Abstract
Anemia is very common in patients with inflammatory disorders. Its prevalence is associated with severity of the underlying disease, and it negatively affects quality of life and cardio-vascular performance of patients. Anemia of inflammation (AI) is caused by disturbances of iron metabolism resulting in iron retention within macrophages, a reduced erythrocyte half-life, and cytokine mediated inhibition of erythropoietin function and erythroid progenitor cell differentiation. AI is mostly mild to moderate, normochromic and normocytic, and characterized by low circulating iron, but normal and increased levels of the storage protein ferritin and the iron hormone hepcidin. The primary therapeutic approach for AI is treatment of the underlying inflammatory disease which mostly results in normalization of hemoglobin levels over time unless other pathologies such as vitamin deficiencies, true iron deficiency on the basis of bleeding episodes, or renal insufficiency are present. If the underlying disease and/or anemia are not resolved, iron supplementation therapy and/or treatment with erythropoietin stimulating agents may be considered whereas blood transfusions are an emergency treatment for life-threatening anemia. New treatments with hepcidin-modifying strategies and stabilizers of hypoxia inducible factors emerge but their therapeutic efficacy for treatment of AI in ill patients needs to be evaluated in clinical trials.
1. Introduction
Anemia is common in patients with acute or chronic inflammatory disorders, and termed as anemia of chronic disease (ACD) or anemia of inflammation (AI) [1,2,3,4]. AI is the most frequent anemia etiology in hospitalized patients and suggested to be the second most prevalent anemia after iron deficiency anemia (IDA), accounting for up to 40% of all anemias worldwide [5]. In patients with AI, disturbances of iron metabolism, dysfunction of erythropoietin, suppression of erythropoiesis and reduction of erythrocyte survival are caused by an activated immune system [2]. Patients with AI typically present with a mild to moderate, normochromic, normocytic anemia accompanied by reduced serum iron and normal to elevated iron stores [1].
Initially linked to infectious diseases, cancer or autoimmune disorders [6,7,8,9,10,11], AI is also common in patients with chronic kidney disease (CKD) [12,13,14,15], coronary artery disease (CAD) [16], chronic heart failure (CHF) [17,18], advanced atherosclerosis [19] or chronic pulmonary disease [20,21,22]. Its occurrence is associated with progression and severity of the underlying disease, decreased quality of life (QoL), a reduced cardio-vascular performance and an adverse outcome [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26]. In cancer patients for instance, anemia is a common comorbidity found in approximately 40–60% of patients with malignancies mostly due to AI but often aggravated by anti-cancer therapy [27,28]. It is associated with fatigue and poor QoL [25,29] as well as impaired local tumor control [30,31]. Moreover, anemia is encountered in approximately 20% of CHF patients [32] and associated with higher NYHA classes, increased NT-proBNP concentrations and mortality [17]. Whether anemia deteriorates the underlying disease or only reflects an advanced disease stage is still controversial. Anemia in the elderly is also suggested to be partly caused by an activated immune system and thus AI shares several features with other pathologies [33,34].
This review will first summarize the physiology of iron metabolism and erythropoiesis, as well as pathophysiology and diagnosis of AI and then focus on established therapies and new therapeutic options, which are currently evaluated in clinical or laboratory studies in patients with anemia and inflammatory disease.
2. Iron Homeostasis and Erythropoiesis
Iron (Fe) is crucially involved in multiple important biochemical pathways, including mitochondrial respiration, metabolic processes, hormone synthesis or deoxyribonucleic acid (DNA) synthesis (reviewed by Muckenthaler et al. [35]). However, its best-known role is the one as the functional component of hemoglobin and myoglobin to enable oxygen binding and transport. At least two million erythrocytes are normally synthesized per second in the adult human bone marrow. Each of these erythrocytes contains about 280 million hemoglobin molecules, which in turn contain four heme-subunits with central iron atoms. These iron atoms represent the binding sites for oxygen thus providing more than a billion oxygen binding sites per erythrocyte. [36,37] The daily iron demand for metabolic processes in adult humans is about 20–30 mg. Most of this daily required iron comes from macrophages, which recycle iron by phagocytosis of senescent erythrocytes, while about 10% of the daily needs are provided by duodenal absorption of iron from the diet. Iron is found in both vegetables and meat, with heme bound iron having a higher bioavailability [38,39]. Iron can also catalyze the generation of radicals which damage cellular macromolecules thus promoting cellular death and tissue damage [40,41]. These reactive radicals can have favorable anti-microbial effects, but usually are causing tissue injury and organ failure as seen in patients with hemochromatosis [42,43]. Therefore, iron homeostasis needs to be tightly regulated in the human body.
2.1. Duodenal Iron Absorption, Circulatory Iron Transport and Cellular Iron Uptake
Iron is absorbed in proximal duodenal endothelial cells either as heme iron via the heme transporter heme carrier protein 1 (HCP1) or as non-heme iron via the divalent metal transporter 1 (DMT1). Luminal non-heme iron (principally Fe3+ complex formations) is first reduced by the duodenal cytochrome B (DCYTB) to Fe2+ before it is transported across the cellular membrane by DMT1 [44,45]. Iron is either stored as mucosal ferritin in duodenal endothelial cells or transported to the bloodstream by ferroportin 1 (FPN1) [46]. FPN1 is the only known cellular iron exporter and not only responsible for iron export from duodenal enterocytes but also for iron mobilization from hepatocytes and erythrocyte recycling macrophages [37,47]. After Fe2+ is exported by FPN1, it is oxidized to Fe3+ by hephaestin (HEPH) at the basolateral membrane of duodenal enterocytes and attached to transferrin [44]. Contrary, mucosal ferritin is lost through shedding of mucosal endothelial cells over time. This is the only route for body iron elimination since there is no regulated iron excretion. [48]
Iron transport in the circulation occurs almost entirely bound to the beta globulin transferrin, which is synthesized in the liver [49]. Iron also binds to lactoferrin, which further regulates the proliferation and activation of monocytes/macrophages, lymphocytes and natural killer cells (NK-cells) [50]. Cells requiring iron express a transferrin receptor (TFR) in their membrane: TFR1 is ubiquitously expressed while TFR2 is only expressed in hepatocytes, duodenal crypt cells, erythroid cells, osteoblasts, neurons and macrophages [51,52]. Moreover, TFR1 is post-transcriptionally regulated by iron via iron regulatory proteins (IRP), while TFR2 is not regulated by IRPs [53,54]. Transferrin, when loaded with two molecules of iron, has an approximately 30-fold higher affinity to TFR1 than to TFR2 [55]. The transferrin-Fe-TFR1 complex is internalized by cells through endocytosis after transferrin has bound to TFR1, while TFR2 has a regulatory function unrelated to iron transport [56,57]. After the endocytic vesicle with the transferrin-Fe-TFR1 complex coalesces with a primary lysosome, Fe3+ is released from transferrin in the acid lysosomal milieu and converted via the action of six transmembrane epithelial antigen of the prostate 3 (STEAP3) to Fe2+ [58] and then transported to the cytosol by DMT1 [59]. Transferrin-Fe binding to TFR2 activates among others ERK1/2 and p38 MAPK pathway [60,61] and further modulates BMP/SMAD pathway (see below) [62].
Most of the iron is stored in hepatocytes and macrophages of the reticuloendothelial system bound to ferritin [63]. Iron stores in humans can usually sustain iron requirements for several years. While hepatocytes acquire iron primarily by transferrin uptake, macrophages retain iron from uptake and digestion of senescent erythrocytes, also named erythrophagocytosis [63]. Hemosiderin, another iron storage protein, is formed when ferritin is partially digested and deposited at lysosomal membranes [64]. It is primarily found in cells with iron overload and mobilizes iron irregularly and slowly [65,66]. Finally, iron can be mobilized from ferritin via nuclear receptor coactivator 4 (NCOA-4) mediated autophagy [67] and then exported from cells by FPN1 [68]. After excretion of Fe2+ by FPN1, in hepatocytes and macrophages the ferroxidase ceruloplasmin oxidizes Fe2+ to Fe3+, which is then again bound to transferrin [68,69]. In addition, ferritin can be secreted by vesicular pathways to the circulation [70].
2.2. Systemic Regulation of Iron Metabolism
Hepcidin is the main systemic regulating hormone of iron metabolism and is primarily synthesized in the liver [71,72]. Hepcidin binds to FPN1, which induces its lysosomal degradation [69]. This reduces circulating iron concentrations by decreasing release of recycled iron from macrophages and stored iron in hepatocytes concomitant to a reduced intestinal iron uptake. [71,73]. Bone morphogenetic protein 6 (BMP6), a member of the tumor growth factor beta (TGF-β) family, is the most potent inducer of hepcidin production and it is expressed in the liver in response to iron overload [62,74,75]. It binds to BMP receptor type 1 (BMPR1) in conjunction with the co-receptor hemojuvelin (HJV) thus upregulating HAMP expression via SMAD1/5/8 signaling [76,77]. More recently, BMP2 was also shown to be critically involved in HAMP expression. It is regulated by dietary iron (although to a lesser extent than BMP6) and binds to HJV with a similar high affinity as BMP6 [78,79]. BMP2/6 preferentially signals via the BMPR1 activin receptor-like kinase 3 (ALK3) for hepcidin induction [80]. BMP2/6 regulation of HAMP gene expression represents a feedback loop to prevent iron overload with consecutive tissue damage.
BMP/SMAD pathway signaling can be modulated by several iron proteins including human hemochromatosis protein (HFE), TFR2 and TMPRSS6 [62]. High concentrations of transferrin-bound iron (TFBI) competitively binds to TFR1 subsequently causing displacement of HFE which then associates with TFR2 [81]. This complex then interacts with HJV and ALK3 thus preventing its degradation, increasing ALK3 expression at the cell surface and inducing HAMP expression [82,83,84]. Contrary, the serine protease matriptase-2 encoded by the TMPRSS6 gene was shown to inhibit the BMP/SMAD pathway related HAMP expression [85]. Because of sex differences found in iron loading disease, several studies investigated the impact of sex hormones on hepcidin regulation [62]. The male sex hormone testosterone was shown to suppress hepcidin production by activating epidermal growth factor receptor (EGFR) and by inhibiting SMAD1/5/8 signaling pathway [86,87]. Actually, testosterone administration was demonstrated to suppress serum hepcidin levels dose-dependent in men with an increase in hemoglobin levels [88]. Contrary, the female sex hormone progesterone was reported to induce hepcidin production independent of BMP/SMAD signaling [89], while data on estrogen are contradictory [90,91]. Interestingly, the recently discovered regulator of phosphate and mineral metabolism, namely fibroblast growth factor 23 (FGF23), was shown to decrease hepcidin expression induced by BMP6 or IL-6 [92]. (Figure 1).
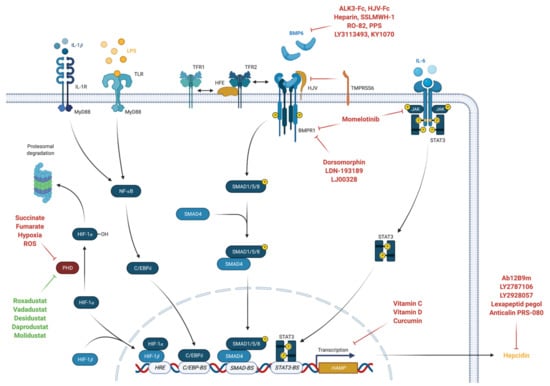
Figure 1.
Regulation of HAMP gene expression (created with BioRender).
2.3. Erythropoiesis, Erythrocyte Life Cycle and Erythrophagocytosis
Erythrocytes are produced in the bone marrow following differentiations and proliferations from multipotent hematopoietic stem and progenitor cells. After seven stages of differentiations from megakaryocyte erythroid progenitor cells (MEP) to proerythroblasts (ProE) and further to orthochromatic erythroblasts (OrthoE), the nucleus is expelled, and cells develop into reticulocytes. After 1–3 days, reticulocytes are released from the bone marrow into the circulation where they develop into mature erythrocytes after additional 1–2 days. Thus, the number of circulating reticulocytes represents recent erythropoiesis and is a good indicator of bone marrow activity. In addition, the hemoglobin content of reticulocytes (CHr) provides an indirect measure of functional iron availability for erythropoiesis within the previous 3–4 days [93,94].
Erythropoiesis is dependent on vitamin B9 (folic acid), vitamin B12 (cobalamin) and iron, and mainly regulated by erythropoietin (EPO), which is produced in the kidney and to a lesser extent in the liver in response to hypoxia [95,96]. In healthy individuals a feedback loop regulates EPO synthesis to maintain production of erythrocytes equable to erythrocyte destruction to sustain sufficient tissue oxygenation [95]. Of note, EPO impacts on iron delivery for erythroid progenitors by stimulating TFR1 mediated iron uptake [97], whereas iron deficiency (ID) results in reduced expression of the erythropoietin receptor (EPOR) component SCRIBBLE and thus in reduced in EPO signaling in erythroblasts [98]. Decreased serum iron reduces plasma membrane TFR2 by its internalization and lysosomal degradation alongside with SCRIBBLE thus decreasing HAMP expression in the liver together with an enhanced EPO sensitivity in erythroid cells [98,99,100]. Erythroblasts further produce erythroferrone (ERFE) in response to EPO via the Janus kinase (JAK)/signaling transducer and activator of transcription 5 (STAT5) signaling pathway [101]. ERFE was shown to suppress hepcidin induction by BMP2/6 thus enhancing iron availability for erythropoiesis [80,102,103]. There are also several other factors that stimulate erythropoiesis including stem cell factor (SCF) insulin, insulin-like growth factor (ILGF), activin A and angiotensin II [93,104,105].
Erythrocytes circulate for about 100–120 days before macrophages, resident in the red pulp of the spleen or in the circulation, internalize them via phagocytosis and liberate iron from heme-subunits via the enzyme heme oxygenase 1 (HMOX1) [47,106,107]. Iron is then transported to the cytosol by DMT1 where it is either stored to ferritin or released into the circulation by FPN1 [108,109]. Interestingly, iron liberated from damaged or senescent erythrocytes first accumulates in cells of the reticuloendothelial system before it is distributed to hepatocytes within one week [63]. In the bone marrow, iron is stored within resident erythroid island macrophages which play an important role in local iron management and further provide iron for erythropoiesis [110].
2.4. Regulation of Iron Metabolism and Erythropoiesis on Cellular Level
Iron metabolism is primarily regulated by hypoxia inducible factors (HIF) and iron regulatory proteins (IRP). HIFs respond to decreased oxygen availability not only by adaptions in iron metabolism, but also by increasing erythropoietin expression and subsequently erythropoiesis. HIF is a transcription factor composed of an alpha and beta subunit. Under normoxic conditions, HIF-𝛼 are hydroxylated, ubiquitinated and proteolytically degraded, while HIF-𝛽 are constitutively expressed. [111] Prolyl hydroxylases (PHD) catalyze the hydroxylation of HIF-𝛼 in the presence of oxygen, iron oxyglutarate and ascorbate [112,113]. Under hypoxic conditions, HIF-𝛼 cannot be hydroxylated, thus translocates to the nucleus and binds to hypoxia responsive elements (HRE) as heterodimer with HIF-𝛽 [114]. HREs occur in several genes including EPO, HAMP, transferrin, TFR1, TFR2, DMT1, DCYTB or vascular endothelial growth factor (VEGF) [115,116,117]. There are three isoforms of HIFs: HIF-1, HIF-2 and HIF-3 [111]. While HIF-2 is the predominant HIF in the regulation of FPN1, DMT1, DYCTB, and EPO transcription [118,119,120,121], HIF-1 is predominant in the regulation of TFR, ceruloplasmin, HMOX1 and hepcidin [122,123,124,125,126].
Cellular iron homeostasis is further orchestrated via the interaction of cytoplasmic iron regulatory proteins (IRP) with messenger ribonucleic acid (mRNA) stem loop structures named iron responsive elements (IRE) [127]. IREs are found within the untranslated regions of mRNAs coding for ferritin, DMT1, TFR1, FPN1 and ALAS2, the latter being a key enzyme in the biosynthesis of heme [128]. Cellular ID increases the binding affinity of IRPs to IREs [129,130,131]. While this inhibits ferritin, FPN1 and ALAS2 expression by blockage of the translation initiation complex, it increases iron uptake by prolonging half-life of TFR1 mRNA [129,132]. Thereby, the cell increases intracellular iron availability by reducing iron storage and increasing iron uptake [133]. Beside cellular ID, other conditions including nitric oxide, hydrogen peroxide and hypoxia also regulate the affinity of IRPs to IREs [134,135,136]. Interestingly, hypoxia suppresses the expression of IRP-1, which increases the translation of HIF-2𝛼 mRNA by decreasing affinity of IRP-1 to IRE in HIF-2𝛼 mRNA [137,138].
Recently, the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) was indicated as another regulator of iron metabolism and erythropoiesis [139]. NRF2 is continuously ubiquitinated and subsequent proteasomal degraded under normoxic conditions dependent on Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (KEAP1) [140]. Oxidative stress causes a disruption of the NRF2-KEAP1 interaction thus preventing NRF2 degradation [139]. NRF2 then translocates to the nucleus where it binds to antioxidant response elements (ARE) found in several genes, involved in heme generation or heme catabolism including hemoglobin subunits, heme-responsive gene 1 (HRG1), HMOX1, BMP6, FPN1 or ferritin, thus inducing their expression [139,141,142,143,144]. NRF2 stimulated ferritin expression increases iron storage capacities thus decreasing labile iron pool (LIP) and iron mediated oxidative stress [143]. NRF2 also induces FPN1 expression and reduces intracellular iron levels within macrophages [145,146]. NRF2 activation is further inhibited by the transcriptional repressor BRCA-1-associated carboxy-terminal helicase (BACH1) [147]. BACH1 acts as heme sensor since it is ubiquitylated and targeted for proteasomal degradation upon binding to heme [148]. Thus erythrophagocytosis with consequently increased heme supply increases NRF2 activity with subsequent upregulation of HMOX1 (increased iron liberation from erythrocytic heme-subunits) and FPN1 (thus increasing iron export to the circulation) [141]. Interestingly, NRF2 suppression further decreases HIF-1𝛼 depicting an interplay between these two transcription pathways [149,150].
3. Pathophysiology of Anemia in Inflammatory Disorders
The pathophysiology of anemia of inflammation (AI) is characterized by increased iron acquisition and retention within macrophages of the reticuloendothelial system. Concurrent intestinal iron absorption is suppressed, with consequently reduced serum iron concentrations thus limiting iron availability for erythropoiesis [1,2]. Erythropoiesis is also directly inhibited by cytokines. They damage senescent erythrocytes together with inflammation driven radicals and reduce erythrocyte half-life which is further reduced by stimulation of erythrophagocytosis [1,2,11,73,151,152,153]. Furthermore, cytokines impact on Epo formation and its biological activity thereby further contributing to anemia development [1,2,154,155]. Figure 2 gives a short overview of the main pathophysiological pathways involved in the development of AI.
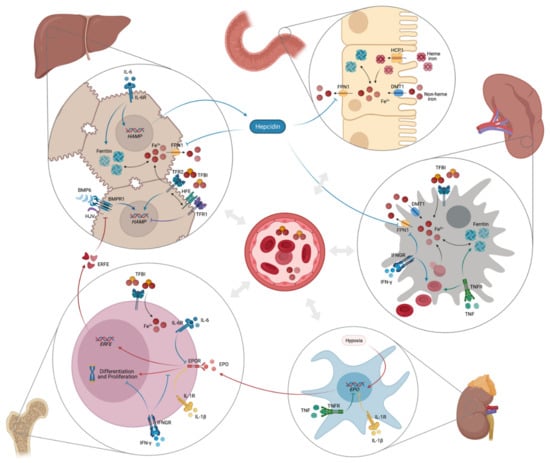
Figure 2.
Pathophysiology of anemia of inflammation (created with BioRender).
3.1. Inflammation-Induced Disturbances of Iron Metabolism
Immune activation in response to microbes, auto-antigens or tumor antigens stimulates release of several pro-inflammatory cytokines by immune cells, which then cause disturbances in iron homeostasis [1]. Interferon gamma (IFN-γ), lipopolysaccharide (LPS) and tumor necrosis factor alpha (TNF-α) upregulate the expression of DMT1 and downregulate the expression of FPN1 with subsequent increased iron uptake and reduced iron release from pro-inflammatory macrophages of the reticuloendothelial system [37,156]. FPN1 is also expressed in duodenal enterocytes which is why under inflammatory conditions its downregulation limits also intestinal iron absorption [37]. Simultaneously, IL-1β, IL-6 but also anti-inflammatory cytokines such as IL-4 and IL-13 and IL-10 increase uptake of TFBI and upregulate synthesis of the iron storage protein ferritin by different mechanisms [1,157,158,159]. Inflammation also upregulates the expression of the iron-regulatory protein hepcidin [160]. IL-6, and to a lesser extent IL-22, stimulate HAMP expression in hepatocytes via the JAK/STAT3 signaling pathway [161,162]. Recent studies demonstrated that ALK3 is also involved in IL-6 mediated hepcidin response, but the exact mechanisms are still unknown [163,164]. Although inflammation suppresses expression of HJV and BMP2/6 in the liver [165,166], an intact BMP/SMAD pathway is essential for maximal inflammation-related hepcidin synthetization [62]. Activin B, a cytokine of the TNF-β family, was demonstrated to upregulate HAMP expression in vitro under inflammatory conditions by activating the BMP/SMAD pathway while the functional significance in vivo remains uncertain [167,168,169]. Finally, hepcidin synthesis by hepatocytes is also stimulated by IL-1β independent of BMP/SMAD or JAK/STAT3 pathway via a CCAAT enhancer-binding protein (C/EBP)-binding site (C/EBP-BS) in the HAMP promoter site [170]. In addition, lipopolysaccharides (LPS) via Toll-like receptor 4 (TLR4) and also IL-6 induce hepcidin expression by macrophages which then target FPN1 in an autocrine fashion [171,172].
3.2. Erythropoiesis Suppression and Decreased Erythrocyte Survival by Inflammation
Beside reduced iron availability for erythropoiesis, inflammation also directly impairs erythropoiesis by reducing the production and activity of EPO and repressing erythroid progenitor cells proliferation and differentiation. EPO levels are rather low in patients with AI despite hypoxia and low serum iron levels [1]. Actually, IFN-γ, IL-1β and TNF-α directly inhibit renal EPO expression [1,154]. This suppresses hypoxia-mediated EPO stimulation and induces oxidative stress thus causing harm to EPO-producing cells [173,174]. In addition, EPO-mediated signaling is impaired by these pro-inflammatory cytokines, while the number of EPOR is unchanged [174]. Reduced signal may be also linked to ID as the functional expression of the EPOR component SCRIBBLE is reduced by ID in erythroid progenitors [98]. EPO and hypoxia further inhibit HAMP expression by inducing HIF-1, ERFE [175], matriptase-2 [176], platelet derived growth factor BB (PDGF-BB) [177] and growth differentiation factor-15 (GDF-15) [178,179]. Inflammation-related EPO reduction thus aggravates hepcidin-mediated iron limitation to erythroid progenitor cells [2]. Moreover, several cytokines including type I interferons, IFN-γ, TNF or IL-1β have been shown to inhibit erythroid progenitor cell proliferation or differentiation directly or via induction of radical formation of ceramide mediated apoptotic processes [180,181,182]. Erythrophagocytosis by hepatic and especially splenic macrophages is also upregulated during inflammation resulting in a decreased erythrocyte half-life [2,47]. IFN-γ and TNF-α promote degradation and phagocytosis of erythrocytes thus reducing erythrocyte survival [183,184,185]. Moreover, inflammation was shown to induce erythrocyte membrane lipid remodeling and oxidative damage of erythrocytes with impaired erythrocyte function and survival [186,187].
3.3. Tryptophan Metabolism in the Pathogenesis of Anemia of Inflammation
IFN-γ, one of the main cytokines of T-helper cell type 1 (Th1) immune response, is centrally involved in the immune response to intracellular microbes and a central activator of anti-microbial immune effector pathways [188]. IFN-γ induces neopterin production in macrophages which reflects the extent of cellular immune activation and oxidative stress, and has thus been used as a clinical marker to monitor cellular immune activation in vivo [189]. Elevated serum concentrations of neopterin were associated with a poor outcome and anemia in patients with cancer [25,190], CAD [16], CHF [191] and pulmonary disease [192,193]. IFN-γ further activates indoleamine 2,3-dioxygenase (IDO) thus promoting tryptophan breakdown [194,195,196]. Tryptophan is not only essential for the growth and proliferation of various cells including hematopoietic stem and progenitor cells, but also for microbes and malignant cells. Therefore, tryptophan depletion is initially also a natural defense mechanism of the human body [197,198]. Interestingly, tryptophan metabolites including kynurenine stimulate hepcidin expression and inhibit EPO production by activating the aryl hydrocarbon receptor (AHR), which competes with HIF-2𝛼 in binding HIF-1𝛽 [199]. Moreover, quinolinic acid (QUIN), another down-stream metabolite of tryptophan, was demonstrated to inhibit EPO production [200] by preventing HIF-1𝛼 activation [201]. Tryptophan is also a crucial amino acid for brain homeostasis including serotonin and melatonin production [197]. Increased inflammation induced tryptophan breakdown via the kynurenine pathway reduces tryptophan availability for serotonin and melatonin synthesis, thus contributing to neurobehavioral symptoms including fatigue or depression [202,203], which are also common symptoms in patients suffering from anemia. Decreased tryptophan concentrations and increased kynurenine formation have been associated with decreased hemoglobin concentrations in patients with HIV-infection [204], with cancer [25,197] and also in patients with anemia of chronic disease [205]. Interestingly, both anemia and tryptophan catabolism correlated significantly with an impaired QoL and depression in patients with solid tumors [25]. Therefore, AI and tryptophan breakdown appear to be interconnected and might both contribute importantly to fatigue, depression and decreased QoL in patients with inflammatory disorders.
4. Diagnosis of Anemia in Patients with Inflammatory Disorders
Anemia is defined according to the criteria of the World Health Organization (WHO) as hemoglobin < 130 g/L in men and < 120 g/L in women [206]: mild anemia is defined as hemoglobin ≥ 110 g/L, moderate anemia as hemoglobin < 110 g/L and severe anemia as hemoglobin < 80 g/L. AI usually presents normocytic, normochromic, as mild to moderate anemia with decreased circulating iron concentrations and normal or increased levels of the iron storage protein ferritin while transferrin levels are reduced [155]. Because, transferrin synthesis is suppressed under inflammatory conditions and by malnutrition [155,207], transferrin saturation (TfS) may in some circumstances appear normal [208]. Therefore, the widely used threshold for TfS of 20% often fails to detect patients with pathophysiological AI as a consequence of inflammation driven Tf suppression [209,210].
The main challenge in the diagnosis is identifying the coexistence of true or absolute ID in patients with AI, because these patients need further specific diagnostics to evaluate the source of blood loss (i.e., gastrointestinal bleeding, iatrogenic blood loss due to blood sampling or other procedures, malnutrition, increased iron requirements during pregnancies or in growing children) and then different treatment as compared to subjects suffering from AI alone [181]. Several biomarkers, alone or in combination, were studied for detection of true ID in patients with ongoing inflammation. Although some studies showed promising results, these biomarkers strongly depend on laboratory equipment and were not evaluated in prospective studies [208,211,212,213]. An early and sensitive biomarker for erythropoiesis is the reticulocyte hemoglobin content (CHr), which is a reliable indicator of IDA and not influenced by inflammation [92,214]. Moreover, serum concentrations of soluble transferrin receptor (sTFR) are elevated in patients with ID reflecting an increased iron requirement of cells expressing TfR including erythroid progenitor cells [215,216]. Although sTFR was shown to be a good diagnostic biomarker for differentiating between AI with or without concomitant IDA [217], it is also affected by inflammation independent of iron status [218]. Calculation of the sTFR/log ferritin ratio, also known as ferritin index, seems to be suited well for the detection of depleted iron stores also in patients with signs of inflammation [217,219]. Currently, a ferritin index > 2 defines patients with AI and IDA, while a ferritin index < 1 defines patients with AI leaving a clinically relevant diagnostic grey zone [2,208,220]. Hepcidin plays a central role in the pathophysiology of AI, which is why its usefulness in the diagnosis of AI has been extensively studied [221,222,223]. While patients with AI have higher serum hepcidin concentrations compared to healthy controls, patients with IDA have significantly lower or even undetectable serum concentrations of hepcidin. Interestingly, patients with AI and IDA present with significantly lower hepcidin levels compared to patients with AI alone since the suppression by ID dominates over inflammation-related stimulation of hepcidin expression. [73,224,225] Nevertheless, there are still no established and clinically verified diagnostic algorithms for detecting absolute ID in patients with anemia and an activated immune system. A combination of ferritin cutoffs, transferrin and hepcidin but also of classical hematological parameters such as mean corpuscular volume (MCV) or mean hemoglobin content of erythrocytes (MCH), the latter two being reduced with ACD and true ID [155], may help in finding the right diagnosis [226].
5. Established Treatments of Anemia of Inflammation
The primary therapeutic approach for AI is detection and treatment of the underlying inflammatory disease, which usually results in resolution of AI over time [2]. Anti-inflammatory treatment in patients with AI based on auto-inflammatory disorders was demonstrated to ameliorate anemia in addition to an improvement of the underlying disease. TNF inhibitors including infliximab, adalimumab and certolizumab pegol play a key role in the management of inflammatory bowel disease (IBD) patients and were shown to increase hemoglobin concentrations especially in IBD patients with severe anemia [227]. Similarly, infliximab significantly improves hemoglobin levels in patients with rheumatoid arthritis [10].
More recently, the IL-1𝛽 inhibitor canakinumab reduced the prevalence of anemia and increased hemoglobin levels in anemic patients with atherosclerotic disease [228]. Accordingly, treatment with the anti-IL-6 receptor antibody toculizumab improved both, the underlying disease and anemia in patients with Castleman disease [229]. Of note, anti-IL-6R treatment of patients with advanced ovarian or lung cancer also ameliorated hemoglobin levels [230,231]. Finally, statins including simvastatin and atorvastatin downregulate hepcidin expression dose-dependently with a concomitant improvement of hemoglobin levels [232,233].
Before considering specific therapeutic options for anemia in patients with inflammatory disease, concomitant pathologies contributing to the severity of AI, including bleeding, vitamin deficiencies, hemolysis, renal insufficiency, side effects of co-medications on hematopoiesis should be evaluated and if possible treated [2,6]. If AI is not resolved by treatment of the underlying diseases or sufficient therapy of the underlying disease is not possible, other therapeutic options can be considered. The therapeutic decision must consider the underlying disease and the severity of anemia. In patients with an active infection, AI is actually a defense strategy of the body against invading microbes to restrict iron availability for pathogens also termed as “nutritional immunity” [234,235]. Therefore, treatment of AI in these patients, specifically with iron supplementation, not only promotes pathogen growth with increased virulence but also impairs the host immune response against pathogens [236,237,238,239]. There are three established therapies for the treatment of AI: iron replacement therapy, treatment with erythropoiesis-stimulating agents (ESAs) and red blood cell transfusion [2].
5.1. Iron Replacemant Therapy
Iron replacement therapy is an established treatment in patients with AI and should be considered, especially in patients with concomitant absolute or true ID. Iron can be supplemented either with oral or parenteral intravenous iron preparations [2]. Oral iron preparations were demonstrated to have a similarly efficacy compared to intravenous preparations in patients with inflammatory bowel disease, concomitant true ID and low disease activity [240] with the advantage of easy self-administration and low costs [241]. However, the bioavailability of oral iron supplementation is influenced by several factors. While vitamin C and overnight fasting were shown to increase iron uptake, proton pump inhibitors or milk products and tea decrease iron bioavailability [242]. Iron should be further taken only once in the morning since more frequent intake can reduce iron uptake by increased iron-related hepcidin production similar to inflammation-related hepcidin production [243]. Low absorption of oral iron increases intestinal iron concentrations, with consecutive gastrointestinal side effects including nausea, vomiting, abdominal pain and constipation, which then further limits compliance of patients [241]. Interestingly, recent studies further showed that iron therapy significantly affects the phylogenetic composition of the microbiome as well as the fecal metabolite landscape with a potential unfavorable effect on the disease [244,245]. Further studies investigating the clinical and longtime effects especially in patients with IBD are of interest, since also intravenous iron administration affects the composition of the microbiome [241].
Inflammation (even low-grade inflammation) impairs intestinal iron absorption due to hepcidin-related internalization of FPN1 making oral iron supplementation ineffective [73,246]. Therefore, parenteral intravenous iron administration is the possible option of iron supplementation in patients with chronic (inflammatory) diseases. Intravenous iron preparations have the advantage of more rapid repletion of iron deficiency. Specifically, newer carbohydrate iron formulations enable the administration of higher dosages up to 1000 mg with one infusion [241]. On the other hand, higher costs and very rare but severe anaphylactic reactions with the need for equipment to manage these life-threating situations have to be taken into consideration [247]. Although, parenteral iron administration has earlier been reserved for patients with intolerance or inadequate response to oral iron supplementation as well as for rapid iron replenishment [241], nowadays it is an established for treatment of AI with concomitant IDA in or even for ID without anemia in patients with several chronic diseases [248,249]. A meta-analysis of patients with IBD and IDA demonstrated a higher efficacy of intravenous iron administration in increasing hemoglobin levels compared to oral iron supplementation with consistently fewer gastrointestinal adverse events [250]. In CHF patients with absolute or functional ID parenteral iron supplementation was shown to improve cardiac performance and QoL even independent of concomitant anemia [251,252,253], however, concerns became evident regarding iron accumulation in the myocardium and endothelia and in regard to the need of outcome data [254]. Moreover, several clinical trials investigating iron treatment in anemic cancer patients demonstrated that intravenous (but not oral) iron administration increases hematopoietic response and decreases rate of blood transfusions with a concomitant increase of QoL [255]. However, essential endpoint data on the effect of such treatment on the course of the malignant disease are missing. Iron carbohydrate complexes are taken up by macrophages where iron is released from the carbohydrate shell and transferred to the circulation via FPN1. This transfer of iron from the macrophages is controlled by hepcidin, and it is questionable whether or not in high grade inflammation enough iron will become available for erythropoiesis, a notion which needs to be evaluated experimentally.
There are different intravenous iron formulations available which have recently been reviewed [241,256]. Although the different intravenous iron formulations are licensed and generally recommended to be used in all patients with ID, there are minor differences in physiochemical properties and recommendations by different medical societies (reviewed in [257,258]).
Despite the benefits of intravenous iron administration, there is a rare risk for serious, life-threatening hypersensitivity reactions with need for emergency response [247]. The pathogenesis of hypersensitivity reactions may differ between iron formulations and pre-existing morbidities, while the clinical presentation is the same. The two primary mechanisms of hypersensitivity reactions are immunological IgE-mediated response (e.g., to the dextran component of iron dextrans) and complement activation-related pseudo-allergy (CARPA) [259,260,261]. A recent analysis showed that the risk of severe infusion reactions is comparable between the different iron formulations and less than 1% [257,262]. Another clinical relevant side effect that was increasingly recognized lately after administration of certain intravenous formulations is hypophosphatemia [117]. Actually, moderate to severe hypophosphatemia (<2.0 mg/dL) affects up to 75.0% of patients treated with ferric carboxymaltose when compared to iron dextran (0%), ferrumoxytol (0.9%) or ferric derisomaltose (7.9%) [263,264]. Ferric carboxymaltose and to a lesser extent also ferric derisomaltose increase the expression of the hormone FGF23, which increases urinary phosphate excretion and inhibits 1-alpha hydroxylase and thus vitamin D activation [117,265]. The following biochemical changes are also described as 6H-Syndrome (High FGF23, Hyperphosphaturia, Hypophosphatemia, Hypovitaminosis D, Hypocalcemia and secondary Hyperparathyroidism) causing osteomalacia, bone fractures, muscular weakness and respiratory failure in rare cases [117]. FGF23 further reduces the secretion of EPO and directly enhances erythrocyte apoptosis [266]. This may negatively affect AI pathogenesis, along with an effect of FGF23 on immune activation and a subsequent increased of hepcidin production [266,267,268,269,270,271].
5.2. Erythropoiesis-Stimulating Agents
Treatment with recombinant human erythropoiesis stimulating agents (ESA) is a widely used therapy for AI and specifically used in patients with CKD or cancer [1,2]. Beside the initially developed recombinant erythropoietins and the extended half-life product darbapoetin, several Epo biosimilars with different half-life and EpoR affinity are now available [272]. Nevertheless, several studies suggest a more restrictive use of ESAs due to side effects including increased mortality in some specific patient groups. Specifically, a Cochrane meta-analysis showed a decreased overall survival with increased risk for thromboembolic complications, hemorrhage, and hypertension in anemic patients with cancer [273]. Moreover, a recent study demonstrated that ESAs might stimulate tumor growth and progression via the ephrin type-B receptor 4 (EPHB4) that triggers downstream signaling via STAT3 [274]. Determining the tumorous expression of EPHB4 might serve as a screening tool to identify patients with low risk of tumor progression after treatment with ESAs [9]. In addition, ESAs are associated with an increased risk of death or cardiovascular events in both patients with CKD receiving dialysis [275], but also patients not receiving dialysis having a poor initial hematopoietic response [276,277]. Interestingly, functional ID with lower TfS and elevated inflammatory markers are associated with a poor hematopoietic response to ESA treatment [277].
Several mechanisms limit the effectiveness of ESAs in patients with AI. First of all, cytokines not only suppress EPO production, they also suppress EPO-mediated signaling [174] and directly impair erythroid cell proliferation and differentiation [180]. Moreover, EPOR expression is inhibited by FGF23 [268], which is elevated in patients with an activated immune system [267,278,279,280], hyperparathyroidism [281] or hyperphosphatemia [282]. Importantly, ID also negatively impacts on EPO signaling in erythroid cells via its effect on Scribble expression [98], but also blunts erythroid differentiation at later stages [283]. Therefore, treatment strategies enabling a reduction of ESAs dosages by supplementing iron or mobilizing iron via anti-hepcidin therapeutic strategies gained increasing interest over the last years [283].
5.3. Blood Transfusions
Blood transfusions are an established and rapid therapy in patients with severe or life-threatening anemia, especially in those with concomitant pathologies including bleeding or chemotherapy induced anemia [1]. Because of the increased risk of allergic reactions, acute hemolytic reactions, acute lung injury and the lack of evidence showing an improvement in outcome, a restrictive blood transfusion strategy is recommended for almost all pathologies [2,284,285]. Actually, two observational studies in anemic HF patients demonstrated that patients receiving blood transfusions had a worse clinical outcome and prognosis [286]. Also in anemic patients with advanced CKD, blood transfusions were shown to cause hyperkalemia and heart failure [287]. Finally, numerous meta-analyses in cancer patients have demonstrated an increased mortality, morbidity and risk for tumor recurrence in patients receiving blood transfusions [255]. Restrictive transfusion protocols seem to be associated with a better outcome [288]. Actually, restrictive use of blood transfusion was shown to be associated with a lower mortality than liberal blood transfusion strategy in critically ill patients [285].
6. Novel Therapeutic Principles
Although iron supplementation remains first line therapy for patients with AI and concomitant IDA, new treatment strategies targeting the restricted iron storages in patients with pure AI gained interest in the last years. Mobilization of stored iron from macrophages of the reticuloendothelial system (RES) is probably more suitable than repeated iron supplementation as in the course of advanced inflammation supplemental iron is mainly caught in the RES based on the inhibitory activity of hepcidin on FPN1 mediated iron egress [69,73]. There are two new main therapeutic strategies of rearranging body iron stores: modification (down-regulation) of hepcidin synthesis and function, and stabilization of HIF via inhibition of PHD.
6.1. Hepcidin-Modifying Treatments
Hepcidin is recognized as master-regulator of iron metabolism and centrally involved in the pathophysiology of AI. Its effects can be modified by neutralization of hepcidin in the circulation, by targeting hepcidin expression in the liver or by antagonizing hepcidin binding to its receptor, FPN1. (see next chapter).
First studies investigated the effects of hepcidin antibodies on disease progression in mice with AI. The human anti-hepcidin monoclonal antibody Ab12B9m was shown to neutralize hepcidin in vitro and in vivo and to improve anemia in mice when combined with ESA [289,290,291]. Recently a phase I clinical trial investigating effects and safety of the fully humanized monoclonal antibody LY2787106 with a high affinity for hepcidin was tested in cancer patients with anemia. LY2787106 dose-dependently increased serum iron concentrations and TfS within 24 h after administration [292]. Another pharmaceutical approach to hepcidin neutralization is the use of the Spiegelmer® lexaptepid pegol. It binds hepcidin with a high affinity and thus inhibits its biological function [293]. A recent placebo-controlled phase I clinical trial demonstrated that lexaptepid pegol increases serum iron concentrations and TfS in a dose-dependent manner [294] and significantly inhibits hepcidin mediated induction of hypoferremia in healthy humans injected with Escherichia coli LPS [295]. In a pilot phase II study in patients with cancer related anemia, lexaptepid pegol increased hemoglobin concentrations and CHr, and decreased sTFR and ferritin concentrations in 5 of 12 patients [296].
Finally, hepcidin effects can be modified by blocking its effector binding site. LY2928057 was recently shown to bind FPN1 thus blocking its interaction with hepcidin and allowing iron efflux. This leads to an increase of serum iron concentrations, TfS and hepcidin with a slower decline of hemoglobin concentrations and a reduction of ferritin concentrations in CKD patients. [297,298] Also, anticalins which are biopharmaceuticals derived from human lipocalins that bind to protein targets, might be effective by mimicking antibody activity [299]. The anticalin PRS-080 decreased hepcidin concentrations with a subsequent increase of serum iron concentrations and TfS in a phase I clinical trial with healthy subjects and CKD patients [300,301].
6.2. Modifying Hepcidin Synthetisation by Targeting the BMP/SMAD Pathway
Considering the importance of BMP/SMAD signaling not only in iron-related regulation of hepcidin expression but also in inflammation-related hepcidin expression, pre-clinical studies investigated the effect of BMP/SMAD pathway inhibitors on disease progression of AI [62].
First, inhibition of BMP/SMAD signaling by the BMPR1 (ALK2/ALK3/ALK6) inhibitor dorsomorphin and its derivate LDN-193189 as well as the human recombinant BMP ligand antagonist ALK3-Fc and the soluble hemojuvelin-Fc (HJV-Fc) were shown to reduce hepcidin expression in animal models of AI thus stimulating mobilization of iron from tissue stores and resolving anemia [164,302,303,304,305]. However, dorsomorphin and LDN-193189 also significantly inhibit TGF-𝛽 receptors (TGF𝛽R) at moderate concentrations with the consequence of more interactions [306]. Thus, a new class BMPR1 inhibitor named LJ00328 with a greater selectivity for BMPRs than for TGF𝛽R was developed. LJ00328 was demonstrated to have the greatest potency against ALK2/ALK3 which are the preferentially BMP2/6 receptors for hepcidin induction in the liver [307]. Indeed, LJ000328 significantly repressed hepcidin production in a mice model of iron refractory IDA [308]. Another animal study revealed that the JAK1/2 inhibitor momelotinib (MMB) ameliorates anemia in a rodent AI model by inhibiting ALK2 [309]. Actually, results from a phase 2 study in patients with myelofibrosis showed an improvement of anemia after MMB treatment [310].
In 2011, commercial heparins were shown to efficiently inhibit hepcidin expression in vitro and in vivo by binding and sequestering BMP6, however, with the unfavorable side effect of its anticoagulant activity [311]. Since BMP6 is one of the two main BMP regulators of HAMP expression, this led to the development of modified heparins with low anti-coagulant and high hepcidin suppressing activity. Modified over-sulfated (SSLMWH-1) or glycol-split heparins (RO-82) as well as the heparinoid pentosan polysulfate (PPS) exert a similar inhibition of hepcidin expression with iron redistribution concomitant to a reduced anticoagulant activity in vitro and in vivo in mice thus depicting a good alternative [312,313,314]. Recently, a low anticoagulant heparin-iron complex was shown to inhibit hepcidin expression and ameliorate AI in mice as well [315], however, it could not inhibit hepcidin formation upon bacterial infection in mice which was also true for dorsomorphin [316]. Interestingly, loss of BMP6 or HJV represses SMAD related HAMP expression in the liver thus causing increased extrahepatic iron loading [317]. This demonstrates the complex regulator networks affecting hepcidin expression suggesting that pharmacological hepcidin repression must be critically analyzed and indicated.
More recently, a fully human anti-BMP6 antibody (KY1070) was shown to counteract the hepcidin-driven iron limitation for erythropoiesis in two-different, well-established, AI rodent models. In addition, combination with darbepoetin alfa further improved the erythroid response when compared to monotherapy [283]. Another study in CKD patients showed that LY3113493 specifically blocked binding of BMP6 to its receptor with an increase of serum iron and TfS and decrease of hepcidin levels [297].
However, BMP signaling is critically involved in normal cardiovascular structure and function, bone homeostasis, and progression of specific cancers [318] which limits the therapeutic approach of broadly effective BMP signaling inhibitors for specific diseases such as AI. Unfortunately, clinical trials with human patients investigating the effects of BMP signaling inhibition in patients with AI are lacking by now.
6.3. Stabilization of Hypoxia-Inducible Factor
Another new promising therapy strategy is the inhibition of prolyl hydroxylase, which stabilizes hypoxia-inducible factor thus promoting erythropoietin production, increasing intestinal iron uptake and mobilization of iron stores [319]. Oral hypoxia-inducible factor prolyl hydroxylase inhibitors (HIF-PHI) including roxadustat, vadadustat, desidustat, daprodustat and molidustat were shown to significantly increase hemoglobin levels and total iron binding capacity concomitant to an decrease in hepcidin and ferritin levels in patients with anemia and chronic kidney disease [320]. Moreover, several preclinical studies have suggested a potential cardiovascular benefit due to direct actions of HIF activation in myocardial infarction, cardiac remodeling and atherosclerosis as well as amelioration of glucose and lipid metabolism [321]. Actually, roxadustat was shown to reduce the average total cholesterol regardless of lipid-lowering treatments [322,323,324].
In a recent meta-analysis by Chen and coworkers including 13,146 patients, HIF-PHI were well tolerated during long-term use with comparable risk of serious adverse events compared to patients treated only with ESAs. However, HIF-PHI treatment was associated with a slightly increased risk for thrombosis events when compared to patients treated only with ESAs [320]. Several phase 3 trials further evaluating the efficacy and safety of HIF-PHI in CKD patients including the long-term cardiovascular risk are currently ongoing. Interestingly, roxadustat also raised hemoglobin levels in CKD patients with immune activation [323,324,325], which suggests that HIF-PHI might be a possible treatment strategy for anemia in patients with other chronic inflammatory disease, a notion that needs to be experimentally and clinically verified.
7. Nutrition in the Treatment of Anemia of Inflammation
Several nutrients including vitamins, amino acids and iron are essential for hemoglobin synthesis and erythropoiesis [326]. Especially vitamin B9 (folic acid) and vitamin B12 (cobalamin) are indispensable for heme synthesis and erythropoiesis. Deficiency of these two vitamins results in (hemolytic) anemia or further aggravates anemia in patients with inflammatory disorders [327]. In patients with AI and concomitant vitamin B9 or B12 deficiency, these vitamins should be supplemented [2]. Interestingly, the synthetic vitamin B1 (thiamine) derivative fursultiamine was shown to inhibit hepcidin-induced Fpn1 internalization thus promoting cellular iron export in vitro [328]. In a mice model of intracerebral hemorrhage, fursultiamine was demonstrated to improve brain iron efflux and reduce oxidative brain injury aggravated by hepcidin [329].
Vitamin D supplementation was shown to inhibit hepcidin expression in patients with CKD and healthy adults [330,331] with a significant increase of hemoglobin levels in critically ill patients [332]. Actually, vitamin D response elements (VDREs) are found in the promoter region of HAMP and have a suppressing effect [330]. Vitamin D receptor signaling further has anti-inflammatory effects, which may positively affect the course of an underlying chronic inflammatory disease [333,334]. In fact, vitamin D plays a crucial role in the modulation of innate and adaptive immune response [335] and can induce a shift from a proinflammatory to a more tolerogenic immune status with diverse effects on T cell subtypes: e.g., it suppresses Th1-cell proliferation, differentiation and cytokine production [336,337]. Furthermore, also Th2-cells [338] and Treg-cells are induced [339], while Th17-mediated autoimmunity can be ameliorated by 1,25-dihydroxyvitamin D3 [340]. As chronic diseases often go along with vitamin D deficiency [341] and low vitamin D levels are correlated with higher immune activation markers like neopterin, e.g., in CAD patients [342], vitamin D supplementation of patients might be very effective to down-regulate underlying inflammatory processes.
Dietary vitamin C (ascorbate) provides reducing capacity for Fe3+ reduction thus enhancing the activity of the enzyme ferric reductase Dcytb and increasing iron absorption [241]. Vitamin C supplementation must be considered as additional treatment especially in patients with concomitant IDA. In addition, vitamin C was demonstrated to increase ferritin concentrations and stimulate transferrin-dependent iron uptake [343,344,345]. Finally, vitamin C directly inhibits hepcidin production and enhances EPOR expression in human liver carcinoma cells [346] thus exerting an additional effect on erythropoiesis in combination with EPO [347]. Vitamin C is also one of the most potent antioxidants and plays an important role for immune function, e.g., it can enhance differentiation and proliferation of B- and T-cells [348]. As higher vitamin C intake and plasma levels were inversely associated with C-reactive protein concentrations [349], and lower vitamin C and E concentrations coincide with higher neopterin concentrations in patients with CAD [350], vitamin C appears to have anti-inflammatory properties [349], which might be beneficial for patients with chronic immune activation. As vitamin C is also a co-factor for the asparagyl and prolyl hydroxylases required for the downregulation of HIF-1α [351], decreased vitamin C availability might also impact erythropoietin formation.
The polyphenol curcumin was also shown to reduce pro-inflammatory cytokine production and directly decrease hepcidin transcription in vitro and in vivo [352,353]. Finally, the Nrf2 activator sulforaphane (SFN), which is a broccoli-derived isothiocyanate (ITC) was shown to counteract the inflammatory induced hepcidin expression with consequent repression of Fpn1 expression in murine macrophages and human cancer cells [146,354].
Finally, diet supplementation with the iron-binding protein lactoferrin seems to be advantageous in the prophylaxis and treatment of mild iron deficiency but may also exert anti-microbial activities [355]. Apart from direct pathogen destruction lactoferrin also regulates cells of innate and adaptive immune system by stimulating the secretion of anti-infectious and anti-tumor cytokines [356,357]. Lactoferrin changes the macrophage phenotype from pro-inflammatory to anti-inflammatory thus decreasing IL-6 and hepcidin expression, while increasing FPN1 and ceruloplasmin expression thereby promoting iron egress from macrophages [358,359].
8. Conclusions
Anemia of inflammation (AI) is a very frequent clinical condition affecting more than a billion people worldwide. A deep understanding of the underlying pathophysiology along with the correct diagnosis of anemia of inflammation and with identification of aggravating factors for anemia severity is vital for selecting the most appropriate therapy. Apart from established treatments including iron supplementation, erythropoiesis stimulating agents or blood transfusions, new therapeutic options, including modulators of hepcidin function or hypoxia inducible factors prolyl hydroxylase inhibitors were identified and evaluated mostly in pre-clinical studies over the last years. However, some of them have entered into clinical trials and those results concerning anemia correction in different inflammatory disorders are awaited.
Author Contributions
Conceptualization, L.L. and G.W.; methodology, L.L. and G.W.; software, L.L.; validation, D.F., K.K. and G.W.; writing—original draft preparation, L.L.; writing—review and editing, D.F., K.K. and G.W.; visualization, L.L.; supervision, K.K. and G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weiss, G.; Goodnough, L.T. Anemia of Chronic Disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Anemia of inflammation. Hematol. Oncol. Clin. N. Am. 2014, 28, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Kassebaum, N.J.; Jasrasaria, R.; Naghavi, M.; Wulf, S.K.; Johns, N.; Lozano, R.; Regan, M.; Weatherall, D.; Chou, D.P.; Eisele, T.P.; et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014, 123, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Schett, G. Anaemia in inflammatory rheumatic diseases. Nat. Rev. Rheumatol. 2013, 9, 205–215. [Google Scholar] [CrossRef]
- Fuchs, D.; Zangerle, R.; Artner-Dworzak, E.; Weiss, G.; Fritsch, P.; Tilz, G.P.; Dierich, M.P.; Wachter, H. Association between immune activation, changes of iron metabolism and anaemia in patients with HIV infection. Eur. J. Haematol. 1993, 50, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Dicato, M.; Plawny, L.; Diederich, M. Anemia in cancer. Ann. Oncol. 2010, 21 (Suppl. 7), vii167–vii172. [Google Scholar] [CrossRef]
- Gilreath, J.A.; Rodgers, G.M. How I treat cancer-associated anemia. Blood 2020, 136, 801–813. [Google Scholar] [CrossRef]
- Papadaki, H.A.; Kritikos, H.D.; Valatas, V.; Boumpas, D.T.; Eliopoulos, G.D. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: Improvement following anti–tumor necrosis factor-α antibody therapy. Blood 2002, 100, 474–482. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Comin-Colet, J.; de Francisco, A.; Dignass, A.; Doehner, W.; Lam, C.S.; Macdougall, I.C.; Rogler, G.; Camaschella, C.; Kadir, R.; et al. Iron deficiency across chronic inflammatory conditions: International expert opinion on definition, diagnosis, and management. Am. J. Hematol. 2017, 92, 1068–1078. [Google Scholar] [CrossRef] [Green Version]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. JASN 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, C.; Tsuchiya, K.; Maeda, K.; Nitta, K. Renal Anemia and Iron Metabolism. Contrib. Nephrol. 2018, 195, 62–73. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kuragano, T. Potential hazards of recent trends in liberal iron use for renal anemia. Clin. Kidney J. 2020, 14, 59–69. [Google Scholar] [CrossRef]
- Coyne, D.W.; Goldsmith, D.; Macdougall, I.C. New options for the anemia of chronic kidney disease. Kidney Int. Suppl. 2017, 7, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Lanser, L.; Fuchs, D.; Scharnagl, H.; Grammer, T.; Kleber, M.E.; März, W.; Weiss, G.; Kurz, K. Anemia of Chronic Disease in Patients With Cardiovascular Disease. Front. Cardiovasc. Med. 2021, 8, 666638. [Google Scholar] [CrossRef]
- Kurz, K.; Lanser, L.; Seifert, M.; Kocher, F.; Pölzl, G.; Weiss, G. Anaemia, iron status, and gender predict the outcome in patients with chronic heart failure. ESC Heart Fail. 2020, 7, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; von Haehling, S.; Anker, S.D.; Macdougall, I.C.; Ponikowski, P. Iron deficiency and heart failure: Diagnostic dilemmas and therapeutic perspectives. Eur. Heart. J. 2013, 34, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnak, M.J.; Tighiouart, H.; Manjunath, G.; MacLeod, B.; Griffith, J.; Salem, D.; Levey, A.S. Anemia as a risk factor for cardiovascular disease in The Atherosclerosis Risk in Communities (ARIC) study. J. Am. Coll. Cardiol. 2002, 40, 27–33. [Google Scholar] [CrossRef]
- Similowski, T.; Agustí, A.; MacNee, W.; Schönhofer, B. The potential impact of anaemia of chronic disease in COPD. Eur. Respir. J. 2006, 27, 390–396. [Google Scholar] [CrossRef]
- Sonnweber, T.; Nairz, M.; Theurl, I.; Petzer, V.; Tymoszuk, P.; Haschka, D.; Rieger, E.; Kaessmann, B.; Deri, M.; Watzinger, K.; et al. The crucial impact of iron deficiency definition for the course of precapillary pulmonary hypertension. PLoS ONE 2018, 13, e0203396. [Google Scholar] [CrossRef]
- Pizzini, A.; Aichner, M.; Sonnweber, T.; Tancevski, I.; Weiss, G.; Löffler-Ragg, J. The Significance of iron deficiency and anemia in a real-life COPD cohort. Int. J. Med. Sci. 2020, 17, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.J.; Salas, M.; Ward, A.; Goss, G. Anemia as an independent prognostic factor for survival in patients with cancer. Cancer 2001, 91, 2214–2221. [Google Scholar] [CrossRef]
- Grammer, T.B.; Kleber, M.E.; Silbernagel, G.; Pilz, S.; Scharnagl, H.; Tomaschitz, A.; König, W.; März, W. Hemoglobin, iron metabolism and angiographic coronary artery disease (The Ludwigshafen Risk and Cardiovascular Health Study). Atherosclerosis 2014, 236, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Kink, P.; Egger, E.M.; Lanser, L.; Klaunzner, M.; Holzner, B.; Willenbacher, W.; Kasseroler, M.T.; Fuchs, D.; Weiss, G.; Kurz, K. Immune Activation and Anemia Are Associated with Decreased Quality of Life in Patients with Solid Tumors. J. Clin. Med. 2020, 9, 3248. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; von Haehling, S.; Doehner, W.; Banasiak, W.; et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J. Card. Fail. 2011, 17, 899–906. [Google Scholar] [CrossRef]
- Gaspar, B.L.; Sharma, P.; Das, R. Anemia in malignancies: Pathogenetic and diagnostic considerations. Hematology 2015, 20, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bohlius, J.; Bohlke, K.; Castelli, R.; Djulbegovic, B.; Lustberg, M.B.; Martino, M.; Mountzios, G.; Peswani, N.; Porter, L.; Tanaka, T.N.; et al. Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update. Blood Adv. 2019, 3, 1197–1210. [Google Scholar] [CrossRef] [Green Version]
- Kallich, J.D.; Tchekmedyian, N.S.; Damiano, A.M.; Shi, J.; Black, J.T.; Erder, M.H. Psychological outcomes associated with anemia-related fatigue in cancer patients. Oncology 2002, 16, 117–124. [Google Scholar]
- Sabbatini, P. The relationship between anemia and quality of life in cancer patients. Oncologist 2000, 5 (Suppl. 2), 19–23. [Google Scholar] [CrossRef]
- Knight, K.; Wade, S.; Balducci, L. Prevalence and outcomes of anemia in cancer: A systematic review of the literature. Am. J. Med. 2004, 116 (Suppl. 7A), 11S–26S. [Google Scholar] [CrossRef]
- Tang, Y.-D.; Katz, S.D. Anemia in Chronic Heart Failure. Circulation 2006, 113, 2454–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodnough, L.T.; Schrier, S.L. Evaluation and management of anemia in the elderly. Am. J. Hematol. 2014, 89, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Stauder, R.; Valent, P.; Theurl, I. Anemia at older age: Etiologies, clinical implications, and management. Blood 2018, 131, 505–514. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. 1), S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G. Iron metabolism in the anemia of chronic disease. Biochim. Biophys. Acta 2009, 1790, 682–693. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Emerit, J.; Beaumont, C.; Trivin, F. Iron metabolism, free radicals, and oxidative injury. Biomed. Pharm. Ther 2001, 55, 333–339. [Google Scholar] [CrossRef]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Haschka, D.; Demetz, E.; Weiss, G. Iron at the interface of immunity and infection. Front. Pharm. 2014, 5, 152. [Google Scholar] [CrossRef] [Green Version]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharm. 2005, 202, 199–211. [Google Scholar] [CrossRef]
- Sharp, P.; Srai, S.-K. Molecular mechanisms involved in intestinal iron absorption. World J. Gastroenterol. 2007, 13, 4716–4724. [Google Scholar] [CrossRef]
- Mackenzie, B.; Garrick, M.D. Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am. J. Physiol.-Gastroint. Liver Physiol. 2005, 289, G981–G986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef]
- Skikne, B.S.; Whittaker, P.; Cooke, A.; Cook, J.D. Ferritin excretion and iron balance in humans. Br. J. Haematol. 1995, 90, 681–687. [Google Scholar] [CrossRef]
- Bomford, A.B.; Munro, H.N. Transferrin and its receptor: Their roles in cell function. Hepatology 1985, 5, 870–875. [Google Scholar] [CrossRef]
- Brock, J.H. The physiology of lactoferrin. Biochem. Cell Biol. 2002, 80, 1–6. [Google Scholar] [CrossRef]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.; Verdier, F. Transferrin Receptors in Erythropoiesis. Int. J. Mol. Sci. 2020, 21, 9713. [Google Scholar] [CrossRef]
- Silvestri, L.; Nai, A.; Pagani, A.; Camaschella, C. The extrahepatic role of TFR2 in iron homeostasis. Front. Pharmacol. 2014, 5, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, R.E.; Migas, M.C.; Holden, C.C.; Waheed, A.; Britton, R.S.; Tomatsu, S.; Bacon, B.R.; Sly, W.S. Transferrin receptor 2: Continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 2000, 97, 2214–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleven, M.D.; Jue, S.; Enns, C.A. Transferrin Receptors TfR1 and TfR2 Bind Transferrin through Differing Mechanisms. Biochemistry 2018, 57, 1552–1559. [Google Scholar] [CrossRef]
- Cao, H.; Schroeder, B.; Chen, J.; Schott, M.B.; McNiven, M.A. The Endocytic Fate of the Transferrin Receptor Is Regulated by c-Abl Kinase. J. Biol. Chem. 2016, 291, 16424–16437. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [Green Version]
- Tabuchi, M.; Yoshimori, T.; Yamaguchi, K.; Yoshida, T.; Kishi, F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 2000, 275, 22220–22228. [Google Scholar] [CrossRef] [Green Version]
- Calzolari, A.; Raggi, C.; Deaglio, S.; Sposi, N.M.; Stafsnes, M.; Fecchi, K.; Parolini, I.; Malavasi, F.; Peschle, C.; Sargiacomo, M.; et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 2006, 119, 4486–4498. [Google Scholar] [CrossRef] [Green Version]
- Maura, P.; Sara, L.; Valentina, G.; Federica, M.; Dario, F.; Laura, S.; Antonella, R.; Paolo, A. Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation. Haematologica 2010, 95, 1832–1840. [Google Scholar] [CrossRef]
- Xiao, X.; Alfaro-Magallanes, V.M.; Babitt, J.L. Bone morphogenic proteins in iron homeostasis. Bone 2020, 138, 115495. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”—How macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflügers Arch. -Eur. J. Physiol. 2017, 469, 397–418. [Google Scholar] [CrossRef] [Green Version]
- Iancu, T.C. Ferritin and hemosiderin in pathological tissues. Electron. Microsc. Rev. 1992, 5, 209–229. [Google Scholar] [CrossRef]
- Saito, H. Metabolism of iron stores. Nagoya J. Med. Sci. 2014, 76, 235–254. [Google Scholar] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Loréal, O.; Haziza-Pigeon, C.; Troadec, M.B.; Detivaud, L.; Turlin, B.; Courselaud, B.; Ilyin, G.; Brissot, P. Hepcidin in iron metabolism. Curr. Protein Pept. Sci. 2005, 6, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Aigner, E.; Theurl, M.; Nairz, M.; Seifert, M.; Schroll, A.; Sonnweber, T.; Eberwein, L.; Witcher, D.R.; Murphy, A.T.; et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: Diagnostic and therapeutic implications. Blood 2009, 113, 5277–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Meynard, D.; Monnier, A.; Darnaud, V.; Bouvet, R.; Wang, R.H.; Deng, C.; Vaulont, S.; Mosser, J.; Coppin, H.; et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008, 112, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Investig. 2005, 115, 2180–2186. [Google Scholar] [CrossRef] [Green Version]
- Rishi, G.; Daniel, F.W.; Subramaniam, V.N. Hepcidin: Regulation of the master iron regulator. Biosci. Rep. 2015, 35, e00192. [Google Scholar] [CrossRef] [Green Version]
- Canali, S.; Wang, C.-Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Xu, Y.; Traeger, L.; Dogan, D.Y.; Xiao, X.; Steinbicker, A.U.; Babitt, J.L. Erythroferrone lowers hepcidin by sequestering BMP2/6 heterodimer from binding to the BMP type I receptor ALK3. Blood 2020, 135, 453–456. [Google Scholar] [CrossRef]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.G.; Wang, Y.; Wu, Q.; Cheng, W.H.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.J.; Yu, P.B.; Wang, F.; et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Traeger, L.; Enns, C.A.; Krijt, J.; Steinbicker, A.U. The hemochromatosis protein HFE signals predominantly via the BMP type I receptor ALK3 in vivo. Commun. Biol. 2018, 1, 65. [Google Scholar] [CrossRef]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Xiao, X.; Bayer, A.; Xu, Y.; Dev, S.; Canali, S.; Nair, A.V.; Masia, R.; Babitt, J.L. Ablation of Hepatocyte Smad1, Smad5, and Smad8 Causes Severe Tissue Iron Loading and Liver Fibrosis in Mice. Hepatology 2019, 70, 1986–2002. [Google Scholar] [CrossRef] [PubMed]
- Latour, C.; Kautz, L.; Besson-Fournier, C.; Island, M.L.; Canonne-Hergaux, F.; Loréal, O.; Ganz, T.; Coppin, H.; Roth, M.P. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 2014, 59, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Bachman, E.; Feng, R.; Travison, T.; Li, M.; Olbina, G.; Ostland, V.; Ulloor, J.; Zhang, A.; Basaria, S.; Ganz, T.; et al. Testosterone Suppresses Hepcidin in Men: A Potential Mechanism for Testosterone-Induced Erythrocytosis. J. Clin. Endocrinol. Metab. 2010, 95, 4743–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Rhee, D.K.; Malhotra, R.; Mayeur, C.; Hurst, L.A.; Ager, E.; Shelton, G.; Kramer, Y.; McCulloh, D.; Keefe, D.; et al. Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J. Clin. Investig. 2016, 126, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jian, J.; Katz, S.; Abramson, S.B.; Huang, X. 17β-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 2012, 153, 3170–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; Tamaki, T. Estrogen Regulates Hepcidin Expression via GPR30-BMP6-Dependent Signaling in Hepatocytes. PLoS ONE 2012, 7, e40465. [Google Scholar] [CrossRef]
- Higashimoto, Y.; Tanaka, K.; Matsui, T.; Sakaguchi, T.; Yamagishi, S.-i.; Motomiya, Y. Fibroblast Growth Factor 23 Contributes to Regulation of Hepcidin/Ferroportin Axis. Austin. J. Pharmacol. Ther. 2020, 8, 5. [Google Scholar]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Mast, A.E.; Blinder, M.A.; Dietzen, D.J. Reticulocyte hemoglobin content. Am. J. Hematol. 2008, 83, 307–310. [Google Scholar] [CrossRef]
- Kuhrt, D.; Wojchowski, D.M. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015, 125, 3536–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koury, M.J.; Ponka, P. New insights into erythropoiesis: The roles of folate, vitamin B12, and iron. Annu. Rev. Nutr. 2004, 24, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Houston, T.; Kastner, S.; Jöhrer, K.; Grünewald, K.; Brock, J.H. Regulation of cellular iron metabolism by erythropoietin: Activation of iron-regulatory protein and upregulation of transferrin receptor expression in erythroid cells. Blood 1997, 89, 680–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, S.; Delehanty, L.; Grado, S.; Holy, M.; White, Z., 3rd; Freeman, K.; Kurita, R.; Nakamura, Y.; Bullock, G.; Goldfarb, A. Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor. J. Exp. Med. 2018, 215, 661–679. [Google Scholar] [CrossRef]
- Pantopoulos, K. TfR2 links iron metabolism and erythropoiesis. Blood 2015, 125, 1055–1056. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.B.; Chen, J.; Murchison, N.; Green, F.A.; Enns, C.A. Transferrin receptor 2: Evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 2007, 18, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, R.; Ganz, T. Erythroferrone: An Erythroid Regulator of Hepcidin and Iron Metabolism. Hemasphere 2018, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Klingmüller, U.; Besmer, P.; Lodish, H.F. Interaction of the erythropoietin and stem-cell-factor receptors. Nature 1995, 377, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Maguer-Satta, V.É.; Bartholin, L.; Jeanpierre, S.; Ffrench, M.; Martel, S.; Magaud, J.-P.; Rimokh, R. Regulation of human erythropoiesis by activin A, BMP2, and BMP4, members of the TGFβ family. Exp. Cell Res. 2003, 282, 110–120. [Google Scholar] [CrossRef]
- Vinchi, F.; Vance, S.Z. Reshaping Erythrophagocytosis and Iron Recycling by Reticuloendothelial Macrophages. Hemasphere 2021, 5, e525. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharm. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Soe-Lin, S.; Sheftel, A.D.; Wasyluk, B.; Ponka, P. Nramp1 equips macrophages for efficient iron recycling. Exp. Hematol. 2008, 36, 929–937. [Google Scholar] [CrossRef]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [Green Version]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Chepelev, N.L.; Willmore, W.G. Regulation of iron pathways in response to hypoxia. Free Radic. Biol. Med. 2011, 50, 645–666. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [Green Version]
- Zoller, H.; Schaefer, B.; Glodny, B. Iron-induced hypophosphatemia: An emerging complication. Curr. Opin. Nephrol. Hypertens. 2017, 26, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Interleukin 6 and its Receptor: Ten Years Later. Int. Rev. Immunol. 1998, 16, 249–284. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Delga, S.; Deschemin, J.C.; Vaulont, S.; Peyssonnaux, C. Deletion of HIF-2α in the enterocytes decreases the severity of tissue iron loading in hepcidin knockout mice. Blood 2012, 119, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Lok, C.N.; Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacchini, L.; Bianchi, L.; Bernelli-Zazzera, A.; Cairo, G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 1999, 274, 24142–24146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, C.K.; Mazumder, B.; Fox, P.L. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000, 275, 21048–21054. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, D.; Pastore, Y.D.; Divoky, V.; Liu, E.; Mlodnicka, A.E.; Rainey, K.; Ponka, P.; Semenza, G.L.; Schumacher, A.; Prchal, J.T. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J. Biol. Chem. 2006, 281, 25703–25711. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Eisenstein, R.S.; Ross, K.L. Novel roles for iron regulatory proteins in the adaptive response to iron deficiency. J. Nutr. 2003, 133, 1510S–1516S. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef]
- Hentze, M.W.; Rouault, T.A.; Harford, J.B.; Klausner, R.D. Oxidation-Reduction and the Molecular Mechanism of a Regulatory RNA-Protein Interaction. Science 1989, 244, 357–359. [Google Scholar] [CrossRef]
- Bettany, A.J.; Eisenstein, R.S.; Munro, H.N. Mutagenesis of the iron-regulatory element further defines a role for RNA secondary structure in the regulation of ferritin and transferrin receptor expression. J. Biol. Chem. 1992, 267, 16531–16537. [Google Scholar] [CrossRef]
- Haile, D.J.; Rouault, T.A.; Tang, C.K.; Chin, J.; Harford, J.B.; Klausner, R.D. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: Role of the iron-sulfur cluster. Proc. Natl. Acad. Sci. USA 1992, 89, 7536–7540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, E.S.; Rawlins, M.L.; Leibold, E.A. Oxygen and iron regulation of iron regulatory protein 2. J. Biol. Chem. 2003, 278, 40337–40342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, G.; Goossen, B.; Doppler, W.; Fuchs, D.; Pantopoulos, K.; Werner-Felmayer, G.; Wachter, H.; Hentze, M.W. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 1993, 12, 3651–3657. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Hentze, M.W. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 1995, 14, 2917–2924. [Google Scholar] [CrossRef]
- Schneider, B.D.; Leibold, E.A. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood 2003, 102, 3404–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Ebert, B.L.; Neil, C.; Brenner, K.; Papaioannou, I.; Melas, A.; Tolliday, N.; Lamb, J.; Pantopoulos, K.; Golub, T.; et al. Small-molecule inhibitors of HIF-2a translation link its 5’UTR iron-responsive element to oxygen sensing. Mol. Cell 2008, 32, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox. Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes. Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Samuele, M.; Deborah, C.; Erika, M.; Jens, S.; Emilia, T.; Emanuela, T.; Martina, U.M. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.-R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homoeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schleicher, U.; Schroll, A.; Sonnweber, T.; Theurl, I.; Ludwiczek, S.; Talasz, H.; Brandacher, G.; Moser, P.L.; Muckenthaler, M.U.; et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J. Exp. Med. 2013, 210, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef]
- Renassia, C.; Peyssonnaux, C. New insights into the links between hypoxia and iron homeostasis. Curr. Opin. Hematol. 2019, 26, 125–130. [Google Scholar] [CrossRef]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress Erythropoiesis is a Key Inflammatory Response. Cells 2020, 9, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, J.W. The anemia of inflammation/malignancy: Mechanisms and management. Hematol. Am. Soc. Hematol. Educ. Program 2008, 1, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Proinflammatory cytokines lowering erythropoietin production. J. Interferon Cytokine Res. 1998, 18, 555–559. [Google Scholar] [CrossRef]
- Theurl, I.; Mattle, V.; Seifert, M.; Mariani, M.; Marth, C.; Weiss, G. Dysregulated monocyte iron homeostasis and erythropoietin formation in patients with anemia of chronic disease. Blood 2006, 107, 4142–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwiczek, S.; Aigner, E.; Theurl, I.; Weiss, G.n. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood 2003, 101, 4148–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Ulmer, H.; Kaser, A.; Weiss, G. Role of IL-10 for induction of anemia during inflammation. J. Immunol. 2002, 169, 2204–2209. [Google Scholar] [CrossRef]
- Weiss, G.; Bogdan, C.; Hentze, M.W. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J. Immunol. 1997, 158, 420–425. [Google Scholar]
- Miller, L.L.; Miller, S.C.; Torti, S.V.; Tsuji, Y.; Torti, F.M. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1991, 88, 4946–4950. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeur, C.; Lohmeyer, L.K.; Leyton, P.; Kao, S.M.; Pappas, A.E.; Kolodziej, S.A.; Spagnolli, E.; Yu, B.; Galdos, R.L.; Yu, P.B.; et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 2014, 123, 2261–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbicker, A.U.; Sachidanandan, C.; Vonner, A.J.; Yusuf, R.Z.; Deng, D.Y.; Lai, C.S.; Rauwerdink, K.M.; Winn, J.C.; Saez, B.; Cook, C.M.; et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011, 117, 4915–4923. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Canali, S.; Bayer, A.; Dev, S.; Agarwal, A.; Babitt, J.L. Iron, erythropoietin, and inflammation regulate hepcidin in Bmp2-deficient mice, but serum iron fails to induce hepcidin in Bmp6-deficient mice. Am. J. Hematol. 2019, 94, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Krijt, J.; Vokurka, M.; Chang, K.-T.; Nečas, E. Expression of Rgmc, the murine ortholog of hemojuvelin gene, is modulated by development and inflammation, but not by iron status or erythropoietin. Blood 2004, 104, 4308–4310. [Google Scholar] [CrossRef]
- Besson-Fournier, C.; Latour, C.; Kautz, L.; Bertrand, J.; Ganz, T.; Roth, M.P.; Coppin, H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012, 120, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, Y.; Sugiyama, M.; Hashimoto, O.; Murakami, M.; Matsui, T.; Funaba, M. Regulation of hepcidin expression by inflammation-induced activin B. Sci. Rep. 2016, 6, 38702. [Google Scholar] [CrossRef] [Green Version]
- Canali, S.; Core, A.B.; Zumbrennen-Bullough, K.B.; Merkulova, M.; Wang, C.-Y.; Schneyer, A.L.; Pietrangelo, A.; Babitt, J.L. Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice. Endocrinology 2016, 157, 1146–1162. [Google Scholar] [CrossRef]
- Kanamori, Y.; Murakami, M.; Sugiyama, M.; Hashimoto, O.; Matsui, T.; Funaba, M. Interleukin-1β (IL-1β) transcriptionally activates hepcidin by inducing CCAAT enhancer-binding protein δ (C/EBPδ) expression in hepatocytes. J. Biol. Chem. 2017, 292, 10275–10287. [Google Scholar] [CrossRef] [Green Version]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Ferla, K.; Reimann, C.; Jelkmann, W.; Hellwig-Bürgel, T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-κB. FASEB J. 2002, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef]
- Latour, C.; Wlodarczyk, M.F.; Jung, G.; Gineste, A.; Blanchard, N.; Ganz, T.; Roth, M.P.; Coppin, H.; Kautz, L. Erythroferrone contributes to hepcidin repression in a mouse model of malarial anemia. Haematologica 2017, 102, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nai, A.; Rubio, A.; Campanella, A.; Gourbeyre, O.; Artuso, I.; Bordini, J.; Gineste, A.; Latour, C.; Besson-Fournier, C.; Lin, H.Y.; et al. Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 2016, 127, 2327–2336. [Google Scholar] [CrossRef] [Green Version]
- Sonnweber, T.; Nachbaur, D.; Schroll, A.; Nairz, M.; Seifert, M.; Demetz, E.; Haschka, D.; Mitterstiller, A.-M.; Kleinsasser, A.; Burtscher, M.; et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 2014, 63, 1951–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Finkenstedt, A.; Bianchi, P.; Theurl, I.; Vogel, W.; Witcher, D.R.; Wroblewski, V.J.; Murphy, A.T.; Zanella, A.; Zoller, H. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br. J. Haematol. 2009, 144, 789–793. [Google Scholar] [CrossRef]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J. Exp. Med. 2021, 218, e20201541. [Google Scholar] [CrossRef]
- Valente de Souza, L.; Hoffmann, A.; Weiss, G. Impact of bacterial infections on erythropoiesis. Expert Rev. Anti-Infect. 2021, 19, 619–633. [Google Scholar] [CrossRef]
- Means, R.T. Recent developments in the anemia of chronic disease. Curr. Hematol. Rep. 2003, 2, 116–121. [Google Scholar]
- Moldawer, L.L.; Marano, M.A.; Wei, H.; Fong, Y.; Silen, M.L.; Kuo, G.; Manogue, K.R.; Vlassara, H.; Cohen, H.; Cerami, A.; et al. Cachectin/tumor necrosis factor-α alters red blood cell kinetics and induces anemia in vivo. FASEB J. 1989, 3, 1637–1643. [Google Scholar] [CrossRef]
- Libregts, S.F.; Gutiérrez, L.; de Bruin, A.M.; Wensveen, F.M.; Papadopoulos, P.; van Ijcken, W.; Ozgür, Z.; Philipsen, S.; Nolte, M.A. Chronic IFN-γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood 2011, 118, 2578–2588. [Google Scholar] [CrossRef] [Green Version]
- Mitlyng, B.L.; Singh, J.A.; Furne, J.K.; Ruddy, J.; Levitt, M.D. Use of breath carbon monoxide measurements to assess erythrocyte survival in subjects with chronic diseases. Am. J. Hematol. 2006, 81, 432–438. [Google Scholar] [CrossRef]
- Dinkla, S.; van Eijk, L.T.; Fuchs, B.; Schiller, J.; Joosten, I.; Brock, R.; Pickkers, P.; Bosman, G.J. Inflammation-associated changes in lipid composition and the organization of the erythrocyte membrane. BBA Clin. 2016, 5, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.M.; Sharpe, M.D.; Singer, M.; Ellis, C.G. The Effect of Sepsis on the Erythrocyte. Int. J. Mol. Sci. 2017, 18, 1932. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murr, C.; Widner, B.; Wirleitner, B.; Fuchs, D. Neopterin as a marker for immune system activation. Curr. Drug. Metab. 2002, 3, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Melichar, B.; Spisarova, M.; Bartouskova, M.; Krcmova, L.K.; Javorska, L.; Studentova, H. Neopterin as a biomarker of immune response in cancer patients. Ann. Transl. Med. 2017, 5, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanser, L.; Pölzl, G.; Fuchs, D.; Weiss, G.; Kurz, K. Neopterin is Associated with Disease Severity and Outcome in Patients with Non-Ischaemic Heart Failure. J. Clin. Med. 2019, 8, 2230. [Google Scholar] [CrossRef] [Green Version]
- Pizzini, A.; Lunger, F.; Sahanic, A.; Nemati, N.; Fuchs, D.; Weiss, G.; Kurz, K.; Bellmann-Weiler, R. Diagnostic and Prognostic Value of Inflammatory Parameters Including Neopterin in the Setting of Pneumonia, COPD, and Acute Exacerbations. COPD 2017, 14, 298–303. [Google Scholar] [CrossRef]
- Smukowska-Gorynia, A.; Marcinkowska, J.; Chmara, E.; Malaczynska-Rajpold, K.; Slawek-Szmyt, S.; Cieslewicz, A.; Janus, M.; Araszkiewicz, A.; Jankiewicz, S.; Komosa, A.; et al. Neopterin as a Biomarker in Patients with Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension. Respiration 2018, 96, 222–230. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug. Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Lemos, H.; Huang, L.; Prendergast, G.C.; Mellor, A.L. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer 2019, 19, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Lanser, L.; Kink, P.; Egger, E.M.; Willenbacher, W.; Fuchs, D.; Weiss, G.; Kurz, K. Inflammation-Induced Tryptophan Breakdown is Related With Anemia, Fatigue, and Depression in Cancer. Front. Immunol. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, J.M.; Ozaki, Y.; Byrne, G.I.; Brown, R.R.; Borden, E.C. Interferons and indoleamine 2,3-dioxygenase: Role in antimicrobial and antitumor effects. Experientia 1989, 45, 535–541. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Kynurenine, by activating aryl hydrocarbon receptor, decreases erythropoietin and increases hepcidin production in HepG2 cells: A new mechanism for anemia of inflammation. Exp. Hematol. 2016, 44, 60–67. [Google Scholar] [CrossRef]
- Pawlak, D.; Koda, M.; Pawlak, S.; Wolczynski, S.; Buczko, W. Contribution of quinolinic acid in the development of anemia in renal insufficiency. Am. J. Physiol. Ren. Physiol. 2003, 284, F693–F700. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.E.; Willmore, W.G.; Gu, J.; Goldberg, M.A.; Bunn, H.F. Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J. Biol. Chem. 1999, 274, 9038–9044. [Google Scholar] [CrossRef] [Green Version]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpe, S.; Maes, M. IDO and interferon-alpha-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Fernstrom, J.D.; Wurtman, R.J. Brain serotonin content: Physiological dependence on plasma tryptophan levels. Science 1971, 173, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Schroecksnadel, K.; Zangerle, R.; Bellmann-Weiler, R.; Garimorth, K.; Weiss, G.; Fuchs, D. Indoleamine-2, 3-dioxygenase and other interferon-gamma-mediated pathways in patients with human immunodeficiency virus infection. Curr. Drug. Metab. 2007, 8, 225–236. [Google Scholar] [CrossRef]
- Weiss, G.; Schroecksnadel, K.; Mattle, V.; Winkler, C.; Konwalinka, G.; Fuchs, D. Possible role of cytokine-induced tryptophan degradation in anaemia of inflammation. Eur. J. Haematol. 2004, 72, 130–134. [Google Scholar] [CrossRef]
- World Health Organization. Haemoglobin Concentrations for the Diagnosis of Anaemia and Assessment of Severity; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10, 1294. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Thomas, L. Anemia of chronic disease: Pathophysiology and laboratory diagnosis. Lab. Hematol. 2005, 11, 14–23. [Google Scholar] [CrossRef]
- Bellmann-Weiler, R.; Lanser, L.; Barket, R.; Rangger, L.; Schapfl, A.; Schaber, M.; Fritsche, G.; Wöll, E.; Weiss, G. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J. Clin. Med. 2020, 9, 2429. [Google Scholar] [CrossRef] [PubMed]
- Lanser, L.; Burkert, F.R.; Bellmann-Weiler, R.; Schroll, A.; Wildner, S.; Fritsche, G.; Weiss, G. Dynamics in Anemia Development and Dysregulation of Iron Homeostasis in Hospitalized Patients with COVID-19. Metabolites 2021, 11, 653. [Google Scholar] [CrossRef]
- van Santen, S.; de Mast, Q.; Oosting, J.D.; van Ede, A.; Swinkels, D.W.; van der Ven, A.J. Hematologic parameters predicting a response to oral iron therapy in chronic inflammation. Haematologica 2014, 99, e171–e173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodnough, L.T.; Nemeth, E.; Ganz, T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood 2010, 116, 4754–4761. [Google Scholar] [CrossRef]
- Brugnara, C.; Adamson, J.; Auerbach, M.; Kane, R.; Macdougall, I.; Mast, A. Iron deficiency: What are the future trends in diagnostics and therapeutics? Clin. Chem. 2013, 59, 740–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alageeli, A.A.; Alqahtany, F.S.; Algahtani, F.H. The Role of Reticulocyte Hemoglobin Content for the Diagnosis of Functional Iron Deficiency in Hemodialyzed patients. Saudi. J. Biol. Sci. 2021, 28, 50–54. [Google Scholar] [CrossRef]
- Kohgo, Y.; Niitsu, Y.; Kondo, H.; Kato, J.; Tsushima, N.; Sasaki, K.; Hirayama, M.; Numata, T.; Nishisato, T.; Urushizaki, I. Serum transferrin receptor as a new index of erythropoiesis. Blood 1987, 70, 1955–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punnonen, K.; Irjala, K.; Rajamäki, A. Iron-deficiency anemia is associated with high concentrations of transferrin receptor in serum. Clin. Chem. 1994, 40, 774–776. [Google Scholar] [CrossRef]
- Skikne, B.S.; Punnonen, K.; Caldron, P.H.; Bennett, M.T.; Rehu, M.; Gasior, G.H.; Chamberlin, J.S.; Sullivan, L.A.; Bray, K.R.; Southwick, P.C. Improved differential diagnosis of anemia of chronic disease and iron deficiency anemia: A prospective multicenter evaluation of soluble transferrin receptor and the sTfR/log ferritin index. Am. J. Hematol. 2011, 86, 923–927. [Google Scholar] [CrossRef]
- Goodnough, L.T.; Skikne, B.; Brugnara, C. Erythropoietin, iron, and erythropoiesis. Blood 2000, 96, 823–833. [Google Scholar] [CrossRef]
- Punnonen, K.; Irjala, K.; Rajamäki, A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood 1997, 89, 1052–1057. [Google Scholar] [CrossRef]
- Weiss, G. Anemia of Chronic Disorders: New Diagnostic Tools and New Treatment Strategies. Semin. Hematol. 2015, 52, 313–320. [Google Scholar] [CrossRef]
- Ganz, T.; Olbina, G.; Girelli, D.; Nemeth, E.; Westerman, M. Immunoassay for human serum hepcidin. Blood 2008, 112, 4292–4297. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Theurl, I.; Eder, S.; Koppelstaetter, C.; Kurz, K.; Sonnweber, T.; Kobold, U.; Mayer, G. Serum hepcidin concentration in chronic haemodialysis patients: Associations and effects of dialysis, iron and erythropoietin therapy. Eur. J. Clin. Invest. 2009, 39, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Kroot, J.J.; Tjalsma, H.; Fleming, R.E.; Swinkels, D.W. Hepcidin in Human Iron Disorders: Diagnostic Implications. Clin. Chem. 2011, 57, 1650–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasocki, S.; Baron, G.; Driss, F.; Westerman, M.; Puy, H.; Boutron, I.; Beaumont, C.; Montravers, P. Diagnostic accuracy of serum hepcidin for iron deficiency in critically ill patients with anemia. Intensive Care Med. 2010, 36, 1044–1048. [Google Scholar] [CrossRef]
- Theurl, I.; Schroll, A.; Nairz, M.; Seifert, M.; Theurl, M.; Sonnweber, T.; Kulaksiz, H.; Weiss, G. Pathways for the regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica 2011, 96, 1761–1769. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Tancevski, I.; Löffler-Ragg, J.; Weiss, G. Anaemia, iron homeostasis and pulmonary hypertension: A review. Intern. Emerg. Med. 2020, 15, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Koutroubakis, I.E.; Ramos-Rivers, C.; Regueiro, M.; Koutroumpakis, E.; Click, B.; Schwartz, M.; Swoger, J.; Baidoo, L.; Hashash, J.G.; Barrie, A.; et al. The Influence of Anti-tumor Necrosis Factor Agents on Hemoglobin Levels of Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1587–1593. [Google Scholar] [CrossRef]
- Vallurupalli, M.; MacFadyen, J.G.; Glynn, R.J.; Thuren, T.; Libby, P.; Berliner, N.; Ridker, P.M. Effects of Interleukin-1β Inhibition on Incident Anemia: Exploratory Analyses From a Randomized Trial. Ann. Intern. Med. 2020, 172, 523–532. [Google Scholar] [CrossRef]
- Song, S.N.; Tomosugi, N.; Kawabata, H.; Ishikawa, T.; Nishikawa, T.; Yoshizaki, K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood 2010, 116, 3627–3634. [Google Scholar] [CrossRef] [Green Version]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-C.; Chiu, P.-F.; Chen, H.-L.; Chang, T.-L.; Chang, Y.-J.; Huang, C.-H. Simvastatin downregulates the expression of hepcidin and erythropoietin in HepG2 cells. Hemodial. Int. 2013, 17, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Masajtis-Zagajewska, A.; Nowicki, M. Effect of atorvastatin on iron metabolism regulation in patients with chronic kidney disease—A randomized double blind crossover study. Ren. Fail. 2018, 40, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, M.P.; Weiss, G. The Iron age of host–microbe interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, E.D. Iron loading and disease surveillance. Emerg. Infect. Dis. 1999, 5, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pieracci, F.M.; Barie, P.S. Iron and the risk of infection. Surg. Infect. 2005, 6 (Suppl. 1), S41–S46. [Google Scholar] [CrossRef]
- Ganz, T.; Aronoff, G.R.; Gaillard, C.A.J.M.; Goodnough, L.T.; Macdougall, I.C.; Mayer, G.; Porto, G.; Winkelmayer, W.C.; Wish, J.B. Iron Administration, Infection, and Anemia Management in CKD: Untangling the Effects of Intravenous Iron Therapy on Immunity and Infection Risk. Kidney Med. 2020, 2, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Haschka, D.; Valente de Souza, L.; Tymoszuk, P.; Seifert, M.; von Raffay, L.; Hilbe, R.; Petzer, V.; Moser, P.L.; Nairz, M.; et al. Baseline iron status and presence of anaemia determine the course of systemic Salmonella infection following oral iron supplementation in mice. EBioMedicine 2021, 71, 103568. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; Staun, M.; Tandon, R.K.; Altorjay, I.; Thillainayagam, A.V.; Gratzer, C.; Nijhawan, S.; Thomsen, L.L. A randomized, open-label, non-inferiority study of intravenous iron isomaltoside 1000 (Monofer) compared with oral iron for treatment of anemia in IBD (PROCEED). Am. J. Gastroenterol. 2013, 108, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, O.H.; Soendergaard, C.; Vikner, M.E.; Weiss, G. Rational Management of Iron-Deficiency Anaemia in Inflammatory Bowel Disease. Nutrients 2018, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Aspuru, K.; Villa, C.; Bermejo, F.; Herrero, P.; López, S.G. Optimal management of iron deficiency anemia due to poor dietary intake. Int. J. Gen. Med. 2011, 4, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Moretti, D.; Goede, J.S.; Zeder, C.; Jiskra, M.; Chatzinakou, V.; Tjalsma, H.; Melse-Boonstra, A.; Brittenham, G.; Swinkels, D.W.; Zimmermann, M.B. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood 2015, 126, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Clavel, T.; Smirnov, K.; Schmidt, A.; Lagkouvardos, I.; Walker, A.; Lucio, M.; Michalke, B.; Schmitt-Kopplin, P.; Fedorak, R.; et al. Oral versus intravenous iron replacement therapy distinctly alters the gut microbiota and metabolome in patients with IBD. Gut 2017, 66, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Wagner, S.J.; Martínez, I.; Walter, J.; Chang, J.-S.; Clavel, T.; Kisling, S.; Schuemann, K.; Haller, D. Depletion of luminal iron alters the gut microbiota and prevents Crohn’s disease-like ileitis. Gut 2011, 60, 325–333. [Google Scholar] [CrossRef]
- Girelli, D.; Ugolini, S.; Busti, F.; Marchi, G.; Castagna, A. Modern iron replacement therapy: Clinical and pathophysiological insights. Int. J. Hematol. 2018, 107, 16–30. [Google Scholar] [CrossRef] [Green Version]
- David, R.; Joergen, F.; Steven, F.; Michael, H.; Stefanie, H.; Francesco, L.; Shalini, P.; Janos, S.; Guenter, W. Hypersensitivity reactions to intravenous iron: Guidance for risk minimization and management. Haematologica 2014, 99, 1671–1676. [Google Scholar] [CrossRef] [Green Version]
- Petzer, V.; Theurl, I.; Weiss, G. Established and Emerging Concepts to Treat Imbalances of Iron Homeostasis in Inflammatory Diseases. Pharmaceuticals 2018, 11, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camaschella, C.; Girelli, D. The changing landscape of iron deficiency. Mol. Asp. Med. 2020, 75, 100861. [Google Scholar] [CrossRef]
- Bonovas, S.; Fiorino, G.; Allocca, M.; Lytras, T.; Tsantes, A.; Peyrin-Biroulet, L.; Danese, S. Intravenous Versus Oral Iron for the Treatment of Anemia in Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Medicine 2016, 95, e2308. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, W.; Xu, Y.; Qian, Z. Iron supplementation improves cardiovascular outcomes in patients with heart failure. Am. J. Med. 2019, 132, 955–963. [Google Scholar] [CrossRef]
- Chopra, V.K.; Anker, S.D. Anaemia, iron deficiency and heart failure in 2020: Facts and numbers. ESC Heart Fail. 2020, 7, 2007–2011. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Punj, S.; Ghafourian, K.; Ardehali, H. Iron deficiency and supplementation in heart failure and chronic kidney disease. Mol. Asp. Med. 2020, 75, 100873. [Google Scholar] [CrossRef] [PubMed]
- Busti, F.; Marchi, G.; Ugolini, S.; Castagna, A.; Girelli, D. Anemia and Iron Deficiency in Cancer Patients: Role of Iron Replacement Therapy. Pharmaceuticals 2018, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, M.; Gafter-Gvili, A.; Macdougall, I.C. Intravenous iron: A framework for changing the management of iron deficiency. Lancet Haematol. 2020, 7, e342–e350. [Google Scholar] [CrossRef]
- Schaefer, B.; Meindl, E.; Wagner, S.; Tilg, H.; Zoller, H. Intravenous iron supplementation therapy. Mol. Asp. Med. 2020, 75, 100862. [Google Scholar] [CrossRef]
- Neiser, S.; Rentsch, D.; Dippon, U.; Kappler, A.; Weidler, P.G.; Göttlicher, J.; Steininger, R.; Wilhelm, M.; Braitsch, M.; Funk, F.; et al. Physico-chemical properties of the new generation IV iron preparations ferumoxytol, iron isomaltoside 1000 and ferric carboxymaltose. BioMetals 2015, 28, 615–635. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Novey, H.S.; Pahl, M.; Haydik, I.; Vaziri, N.D. Immunologic studies of anaphylaxis to iron dextran in patients on renal dialysis. Ann. Allergy 1994, 72, 224–228. [Google Scholar]
- Szebeni, J.; Fishbane, S.; Hedenus, M.; Howaldt, S.; Locatelli, F.; Patni, S.; Rampton, D.; Weiss, G.; Folkersen, J. Hypersensitivity to intravenous iron: Classification, terminology, mechanisms and management. Br. J. Pharm. 2015, 172, 5025–5036. [Google Scholar] [CrossRef] [Green Version]
- Nathell, L.; Gohlke, A.; Wohlfeil, S. Reported Severe Hypersensitivity Reactions after Intravenous Iron Administration in the European Economic Area (EEA) Before and After Implementation of Risk Minimization Measures. Drug. Saf. 2020, 43, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Chertow, G.M.; Macdougall, I.C.; Kaper, R.; Krop, J.; Strauss, W. Randomized trial of intravenous iron-induced hypophosphatemia. JCI Insight 2018, 3, e124486. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Rubin, J.; Achebe, M.; Econs, M.J.; Peacock, M.; Imel, E.A.; Thomsen, L.L.; Carpenter, T.O.; Weber, T.; Brandenburg, V.; et al. Effects of Iron Isomaltoside vs Ferric Carboxymaltose on Hypophosphatemia in Iron-Deficiency Anemia: Two Randomized Clinical Trials. JAMA 2020, 323, 432–443. [Google Scholar] [CrossRef]
- Wolf, M.; Koch, T.A.; Bregman, D.B. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J. Bone Miner Res. 2013, 28, 1793–1803. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanser, L.; Kurz, K.; Nemati, N.; Weiss, G.; Pölzl, G. FGF23 and Immune Activation are Correlated in Chronic Heart Failure and Additive Predictors of Poor Prognosis. Cardiovasc. Disord. Med. 2020, 2, 7. [Google Scholar] [CrossRef]
- Coe, L.M.; Madathil, S.V.; Casu, C.; Lanske, B.; Rivella, S.; Sitara, D. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. J. Biol. Chem. 2014, 289, 9795–9810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agoro, R.; Montagna, A.; Goetz, R.; Aligbe, O.; Singh, G.; Coe, L.M.; Mohammadi, M.; Rivella, S.; Sitara, D. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. 2018, 32, 3752–3764. [Google Scholar] [CrossRef] [Green Version]
- Lewerin, C.; Ljunggren, Ö.; Nilsson-Ehle, H.; Karlsson, M.K.; Herlitz, H.; Lorentzon, M.; Ohlsson, C.; Mellström, D. Low serum iron is associated with high serum intact FGF23 in elderly men: The Swedish MrOS study. Bone 2017, 98, 1–8. [Google Scholar] [CrossRef]
- Mehta, R.; Cai, X.; Hodakowski, A.; Lee, J.; Leonard, M.; Ricardo, A.; Chen, J.; Hamm, L.; Sondheimer, J.; Dobre, M.; et al. Fibroblast Growth Factor 23 and Anemia in the Chronic Renal Insufficiency Cohort Study. Clin. J. Am. Soc. Nephrol. 2017, 12, 1795–1803. [Google Scholar] [CrossRef]
- Portolés, J.; Martín, L.; Broseta, J.J.; Cases, A. Anemia in Chronic Kidney Disease: From Pathophysiology and Current Treatments, to Future Agents. Front. Med. 2021, 8, 642296. [Google Scholar] [CrossRef]
- Tonia, T.; Mettler, A.; Robert, N.; Schwarzer, G.; Seidenfeld, J.; Weingart, O.; Hyde, C.; Engert, A.; Bohlius, J. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst. Rev. 2012, 12, Cd003407. [Google Scholar] [CrossRef] [Green Version]
- Pradeep, S.; Huang, J.; Mora, E.M.; Nick, A.M.; Cho, M.S.; Wu, S.Y.; Noh, K.; Pecot, C.V.; Rupaimoole, R.; Stein, M.A.; et al. Erythropoietin Stimulates Tumor Growth via EphB4. Cancer Cell 2015, 28, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Streja, E.; Kovesdy, C.P.; Greenland, S.; Kopple, J.D.; McAllister, C.J.; Nissenson, A.R.; Kalantar-Zadeh, K. Erythropoietin, Iron Depletion, and Relative Thrombocytosis: A Possible Explanation for Hemoglobin-Survival Paradox in Hemodialysis. Am. J. Kidney Dis. 2008, 52, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdougall, I.C.; Provenzano, R.; Sharma, A.; Spinowitz, B.S.; Schmidt, R.J.; Pergola, P.E.; Zabaneh, R.I.; Tong-Starksen, S.; Mayo, M.R.; Tang, H.; et al. Peginesatide for Anemia in Patients with Chronic Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2013, 368, 320–332. [Google Scholar] [CrossRef]
- Solomon, S.D.; Uno, H.; Lewis, E.F.; Eckardt, K.-U.; Lin, J.; Burdmann, E.A.; de Zeeuw, D.; Ivanovich, P.; Levey, A.S.; Parfrey, P.; et al. Erythropoietic Response and Outcomes in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2010, 363, 1146–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Francis, C.; David, V. Inflammation regulates fibroblast growth factor 23 production. Curr. Opin. Nephrol. Hypertens. 2016, 25, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Lanser, L.; Nemati, N.; Seifert, M.; Fuchs, D.; Weiss, G.; Pölzl, G.; Kurz, K. Inflammation, iron and vitamin D metabolism in different cardiomyopathy aetiologies. Pteridines 2020, 31, 28. [Google Scholar] [CrossRef]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol. 2010, 299, F882–F889. [Google Scholar] [CrossRef] [Green Version]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Min. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Petzer, V.; Tymoszuk, P.; Asshoff, M.; Carvalho, J.; Papworth, J.; Deantonio, C.; Bayliss, L.; Wake, M.S.; Seifert, M.; Brigo, N.; et al. A fully human anti-BMP6 antibody reduces the need for erythropoietin in rodent models of the anemia of chronic disease. Blood 2020, 136, 1080–1090. [Google Scholar] [CrossRef]
- Goodnough, L.T.; Murphy, M.F. Do liberal blood transfusions cause more harm than good? BMJ 2014, 349, g6897. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Colomo, A.; Bosch, A.; Concepción, M.; Hernandez-Gea, V.; Aracil, C.; Graupera, I.; Poca, M.; Alvarez-Urturi, C.; Gordillo, J.; et al. Transfusion Strategies for Acute Upper Gastrointestinal Bleeding. N. Engl. J. Med. 2013, 368, 11–21. [Google Scholar] [CrossRef]
- Kao, D.P.; Kreso, E.; Fonarow, G.C.; Krantz, M.J. Characteristics and outcomes among heart failure patients with anemia and renal insufficiency with and without blood transfusions (public discharge data from California 2000–2006). Am. J. Cardiol. 2011, 107, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Gill, K.; Fink, J.C.; Gilbertson, D.T.; Monda, K.L.; Muntner, P.; Lafayette, R.A.; Petersen, J.; Chertow, G.M.; Bradbury, B.D. Red blood cell transfusion, hyperkalemia, and heart failure in advanced chronic kidney disease. Pharmacoepidemiol. Drug Saf. 2015, 24, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.S.; Taylor, J.S.; Lopez-Olivo, M.A.; Munsell, M.F.; VonVille, H.M.; Lairson, D.R.; Bodurka, D.C. How low should we go: A systematic review and meta-analysis of the impact of restrictive red blood cell transfusion strategies in oncology. Cancer Treat Rev. 2016, 46, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sasu, B.J.; Cooke, K.S.; Arvedson, T.L.; Plewa, C.; Ellison, A.R.; Sheng, J.; Winters, A.; Juan, T.; Li, H.; Begley, C.G.; et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010, 115, 3616–3624. [Google Scholar] [CrossRef]
- Cooke, K.S.; Hinkle, B.; Salimi-Moosavi, H.; Foltz, I.; King, C.; Rathanaswami, P.; Winters, A.; Steavenson, S.; Begley, C.G.; Molineux, G.; et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood 2013, 122, 3054–3061. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.J.; Krzyzanski, W.; Wang, Y.M.; Li, H.; Rose, M.J.; Ma, M.; Wu, Y.; Hinkle, B.; Perez-Ruixo, J.J. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J. 2010, 12, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Vadhan-Raj, S.; Abonour, R.; Goldman, J.W.; Smith, D.A.; Slapak, C.A.; Ilaria, R.L., Jr.; Tiu, R.V.; Wang, X.; Callies, S.; Cox, J.; et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J. Hematol. Oncol. 2017, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Schwoebel, F.; van Eijk, L.T.; Zboralski, D.; Sell, S.; Buchner, K.; Maasch, C.; Purschke, W.G.; Humphrey, M.; Zöllner, S.; Eulberg, D.; et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013, 121, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Warrington, S.; Cortezi, B.; Zöllner, S.; Vauléon, S.; Swinkels, D.W.; Summo, L.; Schwoebel, F.; Riecke, K. Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. Br. J. Pharmacol. 2016, 173, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, L.T.; John, A.S.E.; Schwoebel, F.; Summo, L.; Vauléon, S.; Zöllner, S.; Laarakkers, C.M.; Kox, M.; van der Hoeven, J.G.; Swinkels, D.W.; et al. Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood 2014, 124, 2643–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiev, P.; Lazaroiu, M.; Ocroteala, L.; Grudeva-Popova, J.; Gheorghita, E.; Vasilica, M.; Popescu, S.M.; Cucuianu, A.; Summo, L.; Schwoebel, F.; et al. Abstract 3847: The anti-hepcidin Spiegelmer® Lexaptepid Pegol (NOX-H94) as treatment of anemia of chronic disease in patients with multiple myeloma, low grade lymphoma, and CLL: A phase II pilot study. Cancer Res. 2014, 74, 3847. [Google Scholar] [CrossRef]
- Sheetz, M.; Barrington, P.; Callies, S.; Berg, P.H.; McColm, J.; Marbury, T.; Decker, B.; Dyas, G.L.; Truhlar, S.M.E.; Benschop, R.; et al. Targeting the hepcidin–ferroportin pathway in anaemia of chronic kidney disease. Br. J. Clin. Pharmacol. 2019, 85, 935–948. [Google Scholar] [CrossRef]
- Barrington, P.; Sheetz, M.J.; Callies, S.; Waters, D.G.; Berg, P.H.; Pappas, D.; Marbury, T.C.; Decker, B.S.; Berg, J.K. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of an Anti-Ferroportin Antibody in Patients with Anemia Due to Chronic Renal Failure. Blood 2016, 128, 1280. [Google Scholar] [CrossRef]
- Rothe, C.; Skerra, A. Anticalin(®) Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Moebius, U.; Feuerer, W.; Fenzl, E.; van Swelm, R.; Swinkels, D.W.; Hohlbaum, A. A Phase I Study Investigating the Safety, Tolerability, Pharmacokinetics and Pharmacodynamic Activity of the Hepcidin Antagonist PRS-080#022. Results from a Randomized, Placebo Controlled, Double-Blind Study Following Single Administration to Healthy Subjectsa. Blood 2015, 126, 536. [Google Scholar] [CrossRef]
- Renders, L.; Budde, K.; Rosenberger, C.; van Swelm, R.; Swinkels, D.; Dellanna, F.; Feuerer, W.; Wen, M.; Erley, C.; Bader, B.; et al. First-in-human Phase I studies of PRS-080#22, a hepcidin antagonist, in healthy volunteers and patients with chronic kidney disease undergoing hemodialysis. PLoS ONE 2019, 14, e0212023. [Google Scholar] [CrossRef]
- Sun, C.C.; Vaja, V.; Chen, S.; Theurl, I.; Stepanek, A.; Brown, D.E.; Cappellini, M.D.; Weiss, G.; Hong, C.C.; Lin, H.Y.; et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol. Dial. Transpl. 2013, 28, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeur, C.; Kolodziej, S.A.; Wang, A.; Xu, X.; Lee, A.; Yu, P.B.; Shen, J.; Bloch, K.D.; Bloch, D.B. Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica 2015, 100, e68–e71. [Google Scholar] [CrossRef] [Green Version]
- Theurl, M.; Nairz, M.; Schroll, A.; Sonnweber, T.; Asshoff, M.; Haschka, D.; Seifert, M.; Willenbacher, W.; Wilflingseder, D.; Posch, W.; et al. Hepcidin as a predictive factor and therapeutic target in erythropoiesis-stimulating agent treatment for anemia of chronic disease in rats. Haematologica 2014, 99, 1516–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Schroll, A.; Sonnweber, T.; Nairz, M.; Theurl, M.; Willenbacher, W.; Eller, K.; Wolf, D.; Seifert, M.; Sun, C.C.; et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011, 118, 4977–4984. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Ho, J.N.; Lewis, J.A.; Karim, K.A.; Daniels, R.N.; Gentry, P.R.; Hopkins, C.R.; Lindsley, C.W.; Hong, C.C. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol. 2010, 5, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girelli, D.; Nemeth, E.; Swinkels, D.W. Hepcidin in the diagnosis of iron disorders. Blood 2016, 127, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Belot, A.; Gourbeyre, O.; Fay, A.; Palin, A.; Besson-Fournier, C.; Latour, C.; Hopkins, C.R.; Tidmarsh, G.F.; Coppin, H.; Roth, M.-P.; et al. LJ000328, a novel ALK2/3 kinase inhibitor, represses hepcidin and significantly improves the phenotype of IRIDA. Haematologica 2020, 105, e385–e388. [Google Scholar] [CrossRef] [Green Version]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Laborde, R.R.; Lasho, T.L.; Finke, C.; Begna, K.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Leontovich, A.; Kowalski, M.; et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia 2013, 27, 1322–1327. [Google Scholar] [CrossRef]
- Poli, M.; Girelli, D.; Campostrini, N.; Maccarinelli, F.; Finazzi, D.; Luscieti, S.; Nai, A.; Arosio, P. Heparin: A potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011, 117, 997–1004. [Google Scholar] [CrossRef]
- Poli, M.; Asperti, M.; Ruzzenenti, P.; Mandelli, L.; Campostrini, N.; Martini, G.; Di Somma, M.; Maccarinelli, F.; Girelli, D.; Naggi, A.; et al. Oversulfated heparins with low anticoagulant activity are strong and fast inhibitors of hepcidin expression in vitro and in vivo. Biochem. Pharmacol. 2014, 92, 467–475. [Google Scholar] [CrossRef]
- Poli, M.; Asperti, M.; Naggi, A.; Campostrini, N.; Girelli, D.; Corbella, M.; Benzi, M.; Besson-Fournier, C.; Coppin, H.; Maccarinelli, F.; et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 2014, 123, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Asperti, M.; Denardo, A.; Gryzik, M.; Castagna, A.; Girelli, D.; Naggi, A.; Arosio, P.; Poli, M. Pentosan polysulfate to control hepcidin expression in vitro and in vivo. Biochem. Pharm. 2020, 175, 113867. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X.; Zhang, H.; Chen, Q.; Cui, H. Low anticoagulant heparin-iron complex targeting inhibition of hepcidin ameliorates anemia of chronic disease in rodents. Eur. J. Pharmacol. 2021, 897, 173958. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; de Souza, L.V.; Seifert, M.; von Raffay, L.; Haschka, D.; Grubwieser, P.; Grander, M.; Mitterstiller, A.-M.; Nairz, M.; Poli, M.; et al. Pharmacological Targeting of BMP6-SMAD Mediated Hepcidin Expression Does Not Improve the Outcome of Systemic Infections With Intra-Or Extracellular Gram-Negative Bacteria in Mice. Front. Cell. Infect. Microbiol. 2021, 11, 705087. [Google Scholar] [CrossRef] [PubMed]
- Latour, C.; Besson-Fournier, C.; Gourbeyre, O.; Meynard, D.; Roth, M.-P.; Coppin, H. Deletion of BMP6 worsens the phenotype of HJV-deficient mice and attenuates hepcidin levels reached after LPS challenge. Blood 2017, 130, 2339–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Puerto, M.C.; Iyengar, P.V.; García de Vinuesa, A.; ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef]
- Haase, V.H. Therapeutic targeting of the HIF oxygen-sensing pathway: Lessons learned from clinical studies. Exp. Cell Res. 2017, 356, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cheng, Q.; Wang, J.; Zhao, X.; Zhu, S. Long-term efficacy and safety of hypoxia-inducible factor prolyl hydroxylase inhibitors in anaemia of chronic kidney disease: A meta-analysis including 13,146 patients. J. Clin. Pharm. Ther. 2021, 46, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Eckardt, K.-U. HIF Activation Against CVD in CKD: Novel Treatment Opportunities. Semin. Nephrol. 2018, 38, 267–276. [Google Scholar] [CrossRef]
- Besarab, A.; Provenzano, R.; Hertel, J.; Zabaneh, R.; Klaus, S.J.; Lee, T.; Leong, R.; Hemmerich, S.; Yu, K.-H.P.; Neff, T.B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol. Dial. Transplant. 2015, 30, 1665–1673. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Grzeszczak, W.; Szczyra, D.; Śnit, M. Whether Prolyl Hydroxylase Blocker—Roxadustat—In the Treatment of Anemia in Patients with Chronic Kidney Disease Is the Future? Int. J. Environ. Res. Public Health 2021, 18, 1612. [Google Scholar] [CrossRef] [PubMed]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef] [Green Version]
- Acharya, U.; Gau, J.-T.; Horvath, W.; Ventura, P.; Hsueh, C.-T.; Carlsen, W. Hemolysis and hyperhomocysteinemia caused by cobalamin deficiency: Three case reports and review of the literature. J. Hematol. Oncol. 2008, 1, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, E.; Sugianto, P.; Hsu, J.; Damoiseaux, R.; Ganz, T.; Nemeth, E. High-throughput screening of small molecules identifies hepcidin antagonists. Mol. Pharm. 2013, 83, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.Y.; Liu, L.; Wang, F.X.; Yang, Y.R.; Hao, J.W.; Wang, P.F.; Zhong, Q.; Zhou, K.; Xiong, A.; Zhu, W.Y.; et al. Toll-Like Receptor 4/MyD88-Mediated Signaling of Hepcidin Expression Causing Brain Iron Accumulation, Oxidative Injury, and Cognitive Impairment After Intracerebral Hemorrhage. Circulation 2016, 134, 1025–1038. [Google Scholar] [CrossRef]
- Bacchetta, J.; Zaritsky, J.J.; Sea, J.L.; Chun, R.F.; Lisse, T.S.; Zavala, K.; Nayak, A.; Wesseling-Perry, K.; Westerman, M.; Hollis, B.W.; et al. Suppression of iron-regulatory hepcidin by vitamin D. J. Am. Soc. Nephrol. 2014, 25, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Alvarez, J.A.; Kearns, M.D.; Hao, L.; Sloan, J.H.; Konrad, R.J.; Ziegler, T.R.; Zughaier, S.M.; Tangpricha, V. High-dose vitamin D(3) reduces circulating hepcidin concentrations: A pilot, randomized, double-blind, placebo-controlled trial in healthy adults. Clin. Nutr. 2017, 36, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Jones, J.L.; Han, J.E.; Alvarez, J.A.; Sloan, J.H.; Konrad, R.J.; Zughaier, S.M.; Martin, G.S.; Ziegler, T.R.; Tangpricha, V. High-Dose Vitamin D3 Administration Is Associated With Increases in Hemoglobin Concentrations in Mechanically Ventilated Critically Ill Adults: A Pilot Double-Blind, Randomized, Placebo-Controlled Trial. J. Parenter. Enter. Nutr. 2018, 42, 87–94. [Google Scholar] [CrossRef]
- Fletcher, J.; Cooper, S.C.; Ghosh, S.; Hewison, M. The Role of Vitamin D in Inflammatory Bowel Disease: Mechanism to Management. Nutrients 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, G.; Souberbielle, J.C.; Chazot, C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Prietl, B.; Treiber, G.; Pieber, T.R.; Amrein, K. Vitamin D and immune function. Nutrients 2013, 5, 2502–2521. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.M.; Archer, D.C.; Beck, L.; Spiegelberg, H.L. Immunosuppressive actions of 1,25-dihydroxyvitamin D3: Preferential inhibition of Th1 functions. J. Nutr. 1995, 125, 1704s–1708s. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Gysemans, C.; Mathieu, C. Vitamin D in autoimmune, infectious and allergic diseases: A vital player? Best. Pr. Res. Clin. Endocrinol. Metab. 2011, 25, 617–632. [Google Scholar] [CrossRef]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, L.E.; Burke, F.; Mura, M.; Zheng, Y.; Qureshi, O.S.; Hewison, M.; Walker, L.S.; Lammas, D.A.; Raza, K.; Sansom, D.M. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J. Immunol. 2009, 183, 5458–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Pantalena, L.C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol. Cell. Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef] [Green Version]
- De Martinis, M.; Allegra, A.; Sirufo, M.M.; Tonacci, A.; Pioggia, G.; Raggiunti, M.; Ginaldi, L.; Gangemi, S. Vitamin D Deficiency, Osteoporosis and Effect on Autoimmune Diseases and Hematopoiesis: A Review. Int. J. Mol. Sci. 2021, 22, 8855. [Google Scholar] [CrossRef]
- Murr, C.; Pilz, S.; Grammer Tanja, B.; Kleber Marcus, E.; Meinitzer, A.; Boehm Bernhard, O.; März, W.; Fuchs, D. Vitamin D deficiency parallels inflammation and immune activation, the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin. Chem. Lab. Med. 2012, 50, 2205. [Google Scholar] [CrossRef]
- Toth, I.; Bridges, K.R. Ascorbic Acid Enhances Ferritin mRNA Translation by an IRP/Aconitase Switch *. J. Biol. Chem. 1995, 270, 19540–19544. [Google Scholar] [CrossRef] [Green Version]
- Bridges, K.R. Ascorbic acid inhibits lysosomal autophagy of ferritin. J. Biol. Chem. 1987, 262, 14773–14778. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Chikhani, S.; Richardson, V.; Richardson, D.R. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim. Biophys. Acta. (BBA)-Mol. Cell. Res. 2013, 1833, 1527–1541. [Google Scholar] [CrossRef] [Green Version]
- Chiu, P.F.; Ko, S.Y.; Chang, C.C. Vitamin C affects the expression of hepcidin and erythropoietin receptor in HepG2 cells. J. Ren. Nutr. 2012, 22, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Attallah, N.; Osman-Malik, Y.; Frinak, S.; Besarab, A. Effect of intravenous ascorbic acid in hemodialysis patients with EPO-hyporesponsive anemia and hyperferritinemia. Am. J. Kidney Dis. 2006, 47, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wannamethee, S.G.; Lowe, G.D.; Rumley, A.; Bruckdorfer, K.R.; Whincup, P.H. Associations of vitamin C status, fruit and vegetable intakes, and markers of inflammation and hemostasis. Am. J. Clin. Nutr. 2006, 83, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Murr, C.; Winklhofer-Roob, B.M.; Schroecksnadel, K.; Maritschnegg, M.; Mangge, H.; Böhm, B.O.; Winkelmann, B.R.; März, W.; Fuchs, D. Inverse association between serum concentrations of neopterin and antioxidants in patients with and without angiographic coronary artery disease. Atherosclerosis 2009, 202, 543–549. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: Physiological activity in tumor growth and progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef] [Green Version]
- Fatih, N.; Camberlein, E.; Island, M.L.; Corlu, A.; Abgueguen, E.; Détivaud, L.; Leroyer, P.; Brissot, P.; Loréal, O. Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. J. Mol. Med. 2010, 88, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Lainé, F.; Laviolle, B.; Bardou-Jacquet, E.; Fatih, N.; Jezequel, C.; Collet, N.; Ropert, M.; Morcet, J.; Hamon, C.; Reymann, J.-M.; et al. Curcuma decreases serum hepcidin levels in healthy volunteers: A placebo-controlled, randomized, double-blind, cross-over study. Fundam. Clin. Pharmacol. 2017, 31, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Bakheit, A.a.; Abu-Qatouseh, L. Sulforaphane from broccoli attenuates inflammatory hepcidin by reducing IL-6 secretion in human HepG2 cells. J. Funct. Foods 2020, 75, 104210. [Google Scholar] [CrossRef]
- Artym, J.; Zimecki, M.; Kruzel, M.L. Lactoferrin for Prevention and Treatment of Anemia and Inflammation in Pregnant Women: A Comprehensive Review. Biomedicines 2021, 9, 898. [Google Scholar] [CrossRef]
- Kruzel, M.L.; Zimecki, M.; Actor, J.K. Lactoferrin in a Context of Inflammation-Induced Pathology. Front. Immunol. 2017, 8, 1438. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Debbabi, H.; Dubarry, M.; Boyaka, P.; Tomé, D. Regulation of physiological and pathological Th1 and Th2 responses by lactoferrinThis paper is one of a selection of papers published in this Special Issue, entitled 7th International Conference on Lactoferrin: Structure, Function, and Applications, and has undergone the Journal’s usual peer review process. Biochem. Cell. Biol. 2006, 84, 303–311. [Google Scholar] [CrossRef]
- Cutone, A.; Frioni, A.; Berlutti, F.; Valenti, P.; Musci, G.; Bonaccorsi di Patti, M.C. Lactoferrin prevents LPS-induced decrease of the iron exporter ferroportin in human monocytes/macrophages. BioMetals 2014, 27, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Cutone, A.; Rosa, L.; Lepanto, M.S.; Scotti, M.J.; Berlutti, F.; Bonaccorsi di Patti, M.C.; Musci, G.; Valenti, P. Lactoferrin Efficiently Counteracts the Inflammation-Induced Changes of the Iron Homeostasis System in Macrophages. Front. Immunol. 2017, 8, 705. [Google Scholar] [CrossRef] [Green Version]
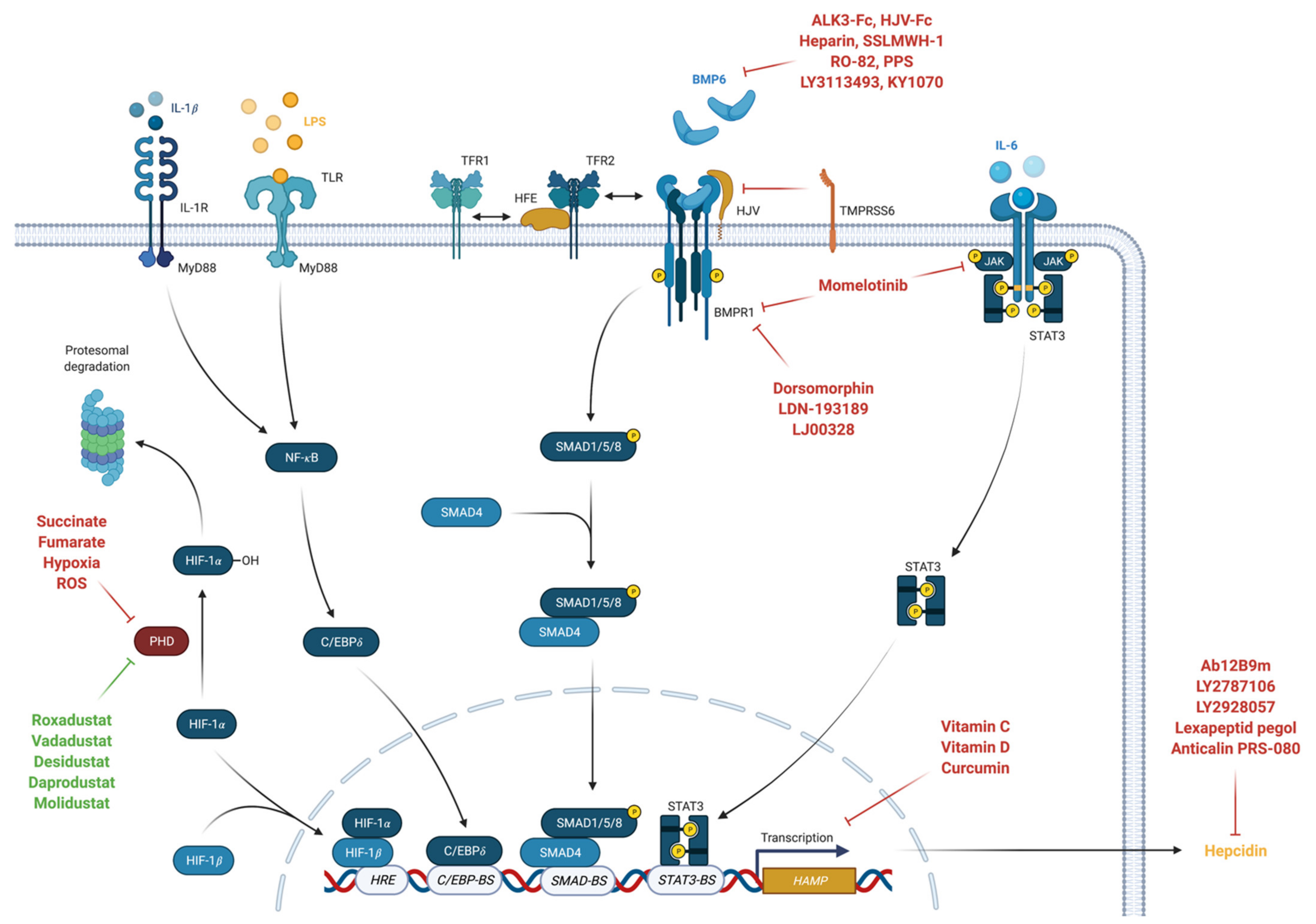
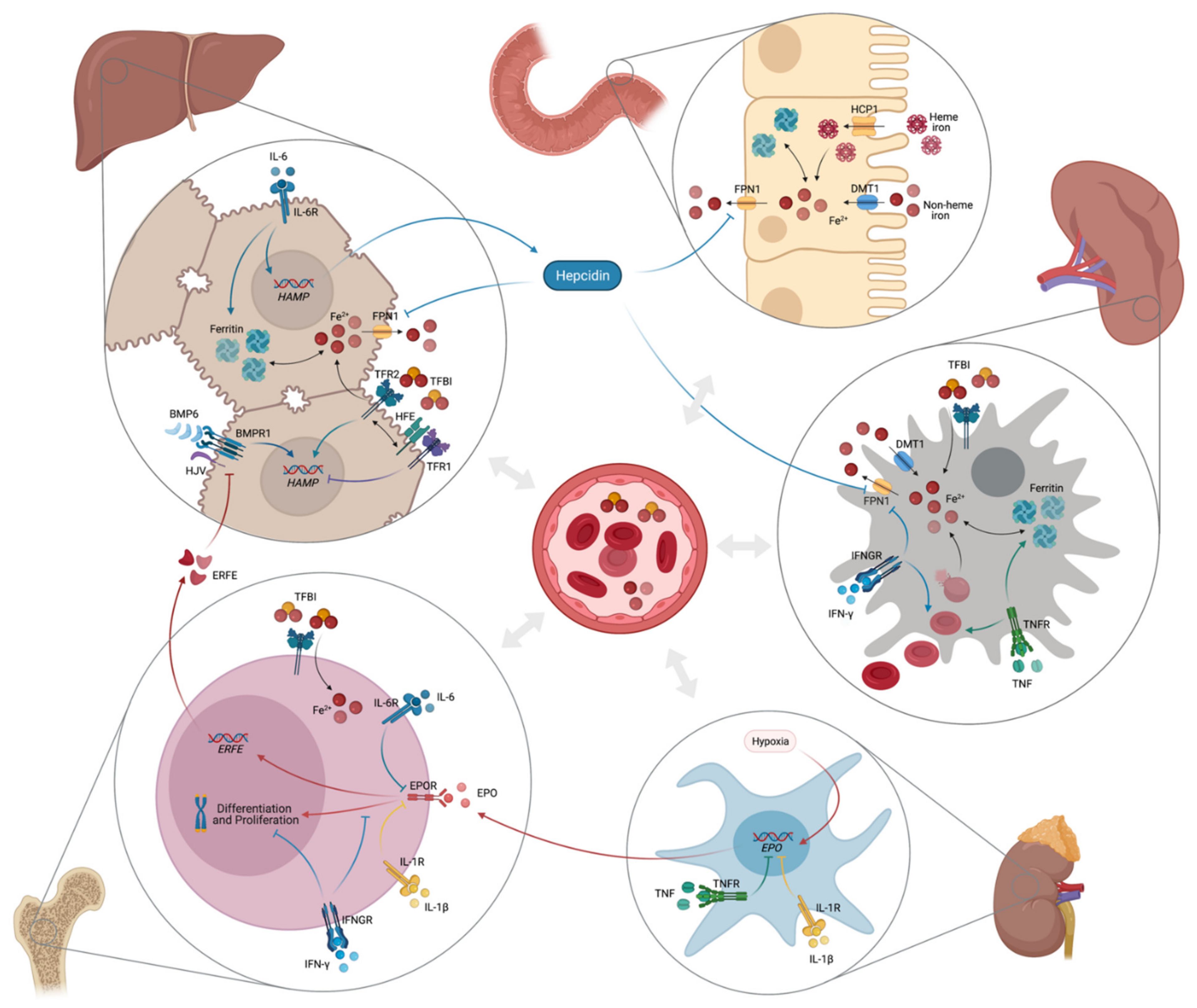
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).