Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Project Design
2.2. Data Collection
2.3. Dietary Assessment
2.4. Dietary Management
- (i)
- Avoidance of high-protein foods including meat, fish, eggs, cheese, nuts and seeds.
- (ii)
- NP allowance (using 1 g protein exchanges), with the amount titrated with blood Tyr concentrations.
- (iii)
- Tyr/Phe-free L-amino acid supplements (protein substitutes) with added vitamins and minerals. Low-Tyr/low-Phe glycomacropeptide-based protein substitutes were not prescribed.
- (iv)
- Provision of low-protein foods, e.g., fruits, some vegetables, oils, sugar and special low-protein foods.
- (v)
- Phe supplements. These were given only if two consecutive Phe levels were below the reference value.
2.5. Blood Tyr and Phe Levels
2.6. Ethical Statement
2.7. Statistics
3. Results
3.1. Patients
3.2. Median Prescribed Protein Equivalent from Total Protein, NP and Protein Substitute
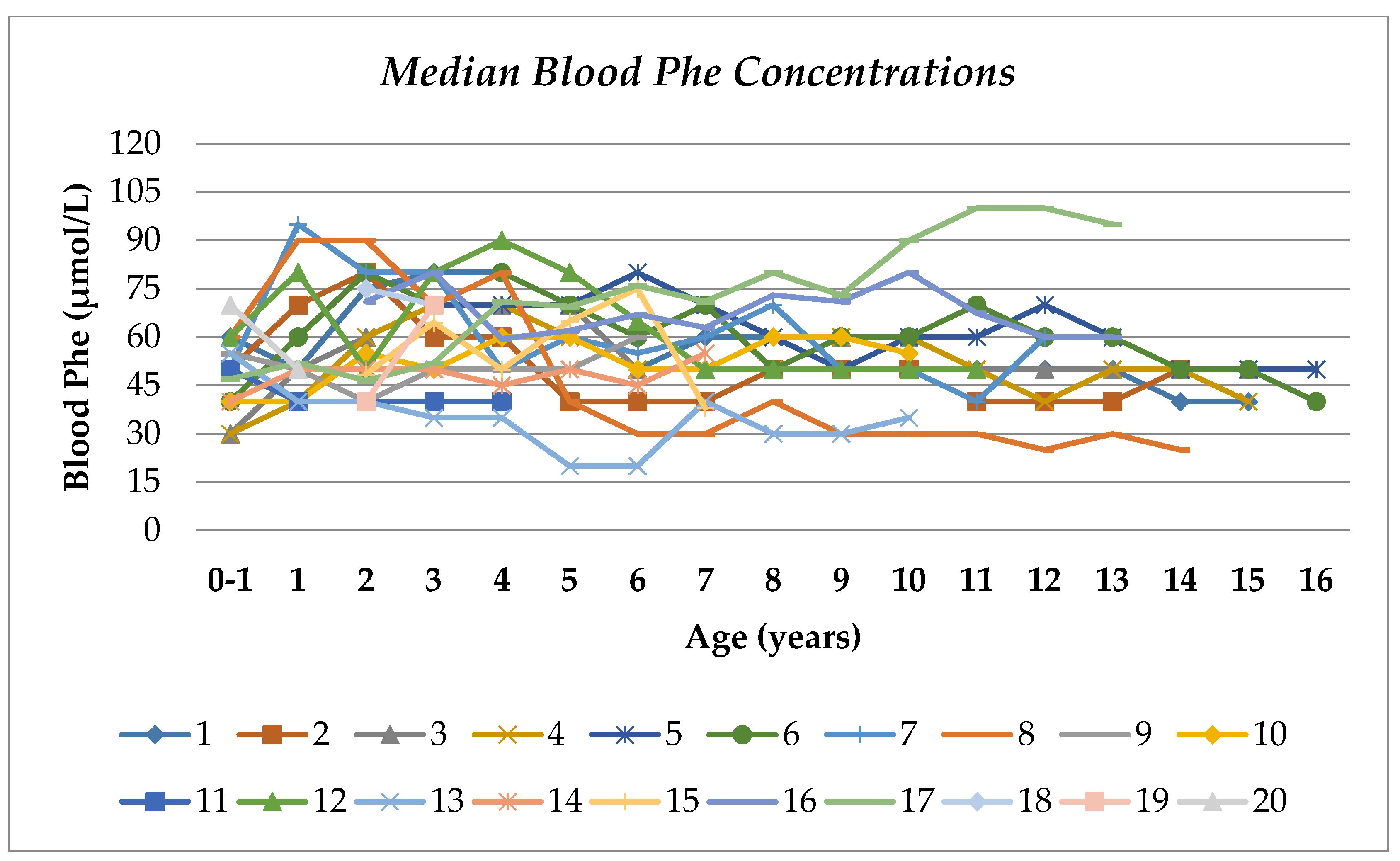
3.3. Blood Phe and Tyr Concentrations
3.4. Consistency of Blood Tyr and Phe Levels Over Time Compared with NP Intake Changes
3.5. Actual Dietary Intake from 24 h Recalls
3.6. NTBC
3.7. Nutritional Support
3.8. Vitamin and Mineral Supplementation
3.9. Phe Supplements
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashorn, M.; Pitkänen, S.; Salo, M.; Heikinheimo, M. Current strategies for the treatment of hereditary tyrosinemia type I. Pediatr. Drugs 2006, 8, 47–54. [Google Scholar] [CrossRef] [PubMed]
- García, M.I.; de la Parra, A.; Arias, C.; Arredondo, M.; Cabello, J.F. Long-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet. Mol. Genet. Metab. Rep. 2017, 11, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Delaet, C.; Dionisi, C.; Leonard, J.V.; McKiernan, P.J.; Mitchell, G.A.; Monti, L.; de Baulny, H.O.; Pintos-Morell, G.; Spiekerkoetter, U. Recommendations for the management of tyrosinaemia type 1. Orphanet J. Rare Dis. 2013, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J.; Preece, M.A.; Chakrapani, A. Outcome of children with hereditary tyrosinaemia following newborn screening. Arch. Dis. Child. 2015, 100, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Chinsky, J.M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C.D.M.; Grompe, M.; Mitchell, G.; Waisbren, S.E.; Gucsavas-Calikoglu, M.; Wasserstein, M.P.; Coakley, K.; et al. Diagnosis and treatment of tyrosinemia type I: A US and Canadian consensus group review and recommendations. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef]
- Mayorandan, S.; Meyer, U.; Gökçay, G.; Segarra, N.G.; de Baulny, H.O.; van Spronsen, F.J.; Zeman, J.; Delaet, C.; Spiekerkoetter, U.; Thimm, E.; et al. Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J. Rare Dis. 2014, 9, 107. [Google Scholar] [CrossRef]
- Couce, M.L.; Dalmau, J.; Del Toro, M.; Pintos-Morell, G.; Aldámiz-Echevarría, L.; Spanish Working Group on Tyrosinemia type1. Tyrosinemia type 1 in Spain: Mutational analysis, treatment and long-term outcome. Pediatr. Int. 2011, 53, 985–989. [Google Scholar] [CrossRef]
- Dixon, M.; Macdonald, A.; White, F.; Stafford, J. Disorders of amino acid metabolism, organic acidaemias and urea cycle disorders. In Clinical Paediatric Dietetics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 381–525. [Google Scholar]
- Daly, A.; Gokmen-Ozel, H.; Macdonald, A.; Preece, M.A.; Davies, P.; Chakrapani, A.; McKiernan, P. Diurnal variation of phenylalanine concentrations in tyrosinaemia type 1: Should we be concerned? J. Hum. Nutr. Diet. 2011, 25, 111–116. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; van Rijn, M.; Meyer, U.; Das, A. Dietary Considerations in Tyrosinemia Type I. In Hereditary Tyrosinemia: Pathogenesis, Screening and Management. Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Cham, Switzerland, 2017; Volume 959, pp. 197–204. [Google Scholar]
- Van Dam, E.; Daly, A.; Venema-Liefaard, G.; van Rijn, M.; Derks, T.; McKiernan, P.J.; Heiner-Fokkema, M.R.; Macdonald, A.; van Spronsen, F.J.; Baumgartner, M.R.; et al. What is the best blood sampling time for metabolic control of phenylalanine and tyrosine concentrations in tyrosinemia type 1 patients? In JIMD Reports; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017; Volume 36, pp. 49–57. [Google Scholar]
- Pinto, A.; Almeida, M.; Macdonald, A.; Ramos, P.C.; Rocha, J.C.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; Jackson, R.; et al. Over restriction of dietary protein allowance: The importance of ongoing reassessment of natural protein tolerance in Phenylketonuria. Nutrients 2019, 11, 995. [Google Scholar] [CrossRef]
- Hanley, W.B.; Linsao, L.; Davidson, W.; Moes, C.A.F. Malnutrition with early treatment of Phenylketonuria. Pediatr. Res. 1970, 4, 318–327. [Google Scholar] [CrossRef]
- Rouse, B.M. Phenylalanine deficiency syndrome. J. Pediatr. 1966, 69, 246–249. [Google Scholar] [CrossRef]
- Van Vliet, D.; Derks, T.; van Rijn, M.; de Groot, M.J.; Macdonald, A.; Heiner-Fokkema, M.; van Spronsen, F.J. Single amino acid supplementation in aminoacidopathies: A systematic review. Orphanet J. Rare Dis. 2014, 9, 7. [Google Scholar] [CrossRef]
- Nutritics, R. Edition (v5. 09) [Computer Software]. Available online: https://www.nutritics.com/p/home (accessed on 26 August 2019).
- The Scientific Advisory Committee on Nutrition. Dietary Reference Values for Energy; SACN: London, UK, 2011. [Google Scholar]
- Crone, M.; van Spronsen, F.J.; Oudshoorn, K.; Bekhof, J.; Van Rijn, G.; Verkerk, P.H. Behavioural factors related to metabolic control in patients with phenylketonuria. J. Inherit. Metab. Dis. 2005, 28, 627–637. [Google Scholar] [CrossRef]
- Walter, J.; White, F.; Hall, S.; Macdonald, A.; Rylance, G.; Boneh, A.; Francis, D.; Shortland, G.; Schmidt, M.; Vail, A. How practical are recommendations for dietary control in phenylketonuria? Lancet 2002, 360, 55–57. [Google Scholar] [CrossRef]
- Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Ozel, H.G.; Lammardo, A.M.; Macdonald, A.; Motzfeldt, K.; Nowacka, M.; Robert, M.; van Rijn, M. Dietary management practices in phenylketonuria across European centres. Clin. Nutr. 2009, 28, 231–236. [Google Scholar] [CrossRef]
- MacLeod, E.L.; Gleason, S.T.; van Calcar, S.C.; Ney, D.M. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol. Genet. Metab. 2009, 98, 331–337. [Google Scholar] [CrossRef]
- El-Shabrawi, M.H.; Kamal, N.M. Current management options for tyrosinemia. Orphan Drugs: Res. Rev. 2013, 3, 1. [Google Scholar] [CrossRef][Green Version]
- Masurel-Paulet, A.; Poggi-Bach, J.; Rolland, M.-O.; Bernard, O.; Guffon, N.; Dobbelaere, D.; Sarles, J.; De Baulny, H.O.; Touati, G. NTBC treatment in tyrosinaemia type I: Long-term outcome in French patients. J. Inherit. Metab. Dis. 2008, 31, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, S.; Holme, E.; Lock, E.; Hjalmarson, O.; Strandvik, B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992, 340, 813–817. [Google Scholar] [CrossRef]
- Cohn, R.M.; Yudkoff, M.; Yost, B.; Segal, S. Phenylalanine-tyrosine deficiency syndrome as a complication of the management of hereditary tyrosinemia. Am. J. Clin. Nutr. 1977, 30, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Van Wyk, K.G.; Leonard, J.V.; Clayton, P.T. Phenylalanine supplementation improves the phenylalanine profile in tyrosinaemia. J. Inherit. Metab. Dis. 2000, 23, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, W.G.; Jahja, R.; Huijbregts, S.; van Spronsen, F.J. Neurological and neuropsychological problems in tyrosinemia type I patients. In Hereditary Tyrosinemia: Pathogenesis, Screening and Management. Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Cham, Switzerland, 2017; Volume 959, pp. 111–122. [Google Scholar]
- Van Ginkel, W.; van Vliet, D.; van der Goot, E.; van Faassen, M.; Vogel, A.; Heiner-Fokkema, M.; van der Zee, E.A.; van Spronsen, F.J. Blood and brain biochemistry and behaviour in ntbc and dietary treated tyrosinemia type 1 Mice. Nutrients 2019, 11, 2486. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Beasley, M.G.; E Ades, A. Intelligence and quality of dietary treatment in phenylketonuria. Arch. Dis. Child. 1990, 65, 472–478. [Google Scholar] [CrossRef]
- Bendadi, F.; de Koning, T.J.; Visser, G.; Prinsen, H.C.; de Sain, M.G.; Verhoeven-Duif, N.; Sinnema, G.; van Spronsen, F.J.; van Hasselt, P.M. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J. Pediatr. 2014, 164, 398–401. [Google Scholar] [CrossRef]
- Hillgartner, M.A.; Coker, S.B.; Koenig, A.E.; Moore, M.E.; Barnby, E.; MacGregor, G.G. Tyrosinemia type I and not treatment with NTBC causes slower learning and altered behavior in mice. J. Inherit. Metab. Dis. 2016, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Barone, H.; Bliksrud, Y.T.; Elgen, I.B.; Szigetvari, P.D.; Kleppe, R.; Ghorbani, S.; Hansen, E.V.; Haavik, J. Tyrosinemia Type 1 and symptoms of ADHD: Biochemical mechanisms and implications for treatment and prognosis. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2019, 183, 95–105. [Google Scholar] [CrossRef]
- Ellaway, C.J.; Holme, E.; Standing, S.; Preece, M.A.; Green, A.; Ploechl, E.; Ugarte, M.; Trefz, F.K.; Leonard, J.V. Outcome of tyrosinaemia type III. J. Inherit. Metab. Dis. 2001, 24, 824–832. [Google Scholar] [CrossRef]
- LaRochelle, J.; Álvarez, F.; Bussières, J.-F.; Chevalier, I.; Dallaire, L.; Dubois, J.; Faucher, F.; Fenyves, D.; Goodyer, P.; Grenier, A.; et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol. Genet. Metab. 2012, 107, 49–54. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Gender | Age at Final Assessment (Years) | Ethnicity | Consanguinity | Siblings with HTI | Age at Diagnosis | Diagnosed by Newborn Screening of PKU or HTI | Presenting Symptoms at Diagnosis | Clinical Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 15.2 | Caucasian | No | No | 2 months | Yes | Pre-symptomatic diagnosis | Dyscalculia, learning difficulties, developmental concerns, anxiety, psychological problems |
| 2 | M | 14.5 | Caucasian | No | No | 3 months | No | Acute liver failure and hernia | Behavioural difficulties, psychological problems, poor school progress. Nasogastric feeds for 3 years |
| 3 | M | 15.2 | Asian | Yes | No | Newborn | Yes | Pre-symptomatic diagnosis | Poor school progress, small splenic cyst |
| 4 | M | 15.1 | Arabic | Yes | Yes | Newborn | Sibling screen | Pre-symptomatic diagnosis | Psychological problems poor school progress, seizures |
| 5 | F | 17.7 | Caucasian | No | Yes | Newborn | Sibling screen | Pre-symptomatic diagnosis | Normal |
| 6 | M | 16.9 | Asian | Yes | Yes | Newborn | Sibling screen | Severe liver dysfunction | Poor school progress, alopecia |
| 7 | M | 13.6 | Asian | Yes | Extended family | Newborn | Family screen | Pre-symptomatic diagnosis | Poor school progress |
| 8 | F | 14.7 | Asian | Yes | Yes | 4 months | No | Acute liver failure and gastrointestinal bleeding | Normal |
| 9 | M | 8.3 | Caucasian | No | No | 3 weeks | Yes | Pre-symptomatic diagnosis | Normal |
| 10 | F | 10.2 | Asian | Yes | No | Newborn | Yes | Pre-symptomatic diagnosis | Poor school progress. Nasogastric feeds for 5 years |
| 11 | F | 4.7 | Caucasian | No | No | 3 months | No | Infantile seizures and acute liver failure | Poor school progress |
| 12 | M | 11.1 | Indian | Yes | Yes | Newborn | Sibling screen | Pre-symptomatic diagnosis | Normal |
| 13 | M | 10.3 | Asian | Yes | Yes | Newborn | Sibling screen | Pre-symptomatic diagnosis | Poor school progress. Early coagulopathy |
| 14 | F | 8.3 | Asian | Yes | Yes | Newborn | Sibling screen | Pre-symptomatic diagnosis | Poor school progress. Early coagulopathy |
| 15 | F | 6.3 * | Caucasian | No | No | 22 months | No | Rickets, hepatomegaly, glycosuria, myopathy, respiratory muscle weakness | Cirrhosis |
| 16 | M | 13.3 * | Caucasian | No | No | 20 months | No | Cirrhosis and portal hypertension | Hepatocellular carcinoma with progression of tumour |
| 17 | F | 12.7 * | Indian | No | No | 3 months | No | Liver failure, hyperinsulinism, feeding problems, congenital cytomegalovirus infection | Compensated cirrhosis |
| 18 | F | 3.4 * | Asian | Yes | No | 24 months | No | Acute liver failure, vomiting, mild fever | Cardiomyopathy |
| 19 | F | 3.2 * | Arabic | No | No | 3 months | No | Hepatosplenomegaly, abdominal distension | Lymphadenopathy |
| 20 | F | Died | Caucasian | No | No | 10 months | No | N/A | Died at age of 3 years due to hepatocellular carcinoma |
| Total Protein | Natural Protein | Protein Equivalent from Protein Substitutes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g/day | g/kg/day | g/day | g/kg/day | g/day | g/kg/day | ||||||||
| Age (years) | No. Subjects Each Age Group | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR |
| 0–1 | 7 | 15 | 3 | 2.4 | 0.8 | 5 | 2 | 0.8 | 0.4 | 11 | 3 | 1.5 | 0.6 |
| 1 | 13 | 28 | 9 | 3.4 | 0.5 | 4 | 2 | 0.4 | 0.2 | 25 | 9 | 2.9 | 0.9 |
| 2 | 19 | 34 | 9 | 2.9 | 0.4 | 5 | 3 | 0.5 | 0.3 | 29 | 5 | 2.4 | 0.3 |
| 3 | 19 | 38 | 9 | 2.8 | 0.6 | 5 | 5 | 0.4 | 0.4 | 31 | 12 | 2.4 | 0.7 |
| 4 | 15 | 44 | 8 | 2.5 | 0.9 | 6 | 3 | 0.4 | 0.3 | 38 | 10 | 2.2 | 0.8 |
| 5 | 15 | 47 | 8 | 2.6 | 0.6 | 8 | 5 | 0.4 | 0.4 | 38 | 9 | 2.3 | 0.4 |
| 6 | 14 | 50 | 9 | 2.4 | 0.6 | 8 | 5 | 0.4 | 0.2 | 42 | 8 | 2.1 | 0.6 |
| 7 | 14 | 53 | 8 | 2.4 | 0.6 | 9 | 7 | 0.4 | 0.2 | 45 | 5 | 1.9 | 0.6 |
| 8 | 14 | 55 | 9 | 2.1 | 0.4 | 10 | 5 | 0.4 | 0.2 | 45 | 2 | 1.8 | 0.3 |
| 9 | 12 | 57 | 11 | 1.9 | 0.4 | 12 | 9 | 0.4 | 0.2 | 45 | 5 | 1.6 | 0.3 |
| 10 | 12 | 63 | 13 | 1.8 | 0.6 | 17 | 9 | 0.5 | 0.2 | 45 | 5 | 1.4 | 0.4 |
| 11 | 10 | 66 | 16 | 1.6 | 0.6 | 17 | 7 | 0.4 | 0.2 | 48 | 15 | 1.3 | 0.3 |
| 12 | 9 | 76 | 18 | 1.4 | 0.7 | 17 | 3 | 0.4 | 0.1 | 60 | 15 | 1.1 | 0.5 |
| 13 | 8 | 78 | 11 | 1.4 | 0.4 | 17 | 3 | 0.3 | 0.1 | 60 | 0 | 1.0 | 0.3 |
| 14 | 7 | 79 | 9 | 1.2 | 0.5 | 17 | 4 | 0.4 | 0.1 | 60 | 0 | 1.0 | 0.3 |
| 15 | 4 | 86 | 12 | 1.6 | 0.4 | 19 | 7 | 0.4 | 0.2 | 60 | 5 | 1.1 | 0.4 |
| 16 | 1 | 84 | 0 | 1.7 | 0.0 | 24 | 0 | 0.5 | 0.0 | 60 | 0 | 1.2 | 0.0 |
| Age (years) | No. Subjects in Each Age Group | Blood Tyr (µmol/L) | % of Blood Tyr Concentrations | Blood Phe (µmol/L) | % of Blood Phe Concentrations | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <200 µmol/L | 200–400 µmol/L | >400 µmol/L | >500 µmol/L | >600 µmol/L | ≤30 µmol/L | ≤ 50 µmol/L | ≤ 100 µmol/L | ||||||||||||||
| Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | ||
| 0–1 | 16 | 280 | 60 | 20 | 16 | 58 | 15 | 12 | 16 | 3 | 6 | 0 | 4 | 50 | 16 | 32 | 23 | 64 | 33 | 98 | 5 |
| 1 | 16 | 320 | 79 | 12 | 14 | 60 | 16 | 28 | 27 | 7 | 12 | 1 | 7 | 50 | 23 | 20 | 22 | 58 | 37 | 96 | 17 |
| 2 | 19 | 340 | 113 | 0 | 6 | 66 | 50 | 20 | 51 | 3 | 22 | 0 | 1 | 55 | 32 | 13 | 12 | 50 | 50 | 97 | 10 |
| 3 | 19 | 405 | 116 | 0 | 2 | 52 | 45 | 46 | 48 | 13 | 37 | 0 | 13 | 70 | 19 | 9 | 11 | 29 | 40 | 97 | 6 |
| 4 | 17 | 420 | 163 | 0 | 2 | 44 | 52 | 56 | 55 | 21 | 40 | 8 | 14 | 60 | 21 | 6 | 10 | 32 | 42 | 100 | 4 |
| 5 | 16 | 360 | 107 | 0 | 0 | 62 | 51 | 38 | 57 | 12 | 22 | 0 | 5 | 61 | 20 | 10 | 14 | 34 | 57 | 99 | 5 |
| 6 | 16 | 400 | 135 | 0 | 1 | 54 | 65 | 46 | 68 | 12 | 31 | 1 | 10 | 53 | 17 | 7 | 9 | 52 | 54 | 100 | 3 |
| 7 | 15 | 400 | 113 | 0 | 0 | 68 | 51 | 32 | 56 | 6 | 38 | 0 | 13 | 50 | 17 | 0 | 14 | 53 | 56 | 100 | 1 |
| 8 | 13 | 370 | 75 | 0 | 2 | 67 | 40 | 33 | 41 | 6 | 17 | 0 | 2 | 50 | 10 | 2 | 7 | 55 | 40 | 100 | 0 |
| 9 | 13 | 380 | 55 | 0 | 0 | 60 | 23 | 40 | 23 | 3 | 14 | 0 | 0 | 60 | 10 | 0 | 2 | 36 | 56 | 100 | 0 |
| 10 | 13 | 380 | 60 | 0 | 0 | 75 | 22 | 25 | 22 | 8 | 10 | 0 | 3 | 60 | 10 | 0 | 8 | 46 | 44 | 100 | 0 |
| 11 | 11 | 365 | 70 | 0 | 0 | 66 | 23 | 34 | 27 | 8 | 16 | 0 | 0 | 50 | 19 | 0 | 4 | 72 | 52 | 100 | 0 |
| 12 | 10 | 358 | 71 | 0 | 0 | 78 | 24 | 22 | 28 | 1 | 6 | 0 | 0 | 55 | 18 | 1 | 8 | 63 | 67 | 100 | 0 |
| 13 | 9 | 370 | 40 | 0 | 0 | 71 | 18 | 29 | 18 | 2 | 3 | 0 | 0 | 50 | 10 | 0 | 4 | 83 | 51 | 100 | 0 |
| 14 | 7 | 380 | 83 | 0 | 2 | 67 | 37 | 33 | 45 | 11 | 15 | 0 | 5 | 50 | 5 | 0 | 10 | 82 | 13 | 100 | 0 |
| 15 | 5 | 385 | 70 | 0 | 0 | 60 | 32 | 40 | 32 | 11 | 20 | 0 | 7 | 50 | 10 | 0 | 0 | 79 | 20 | 100 | 0 |
| 16 | 2 | 318 | 8 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 | 5 | 0 | 0 | 98 | 3 | 100 | 0 |
| Patient No. | Gender | Age at Dietary Assessment (years) | Energy (kcal/d) (EAR %) | Protein | Carbohydrate | Fat | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total (g/day) | Total (g/kg/day) | Natural (g/day) | Natural (g/kg/day) | PE from PS (g/day) | PE from PS (g/kg/day) | % of Energy | (g/day) | (g/kg/day) | % of Energy | (g/day) | (g/kg/day) | % of Energy | ||||
| 1 | F | 15.2 | 1871 (78) | 83 | 1.4 | 23 | 0.4 | 60 | 1.0 | 18 | 242 | 3.8 | 52 | 64 | 1.0 | 30 |
| 2 | M | 14.5 | 2590 (121) | 86 | 1.7 | 26 | 0.5 | 60 | 1.2 | 13 | 386 | 7.5 | 60 | 78 | 1.5 | 27 |
| 3 | M | 15.2 | 2475 (87) | 87 | 1.3 | 27 | 0.4 | 60 | 0.9 | 14 | 286 | 4.2 | 46 | 109 | 1.6 | 40 |
| 4 | M | 15.1 | 1776 (96) | 91 | 1.5 | 30 | 0.5 | 60 | 1.0 | 21 | 263 | 4.3 | 59 | 40 | 0.6 | 20 |
| 5 | F | 17.7 | 1638 (66) | 91 | 1.8 | 31 | 0.6 | 60 | 1.2 | 22 | 207 | 4.2 | 51 | 50 | 1.0 | 27 |
| 6 | M | 16.9 | 2303 (74) | 104 | 1.9 | 44 | 0.8 | 60 | 1.1 | 18 | 322 | 5.9 | 56 | 66 | 1.2 | 26 |
| 7 | M | 13.6 | 2252 (93) | 95 | 2.1 | 35 | 0.8 | 60 | 1.3 | 17 | 376 | 8.3 | 67 | 40 | 1.0 | 16 |
| 8 | F | 14.7 | 2223 (94) | 71 | 1.4 | 26 | 0.5 | 45 | 0.9 | 13 | 320 | 6.5 | 58 | 71 | 1.4 | 29 |
| 9 | M | 8.3 | 1887 (108) | 65 | 2.5 | 25 | 1.0 | 40 | 1.5 | 14 | 256 | 9.7 | 54 | 67 | 2.5 | 32 |
| 10 | F | 10.2 | 1957 (101) | 79 | 1.6 | 30 | 0.6 | 49 | 1.0 | 16 | 269 | 5.7 | 55 | 62 | 1.3 | 29 |
| 11 | F | 4.7 | 1556 (114) | 72 | 4.2 | 27 | 1.6 | 45 | 2.6 | 19 | 194 | 11.4 | 50 | 54 | 3.2 | 31 |
| 12 | M | 11.1 | 1919 (90) | 75 | 2.5 | 32 | 1.1 | 43 | 1.4 | 15 | 286 | 9.5 | 60 | 53 | 1.8 | 25 |
| 13 | M | 10.3 | 1703 (83) | 71 | 2.9 | 26 | 1.1 | 45 | 1.8 | 17 | 226 | 9.2 | 53 | 57 | 2.3 | 30 |
| 14 | F | 8.3 | 1755 (108) | 72 | 3.4 | 27 | 1.3 | 45 | 2.1 | 16 | 235 | 11.0 | 54 | 58 | 2.7 | 30 |
| 15 * | F | 6.3 | Liver transplant | |||||||||||||
| 16 * | M | 13.2 | Liver transplant | |||||||||||||
| 17 * | F | 12.7 | Liver transplant | |||||||||||||
| 18 | F | 3.4 | 961 (89) Liver transplant | 41 | 2.9 | 11 | 0.8 | 30 | 2.1 | 17 | 144 | 10.1 | 60 | 25 | 1.7 | 23 |
| 19 * | F | 3.2 | Liver transplant | |||||||||||||
| 20 | F | Died 3years | 906 (84) | 15 | 1.4 | 8 | 0.7 | 7 | 0.7 | 7 | 119 | 10.8 | 52 | 42 | 3.8 | 41 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, O.; Daly, A.; Pinto, A.; Ashmore, C.; Evans, S.; Gupte, G.; Santra, S.; Preece, M.A.; Mckiernan, P.; Kitchen, S.; et al. Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1. Nutrients 2020, 12, 1148. https://doi.org/10.3390/nu12041148
Yilmaz O, Daly A, Pinto A, Ashmore C, Evans S, Gupte G, Santra S, Preece MA, Mckiernan P, Kitchen S, et al. Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1. Nutrients. 2020; 12(4):1148. https://doi.org/10.3390/nu12041148
Chicago/Turabian StyleYilmaz, Ozlem, Anne Daly, Alex Pinto, Catherine Ashmore, Sharon Evans, Girish Gupte, Saikat Santra, Mary Anne Preece, Patrick Mckiernan, Steve Kitchen, and et al. 2020. "Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1" Nutrients 12, no. 4: 1148. https://doi.org/10.3390/nu12041148
APA StyleYilmaz, O., Daly, A., Pinto, A., Ashmore, C., Evans, S., Gupte, G., Santra, S., Preece, M. A., Mckiernan, P., Kitchen, S., Yabanci Ayhan, N., & MacDonald, A. (2020). Natural Protein Tolerance and Metabolic Control in Patients with Hereditary Tyrosinaemia Type 1. Nutrients, 12(4), 1148. https://doi.org/10.3390/nu12041148