Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product

 , , ,
, , , 
Abstract
1. Introduction
- to determine whether high-resolution 1H NMR analysis can be successfully applied to determine and monitor the FA composition, antioxidant levels, and chain-breaking antioxidant status of CLO products, and the ability of this technique to provide valuable supporting information to that obtained from established, more conventional analytical methods that are available for these purposes;
- to investigate whether the pre-fermentation of cod liver sources successfully fortifies the antioxidant composition of CLO products arising therefrom, and the molecular basis of this fortification; and,
- to apply the newly-developed NMR-based TORA TSE and TDSE approaches to determine the relative abilities of CLO product unsaturated fatty acids (UFAs) to resist peroxidation, and determine whether antioxidant-fortifying, pre-fermentation of their cod liver sources renders them more resistant to such oxidation.
2. Materials and Methods
2.1. CLO Samples
2.2. Authentic Reference Aldehydic LOPs
2.3. Exposure of CLO Samples to TORA Thermal-stressing and Temperature-dependent Storage Episodes (TSEs and TDSEs Respectively), and Their Preparation for 1H NMR Analysis
2.4. Preparation of 2H2O and C 2H3O2H Extracts of CLO Products 1–4
2.5. H NMR Analysis
2.6. Experimental Design and Statistical Analysis
2.7. Analysis of Antioxidants (including Tocopherols), Phenolics, Flavonoids and Flavonones, Anthocyanins, Tannins, Chlorophylls, Carotenes, and Retinol in CLOs
2.8. Determination of the Oxygen Radical Absorbance Capacities (ORACs) of CLO Products
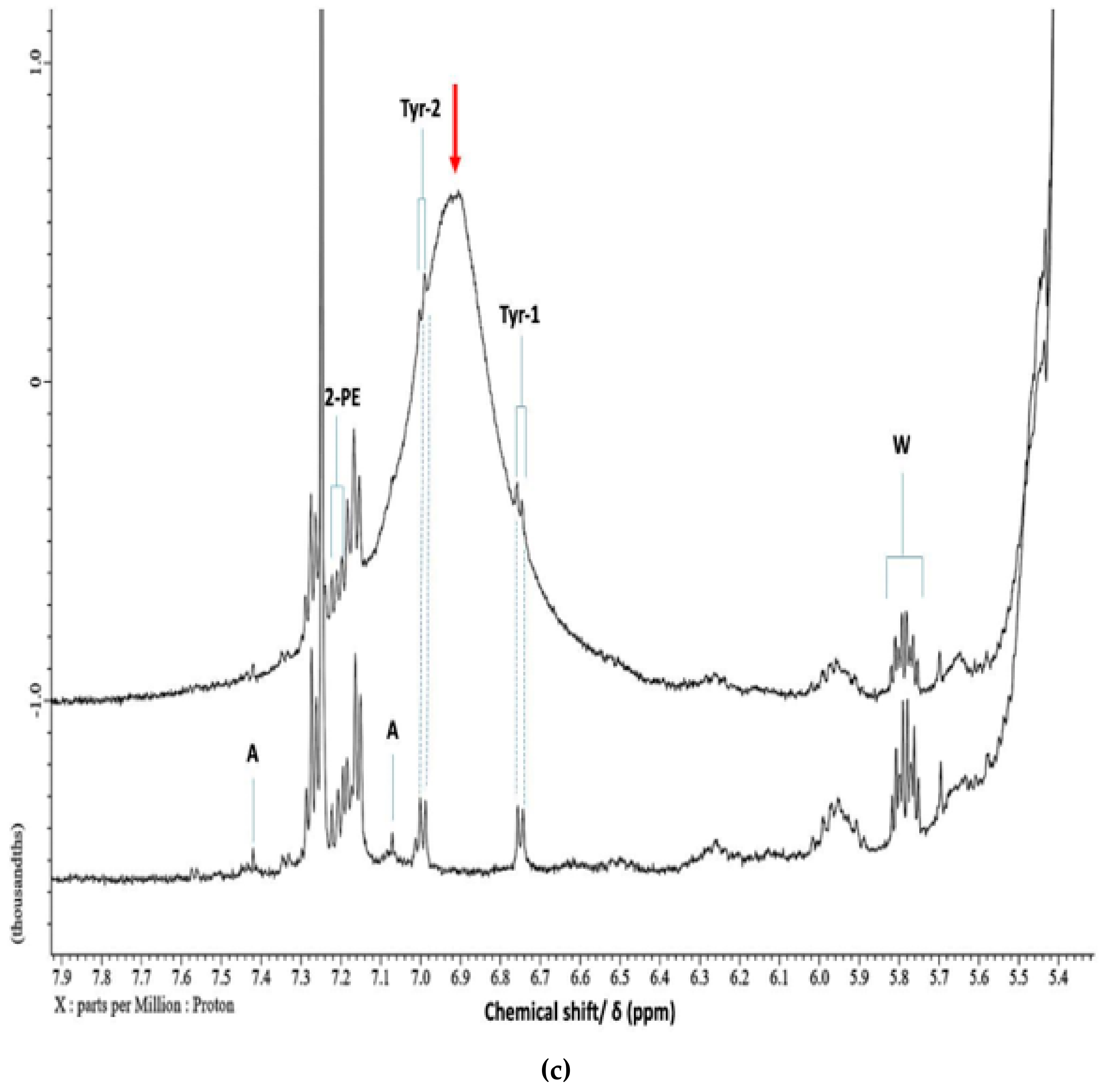
2.9. Biogenic Amine, Total Protein, Collagen, Ammonia and Water (moisture) Analyses
3. Results
3.1. ORAC Values and Antioxidant Status of CLO Products
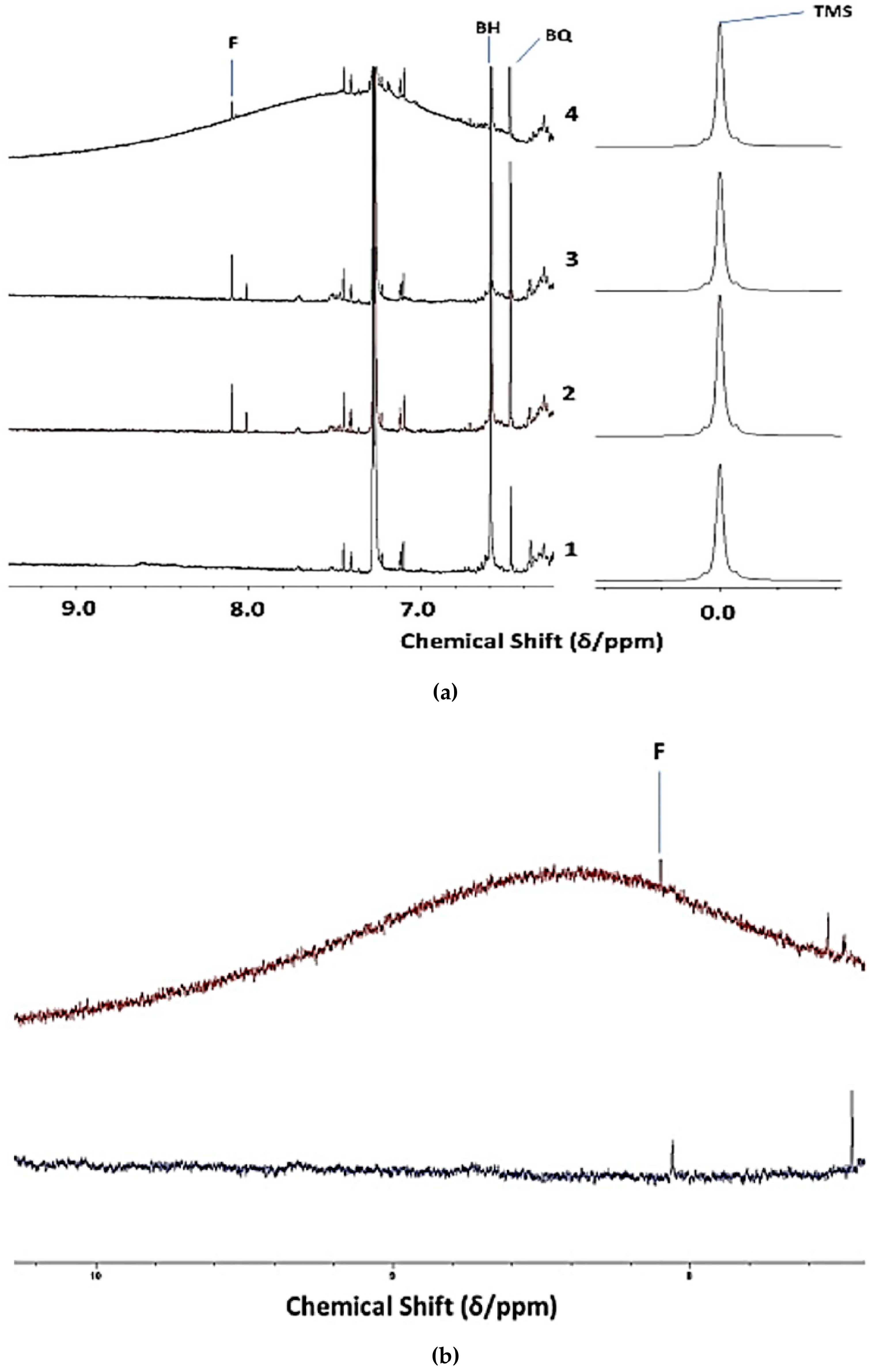
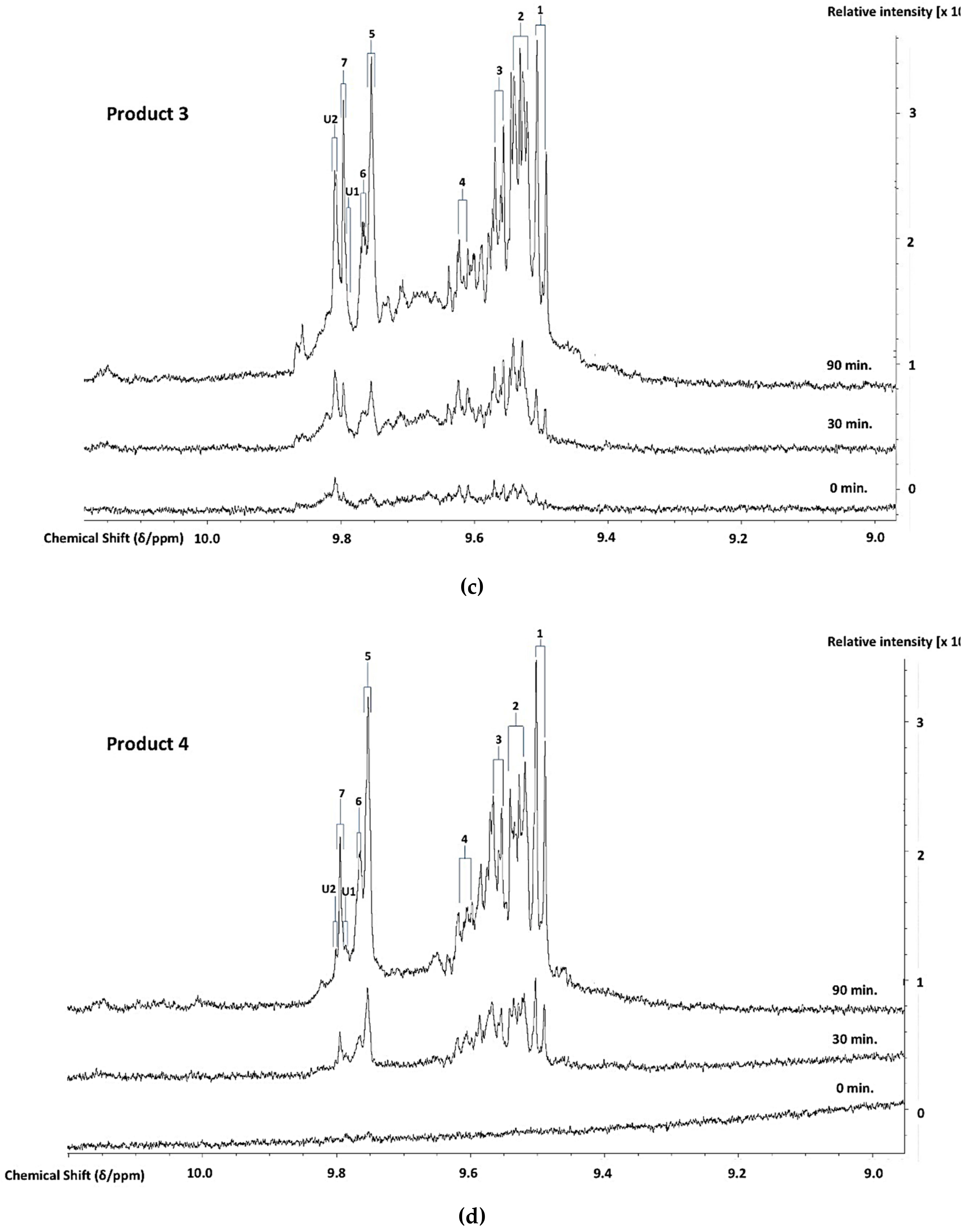
3.2. Investigation of an Unusual Broad 1H NMR Resonance in 1H NMR Spectral Profiles of a CLO Product: Assignment to Collagen/Collagen Hydrolysate Products and Ammonia
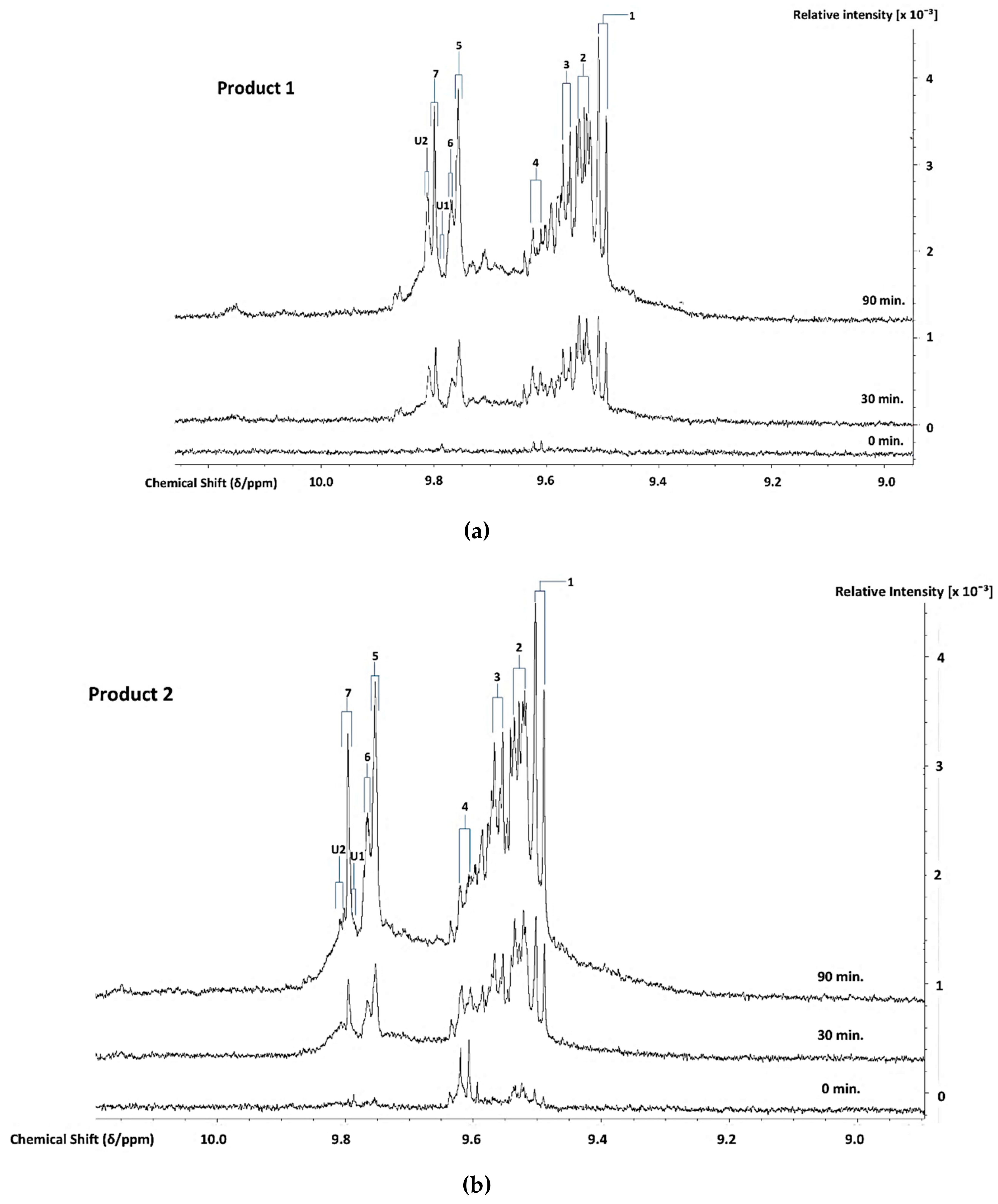
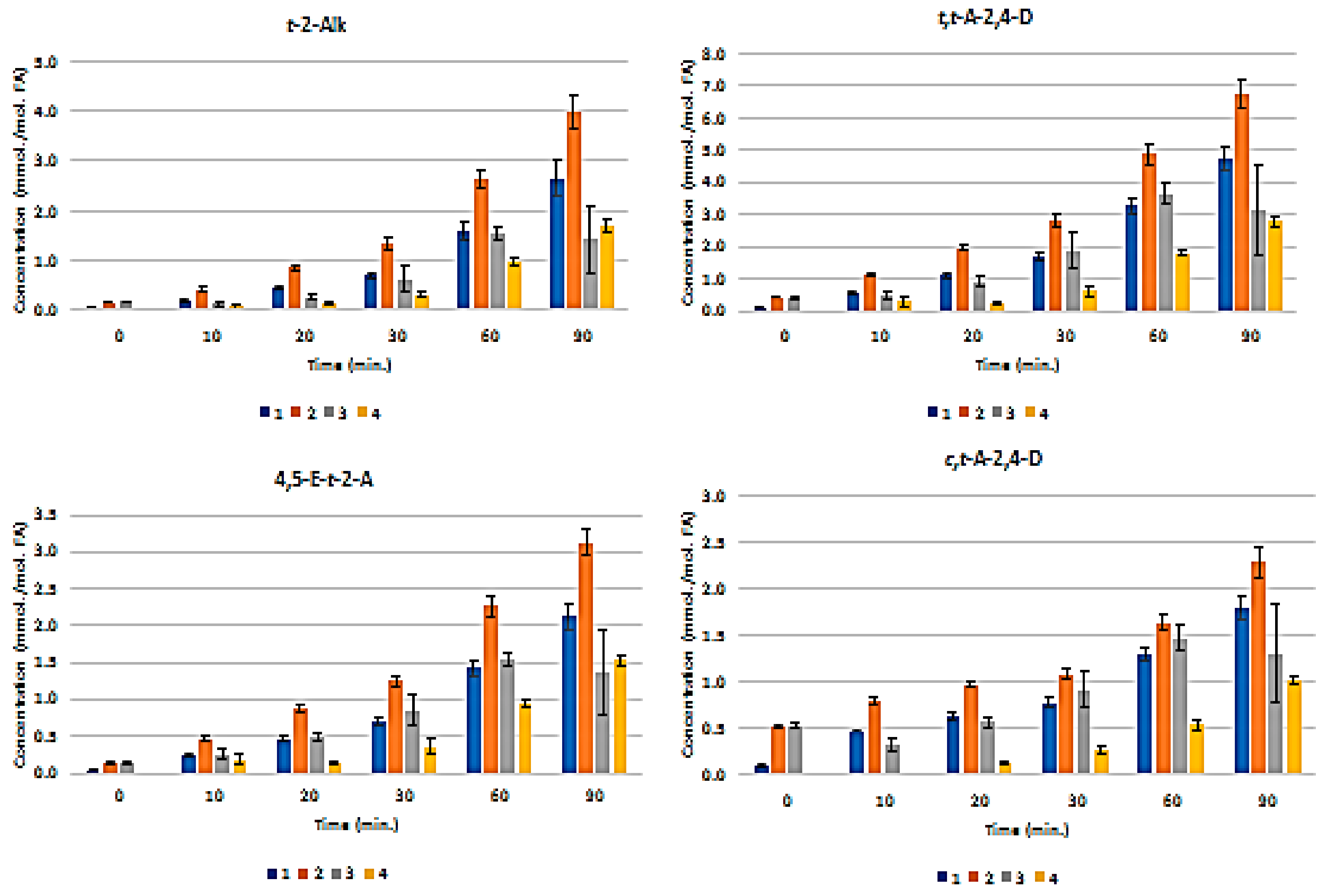
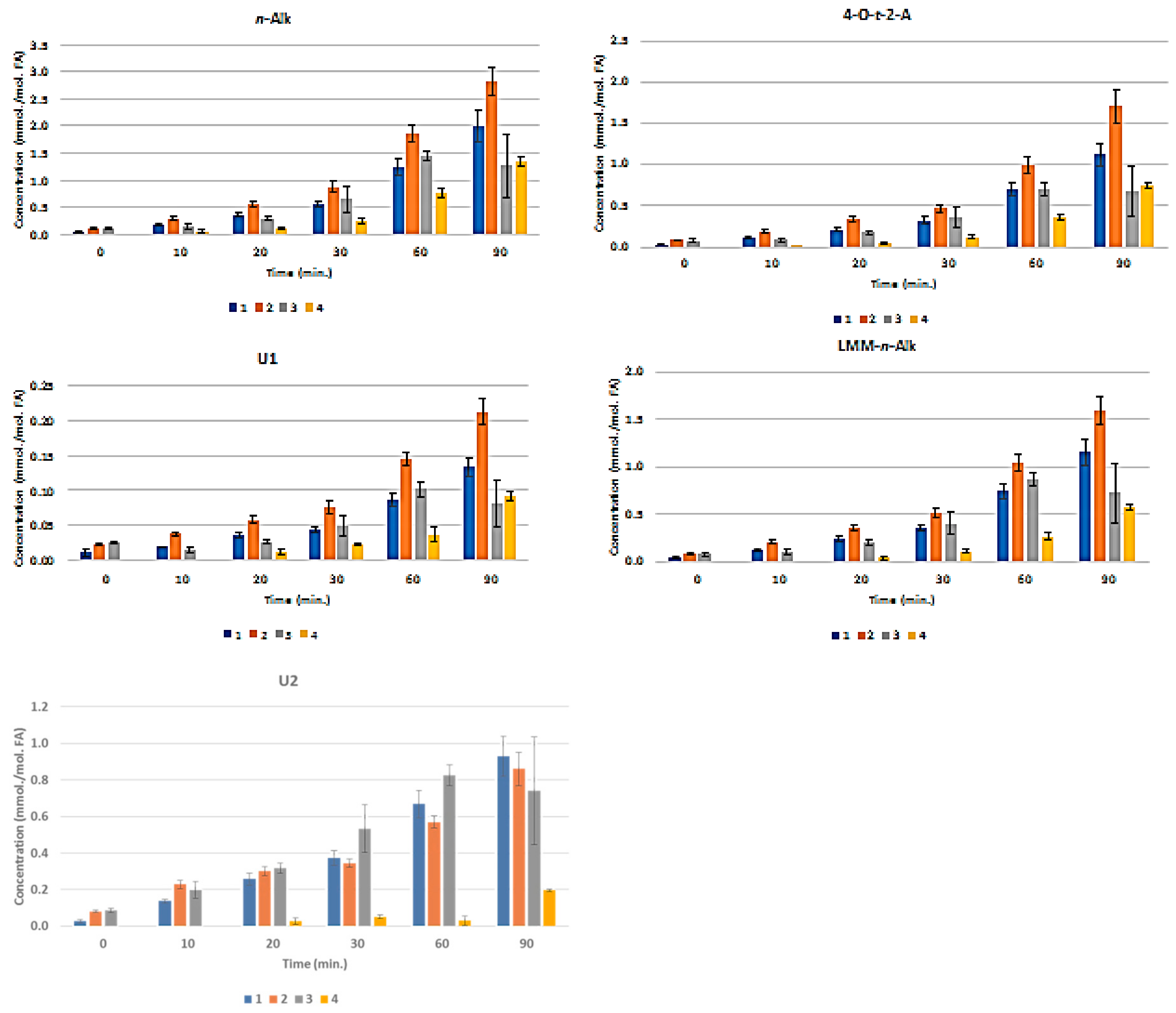
3.3. H NMR Analysis and Time-dependent Monitoring of Secondary Aldehydic LOPs in Thermally-stressed CLOs
3.4. H NMR Analysis and Time-dependent Monitoring of Secondary Aldehydic LOPs in Long-term Stored CLOs
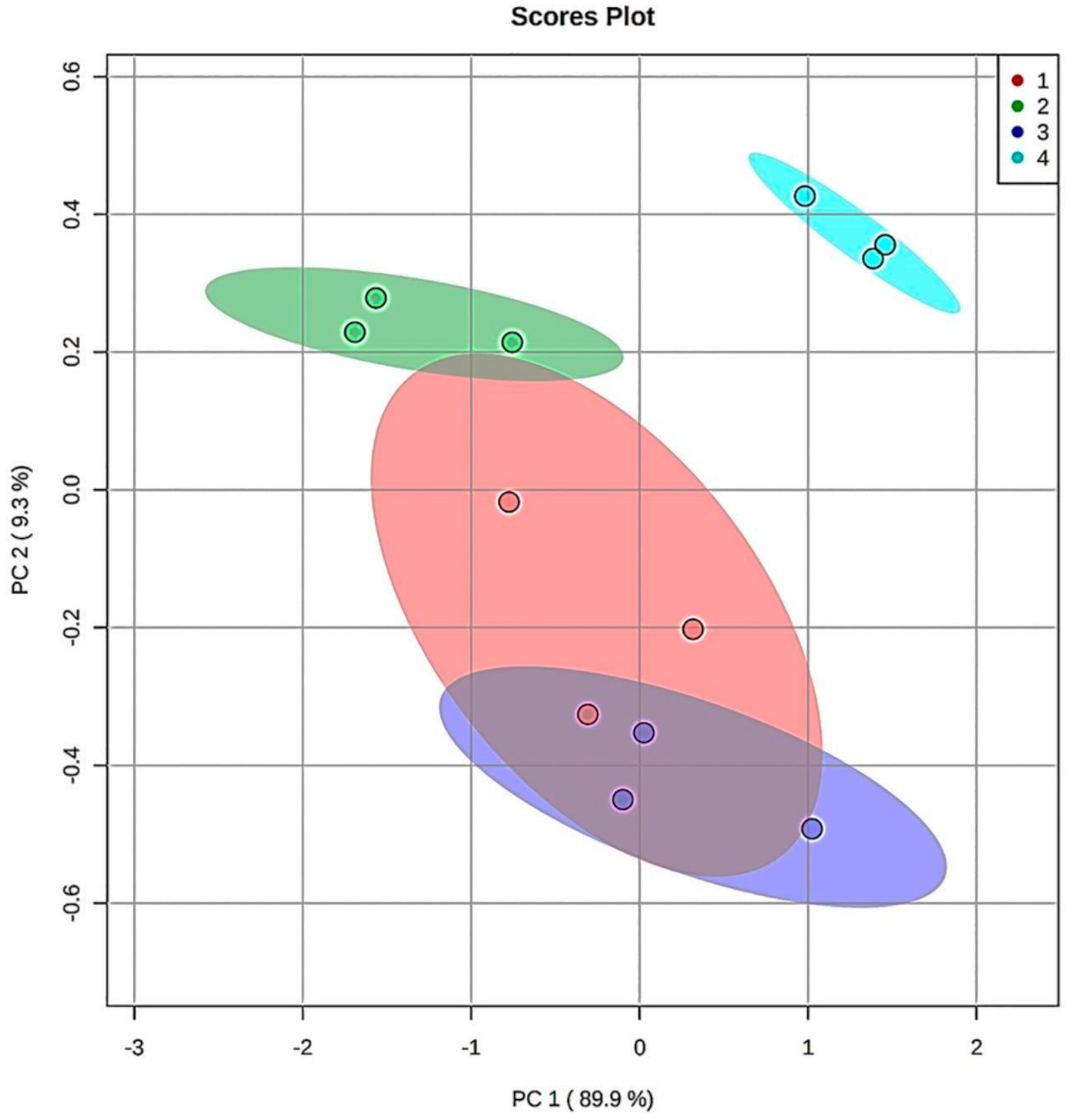
3.5. Chemometric Principal Component Analysis (PCA) of the Aldehydic LOP Concentration Patterns Observed at the Extreme (90 min.) TSE Heating Time-point
4. Discussion
4.1. Antioxidant Potentials of Fermented and Non-fermented CLO Products
4.2. Detection of Collagen and Its Hydrolytic Degradation Products in Fermented CLO: Roles as Antioxidants
4.3. Detection and Antioxidant Potential of Ammonia in the Fermented CLO Product
4.4. Multifunctional Mechanisms of Actions of Fermented CLO Antioxidants and Aldehyde-consuming Agents
4.5. Advantages Offered by NMR-based TORA Analysis of CLO LOPs
4.6. Nutritional and Health Significance of CLO LOPs and Chain-breaking Antioxidants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alliedmarketresearch. Available online: https://www.alliedmarketresearch.com/fish-oil-market (accessed on 19 July 2019).
- Kalra, E.K. Nutraceutical—definition and introduction. AAPS Pharm. Sci. 2003, 5, 25. [Google Scholar] [CrossRef]
- Hamed, I.; Ozogul, F.; Ozogul, Y.; Regenstein, J.M. Marine bioactive compounds and their health benefits: A review. Comprehen. Rev. Food Sci. Food Saf. 2015, 14, 446–465. [Google Scholar] [CrossRef]
- Daliu, P.; Santini, A.; Novellino, E. A decade of nutraceutical patents: Where are ee now in 2018? Expert Opin. Ther. Pat. 2018, 28, 875–882. [Google Scholar] [CrossRef]
- Durazzo, A.; D’Addezio, L.; Camilli, E.; Piccinelli, R.; Turrini, A.; Marletta, L.; Marconi, S.; Lucarini, M.; Lisciani, S.; Gabrielli, P.; et al. From plant compounds to botanicals and back: A current snapshot. Molecules 2018, 23, 1844. [Google Scholar] [CrossRef]
- Siddiqui, N.; Sim, J.; Silwood, C.J.L.; Toms, H.; Iles, R.A.; Grootveld, M. Multicomponent analysis of encapsulated marine oil supplements using high resolution 1H and 13C NMR nuclear magnetic resonance (NMR) techniques. J. Lipid Res. 2003, 44, 2406–2427. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Review. Phospholipids of animal and marine origin: Structure, function, and anti-inflammatory properties. Molecules 2017, 22, 1964. [Google Scholar] [CrossRef]
- Hidalgo, F.J.; Zamora, R. Edible oil analysis by high-resolution nuclear magnetic resonance spectroscopy: Recent advances and future perspectives. Trends Food Sci. Technol. 2003, 14, 499–506. [Google Scholar] [CrossRef]
- Mannina, L.; Sobolev, A.P.; Viel, S. Liquid state 1H high field NMR in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 66, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Marcone, M.F.; Wang, S.; Albabish, W.; Nie, S.; Somnarain, D.; Hill, A. Diverse food-based applications of nuclear magnetic resonance (NMR) technology. Food Res. Int. 2013, 51, 729–747. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. High resolution 1H nuclear magnetic resonance in the study of edible oils and fats. Trends Food Sci. Technol. 2001, 12, 328–338. [Google Scholar] [CrossRef]
- Martínez-Yusta, A.; Guillén, M.D. A study by 1H nuclear magnetic resonance of the influence on the frying medium composition of some soybean oil-food combinations in deep-frying. Food Res. Int. 2014, 55, 347–355. [Google Scholar] [CrossRef]
- Satyarthi, J.K.; Srinivas, D.; Ratnasamy, P. Estimation of free fatty acid content in oils, fats, and biodiesel by 1H NMR spectroscopy. Energy Fuels 2009, 23, 2273–2277. [Google Scholar] [CrossRef]
- Haywood, R.M.; Claxson, A.W.D.; Hawkes, G.E.; Richardson, D.P.; Naughton, D.P.; Coumbarides, G.; Hawkes, J.; Lynch, E.J.; Grootveld, M.C. Detection of aldehydes and their conjugated hydroperoxydiene precursors in thermally-stressed culinary oils and fats: Investigations using high resolution proton NMR spectroscopy. Free Radic. Res. 1995, 22, 441–482. [Google Scholar] [CrossRef] [PubMed]
- Silwood, C.J.L.; Grootveld, M. Application of high-resolution two-dimensional 1H and 13C nuclear magnetic resonance techniques to the characterization of lipid oxidation products in autoxidized linoleoyl/linolenoyglycerols. Lipids 1999, 34, 741–756. [Google Scholar] [CrossRef]
- Alonso-Salces, R.M.; Héberger, K.; Holland, M.V.; Moreno-Rojas, J.M.; Mariani, C.; Bellan, G.; Reniero, F.; Guillou, C. Multivariate analysis of NMR fingerprint of the unsaponifiable fraction of virgin olive oils for authentication purposes. Food Chem. 2010, 118, 956–965. [Google Scholar] [CrossRef]
- Sacchi, R.; Addeo, F.; Paolillo, L. 1H and 13C NMR of virgin olive oil. An overview. Magn. Reson. Chem. 1997, 35, S133–S145. [Google Scholar] [CrossRef]
- Claxson, A.W.D.; Hawkes, G.E.; Richardson, D.P.; Naughton, D.P.; Haywood, R.M.; Chander, C.L.; Atherton, M.; Lynch, E.J.; Grootveld, M.C. Generation of lipid peroxidation products in culinary oils and fats during episodes of thermal stressing: A high field 1H NMR study. FEBS Lett. 1994, 355, 81–90. [Google Scholar] [CrossRef]
- Guillén, M.D.; Uriarte, P.S. Simultaneous control of the evolution of the percentage in weight of polar compounds, iodine value, acyl groups proportions and aldehydes concentrations in sunflower oil submitted to frying temperature in an industrial fryer. Food Cont. 2012, 24, 50–56. [Google Scholar] [CrossRef]
- Parker, T.; Limer, E.; Watson, A.D.; Defernez, M.; Williamson, D.; Kemsley, E.K. 60 MHz 1H-NMR spectroscopy for the analysis of edible oils. Trends Anal. Chem. 2014, 57, 147–158. [Google Scholar] [CrossRef]
- Tyl, C.E.; Brecker, L.; Wagner, K.-H. 1H NMR spectroscopy as tool to follow changes in the fatty acids of fish oils. Lipid Sci. Technol. 2008, 2, 141–148. [Google Scholar] [CrossRef]
- Anihouvi, V.B.; Kindossi, J.M.; Hounhouigan, J.D. Processing and quality characteristics of some major fermented fish products from Africa: A critical review. Int. Res. J. Biol. Sci. 2012, 1, 72–84, ISSN 2278-3202. [Google Scholar]
- Hur, S.J.; Lee, S.Y.; Kim, Y.-C.; Choi, I.; Kim, G.-B. Review. Effect of fermentation on the antioxidant activity in plant-based foods. Food Chem. 2014, 160, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Tamang, J.P.; Shin, D.H.; Jung, S.J.; Chae, S.W. Functional properties of microorganisms in fermented foods. Front. Microbiol. 2016, 7, 578. [Google Scholar] [CrossRef] [PubMed]
- Mateos, R.; Uceda, M.; Aguilera, M.P.; Escuderos, M.E.; Maza, G.B. Relationship of Rancimat method values at varying temperatures for virgin olive oils. Eur. Food Res. Technol. 2006, 223, 246–252. [Google Scholar] [CrossRef]
- Moumtaz, S.; Percival, B.C.; Parmar, D.; Grootveld, K.L.; Jansson, P.; Grootveld, M. Generation of toxic α,β-unsaturated and saturated aldehydes during simulated shallow frying episodes: Comparisons of common frying oils with a novel high-stability algae oil product. Sci. Rep. 2019, 9, 1–21. [Google Scholar]
- Martınez-Yusta, A.; Goicoechea, E.; Guillen, M.D. A Review of thermo-oxidative degradation of food lipids studied by 1H NMR spectroscopy: Influence of degradative conditions and food lipid nature. Compr. Rev. Food Sci. Food Safety. 2014, 13, 838–859. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampsch-Woodill, M.; Flanagan, J.A.; Prior, R.L. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar] [CrossRef]
- Ninfali, P.; Aluigi, G.; Bacchiocca, M.; Magnani, M. Antioxidant capacity of extra-virgin olive oils. J. Am. Oil Chem. Soc. 2001, 78, 243–247. [Google Scholar] [CrossRef]
- Keeler, K. Understanding NMR Spectroscopy, 2nd ed.; Wiley: Hoboken, NJ, USA, 2010; ISBN 978-0-470-78609-7. [Google Scholar]
- Hinds, M.G.; Norton, R.S. NMR spectroscopy of peptides and proteins. Mol. Biotechnol. 1997, 7, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.J.; Radda, G.K.; Kichards, R.E. A hydrogen-deuterium exchange study of the amide protons of polymyxin B by nuclear-magnetic-resonance spectroscopy. Eur. J. Biochem. 1978, 82, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Chemicalbook. Available online: https://www.chemicalbook.com/SpectrumEN_64-04-0_1HNMR.htm (accessed on 13 May 2019).
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 4, 608–617. [Google Scholar] [CrossRef]
- Forino, M.; Tartaglione, L.; Dell’Aversano, C.; Ciminiello, P. NMR-based identification of the phenolic profile of fruits of Lycium barbarum (goji berries). Isolation and structural determination of a novel N-feruloyl tyramine dimer as the most abundant antioxidant polyphenol of goji berries. Food Chem. 2016, 194, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S. 1H NMR Spectroscopy for Assessment of Lipid Oxidation. Adv. NMR Spectrosc. Lipid Oxid. Assess. 2017, 15–31. [Google Scholar]
- LeGresley, A.; Ampem, G.; Grootveld, M.; Percival, B.C.; Naughton, D.P. Characterisation of peroxidation products arising from culinary oils exposed to continuous and discontinuous thermal degradation processes. Food Funct. 2019, 10, 7952–7966. [Google Scholar] [CrossRef]
- Lee, H.; Kitizio, S.A.; Weese, S.J.; Craig-Schmidt, M.C.; Lee, Y.; Wei, C.-I.; An, H. Analysis of headspace volatile and oxidized volatile compounds in DHA-enriched fish oil on accelerated oxidative storage. J. Food Sci. 2003, 68, 2169–2177. [Google Scholar] [CrossRef]
- Benedetti, A.; Comporti, M.; Fulceri, R.; Esterbauer, H. Cytotoxic aldehydes originating from the peroxidation of liver microsomal lipids. Identification of 4,5-dihydroxydecenal. Biochim. Biophys. Acta 1984, 792, 172–181. [Google Scholar] [CrossRef]
- Lodge, J.K.; Patel, S.U.; Sadler, P.J. Aldehydes from metal ion- and lipoxygenase-induced lipid peroxidation: Detection by 1H-n.m.r. spectroscopy. Biochem. J. 1993, 289, 149–153. [Google Scholar] [CrossRef]
- Bertz, S.H.; Dabbagh, G. NMR spectroscopy of malondialdehyde. J. Org. Chem. 1990, 55, 5161–5165. [Google Scholar] [CrossRef]
- Slatter, D.A.; Bolton, C.H.; Bailey, A.J. The importance of lipid-derived malondialdehyde in diabetes mellitus. Diabetologia 2000, 43, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Ninfali, P.; Aluigi, G. Variability of oxygen radical absorbance capacity (ORAC) in different animal species. Free Rad. Res. 1998, 29, 399–408. [Google Scholar] [CrossRef]
- Cao, G.; Russell, R.M.; Lischner, N.; Prior, R.L. Serum antioxidant capacity is increased by consumption of strawberries, spinach, red wine or vitamin C in elderly women. J. Nutr. 1998, 128, 2383–2390. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M. Extractable and non-extractable antioxidants. Molecules 2019, 24, 1933. [Google Scholar] [CrossRef] [PubMed]
- Nemes, A.; Szo˝llo˝si, E.; Stündl, L.; Biró, A.; Homoki, J.R.; Szarvas, M.M.; Balogh, P.; Cziáky, Z.; Remenyik, J. Determination of flavonoid and proanthocyanidin profile of hungarian sour cherry. Molecules 2018, 23, 3278. [Google Scholar] [CrossRef] [PubMed]
- Fraisse, D.; Degerine-Roussel, A.; Bred, A.; Ndoye, S.F.; Vivier, M.; Felgines, C.; Senejoux, F. A Novel HPLC method for direct detection of nitric oxide scavengers from complex plant matrices and its application to Aloysia triphylla Leaves. Molecules 2018, 23, 1574. [Google Scholar] [CrossRef]
- Koch, W.; Kukula-Koch, W.; Komsta, L. Black tea samples origin discrimination using analytical investigations of secondary metabolites, antiradical scavenging activity and chemometric approach. Molecules 2018, 23, 513. [Google Scholar] [CrossRef]
- Anokwuru, C.; Sigidi, M.; Boukandou, M.; Tshisikhawe, P.; Traore, A.; Potgieter, N. Antioxidant activity and spectroscopic characteristics of extractable and non-extractable phenolics from Terminalia sericea Burch. ex DC. Molecules 2018, 23, 1303. [Google Scholar] [CrossRef]
- Thermofisher. Available online: https://www.thermofisher.com/uk/en/home/industrial/spectroscopy-elemental-isotope-analysis/spectroscopy-elemental-isotope-analysis-learning-center/spectroscopy-elemental-isotope-analysis-resource-library/nmr-tech-talk/nmr-tech-talk-february-2015/nmr-spectrum-phenacetin.html (accessed on 7 May 2019).
- Tamiola, K.; Mulder, F.A.A. Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem. Soc. Trans. 2012, 40, 1014–1020. [Google Scholar] [CrossRef]
- Ferris, T.D.; Lee, P.T.; Farrar, T.C. Synthesis of propiolamide and 1H, 13C and 15N NMR spectra of formamide, acetamide and propiolamide. Mag. Res. Chem. 1997, 35, 571–576. [Google Scholar] [CrossRef]
- Haytowitz, D.B.; Bhagwat, S. USDA Database for the Oxygen Radical Absorbance Capacity (ORAC) of Selected Foods, Release 2. Nutrient Data Laboratory Beltsville Human Nutrition Research Center (BHNRC); Agricultural Research Service (ARS) U.S. Department of Agriculture (USDA): Beltsville, MD, USA, May 2010. Available online: http://www.ars.usda.gov/nutrientdata (accessed on 1 February 2020).
- Nagatsuka, N.; Sato, K.; Harada, K.; Nagao, K. Radical scavenging activity of ‘Nikogori’ gelatin gel food made from head, bone, skin, tail and scales of fishes measured using the chemiluminescence method. Int. J. Molec. Med. 2007, 20, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Deyl, Z.; Rosmus, J.; Adam, M. Peptide fragments released from the collagen molecule by pronase. FEBS Lett. 1971, 12, 176–178. [Google Scholar] [CrossRef]
- Smith, J.F.; Li, C.; Roth, M.; Helpe, L.G. Solubility of ammonia in chloroform: Analysis in terms of Henry’s law and the equilibrium constant for hydrogen-bonded complex formation. Can. J. Chem. 1989, 67, 2213–2217. [Google Scholar] [CrossRef]
- Malki, F.; Touati, A.; Hamza, K.; Moulay, S.; Baltas, M. Antioxidant activity of a series of amides. J. Mater. Environ. Sci. 2016, 7, 936–941, ISSN 2028-2508. [Google Scholar]
- Jia, Y.; Li, J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem. Rev. 2015, 115, 1597–1621. [Google Scholar] [CrossRef] [PubMed]
- Poon, T.; Mundy, B.P.; Shattuck, T.W. The Michael reaction. J. Chem. Educ. 2002, 79, 264–267. [Google Scholar] [CrossRef]
- Cortinas, L.; Galobart, J.; Barroeta, A.C.; Baucells, M.D.; Grashorn, M.A. Change in α-tocopherol contents, lipid oxidation and fatty acid profile in eggs enriched with linolenic acid or very long-chain w-3 polyunsaturated fatty acids after different processing methods. J. Sci. Food Agric. 2003, 83, 820–829. [Google Scholar] [CrossRef]
- Rizos, E.C.; Elisaf, M.S. Does supplementation with omega-3 PUFAs add to the prevention of cardiovascular disease? Curr. Cardiol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef]
- Alexander, D.D.; Miller, P.E.; Van Elswyk, M.E.; Kuratko, C.N.; Bylsma, L.C. A meta-analysis of randomized controlled trials and prospective cohort studies of eicosapentaenoic and docosahexaenoic long-chain omega-3 fatty acids and coronary heart disease risk. Mayo Clinic Proc. 2017, 92, 15–29. [Google Scholar] [CrossRef]
- Sargent, J.R.; Tacon, A.G.J. Development of farmed fish: A nutritionally necessary alternative to meat. P. Nutr. Soc. 1999, 58, 377–383. [Google Scholar] [CrossRef]
- Subasinghe, R.; Soto, D.; Jia, J. Global aquaculture and its role in sustainable development. Rev. Aqua. 2009, 1, 2–9. [Google Scholar] [CrossRef]
- Harris, W.S. Fish oil supplementation: Evidence for health benefits. Cleveland Clin. J. Med. 2004, 71, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Percival, B.C.; Zbasnik, R.; Schlegel, V.; Edgar, M.; Zhang, J.; Grootveld, M. Determinations of the peroxidative susceptibilities of cod liver oils by a newly-developed 1H NMR-based method: resistance of an antioxidant-fortified product isolated from pre-fermented sources. BMC Res. Notes 2020, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, S.; Zimmermann, T.E.G.; Salgado, H.R.N. FTIR Spectrophotometry as a green tool for quantitative analysis of drugs: Practical application to amoxicillin. J. Chem. 2018, 3920810. [Google Scholar] [CrossRef]
- Giese, E.; Rohn, S.; Fritsche, J. Chemometric tools for the authentication of cod liver oil based on nuclear magnetic resonance and infrared spectroscopy data. Anal. Bioanal. Chem. 2019, 411, 6931–6942. [Google Scholar] [CrossRef]
- Xu, L.; Fei, T.; Yu, X.; Li, Q.; Liu, L. Qualitative analysis of edible oil oxidation by FTIR spectroscopy using mesh “cell”. Anal. Methods 2015, 7. [Google Scholar] [CrossRef]
- Grootveld, M.; Percival, B.C.; Gibson, M.; Osman, Y.; Edgar, M.; Molinari, M.; Mather, M.L.; Casanova, F.; Wilson, P.B. Progress in low-field benchtop NMR spectroscopy in chemical and biochemical Analysis—A review. Anal. Chim. Acta 2019, 1067, 11–30. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrient/Antioxidant | Concentration Units | Product 1 (n = 4) | Product 2 (n = 4) | Product 3 (n = 4) | Product 4 (n = 5) |
|---|---|---|---|---|---|
| Total Flavonoids* | mg/kg | nd | nd | nd | nd |
| Total Flavanones* | mg/kg | nd | nd | nd | 896 ± 444 |
| Total Phenolics* | mg/kg | nd | nd | nd | 79.7 ± 9.5 |
| Tannins* | mg/kg | nd | nd | nd | 112.2 ± 150.5 |
| Anthocyanins* | mg/kg | nd | nd | nd | nd |
| α-Tocopherol*† | mmol./kg | 0.09 ± 0.03 | 0.12 ± 0.02 | 0.12 ± 0.01 | 0.13 ± 0.09 |
| γ-Tocopherol* | mg/kg | na | na | na | trace |
| β-Carotene* | µmol./kg | na | na | na | 1.49 ± 1.75 |
| Total Carotenoids*† | mg/kg | 0.79 ± 0.07 | 0.73 ± 0.02 | 0.77 ± 0.02 | 1.86 ± 2.04 |
| Total Chlorophyll | mg/kg | na | na | na | 2.04 ± 1.53 |
| Chlorophyll A† | µmol./kg | 0.23 ± 0.01 | 0.23 ± 0.02 | 0.20 ± 0.02 | 0.23 ± 0.03 |
| Chlorophyll B | µmol./kg | nd | nd | nd | 0.42 ± 0.03 |
| Retinol† | mmol./kg | 0.10 ± 0.01 | 0.09 ± 0.03 | 0.16 ± 0.02 | 0.10 ± 0.06 |
| Batch Number | ||||||
|---|---|---|---|---|---|---|
| Biogenic Amine | 1 | 2 | 3 | 4 | 5 | Mean ± SD (Product 4) |
| 2-Phenylethylamine (mmol./kg) | 0.71 | 0.85 | 0.41 | 0.14 | 0.63 | 0.55 ± 0.28 |
| Tyramine* (mmol./kg) | 0.51 | 0.64 | 0.31 | 0.06 | 0.23 | 0.36 ± 0.23 |
| Tryptamine (mmol./kg) | 0.22 | 0.15 | 0.16 | 0.02 | 0.05 | 0.12 ± 0.08 |
| Cadaverine (mmol./kg) | 0.13 | 0.06 | 0.14 | nd | 0.04 | 0.07 ± 0.06 |
| Putrescine (mmol./kg) | 0.08 | 0.06 | 0.08 | nd | nd | 0.04 ± 0.04 |
| Spermidine (mmol./kg) | nd | 0.03 | nd | nd | nd | 0.01 ± 0.01 |
| Total Biogenic Amine Functions** (mmol./kg) | 2.1 | 2.1 | 1.5 | 0.2 | 1.0 | 1.39 ± 0.78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Percival, B.C.; Wann, A.; Zbasnik, R.; Schlegel, V.; Edgar, M.; Zhang, J.; Ampem, G.; Wilson, P.; Le Gresley, A.; Naughton, D.; et al. Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product. Nutrients 2020, 12, 753. https://doi.org/10.3390/nu12030753
Percival BC, Wann A, Zbasnik R, Schlegel V, Edgar M, Zhang J, Ampem G, Wilson P, Le Gresley A, Naughton D, et al. Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product. Nutrients. 2020; 12(3):753. https://doi.org/10.3390/nu12030753
Chicago/Turabian StylePercival, Benita C., Angela Wann, Richard Zbasnik, Vicki Schlegel, Mark Edgar, Jie Zhang, Gilbert Ampem, Philippe Wilson, Adam Le Gresley, Declan Naughton, and et al. 2020. "Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product" Nutrients 12, no. 3: 753. https://doi.org/10.3390/nu12030753
APA StylePercival, B. C., Wann, A., Zbasnik, R., Schlegel, V., Edgar, M., Zhang, J., Ampem, G., Wilson, P., Le Gresley, A., Naughton, D., & Grootveld, M. (2020). Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product. Nutrients, 12(3), 753. https://doi.org/10.3390/nu12030753