Changes in Intestinal Microbiota and Predicted Metabolic Pathways During Colonic Fermentation of Mango (Mangifera indica L.)—Based Bar Indigestible Fraction

 ,
,  ,
,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mango-Based Bar Sample
2.2. Isolation of Indigestible Fraction (IF) in Mango Snack
2.3. Fecal Microbiota Standardization
2.4. In Vitro Colonic Fermentation
2.5. DNA Extraction and Library Construction
2.6. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results and Discussion
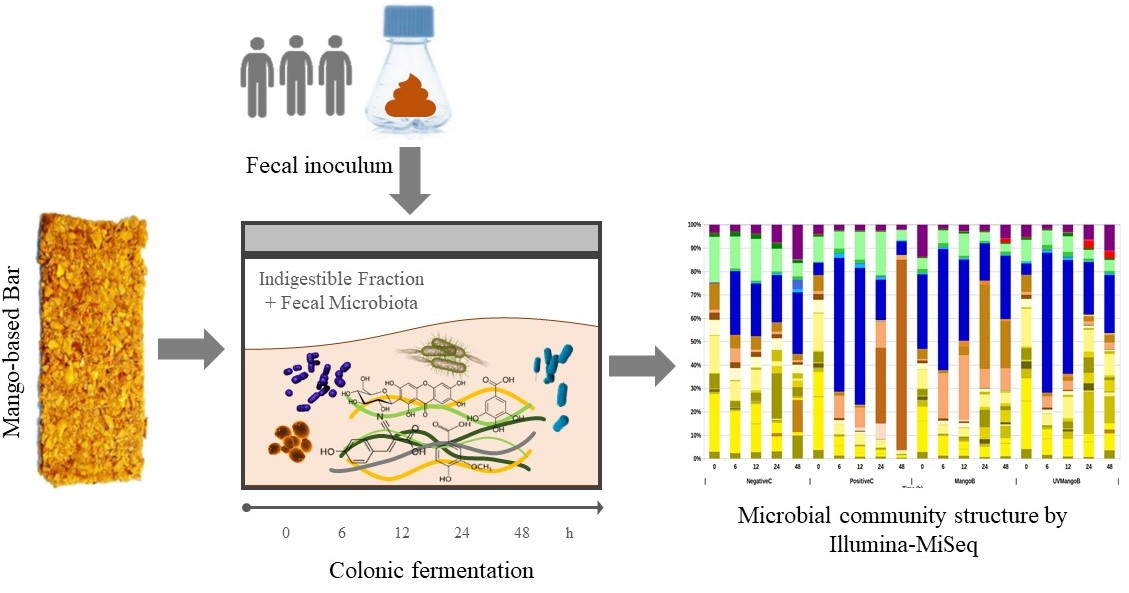
3.1. Extracted DNA and Amplicon Sequence Variants (ASVs) Results
3.2. Bacterial Community Diversity in Colonic Intestinal Fermentation
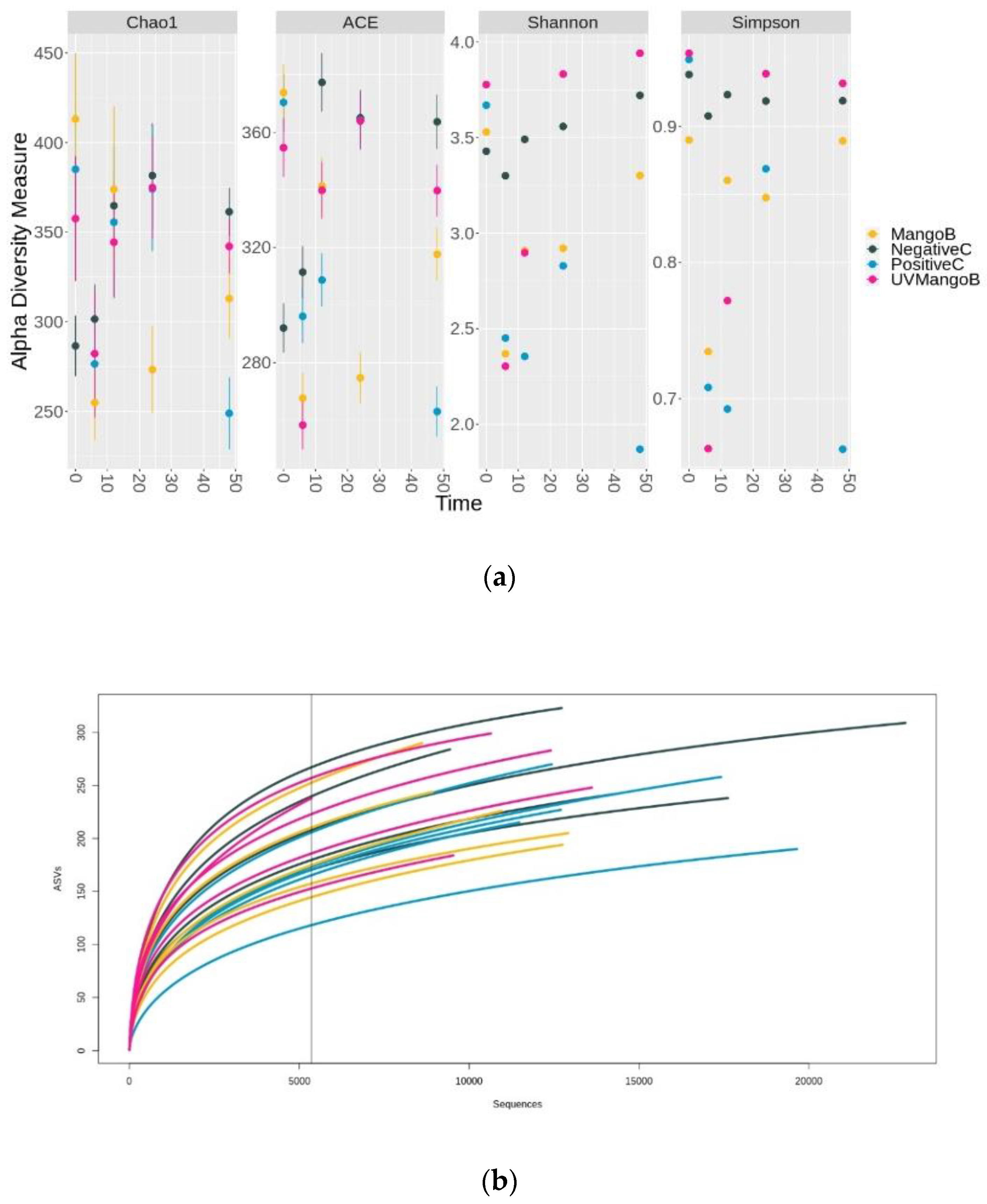
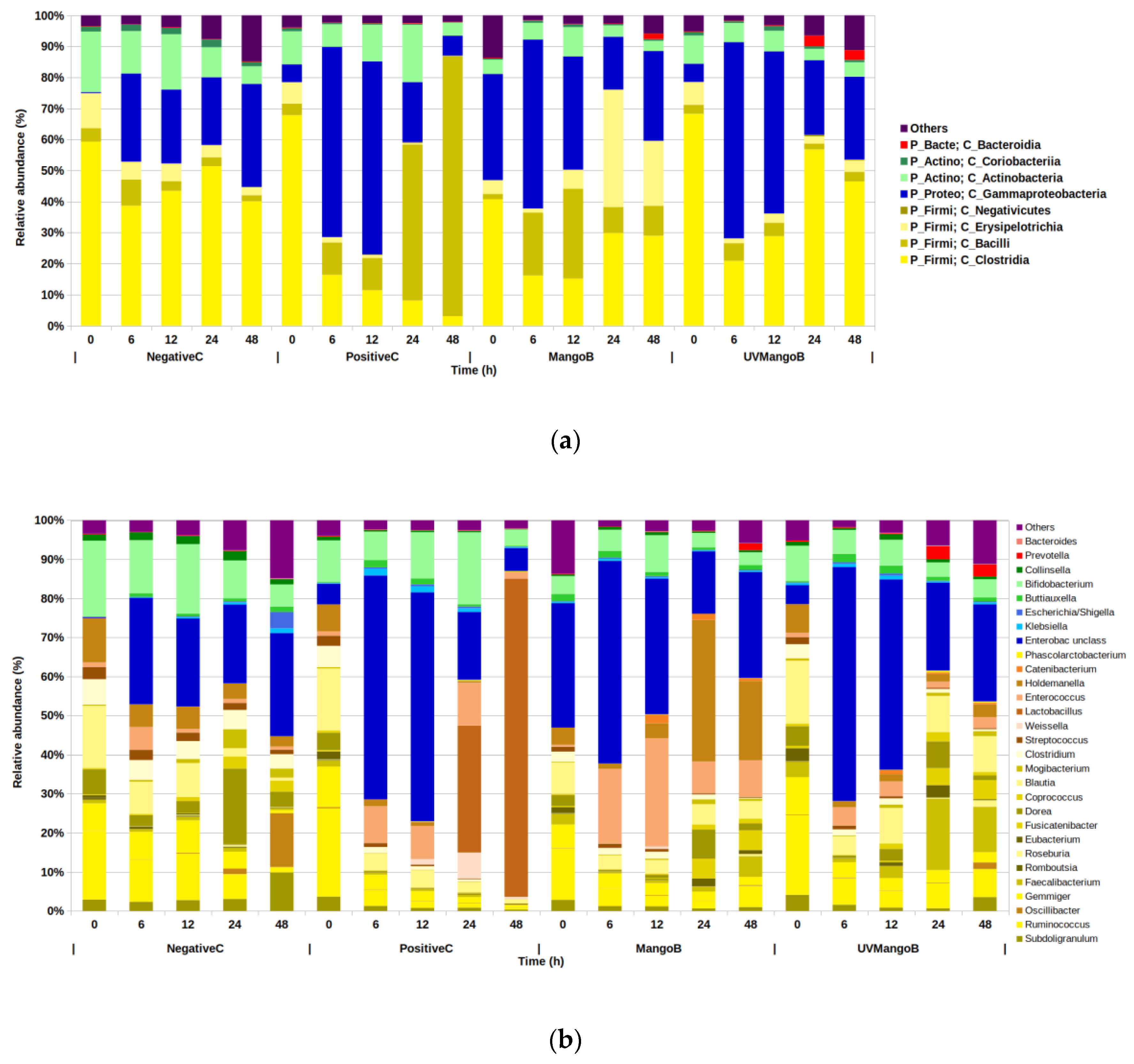
3.3. Changes of Microbial Composition during Mango (Mangifera indica L.) IF Colonic Fermentation
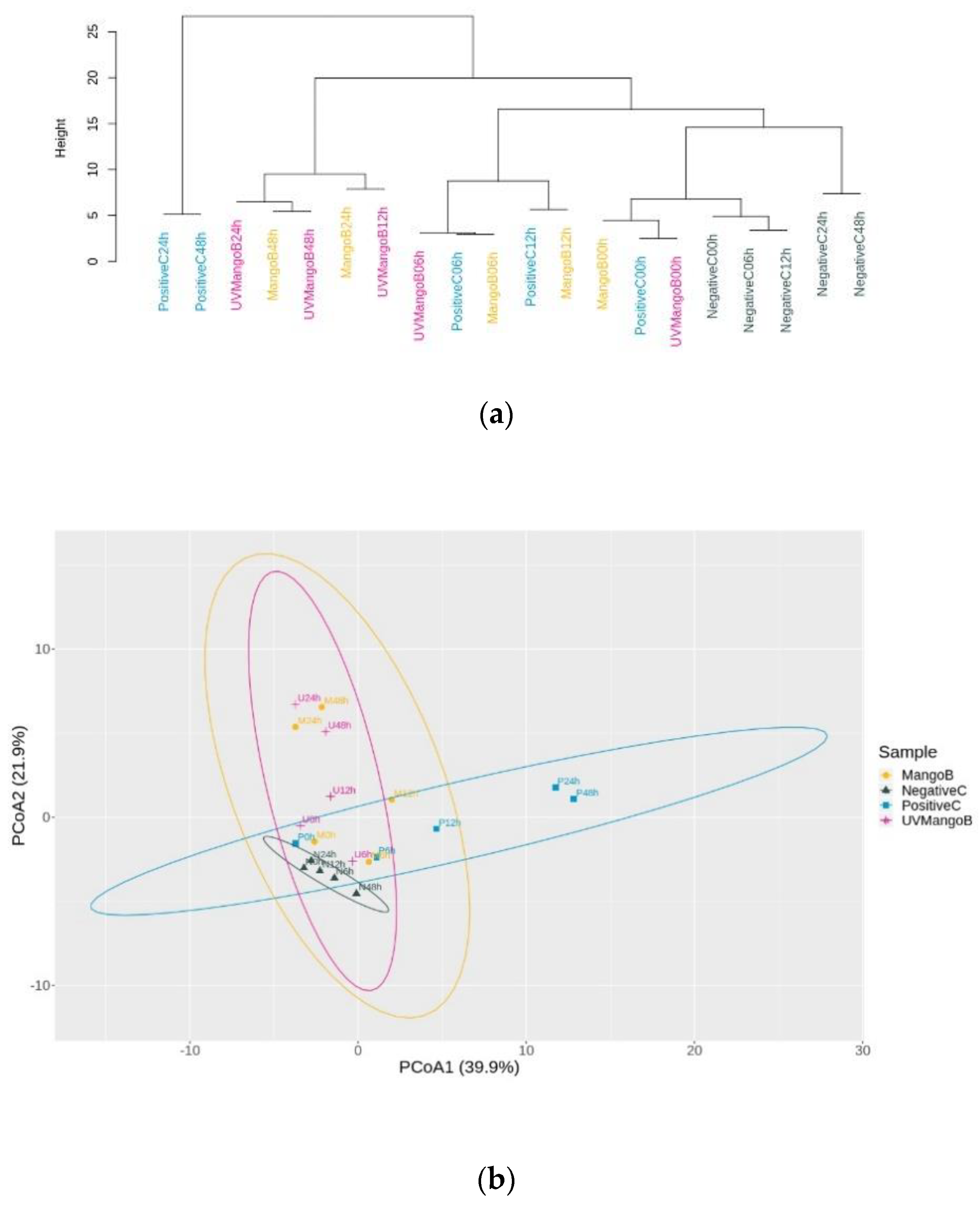
3.4. Sample Clustering and Principal Coordinate Analysis (PCoA) Analysis
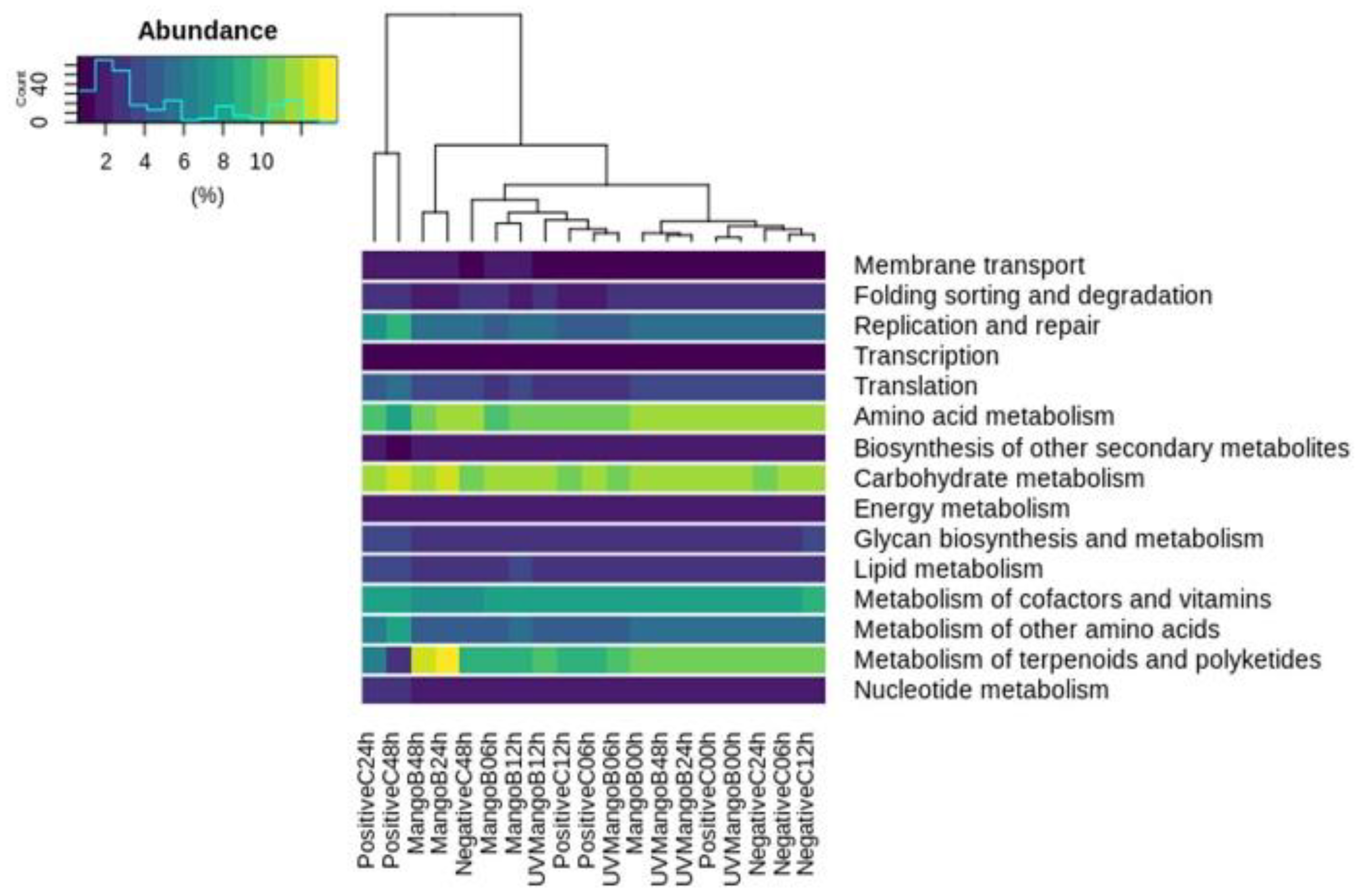
3.5. Metabolic Prediction: Phylogenetic Investigation of Communities by Reconstruction of Unobserved State (PICRUSt) Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Costa, A.G.V.; Garcia-Diaz, D.F.; Jimenez, P.; Silva, P.I. Bioactive compounds and health benefits of exotic tropical red–black berries. J. Funct. Foods 2013, 5, 539–549. [Google Scholar] [CrossRef]
- Instituto Mexicano de la Propiedad Industrial. Declaración General de Protección de la Denominación de Origen Mango Ataulfo del Soconusco Chiapas. 2003. Available online: http://www.impi.gob.mx/TemasInteres/Documents/Declaratoria_Mango_Ataulfo.pdf (accessed on 15 March 2019).
- MOK, C. Biotechnological potentialities and valorization of mango peel waste: A review. Sains Malays. 2014, 43, 1901–1906. [Google Scholar] [CrossRef]
- de Lourdes García-Magaña, M.; García, H.S.; Bello-Pérez, L.A.; Sáyago-Ayerdi, S.G.; de Oca Mata-Montes, M. Functional properties and dietary fiber characterization of mango processing by-products (Mangifera indica L., cv Ataulfo and Tommy Atkins). Plant Foods Hum. Nutr. 2013, 68, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Benitez, F.J.; Pérez-Jiménez, J.; Montalvo-González, E.; González-Aguilar, G.A.; Sáyago-Ayerdi, S.G. In vitro evaluation of the kinetics of the release of phenolic compounds from guava (Psidium guajava L.) fruit. J. Funct. Foods 2018, 43, 139–145. [Google Scholar] [CrossRef]
- Saura-Calixto, F.; Garcia-Alonso, A.; Goni, I.; Bravo, L. In vitro determination of the indigestible fraction in foods: An alternative to dietary fiber analysis. J. Agric. Food Chem. 2000, 48, 3342–3347. [Google Scholar] [CrossRef]
- Zamora-Gasga, V.M.; Cárdenas-Castro, A.P.; Montalvo-González, E.; Loarca-Piña, M.G.F.; Pedro Alberto, V.L.; Tovar, J.; Sáyago-Ayerdi, S.G. In vitro human colonic fermentation of indigestible fraction isolated from lunch menus: Impact on the gut metabolites and antioxidant capacity. Int. J. Food Sci. Nutr. 2018, 69, 718–728. [Google Scholar] [CrossRef]
- Sáyago-Ayerdi, S.G.; Zamora-Gasga, V.M.; Venema, K. Prebiotic effect of predigested mango peel on gut microbiota assessed in a dynamic in vitro model of the human colon (TIM-2). Food Res. Int. 2019, 118, 89–95. [Google Scholar] [CrossRef]
- Cárdenas-Castro, A.P.; Bianchi, F.; Tallarico-Adorno, M.A.; Montalvo-González, E.; Sáyago-Ayerdi, S.G.; Sivieri, K. In vitro colonic fermentation of Mexican “taco” from corn-tortilla and black beans in a Simulator of Human Microbial Ecosystem (SHIME®) system. Food Res. Int. 2019, 118, 81–88. [Google Scholar] [CrossRef]
- Hidalgo, M.; Oruna-Concha, M.J.; Kolida, S.; Walton, G.E.; Kallithraka, S.; Spencer, J.P.; de Pascual-Teresa, S. Metabolism of anthocyanins by human gut microflora and their influence on gut bacterial growth. J. Agric. Food Chem. 2012, 60, 3882–3890. [Google Scholar] [CrossRef]
- Koutsos, A.; Lima, M.; Conterno, L.; Gasperotti, M.; Bianchi, M.; Fava, F.; Vrhovsek, U.; Lovegrove, J.A.; Tuohy, K.M. Effects of commercial apple varieties on human gut microbiota composition and metabolic output using an in vitro colonic model. Nutrients 2017, 9, 533. [Google Scholar] [CrossRef]
- Blancas-Benitez, F.J.; Mercado-Mercado, G.; Quirós-Sauceda, A.E.; Montalvo-González, E.; Gonzalez-Aguilar, G.A.; Sáyago-Ayerdi, S.G. Bioaccesibility of polyphenols associated with dietary fiber and in vitro kinetics release of polyphenols in Mexican ‘Ataulfo’ mango (Mangifera indica L.) by-products. Food Funct. 2015, 6, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Maldonado, L.M.; Blancas-Benítez, F.J.; Zamora-Gasga, V.M.; Cárdenas-Castro, A.P.; Tovar, J.; Sáyago-Ayerdi, S.G. In Vitro Gastrointestinal Digestion and Colonic Fermentation of High Dietary Fiber and Antioxidant-Rich Mango (Mangifera indica L.) “Ataulfo”-Based Fruit Bars. Nutrients 2019, 11, 1564. [Google Scholar] [CrossRef] [PubMed]
- Kemperman, R.A.; Gross, G.; Mondot, S.; Possemiers, S.; Marzorati, M.; Van de Wiele, T.; Doré, J.; Vaughan, E.E. Impact of polyphenols from black tea and red wine/grape juice on a gut model microbiome. Food Res. Int. 2013, 53, 659–669. [Google Scholar] [CrossRef]
- Blatchford, P.; Stoklosinski, H.; Walton, G.; Swann, J.; Gibson, G.; Gearry, R.; Ansell, J. Kiwifruit fermentation drives positive gut microbial and metabolic changes irrespective of initial microbiota composition. Bioact. Carbohydr. Diet. Fibre 2015, 6, 37–45. [Google Scholar] [CrossRef]
- Mosele, J.I.; Macià, A.; Motilva, M.J. Metabolic and microbial modulation of the large intestine ecosystem by non-absorbed diet phenolic compounds: A review. Molecules 2015, 20, 17429–17468. [Google Scholar] [CrossRef] [PubMed]
- Pelpolage, S.W.; Goto, Y.; Nagata, R.; Fukuma, N.; Furuta, T.; Mizu, M.; Han, K.-H.; Fukushima, M. Colonic fermentation of water soluble fiber fraction extracted from sugarcane (Sacchurum officinarum L.) bagasse in murine models. Food Chem. 2019, 292, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Ooga, T.; Kibe, R.; Aiba, Y.; Koga, Y.; Benno, Y. Colonic absorption of low-molecular-weight metabolites influenced by the intestinal microbiome: A pilot study. PLoS ONE 2017, 12, e0169207. [Google Scholar] [CrossRef]
- Ferrocino, I.; Di Cagno, R.; De Angelis, M.; Turroni, S.; Vannini, L.; Bancalari, E.; Rantsiou, K.; Cardinali, G.; Neviani, E.; Cocolin, L. Fecal microbiota in healthy subjects following omnivore, vegetarian and vegan diets: Culturable populations and rRNA DGGE profiling. PLoS ONE 2015, 10, e0128669. [Google Scholar] [CrossRef]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shikari, K.; Ito, M.; Takei, Y.; et al. Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: Terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol. Rep. 2016, 35, 325–333. [Google Scholar] [CrossRef]
- Mego, M.; Manichanh, C.; Accarino, A.; Campos, D.; Pozuelo, M.; Varela, E.; Vulevic, J.; Tzortzis, G.; Gibson, G.; Guarner, F.; et al. Metabolic adaptation of colonic microbiota to galactooligosaccharides: A proof-of-concept-study. Aliment. Pharmacol. Ther. 2017, 45, 670–680. [Google Scholar] [CrossRef]
- Swidsinski, A.; Dörffel, Y.; Loening-Baucke, V.; Gille, C.; Göktas, Ö.; Reißhauer, A.; Neuhaus, J.; Weylandt, K.H.; Guschin, A.; Bock, M. Reduced mass and diversity of the colonic microbiome in patients with multiple sclerosis and their improvement with ketogenic diet. Front. Microbiol. 2017, 8, 1141. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.P.N.; Troise, A.D.; Fogliano, V.; de Vos, W.M. Anaerobic Degradation of N-ε-Carboxymethyllysine, A Major Glycation End Product, By Human Intestinal Bacteria. J. Agric. Food Chem. 2019, 67, 6594–6602. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkilä, J.; Marchesi, J.R.; Smidt, H.; de Vos, M.W.; Ross, R.P.; O’Toole, P.W. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, M.; Venema, K.; Maathuis, A.J.H.; Saura-Calixto, F.D. Metabolite production during in vitro colonic fermentation of dietary fiber: Analysis and comparison of two European diets. J. Agric. Food Chem. 2011, 59, 8968–8975. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 2016, 1, e00009-15. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed on 15 March 2019).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Lee, M. Happy Belly Bioinformatics: An open-source resource dedicated to helping biologists utilize bioinformatics. J. Open Sour. Educ. 2019, 4, 53. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Beiko, R.G.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R. Vegan: Community Ecology Package. R Package Version 2.5–6. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 30 October 2019).
- Galili, T.; Benjamini, Y.; Jefferis, G.; Gallotta, M.; The R Core Team; Hornik, K.; Ligges, U.; Spiess, A.-N.; Horvath, S.; Langfelder, P.; et al. Extending ‘Dendrogram’ Functionality. R Package Dendextend Version 1.13.2. Available online: https://cran.r-project.org/web/packages/dendextend/index.html (accessed on 20 December 2019).
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Semenov, M.; Blagodatskaya, E.; Stepanov, A.; Kuzyakov, Y. DNA-based determination of soil microbial biomass in alkaline and carbonaceous soils of semi-arid climate. J. Arid Environ. 2018, 150, 54–61. [Google Scholar] [CrossRef]
- Grant, L.J.; Mikkelsen, D.; Ouwerkerk, D.; Klieve, A.V.; Gidley, M.J.; Williams, B.A. Whole fruit pulp (mango) and a soluble fibre (pectin) impact bacterial diversity and abundance differently within the porcine large intestine. Bioact. Carbohydr. Diet. Fibre 2019, 100192. [Google Scholar] [CrossRef]
- Serra, J.L.; Moura, F.G.; de Melo Pereira, G.V.; Soccol, C.R.; Rogez, H.; Darnet, S. Determination of the microbial community in Amazonian cocoa bean fermentation by Illumina-based metagenomic sequencing. LWT 2019, 106, 229–239. [Google Scholar] [CrossRef]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: A review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef]
- Grine, G.; Boualam, M.A.; Drancourt, M. Methanobrevibacter smithii, a methanogen consistently colonising the newborn stomach. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2449–2455. [Google Scholar] [CrossRef]
- Adamberg, K.; Adamberg, S.; Ernits, K.; Larionova, A.; Voor, T.; Jaagura, M.; Visnapuu, T.; Alamäe, T. Composition and metabolism of fecal microbiota from normal and overweight children are differentially affected by melibiose, raffinose and raffinose-derived fructans. Anaerobe 2018, 52, 100–110. [Google Scholar] [CrossRef]
- Heir, E.; Liland, K.H.; Carlehög, M.; Holck, A.L. Reduction and inhibition of Listeria monocytogenes in cold-smoked salmon by Verdad N6, a buffered vinegar fermentate, and UV-C treatments. Int. J. Food Microbiol. 2019, 291, 48–58. [Google Scholar] [CrossRef]
- Teoh, L.S.; Lasekan, O.; Adzahan, N.M.; Hashim, N. The effect of ultraviolet treatment on enzymatic activity and total phenolic content of minimally processed potato slices. J. Food Sci. Technol. 2016, 53, 3035–3042. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Braune, A.; Blaut, M. Deglycosylation of puerarin and other aromatic C-glucosides by a newly isolated human intestinal bacterium. Environ. Microbiol. 2011, 13, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Zhu, S.; Komatsu, K.; Hattori, M.; Iwashima, M. Deglycosylation of isoflavone C–glucoside puerarin by combination of two recombinant bacterial enzymes and 3–oxo–glucose. bioRxiv 2019, 816074. [Google Scholar] [CrossRef]
- Gómez, B.; Gullón, B.; Yáñez, R.; Schols, H.; Alonso, J.L. Prebiotic potential of pectins and pectic oligosaccharides derived from lemon peel wastes and sugar beet pulp: A comparative evaluation. J. Funct. Foods 2016, 20, 108–121. [Google Scholar] [CrossRef]
- Koh, A.; de Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, Y.; Wang, J.; Wu, G.; Long, W.; Xue, Z.; Wang, L.; Zhang, X.; Pang, X.; Zhao, Y.; et al. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci. Rep. 2016, 6, 27572. [Google Scholar] [CrossRef]
- Mao, B.; Tang, H.; Gu, J.; Li, D.; Cui, S.; Zhao, J.; Zhang, H.; Chen, W. In vitro fermentation of raffinose by the human gut bacteria. Food Funct. 2018, 9, 5824–5831. [Google Scholar] [CrossRef]
- Zartl, B.; Silberbauer, K.; Loeppert, R.; Viernstein, H.; Praznik, W.; Mueller, M. Fermentation of non-digestible raffinose family oligosaccharides and galactomannans by probiotics. Food Funct. 2018, 9, 1638–1646. [Google Scholar] [CrossRef]
- Panthee, S.; Paudel, A.; Blom, J.; Hamamoto, H.; Sekimizu, K. Complete genome sequence of Weissella hellenica 0916-4-2 and its comparative genomic analysis. Front. Microbiol. 2019, 10, 1619. [Google Scholar] [CrossRef]
- Grand, M.; Aubourg, M.; Pikis, A.; Thompson, J.; Deutscher, J.; Hartke, A.; Sauvageot, N. Characterization of the gen locus involved in β-1,6-oligosaccharide utilization by Enterococcus faecalis. Mol. Microbiol. 2019, 112, 1744–1756. [Google Scholar] [CrossRef]
- Anagnostopoulos, D.; Bozoudi, D.; Tsaltas, D. Enterococci isolated from cypriot green table olives as a new source of technological and probiotic properties. Fermentation 2018, 4, 48. [Google Scholar] [CrossRef]
- Wei, B.; Wang, Y.K.; Qiu, W.H.; Wang, S.J.; Wu, Y.H.; Xu, X.W.; Wang, H. Discovery and mechanism of intestinal bacteria in enzymatic cleavage of C–C glycosidic bonds. Appl. Microbiol. Biotechnol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Martínez, I.; Walter, J.; Keshavarzian, A.; Rose, D.J. In vitro characterization of the impact of selected dietary fibers on fecal microbiota composition and short chain fatty acid production. Anaerobe 2013, 23, 74–81. [Google Scholar] [CrossRef]
- Liu, S.; Yu, Q.; Huang, H.; Hou, K.; Dong, R.; Chen, Y.; Xie, J.; Nie, S.; Xie, M.Y. The effect of bound polyphenols on the fermentation and antioxidant properties of carrot dietary fiber in vivo and in vitro. Food Funct. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Larsen, N.; de Mello Tieghi, T.; Adorno, M.A.T.; Kot, W.; Saad, S.M.I.; Jespersen, L.; Sivieri, K. Modulation of gut microbiota from obese individuals by in vitro fermentation of citrus pectin in combination with Bifidobacterium longum BB-46. Appl. Microbiol. Biotechnol. 2018, 102, 8827–8840. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tapia, M.; Tovar, A.R.; Torres, N. Diet as regulator of gut microbiota and its role in health and disease. Arch. Med. Res. 2019, 50, 259–268. [Google Scholar] [CrossRef]
- Dersjant-Li, Y.; Dusel, G. Increasing the dosing of a Buttiauxella phytase improves phytate degradation, mineral, energy, and amino acid digestibility in weaned pigs fed a complex diet based on wheat, corn, soybean meal, barley, and rapeseed meal. J. Anim. Sci. 2019, 97, 2524–2533. [Google Scholar] [CrossRef]
- Romelle, F.D.; Rani, P.A.; Manohar, R.S. Chemical composition of some selected fruit peels. Eur. J. Food Sci. Technol. 2016, 4, 12–21. [Google Scholar]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Qin, P.; Zou, Y.; Dai, Y.; Luo, G.; Zhang, X.; Xiao, L. Characterization a Novel Butyric Acid-Producing Bacterium Collinsella aerofaciens Subsp. Shenzhenensis Subsp. Nov. Microorganisms 2019, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Meex, R.C.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment/ Time (h) | DNA (ng/µL of sample) 2 | Ratio 260/280 | Paired-End Reads | Number of ASVs | Number of Reads into ASVs |
|---|---|---|---|---|---|
| NegativeC | |||||
| 0 | 3.33 ± 1.26 A, c | 1.36 ± 0.11 | 37,369 ± 44,721 | 186 ± 75 | 17,634 ± 22,852 |
| 6 | 4.12 ± 1.26 A, b | 1.50 ± 0.05 | 36,373 ± 12,886 | 192 ± 82 | 14,331 ± 3447 |
| 12 | 6.51 ± 3.65 A, c | 1.61 ± 0.32 | 46,199 ± 9255 | 256 ± 88 | 22,897 ± 27,685 |
| 24 | 5.69 ± 1.25 A, b | 1.46 ± 0.06 | 23,887 ± 9255 | 257 ± 39 | 9459 ± 3742 |
| 48 | 4.46 ± 1.43 A, b | 1.50 ± 0.01 | 32,985 ± 9351 | 290 ± 26 | 12,738 ± 3886 |
| PositiveC | |||||
| 0 | 8.10 ± 2.69 C, a | 1.79 ± 0.20 | 33,125 ± 18,765 | 239 ± 31 | 12,461 ± 7373 |
| 6 | 33.24 ± 20.33 B,C, a | 1.99 ± 0.22 | 26,111 ± 15,951 | 191 ± 32 | 11,510 ± 8355 |
| 12 | 22.56 ± 8.57 C, b,c | 1.99 ± 0.08 | 28,090 ± 10,072 | 210 ± 27 | 12,720 ± 4653 |
| 24 | 77.84 ± 41.85 B, a,b | 2.09 ± 0.04 | 40,410 ± 5110 | 241 ± 18 | 17,432 ± 3531 |
| 48 | 173.80 ± 37.93 A, a | 2.10 ± 0.07 | 37,155 ± 4676 | 175 ± 24 | 19,665 ± 2817 |
| MangoB | |||||
| 0 | 3.02 ± 0.46 C, c | 1.51 ± 0.42 | 21,467 ± 13,471 | 212 ± 84 | 9405 ± 5675 |
| 6 | 27.21 ± 3.04 C, a | 2.01 ± 0.05 | 25,853 ± 9343 | 184 ± 16 | 12,780 ± 4895 |
| 12 | 70.015 ± 37.54 B,C, a | 2.09 ± 0.11 | 23,745 ± 14,122 | 196 ± 26 | 10,960 ± 6581 |
| 24 | 155.96 ± 65.02 B, a | 2.18 ± 0.05 | 30,332 ± 7471 | 189 ± 23 | 12,939 ± 3135 |
| 48 | 249.02 ± 83.25 A, a | 2.19 ± 0.02 | 22,516 ± 7369 | 217 ± 34 | 8927 ± 3325 |
| UVMangoB | |||||
| 0 | 6.37 ± 3.55 C, b,c | 1.78 ± 0.11 | 13,274 ± 1153 | 129 ± 112 | 3538 ± 3080 |
| 6 | 24.30 ± 5.19 C, a | 7.80 ± 0.11 | 20,042 ± 11,747 | 179 ± 27 | 9578 ± 5704 |
| 12 | 53.27 ± 14.96 C, a,b | 1.84 ± 0.22 | 28,441 ± 5716 | 225 ± 17 | 13,631 ± 2283 |
| 24 | 134.00 ± 64.64 B, a | 2.15 ± 0.07 | 29,500 ± 3691 | 262 ± 8 | 12,447 ± 1313 |
| 48 | 196.82 ± 21.88 A, a | 2.18 ± 0.02 | 25,614 ± 2438 | 285 ± 8 | 10,648 ± 1041 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Sarmiento, W.; Sáyago-Ayerdi, S.G.; Goñi, I.; Gutiérrez-Miceli, F.A.; Abud-Archila, M.; Rejón-Orantes, J.d.C.; Rincón-Rosales, R.; Peña-Ocaña, B.A.; Ruíz-Valdiviezo, V.M. Changes in Intestinal Microbiota and Predicted Metabolic Pathways During Colonic Fermentation of Mango (Mangifera indica L.)—Based Bar Indigestible Fraction. Nutrients 2020, 12, 683. https://doi.org/10.3390/nu12030683
Gutiérrez-Sarmiento W, Sáyago-Ayerdi SG, Goñi I, Gutiérrez-Miceli FA, Abud-Archila M, Rejón-Orantes JdC, Rincón-Rosales R, Peña-Ocaña BA, Ruíz-Valdiviezo VM. Changes in Intestinal Microbiota and Predicted Metabolic Pathways During Colonic Fermentation of Mango (Mangifera indica L.)—Based Bar Indigestible Fraction. Nutrients. 2020; 12(3):683. https://doi.org/10.3390/nu12030683
Chicago/Turabian StyleGutiérrez-Sarmiento, Wilbert, Sonia Guadalupe Sáyago-Ayerdi, Isabel Goñi, Federico Antonio Gutiérrez-Miceli, Miguel Abud-Archila, José del Carmen Rejón-Orantes, Reiner Rincón-Rosales, Betsy Anaid Peña-Ocaña, and Víctor Manuel Ruíz-Valdiviezo. 2020. "Changes in Intestinal Microbiota and Predicted Metabolic Pathways During Colonic Fermentation of Mango (Mangifera indica L.)—Based Bar Indigestible Fraction" Nutrients 12, no. 3: 683. https://doi.org/10.3390/nu12030683
APA StyleGutiérrez-Sarmiento, W., Sáyago-Ayerdi, S. G., Goñi, I., Gutiérrez-Miceli, F. A., Abud-Archila, M., Rejón-Orantes, J. d. C., Rincón-Rosales, R., Peña-Ocaña, B. A., & Ruíz-Valdiviezo, V. M. (2020). Changes in Intestinal Microbiota and Predicted Metabolic Pathways During Colonic Fermentation of Mango (Mangifera indica L.)—Based Bar Indigestible Fraction. Nutrients, 12(3), 683. https://doi.org/10.3390/nu12030683