisms and clinical implications on obesity. Int. J. Food Sci. Nutr. 2018, 69, 125–143. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Fernandes, K.J.L. Neural stem cells and adult brain fatty acid metabolism: Lessons from the 3xTg model of Alzheimer’s disease. Biol. Cell 2018, 110, 6–25. [Google Scholar] [CrossRef]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Reviews. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Giménez, E.; Toscani, A.M. Fatty Acid Signaling Mechanisms in Neural Cells: Fatty Acid Receptors. Front. Cell Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef]
- Feng, L.; Hatten, M.E.; Heintz, N. Brain lipid-binding protein (BLBP): A novel signaling system in the developing mammalian CNS. Neuron 1994, 12, 895–908. [Google Scholar] [CrossRef]
- Arai, Y.; Funatsu, N.; Numayama-Tsuruta, K.; Nomura, T.; Nakamura, S.; Osumi, N. Role of Fabp7, a downstream gene of Pax6, in the maintenance of neuroepithelial cells during early embryonic development of the rat cortex. J. Neurosci. 2005, 25, 9752–9761. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Kersten, S. Mechanisms of gene regulation by fatty acids. Adv. Nutr. 2012, 3, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Fanelli, F.; Ceru, M.P.; Moreno, S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARalpha) and its lipid ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 253–264. [Google Scholar] [CrossRef]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef]
- Basak, S.; Mallick, R.; Banerjee, A.; Pathak, S.; Duttaroy, A.K. Maternal Supply of Both Arachidonic and Docosahexaenoic Acids Is Required for Optimal Neurodevelopment. Nutrients 2021, 13, 2061. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S. Critical periods of vulnerabiliy for the developing nervpus system: Evidence from humans and animal models. Environ. Health Perspect. 2010, 108, 511–533. [Google Scholar]
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Kostović, I.; Sedmak, G.; Judaš, M. Neural histology and neurogenesis of the human fetal and infant brain. Neuroimage 2019, 188, 743–773. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Seki, T.; Imayoshi, I.; Tamamaki, N.; Hayashi, Y.; Tatebayashi, Y.; Hitoshi, S. Neural stem cells and neuro/gliogenesis in the central nervous system: Understanding the structural and functional plasticity of the developing, mature, and diseased brain. J. Physiol. Sci. 2016, 66, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.; Tornoczky, T.; Kosztolanyi, G.; Seress, L. Cell formation in the cortical layers of the developing human cerebellum. Int. J. Dev. Neurosci. 2001, 19, 53–62. [Google Scholar] [CrossRef]
- Seress, L.; Abraham, H.; Tornoczky, T.; Kosztolanyi, G. Cell formation in the human hippocampal formation from mid-gestation to the late postnatal period. Neuroscience 2001, 105, 831–843. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; John Mann, J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Bostrom, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Terreros-Roncal, J.; Flor-García, M.; Rábano, A.; Llorens-Martín, M. Evidences for Adult Hippocampal Neurogenesis in Humans. J. Neurosci. 2021, 41, 2541–2553. [Google Scholar] [CrossRef]
- Wilkinson, G.; Dennis, D.; Schuurmans, C. Proneural genes in neocortical development. Neuroscience 2013, 253, 256–273. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Jessberger, S. Metabolism and neurogenesis. Curr. Opin. Neurobiol. 2017, 42, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Stoll, E.A.; Makin, R.; Sweet, I.R.; Trevelyan, A.J.; Miwa, S.; Horner, P.J.; Turnbull, D.M. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem. Cells 2015, 33, 2306–2319. [Google Scholar] [CrossRef]
- Chorna, N.E.; Santos-Soto, I.J.; Carballeira, N.M.; Morales, J.L.; de la Nuez, J.; Cátala-Valentin, A.; Chornyy, A.P.; Vázquez-Montes, A.; De Ortiz, S.P. Fatty Acid Synthase as a Factor Required for Exercise-Induced Cognitive Enhancement and Dentate Gyrus Cellular Proliferation. PLoS ONE 2013, 8, e77845. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.G.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, Ö.; Suter, U.; Machado, R.A.C.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef]
- Jackowski, S. Cell cycle regulation of membrane phospholipid metabolism. J. Biol. Chem. 1996, 271, 20219–20222. [Google Scholar] [CrossRef]
- Katakura, M.; Hashimoto, M.; Shahdat, H.M.; Gamoh, S.; Okui, T.; Matsuzaki, K.; Shido, O. Docosahexaenoic acid promotes neuronal differentiation by regulating basic helix-loop-helix transcription factors and cell cycle in neural stem cells. Neuroscience 2009, 160, 651–660. [Google Scholar] [CrossRef]
- Insua, M.F.; Garelli, A.; Rotstein, N.P.; German, O.L.; Arias, A.; Politi, L.E. Cell cycle regulation in retinal progenitors by glia-derived neurotrophic factor and docosahexaenoic acid. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2235–2244. [Google Scholar] [CrossRef]
- Okui, T.; Hashimoto, M.; Katakura, M.; Shido, O. Cis-9,trans-11-conjugated linoleic acid promotes neuronal differentiation through regulation of Hes6 mRNA and cell cycle in cultured neural stem cells. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 163–169. [Google Scholar] [CrossRef]
- Hejr, H.; Ghareghani, M.; Zibara, K.; Ghafari, M.; Sadri, F.; Salehpour, Z.; Hamedi, A.; Negintaji, K.; Azari, H.; Ghanbari, A. The ratio of 1/3 linoleic acid to alpha linolenic acid is optimal for oligodendrogenesis of embryonic neural stem cells. Neurosci. Lett. 2017, 651, 216–225. [Google Scholar] [CrossRef]
- Das, S.K.; Mondal, A.K.; Elbein, S.C. Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J. Lipid Res. 2010, 51, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Vesga-Jiménez, D.J.; Martin, C.; Barreto, G.E.; Aristizábal-Pachón, A.F.; Pinzón, A.; González, J. Fatty Acids: An Insight into the Pathogenesis of Neurodegenerative Diseases and Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 2577. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, D.; Zhang, Q.; Wang, J.; Zhan, J.; Xian, X.; Du, Z.; Wang, X.; Hao, A. Palmitic acid affects proliferation and differentiation of neural stem cells in vitro. J. Neurosci. Res. 2014, 92, 574–586. [Google Scholar] [CrossRef]
- Ardah, M.T.; Parween, S.; Varghese, D.S.; Emerald, B.S.; Ansari, S.A. Saturated fatty acid alters embryonic cortical neurogenesis through modulation of gene expression in neural stem cells. J. Nutr. Biochem. 2018, 62, 230–246. [Google Scholar] [CrossRef]
- Kandel, P.; Semerci, F.; Mishra, R.; Choi, W.; Bajic, A.; Baluya, D.; Ma, L.; Chen, K.; Cao, A.C.; Phongmekhin, T.; et al. Oleic acid is an endogenous ligand of TLX/NR2E1 that triggers hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2023784119. [Google Scholar] [CrossRef]
- Murai, K.; Qu, Q.; Sun, G.; Ye, P.; Li, W.; Asuelime, G.; Sun, E.; Tsai, G.E.; Shi, Y. Nuclear receptor TLX stimulates hippocampal neurogenesis and enhances learning and memory in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2014, 111, 9115–9120. [Google Scholar] [CrossRef]
- Freeman, B.A.; Baker, P.R.; Schopfer, F.J.; Woodcock, S.R.; Napolitano, A.; d’Ischia, M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008, 283, 15515–15519. [Google Scholar] [CrossRef]
- Pereckova, J.; Pekarova, M.; Szamecova, N.; Hoferova, Z.; Kamarytova, K.; Falk, M.; Perecko, T. Nitro-Oleic Acid Inhibits Stemness Maintenance and Enhances Neural Differentiation of Mouse Embryonic Stem Cells via STAT3 Signaling. Int. J. Mol. Sci. 2021, 22, 9981. [Google Scholar] [CrossRef]
- Lindqvist, A.; Mohapel, P.; Bouter, B.; Frielingsdorf, H.; Pizzo, D.; Brundin, P.; Erlanson-Albertsson, C. High-fat diet impairs hippocampal neurogenesis in male rats. Eur. J. Neurol. 2006, 13, 1385–1388. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, M.O.; Kim, Y.H.; Kim, J.S.; Han, H.J. Linoleic acid induces mouse embryonic stem cell proliferation via Ca2+/PKC, PI3K/Akt, and MAPKs. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2009, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sakayori, N.; Osumi, N. Polyunsaturated Fatty Acids and their Metabolites in Neural Development and Implications for Psychiatric Disorders. Curr. Psychopharmacol. 2013, 2, 73–83. [Google Scholar] [CrossRef]
- Ghareghani, M.; Zibara, K.; Azari, H.; Hejr, H.; Sadri, F.; Jannesar, R.; Ghalamfarsa, G.; Delaviz, H.; Nouri, E.; Ghanbari, A. Safflower Seed Oil, Containing Oleic Acid and Palmitic Acid, Enhances the Stemness of Cultured Embryonic Neural Stem Cells through Notch1 and Induces Neuronal Differentiation. Front. Neurosci. 2017, 11, 446. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, R.; Ghareghani, M.; Zibara, K.; Tajali Ardakani, M.; Jand, Y.; Azari, H.; Nikbakht, J.; Ghanbari, A. Alyssum homolocarpum seed oil (AHSO), containing natural alpha linolenic acid, stearic acid, myristic acid and β-sitosterol, increases proliferation and differentiation of neural stem cells in vitro. BMC Complementary Altern. Med. 2019, 19, 113. [Google Scholar] [CrossRef]
- Kawakita, E.; Hashimoto, M.; Shido, O. Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 2006, 139, 991–997. [Google Scholar] [CrossRef]
- He, C.; Qu, X.; Cui, L.; Wang, J.; Kang, J.X. Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc. Natl. Acad. Sci. USA 2009, 106, 11370–11375. [Google Scholar] [CrossRef]
- Zhao, W.N.; Hylton, N.K.; Wang, J.; Chindavong, P.S.; Alural, B.; Kurtser, I.; Subramanian, A.; Mazitschek, R.; Perlis, R.H.; Haggarty, S.J. Activation of WNT and CREB signaling pathways in human neuronal cells in response to the Omega-3 fatty acid docosahexaenoic acid (DHA). Mol. Cell Neurosci. 2019, 99, 103386. [Google Scholar] [CrossRef]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.Y. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef]
- Maekawa, M.; Takashima, N.; Matsumata, M.; Ikegami, S.; Kontani, M.; Hara, Y.; Kawashima, H.; Owada, Y.; Kiso, Y.; Yoshikawa, T.; et al. Arachidonic acid drives postnatal neurogenesis and elicits a beneficial effect on prepulse inhibition, a biological trait of psychiatric illnesses. PLoS ONE 2009, 4, e5085. [Google Scholar] [CrossRef]
- Tokuda, H.; Kontani, M.; Kawashima, H.; Kiso, Y.; Shibata, H.; Osumi, N. Differential effect of arachidonic acid and docosahexaenoic acid on age-related decreases in hippocampal neurogenesis. Neurosci. Res. 2014, 88, 58–66. [Google Scholar] [CrossRef]
- Sakayori, N.; Maekawa, M.; Numayama-Tsuruta, K.; Katura, T.; Moriya, T.; Osumi, N. Distinctive effects of arachidonic acid and docosahexaenoic acid on neural stem/progenitor cells. Genes Cells 2011, 16, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Coti Bertrand, P.; O’Kusky, J.R.; Innis, S.M. Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J. Nutr. 2006, 136, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Beltz, B.S.; Tlusty, M.F.; Benton, J.L.; Sandeman, D.C. Omega-3 fatty acids upregulate adult neurogenesis. Neurosci. Lett. 2007, 415, 154–158. [Google Scholar] [CrossRef]
- Fan, C.; Sun, W.; Fu, H.; Dong, H.; Xia, L.; Lu, Y.; Deckelbaum, R.J.; Qi, K. Dietary Ratios of N-6/N-3 Polyunsaturated Fatty Acids during Maternal Pregnancy Affect Hippocampal Neurogenesis and Apoptosis in Mouse Offspring. Nutr. Hosp. 2015, 32, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, M.; Cai, H.; Li, H.; Jiang, P.; Dang, R.; Liu, Y.; He, X.; Xue, Y.; Cao, L.; et al. Maternal diet of polyunsaturated fatty acid altered the cell proliferation in the dentate gyrus of hippocampus and influenced glutamatergic and serotoninergic systems of neonatal female rats. Lipids Health Dis. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Mrzljak, L.; Uylings, H.B.; Kostovic, I.; Van Eden, C.G. Prenatal development of neurons in the human prefrontal cortex: I. A qualitative Golgi study. J. Comp. Neurol. 1988, 271, 355–386. [Google Scholar] [CrossRef] [PubMed]
- Mrzljak, L.; Uylings, H.B.; Kostovic, I.; van Eden, C.G. Prenatal development of neurons in the human prefrontal cortex. II. A quantitative Golgi study. J. Comp. Neurol. 1992, 316, 485–496. [Google Scholar] [CrossRef]
- Kostovic, I.; Judas, M. The development of the subplate and thalamocortical connections in the human foetal brain. Acta Paediatr. 2010, 99, 1119–1127. [Google Scholar] [CrossRef]
- Becker, L.E.; Armstrong, D.L.; Chan, F.; Wood, M.M. Dendritic development in human occipital cortical neurons. Brain Res. 1984, 315, 117–124. [Google Scholar] [CrossRef]
- Lu, D.; He, L.; Xiang, W.; Ai, W.-M.; Cao, Y.; Wang, X.-S.; Pan, A.; Luo, X.-G.; Li, Z.; Yan, X.-X. Somal and Dendritic Development of Human CA3 Pyramidal Neurons F rom Midgestation to Middle Childhood: A Quantitative Golgi Study. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2013, 296, 123–132. [Google Scholar] [CrossRef]
- Uguagliati, B.; Stagni, F.; Emili, M.; Giacomini, A.; Russo, C.; Guidi, S.; Bartesaghi, R. Early Appearance of Dendritic Alterations in Neocortical Pyramidal Neurons of the Ts65Dn Model of Down Syndrome. Dev. Neurosci. 2022, 44, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.; Yamamoto, T. Postnatal ontogenesis of hippocampal CA1 area in rats. I. Development of dendritic arborisation in pyramidal neurons. Brain Res. Bull. 1981, 7, 113–120. [Google Scholar] [CrossRef]
- Juraska, J.M.; Fifkova, E. A Golgi study of the early postnatal development of the visual cortex of the hooded rat. J. Comp. Neurol. 1979, 183, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Uguagliati, B.; Al-Absi, A.R.; Stagni, F.; Emili, M.; Giacomini, A.; Guidi, S.; Nyengaard, J.R.; Bartesaghi, R. Early appearance of developmental alterations in the dendritic tree of the hippocampal granule cells in the Ts65Dn model of Down syndrome. Hippocampus 2021, 31, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Yuste, R.; Bonhoeffer, T. Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 2004, 5, 24–34. [Google Scholar] [CrossRef]
- Meller, K.; Breipohl, W.; Glees, P. Ontogeny of the mouse motor cortex. The polymorph layer or layer VI. A Golgi and electronmicroscopical study. Z Zellforsch. Mikrosk. Anat. 1969, 99, 443–458. [Google Scholar] [CrossRef]
- Romand, S.; Wang, Y.; Toledo-Rodriguez, M.; Markram, H. Morphological development of thick-tufted layer v pyramidal cells in the rat somatosensory cortex. Front. Neuroanat. 2011, 5, 5. [Google Scholar] [CrossRef]
- Medina, J.M.; Tabernero, A. Astrocyte-synthesized oleic acid behaves as a neurotrophic factor for neurons. J. Physiol. Paris 2002, 96, 265–271. [Google Scholar] [CrossRef]
- Tabernero, A.; Lavado, E.M.; Granda, B.; Velasco, A.; Medina, J.M. Neuronal differentiation is triggered by oleic acid synthesized and released by astrocytes. J. Neurochem. 2001, 79, 606–616. [Google Scholar] [CrossRef]
- Bento-Abreu, A.; Tabernero, A.; Medina, J.M. Peroxisome proliferator-activated receptor-alpha is required for the neurotrophic effect of oleic acid in neurons. J. Neurochem. 2007, 103, 871–881. [Google Scholar] [CrossRef]
- Polo-Hernández, E.; De Castro, F.; García-García, A.G.; Tabernero, A.; Medina, J.M. Oleic acid synthesized in the periventricular zone promotes axonogenesis in the striatum during brain development. J. Neurochem. 2010, 114, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.; Tabernero, A.; Medina, J.M. Role of oleic acid as a neurotrophic factor is supported in vivo by the expression of GAP-43 subsequent to the activation of SREBP-1 and the up-regulation of stearoyl-CoA desaturase during postnatal development of the brain. Brain Res. 2003, 977, 103–111. [Google Scholar] [CrossRef]
- Rumora, A.E.; LoGrasso, G.; Hayes, J.M.; Mendelson, F.E.; Tabbey, M.A.; Haidar, J.A.; Lentz, S.I.; Feldman, E.L. The Divergent Roles of Dietary Saturated and Monounsaturated Fatty Acids on Nerve Function in Murine Models of Obesity. J. Neurosci. 2019, 39, 3770–3781. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Xue, R.; Xu, J.; Liu, Z. Effects of docosahexaenoic acid on the survival and neurite outgrowth of rat cortical neurons in primary cultures. J. Nutr. Biochem. 2005, 16, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Mayanagi, T.; Ichijo, H.; Fukumoto, K.; Otsuka, K.; Sakai, A.; Sobue, K. Docosahexaenoic Acid Promotes Axon Outgrowth by Translational Regulation of Tau and Collapsin Response Mediator Protein 2 Expression. J. Biol. Chem. 2016, 291, 4955–4965. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, R.A.; Tabernero, A.; Velasco, A.; Lavado, E.M.; Medina, J.M. The neurotrophic effect of oleic acid includes dendritic differentiation and the expression of the neuronal basic helix-loop-helix transcription factor NeuroD2. J. Neurochem. 2004, 88, 1041–1051. [Google Scholar] [CrossRef]
- Calderon, F.; Kim, H.Y. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004, 90, 979–988. [Google Scholar] [CrossRef]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.S.; Jun, S.B.; Lovinger, D.; Kim, H.Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef]
- Calderon, F.; Kim, H.Y. Role of RXR in neurite outgrowth induced by docosahexaenoic acid. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 227–232. [Google Scholar] [CrossRef]
- Kan, I.; Melamed, E.; Offen, D.; Green, P. Docosahexaenoic acid and arachidonic acid are fundamental supplements for the induction of neuronal differentiation. J. Lipid Res. 2007, 48, 513–517. [Google Scholar] [CrossRef]
- Robson, L.G.; Dyall, S.; Sidloff, D.; Michael-Titus, A.T. Omega-3 polyunsaturated fatty acids increase the neurite outgrowth of rat sensory neurones throughout development and in aged animals. Neurobiol. Aging 2010, 31, 678–687. [Google Scholar] [CrossRef]
- Ikemoto, A.; Kobayashi, T.; Watanabe, S.; Okuyama, H. Membrane fatty acid modifications of PC12 cells by arachidonate or docosahexaenoate affect neurite outgrowth but not norepinephrine release. Neurochem. Res. 1997, 22, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Almaguel, F.G.; Bu, L.; De Leon, D.D.; De Leon, M. Expression of E-FABP in PC12 cells increases neurite extension during differentiation: Involvement of n-3 and n-6 fatty acids. J. Neurochem. 2008, 106, 2015–2029. [Google Scholar] [CrossRef] [PubMed]
- Dotti, C.G.; Esteban, J.A.; Ledesma, M.D. Lipid dynamics at dendritic spines. Front. Neuroanat. 2014, 8, 76. [Google Scholar] [CrossRef]
- Polo-Hernández, E.; Tello, V.; Arroyo, A.A.; Domínguez-Prieto, M.; de Castro, F.; Tabernero, A.; Medina, J.M. Oleic acid synthesized by stearoyl-CoA desaturase (SCD-1) in the lateral periventricular zone of the developing rat brain mediates neuronal growth, migration and the arrangement of prospective synapses. Brain Res. 2014, 1570, 13–25. [Google Scholar] [CrossRef][Green Version]
- Cansev, M.; Marzloff, G.; Sakamoto, T.; Ulus, I.H.; Wurtman, R.J. Giving uridine and/or docosahexaenoic acid orally to rat dams during gestation and nursing increases synaptic elements in brains of weanling pups. Dev. Neurosci. 2009, 31, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Cansev, M.; Wurtman, R.J. Oral supplementation with docosahexaenoic acid and uridine-5’-monophosphate increases dendritic spine density in adult gerbil hippocampus. Brain Res. 2007, 1182, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Cansev, M.; Wurtman, R.J. Chronic administration of docosahexaenoic acid or eicosapentaenoic acid, but not arachidonic acid, alone or in combination with uridine, increases brain phosphatide and synaptic protein levels in gerbils. Neuroscience 2007, 148, 421–431. [Google Scholar] [CrossRef]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef]
- Jeng, J.Y.; Yeh, T.S.; Chiu, Y.H.; Lee, Y.C.; Cheng, H.H.; Hsieh, R.H. Linoleic acid promotes mitochondrial biogenesis and maintains mitochondrial structure for prevention of streptozotocin damage in RIN-m5F cells. Biosci. Biotechnol. Biochem. 2009, 73, 1262–1267. [Google Scholar] [CrossRef]
- Kitajka, K.; Puskás, L.G.; Zvara, A.; Hackler, L., Jr.; Barceló-Coblijn, G.; Yeo, Y.K.; Farkas, T. The role of n-3 polyunsaturated fatty acids in brain: Modulation of rat brain gene expression by dietary n-3 fatty acids. Proc. Natl. Acad. Sci. USA 2002, 99, 2619–2624. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.; Lee, H.K.; Querfurth, H.W. Oleate prevents palmitate-induced mitochondrial dysfunction, insulin resistance and inflammatory signaling in neuronal cells. Biochim. Biophys. Acta 2014, 1843, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Brenna, J.T.; Diau, G.Y. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 fatty acids in neurodegenerative diseases: Focus on mitochondria. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Manabe, S.; Wada, O.; Crawford, M.A. Rapid incorporation of docosahexaenoic acid from dietary sources into brain microsomal, synaptosomal and mitochondrial membranes in adult mice. Int. J. Vitam. Nutr. Research. Int. Z. Fur Vitam.-Und Ernahrungsforschung. J. Int. De Vitaminol. Et De Nutr. 1997, 67, 272–278. [Google Scholar]
- Harper, M.E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331. [Google Scholar] [CrossRef]
- Müller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease--therapeutic aspects. Mol. Neurobiol. 2010, 41, 159–171. [Google Scholar] [CrossRef]
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000, 468, 1–5. [Google Scholar] [CrossRef]
- Ruggiero, F.M.; Cafagna, F.; Petruzzella, V.; Gadaleta, M.N.; Quagliariello, E. Lipid composition in synaptic and nonsynaptic mitochondria from rat brains and effect of aging. J. Neurochem. 1992, 59, 487–491. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Cini, C.; Bellia, F.; Cambria, M.T.; Cornelius, C.; Butterfield, D.A.; et al. Redox proteomics in aging rat brain: Involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J. Neurosci. Res. 2010, 88, 3498–3507. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Ingram, A.; Beckman, J.S.; Magnusson, K.R.; Hagen, T.M. Strategies to protect against age-related mitochondrial decay: Do natural products and their derivatives help? Free Radic Biol. Med. 2022, 178, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Pamplona, R.; Ramirez-Tortosa, M.C.; Granados-Principal, S.; Perez-Lopez, P.; Naudí, A.; Portero-Otin, M.; López-Frías, M.; Battino, M.; Quiles, J.L. Age-related changes in brain mitochondrial DNA deletion and oxidative stress are differentially modulated by dietary fat type and coenzyme Q10. Free Radic Biol. Med. 2011, 50, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.M.; Ding, L.; Wen, M.; Che, H.X.; Huang, J.Q.; Zhang, T.T.; Xue, C.H.; Mao, X.Z.; Wang, Y.M. Mechanisms of DHA-enriched phospholipids in improving cognitive deficits in aged SAMP8 mice with high-fat diet. J. Nutr. Biochem. 2018, 59, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.I.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Lonergan, P.E.; Martin, D.S.; Horrobin, D.F.; Lynch, M.A. Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J. Biol. Chem. 2002, 277, 20804–20811. [Google Scholar] [CrossRef]
- Delerive, P.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef]
- Feng, Z.; Zou, X.; Jia, H.; Li, X.; Zhu, Z.; Liu, X.; Bucheli, P.; Ballevre, O.; Hou, Y.; Zhang, W.; et al. Maternal docosahexaenoic acid feeding protects against impairment of learning and memory and oxidative stress in prenatally stressed rats: Possible role of neuronal mitochondria metabolism. Antioxid. Redox Signal. 2012, 16, 275–289. [Google Scholar] [CrossRef]
- Sampath, H.; Ntambi, J.M. Polyunsaturated fatty acid regulation of gene expression. Nutr. Rev. 2004, 62, 333–339. [Google Scholar] [CrossRef]
- Basak, S.; Vilasagaram, S.; Duttaroy, A.K. Maternal dietary deficiency of n-3 fatty acids affects metabolic and epigenetic phenotypes of the developing fetus. Prostaglandins Leukot. Essent. Fat. Acids 2020, 158, 102109. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Duttaroy, A.K. Maternal PUFAs, Placental Epigenetics, and Their Relevance to Fetal Growth and Brain Development. Reprod Sci. 2022, 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.O.; Taha, A.Y.; Tock, J.L.; Totah, N.K.; Cheon, Y.; Torres, G.E.; Rapoport, S.I.; Moghaddam, B. Adolescent behavior and dopamine availability are uniquely sensitive to dietary omega-3 fatty acid deficiency. Biol. Psychiatry 2014, 75, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gil-Sánchez, A.; Koletzko, B.; Larqué, E. Current understanding of placental fatty acid transport. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 265–272. [Google Scholar] [CrossRef]
- Helland, I.B.; Smith, L.; Saarem, K.; Saugstad, O.D.; Drevon, C.A. Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children’s IQ at 4 years of age. Pediatrics 2003, 111, e39–e44. [Google Scholar] [CrossRef]
- Cheruku, S.R.; Montgomery-Downs, H.E.; Farkas, S.L.; Thoman, E.B.; Lammi-Keefe, C.J. Higher maternal plasma docosahexaenoic acid during pregnancy is associated with more mature neonatal sleep-state patterning. Am. J. Clin. Nutr. 2002, 76, 608–613. [Google Scholar] [CrossRef]
- Colombo, J.; Kannass, K.N.; Shaddy, D.J.; Kundurthi, S.; Maikranz, J.M.; Anderson, C.J.; Blaga, O.M.; Carlson, S.E. Maternal DHA and the development of attention in infancy and toddlerhood. Child Dev. 2004, 75, 1254–1267. [Google Scholar] [CrossRef]
- Freemantle, E.; Vandal, M.; Tremblay-Mercier, J.; Tremblay, S.; Blachère, J.C.; Bégin, M.E.; Brenna, J.T.; Windust, A.; Cunnane, S.C. Omega-3 fatty acids, energy substrates, and brain function during aging. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 213–220. [Google Scholar] [CrossRef]
- Montgomery, P.; Burton, J.R.; Sewell, R.P.; Spreckelsen, T.F.; Richardson, A.J. Low blood long chain omega-3 fatty acids in UK children are associated with poor cognitive performance and behavior: A cross-sectional analysis from the DOLAB study. PLoS ONE 2013, 8, e66697. [Google Scholar] [CrossRef]
- Tanabe, Y.; Hashimoto, M.; Sugioka, K.; Maruyama, M.; Fujii, Y.; Hagiwara, R.; Hara, T.; Hossain, S.M.; Shido, O. Improvement of spatial cognition with dietary docosahexaenoic acid is associated with an increase in Fos expression in rat CA1 hippocampus. Clin. Exp. Pharm. Physiol. 2004, 31, 700–703. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, W.; Liu, T.; Xiao, H.; Liang, W.; Xia, W.; Zhang, J. Perinatal supplementation with omega-3 polyunsaturated fatty acids improves sevoflurane-induced neurodegeneration and memory impairment in neonatal rats. PLoS ONE 2013, 8, e70645. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.A.; Shaikh, I.A.; Khateeb, M.M.; Habeeb, S.M. Omega 3 polyunsaturated fatty acids enhance the protective effect of levetiracetam against seizures, cognitive impairment and hippocampal oxidative DNA damage in young kindled rats. Pharm. Biochem. Behav. 2015, 135, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Choi, J.M.; Lee, J.; Lee, M.H.; Lee, S.; Cho, E.J. Effects of Vegetable Oils with Different Fatty Acid Compositions on Cognition and Memory Ability in Aβ(25–35)-Induced Alzheimer’s Disease Mouse Model. J. Med. Food 2016, 19, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, D. Functional and Structural Benefits Induced by Omega-3 Polyunsaturated Fatty Acids During Aging. Curr. Neuropharmacol. 2017, 15, 534–542. [Google Scholar] [CrossRef]
- Cutuli, D.; Pagani, M.; Caporali, P.; Galbusera, A.; Laricchiuta, D.; Foti, F.; Neri, C.; Spalletta, G.; Caltagirone, C.; Petrosini, L.; et al. Effects of Omega-3 Fatty Acid Supplementation on Cognitive Functions and Neural Substrates: A Voxel-Based Morphometry Study in Aged Mice. Front. Aging Neurosci. 2016, 8, 38. [Google Scholar] [CrossRef]
- Yehuda, S.; Carasso, R.L. Modulation of learning, pain thresholds, and thermoregulation in the rat by preparations of free purified alpha-linolenic and linoleic acids: Determination of the optimal omega 3-to-omega 6 ratio. Proc. Natl. Acad. Sci. USA 1993, 90, 10345–10349. [Google Scholar] [CrossRef]
- Brenna, J.T. Animal studies of the functional consequences of suboptimal polyunsaturated fatty acid status during pregnancy, lactation and early post-natal life. Matern. Child Nutr. 2011, 7, 59–79. [Google Scholar] [CrossRef]
- Brainard, J.S.; Jimoh, O.F.; Deane, K.H.O.; Biswas, P.; Donaldson, D.; Maas, K.; Abdelhamid, A.S.; Hooper, L. Omega-3, Omega-6, and Polyunsaturated Fat for Cognition: Systematic Review and Meta-analysis of Randomized Trials. J. Am. Med. Dir. Assoc. 2020, 21, 1439–1450.e1421. [Google Scholar] [CrossRef]
- Jasbi, P.; Shi, X.; Chu, P.; Elliott, N.; Hudson, H.; Jones, D.; Serrano, G.; Chow, B.; Beach, T.G.; Liu, L.; et al. Metabolic Profiling of Neocortical Tissue Discriminates Alzheimer’s Disease from Mild Cognitive Impairment, High Pathology Controls, and Normal Controls. J. Proteome Res. 2021, 20, 4303–4317. [Google Scholar] [CrossRef]
- Mett, J. The Impact of Medium Chain and Polyunsaturated ω-3-Fatty Acids on Amyloid-β Deposition, Oxidative Stress and Metabolic Dysfunction Associated with Alzheimer’s Disease. Antioxidants 2021, 10, 1991. [Google Scholar] [CrossRef]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Tully, A.M.; Roche, H.M.; Doyle, R.; Fallon, C.; Bruce, I.; Lawlor, B.; Coakley, D.; Gibney, M.J. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: A case-control study. Br. J. Nutr. 2003, 89, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; García-Gómez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Loef, M.; Walach, H. The omega-6/omega-3 ratio and dementia or cognitive decline: A systematic review on human studies and biological evidence. J. Nutr. Gerontol. Geriatr. 2013, 32, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191–192. [Google Scholar] [CrossRef]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann. Neurol. 1997, 42, 776–782. [Google Scholar] [CrossRef]
- Barberger-Gateau, P.; Letenneur, L.; Deschamps, V.; Pérès, K.; Dartigues, J.F.; Renaud, S. Fish, meat, and risk of dementia: Cohort study. BMJ 2002, 325, 932–933. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Qiu, J.; Li, Y.; Wang, J.; Jiao, J. Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: A dose-response meta-analysis of 21 cohort studies. Am. J. Clin. Nutr. 2016, 103, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Su, K.P.; Cheng, T.C.; Liu, H.C.; Chang, C.J.; Dewey, M.E.; Stewart, R.; Huang, S.Y. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: A preliminary randomized double-blind placebo-controlled study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1538–1544. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxén-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.O.; Palmblad, J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: A randomized double-blind trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Sakaguchi, E.; Warashina, S.; Matsukawa, N.; Ishikura, Y.; Kiso, Y.; Sakakibara, M.; Yoshimoto, T.; Guo, J.; Yamashima, T. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci. Res. 2006, 56, 159–164. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; Van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Diet and risk of dementia: Does fat matter?: The Rotterdam Study. Neurology 2002, 59, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Kröger, E.; Verreault, R.; Carmichael, P.H.; Lindsay, J.; Julien, P.; Dewailly, E.; Ayotte, P.; Laurin, D. Omega-3 fatty acids and risk of dementia: The Canadian Study of Health and Aging. Am. J. Clin. Nutr. 2009, 90, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R., Jr.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef]
- Yanai, H. Effects of N-3 Polyunsaturated Fatty Acids on Dementia. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Dierssen, M.; Fructuoso, M.; Martínez de Lagrán, M.; Perluigi, M.; Barone, E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020, 14, 670. [Google Scholar] [CrossRef]
- Yu, Q.; He, Z.; Zubkov, D.; Huang, S.; Kurochkin, I.; Yang, X.; Halene, T.; Willmitzer, L.; Giavalisco, P.; Akbarian, S.; et al. Lipidome alterations in human prefrontal cortex during development, aging, and cognitive disorders. Mol. Psychiatry 2020, 25, 2952–2969. [Google Scholar] [CrossRef]
- de Almeida, E.W.; Greguol, M. Lipid profile in people with Down syndrome: A literature review. J. Hum. Growth Dev. 2020, 30, 197–208. [Google Scholar] [CrossRef]
- Seven, M.; Cengiz, M.; Tüzgen, S.; Iscan, M.Y. Plasma carnitine levels in children with Down syndrome. Am. J. Hum. Biol. Off. J. Hum. Biol. Counc. 2001, 13, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.S.; Kim, S.H.; Fountoulakis, M.; Lubec, G. Heart type fatty acid binding protein (H-FABP) is decreased in brains of patients with Down syndrome and Alzheimer’s disease. J. Neural. Transmission. Suppl. 2003, 67, 225–234. [Google Scholar] [CrossRef]
- Tall, A.R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 2008, 263, 256–273. [Google Scholar] [CrossRef]
- Lorkowski, S.; Rust, S.; Engel, T.; Jung, E.; Tegelkamp, K.; Galinski, E.A.; Assmann, G.; Cullen, P. Genomic sequence and structure of the human ABCG1 (ABC8) gene. Biochem. Biophys. Res. Commun. 2001, 280, 121–131. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, J.; Ren, C.; Chen, H.; Zhang, J. DYRK1A inhibitors for disease therapy: Current status and perspectives. European journal of medicinal chemistry 2022, 229, 114062. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.; Benhaddou, S.; Dard, R.; Tolu, S.; Hamzé, R.; Vialard, F.; Movassat, J.; Janel, N. Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines 2021, 9, 221. [Google Scholar] [CrossRef]
- Pinto, J.; Almeida, L.M.; Martins, A.S.; Duarte, D.; Domingues, M.R.; Barros, A.S.; Galhano, E.; Pita, C.; Almeida Mdo, C.; Carreira, I.M.; et al. Impact of fetal chromosomal disorders on maternal blood metabolome: Toward new biomarkers? Am. J. Obstet. Gynecol. 2015, 213, e841. [Google Scholar] [CrossRef]
- Brooksbank, B.W.; Martinez, M.; Balazs, R. Altered composition of polyunsaturated fatty acyl groups in phosphoglycerides of Down’s syndrome fetal brain. J. Neurochem. 1985, 44, 869–874. [Google Scholar] [CrossRef]
- Tabernero, A.; Velasco, A.; Granda, B.; Lavado, E.M.; Medina, J.M. Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J. Biol. Chem. 2002, 277, 4240–4246. [Google Scholar] [CrossRef]
- Møllgård, K.; Dziegielewska, K.M.; Saunders, N.R.; Zakut, H.; Soreq, H. Synthesis and localization of plasma proteins in the developing human brain. Integrity of the fetal blood-brain barrier to endogenous proteins of hepatic origin. Dev. Biol. 1988, 128, 207–221. [Google Scholar] [CrossRef]
- Hijazi, M.; Medina, J.M.; Velasco, A. Restrained Phosphatidylcholine Synthesis in a Cellular Model of Down’s Syndrome is Associated with the Overexpression of Dyrk1A. Mol. Neurobiol. 2017, 54, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.S.; Bannon, F.J. Serum albumin in Down Syndrome with and without Alzheimer’s Disease. Ir. J. Med. Sci. 2005, 174, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.L. Serum protein and lipoprotein fractions in mongolism. Am. J. Dis. Child. 1961, 102, 369–374. [Google Scholar] [CrossRef]
- Shah, S.N. Fatty acid composition of lipids of human brain myelin and synaptosomes: Changes in phenylketonuria and Down’s syndrome. Int. J. Biochem. 1979, 10, 477–482. [Google Scholar] [CrossRef]
- Murphy, E.J.; Schapiro, M.B.; Rapoport, S.I.; Shetty, H.U. Phospholipid composition and levels are altered in Down syndrome brain. Brain Res. 2000, 867, 9–18. [Google Scholar] [CrossRef]
- Brooksbank, B.W.; Martinez, M. Lipid abnormalities in the brain in adult Down’s syndrome and Alzheimer’s disease. Mol. Chem. Neuropathol. 1989, 11, 157–185. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Dufresne, M.; Joppé, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabé-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheimer’s Disease. Cell Stem. Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [PubMed]
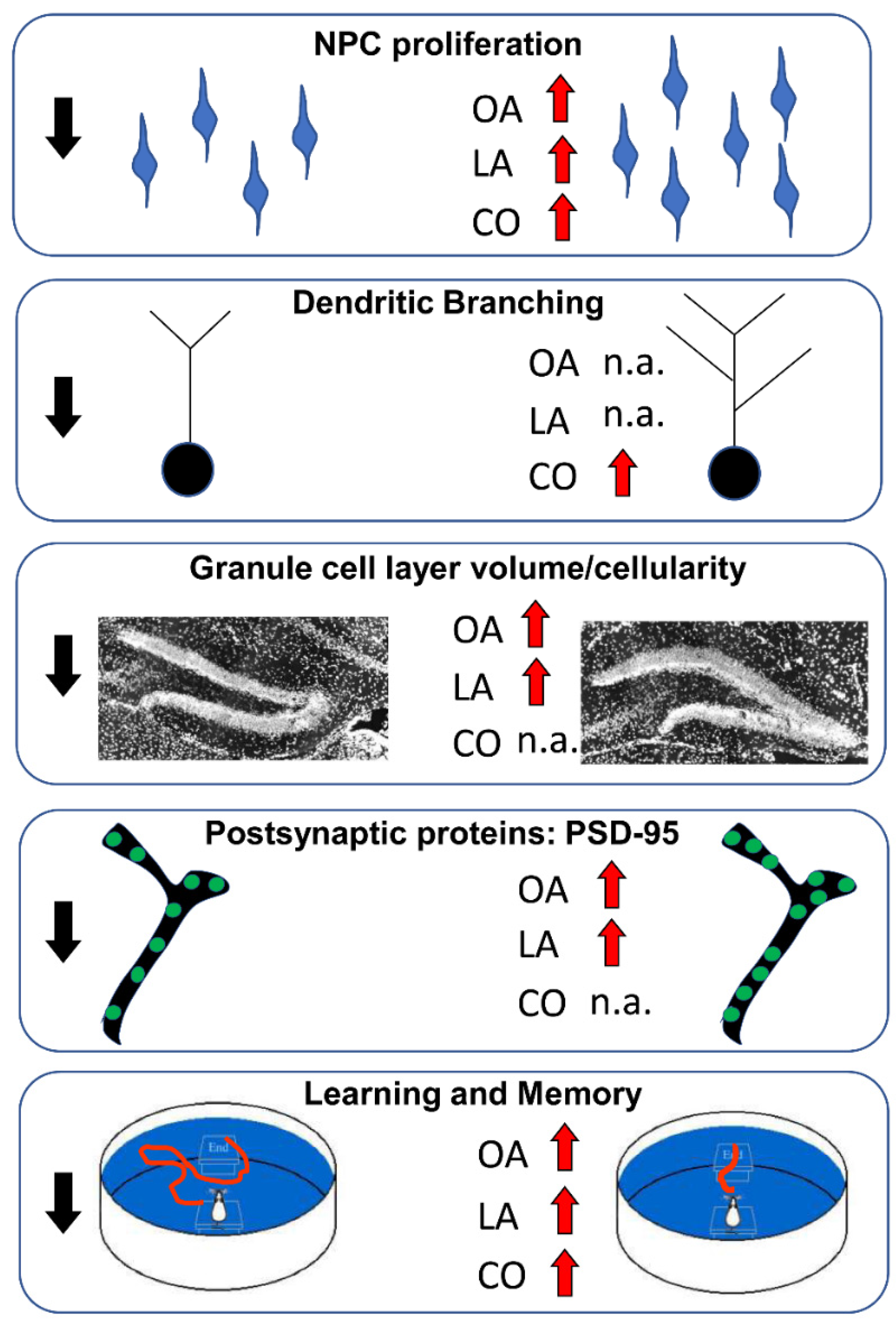
- Garcia-Cerro, S.; Rueda, N.; Vidal, V.; Puente, A.; Campa, V.; Lantigua, S.; Narcis, O.; Velasco, A.; Bartesaghi, R.; Martinez-Cue, C. Prenatal Administration of Oleic Acid or Linolenic Acid Reduces Neuromorphological and Cognitive Alterations in Ts65dn Down Syndrome Mice. J. Nutr. 2020, 150, 1631–1643. [Google Scholar] [CrossRef]
- García-García, A.G.; Polo-Hernández, E.; Tabernero, A.; Medina, J.M. Alpha-fetoprotein (AFP) modulates the effect of serum albumin on brain development by restraining the neurotrophic effect of oleic acid. Brain Res. 2015, 1624, 45–58. [Google Scholar] [CrossRef]
- Kronquist, K.E.; Dreazen, E.; Keener, S.L.; Nicholas, T.W.; Crandall, B.F. Reduced fetal hepatic alpha-fetoprotein levels in Down’s syndrome. Prenat. Diagn. 1990, 10, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Brooksbank, B.W.; Walker, D.; Balazs, R.; Jorgensen, O.S. Neuronal maturation in the foetal brain in Down’s syndrome. Early Hum. Dev. 1989, 18, 237–246. [Google Scholar] [CrossRef]
- Fukaya, M.; Watanabe, M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: A study in the adult mouse brain. J. Comp. Neurol. 2000, 426, 572–586. [Google Scholar] [CrossRef]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.; House, D.R.; Bredt, D.S.; Nicoll, R.A. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci. 2003, 23, 5503–5506. [Google Scholar] [CrossRef]
- Martinez-Cue, C.; Delatour, B.; Potier, M.C. Treating enhanced GABAergic inhibition in Down syndrome: Use of GABA alpha5-selective inverse agonists. Neurosci. Biobehav. Rev. 2014, 46, 218–227. [Google Scholar] [CrossRef]
- Vidal, V.; García-Cerro, S.; Rueda, N.; Puente, A.; Bartesaghi, R.; Martínez-Cué, C. Early postnatal oleic acid administration enhances synaptic development and cognitive abilities in the Ts65Dn mouse model of Down syndrome. Nutr. Neurosci. 2020, 25, 1400–1412. [Google Scholar] [CrossRef]
- Giacomini, A.; Stagni, F.; Emili, M.; Guidi, S.; Salvalai, M.E.; Grilli, M.; Vidal-Sanchez, V.; Martinez-Cue, C.; Bartesaghi, R. Treatment with corn oil improves neurogenesis and cognitive performance in the Ts65Dn mouse model of Down syndrome. Brain Res. Bull. 2018, 140, 378–391. [Google Scholar] [CrossRef]
- Ciaccio, M.; Piccione, M.; Giuffrè, M.; Macaione, V.; Vocca, L.; Bono, A.; Corsello, G. Aminoacid profile and oxidative status in children affected by Down syndrome before and after supplementary nutritional treatment. Ital. J. Biochem. 2003, 52, 72–79. [Google Scholar]
- Vacca, R.A.; Valenti, D. Green tea EGCG plus fish oil omega-3 dietary supplements rescue mitochondrial dysfunctions and are safe in a Down’s syndrome child. Clin. Nutr. 2015, 34, 783–784. [Google Scholar] [CrossRef]
- Scala, I.; Valenti, D.; Scotto D’Aniello, V.; Marino, M.; Riccio, M.P.; Bravaccio, C.; Vacca, R.A.; Strisciuglio, P. Epigallocatechin-3-Gallate Plus Omega-3 Restores the Mitochondrial Complex I and F(0)F(1)-ATP Synthase Activities in PBMCs of Young Children with Down Syndrome: A Pilot Study of Safety and Efficacy. Antioxidants 2021, 10, 469. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; de Lagran, M.M.; Farre, M.; Fito, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Stringer, M.; LaCombe, J.; Patel, R.; Wallace, J.M.; Roper, R.J. Evaluation of the therapeutic potential of Epigallocatechin-3-gallate (EGCG) via oral gavage in young adult Down syndrome mice. Sci. Rep. 2020, 10, 10426. [Google Scholar] [CrossRef] [PubMed]
- Jamal, R.; LaCombe, J.; Patel, R.; Blackwell, M.; Thomas, J.R.; Sloan, K.; Wallace, J.M.; Roper, R.J. Increased dosage and treatment time of Epigallocatechin-3-gallate (EGCG) negatively affects skeletal parameters in normal mice and Down syndrome mouse models. PLoS ONE 2022, 17, e0264254. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, K.; Jing, N.; Zhao, Y.; Yang, X. EGCG regulates fatty acid metabolism of high-fat diet-fed mice in association with enrichment of gut Akkermansia muciniphila. J. Funct. Foods 2020, 75, 104261. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef]
- Zmijewski, P.A.; Gao, L.Y.; Saxena, A.R.; Chavannes, N.K.; Hushmendy, S.F.; Bhoiwala, D.L.; Crawford, D.R. Fish oil improves gene targets of Down syndrome in C57BL and BALB/c mice. Nutr. Res. 2015, 35, 440–448. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short-chain fatty acids | <8 carbons |
| Medium-chain fatty acids | 8–14 carbons |
| Long-chain fatty acid | >16 carbons |
| Very-long-chain fatty acids | >22 carbons |
| Common Name | Abbreviation | C:D n-x |
|---|---|---|
| Saturated fatty acids (SFAs) | ||
| Butyric | 04:00 | |
| Caproic | 06:00 | |
| Caprylic | 08:00 | |
| Capric | 10:00 | |
| Lauric | 12:00 | |
| Myristic | 14:00 | |
| Palmitic | PA | 16:00 |
| Stearic | 18:00 | |
| Arachidic | 20:00 | |
| Mono-unsaturated fatty acids (MUFAs) | ||
| Palmitoleic | 16:01 n-7 | |
| Oleic | OA | 18:01 n-9 |
| Erucic | 22:01 n-9 | |
| Polyunsaturated fatty acids (PUFAs) | ||
| α-Linolenic * | ALA | 18:3 n-3 |
| Stearidonic | SDA | 18:4 n-3 |
| Eicosatetraenoic | ETE | 20:4 n-3 |
| Eicosapentaenoic | EPA | 20:5 n-3 |
| Docosapentaenoic | DPA | 22:5 n-3 |
| Docosahexaenoic | DHA | 22:6 n-3 |
| Linoleic * | LA | 18:2 n-6 |
| γ-Linolenic | GLA | 18:3 n-6 |
| Arachidonic | ARA | 20:4 n-6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cué, C.; Bartesaghi, R. Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients 2022, 14, 2880. https://doi.org/10.3390/nu14142880
Martínez-Cué C, Bartesaghi R. Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients. 2022; 14(14):2880. https://doi.org/10.3390/nu14142880
Chicago/Turabian StyleMartínez-Cué, Carmen, and Renata Bartesaghi. 2022. "Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome?" Nutrients 14, no. 14: 2880. https://doi.org/10.3390/nu14142880
APA StyleMartínez-Cué, C., & Bartesaghi, R. (2022). Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients, 14(14), 2880. https://doi.org/10.3390/nu14142880